

Abstract
The reemergence of deadly pandemic influenza virus strains has necessitated the development of improved methods for rapid detection and subtyping of influenza viruses that will enable more strains to be characterized at the molecular level. Representative circulating strains of human influenza viruses from primary clinical specimens were grown in cell culture, purified through polyethylene glycol precipitation, proteolytically digested with an endoproteinase, and analyzed and identified by high-resolution mass spectrometry using unique signature peptides that are characteristic of type A H1N1 and H3N2 and type B influenza viruses. This proteotyping approach enabled circulating strains of type A influenza virus to be typed and subtyped, cocirculating seasonal and pandemic H1N1 viruses to be differentiated, and the lineage of type B viruses to be determined through single-ion detection by high-resolution mass spectrometry. Results were obtained using virus titers comparable to those used in reverse transcription (RT)-PCR assays with clinical specimens grown in cell cultures. The methodology represents a more rapid and direct approach than RT-PCR and can be integrated into existing procedures currently used for the surveillance of emerging pandemic and seasonal influenza viruses.
INTRODUCTION
The early detection and laboratory diagnosis of the influenza virus in human populations are important for managing the virus, either in the context of annual seasonal outbreaks or following the emergence of pandemic strains. Rapid reliable surveillance of influenza virus is essential for the implementation of infection control strategies, the administration of antiviral therapies, and the production of effective vaccines to control the spread of the virus (1).
Sixty years ago, the World Health Organization established the Global Influenza Surveillance Network (GISN) to monitor human influenza viruses in circulation around the world throughout the year. The primary goal of the system, now known as the Global Influenza Surveillance and Response System (GISRS), is to identify and to characterize emerging influenza virus strains in a timely manner and then recommend the composition of seasonal vaccines against the virus. However, a recent report (2) has highlighted limitations in the current surveillance strategy, which has been described as ad hoc, reactive to outbreaks and pandemics, and lacking full geographic representation.
The surveillance of animal influenza viruses is also critical, given the ability of the virus to cross from animal hosts to human hosts. Such viral reassortment leads to the evolution of strains that pose the greatest pandemic risk. This was illustrated by the unexpected emergence of the pandemic, swine-originating, H1N1 influenza virus in humans in Mexico and California in April 2009 (3). Recent studies have shown that this pandemic strain circulated undetected within swine herds for at least a decade prior to human transmission (4).
Given that influenza infection cannot be reliably diagnosed based on clinical symptoms alone, point-of-care tests followed by laboratory-based analyses are required to identify the presence of and to characterize the virus. Recovery of virus from patients is usually achieved through nasopharyngeal aspirate, nasal wash fluid, and bronchoalveolar lavage fluid samples over nasal and throat swabs, as the latter contain less virus and more contaminating epithelial cells. The virus also is more readily detected in lower versus upper respiratory tract samples.
Although antigen detection assays and direct amplification of virus by reverse transcription (RT)-PCR can be performed with high-quality primary clinical specimens, strains are most often grown first in cell culture. This facilitates the production of sufficient quantities of virus for antigenic and molecular characterization and for testing of strains for their antiviral drug susceptibility. Given the time-consuming and labor-intensive nature of current molecular methods for characterizing influenza viruses, relatively few strains have been fully characterized. During the 2004-2005 influenza season, for example, the WHO Collaborating Centers characterized approximately 6,000 human influenza virus strains, or only 1 per 100,000 isolated (5). Although real-time PCR methods for typing influenza A and B viruses and subtyping H1, H3, H5, and H7 strains (6, 7) have been reported, there is no PCR approach that can simultaneously detect every known hemagglutinin (HA) and neuraminidase (NA) subtype circulating within human, avian, and swine hosts.
A new rapid direct proteotyping approach, using high-resolution mass spectrometry, that allows the influenza virus to be typed and subtyped (8, 9) at the molecular level within whole virus digests has recently been reported. The approach has been shown to be able to distinguish seasonal and pandemic influenza strains (10), to determine strain lineages (11), and to identify reassortant viruses that pose the greatest pandemic risk (12). The practicality of this proteotyping approach for the surveillance of circulating strains of the virus, in terms of primary clinical specimens grown in cell culture, is demonstrated here for the first time. This approach has enabled circulating strains of type A influenza virus to be typed and subtyped, cocirculating seasonal and pandemic H1N1 viruses to be differentiated, and the lineage of type B viruses to be determined through single-ion detection with high-resolution mass spectrometry.
MATERIALS AND METHODS
Growth of clinical virus specimens.
Virus strains characterized in this study were isolated from patients at Westmead Hospital (Sydney, Australia) during the 2011 influenza season and were provided by Janette Taylor and Dominic Dwyer. A real-time multiplex RT-PCR assay was used to identify specimens containing influenza virus, and these positive specimens were typed and then subtyped using primers for H1 and H3 viruses.
The nasopharyngeal aspirate specimens, containing 10 mg/ml gentamicin, were grown in Madin-Darby canine kidney (MDCK) cells using established procedures (13) with minor modifications. Briefly, 5 μl of the primary clinical specimen was diluted in 3 ml of virus growth medium (Dulbecco's modified Eagle's medium containing 100 U/ml penicillin, 10 μg/ml streptomycin, 2 μg/ml N-tosyl l-phenylalanyl chloromethyl ketone [TPCK]-treated trypsin, and 0.2% bovine serum albumin) and inoculated into MDCK cells. Prior to infection, the cell monolayers were washed three times with phosphate-buffered saline. The 175-cm2 flasks were incubated for 30 min to facilitate virus adsorption, after which growth medium was added to increase the volume to 25 ml. Flasks were incubated at 37°C in a humidified incubator in 5% CO2 for 2 to 3 days until a pronounced cytopathic effect was observed. The cell culture medium was then centrifuged at 600 × g, and the supernatant was passed through a 0.22-μm-pore-size filter (Millipore, Australia) to remove cellular debris.
Polyethylene glycol precipitation.
Virus from clarified cell culture supernatants was precipitated for at least 2 h at 4°C with molecular weight 6,000 polyethylene glycol (Millipore, Australia), at a final concentration of 8% (14, 15). Following precipitation, virus was recovered by centrifugation at 9,000 × g for 15 min. The insoluble virus pellet was resuspended in 50 mM ammonium bicarbonate and centrifuged at 9,000 × g for 3 min. This step was repeated three times, in order to remove salts and polyethylene glycol that might interfere with analysis. The pellet was finally resuspended in 70% ethanol for 5 min to inactivate the virus (16), after which the pellet was dried in a SpeedVac concentrator (Labconco Co., Kansas City, MO) and stored at −20°C. All manipulations with live virus were performed in a class 2 biological safety cabinet within a physical containment level 2 (PC2) microbiological laboratory.
In-solution whole virus tryptic digests.
Virus pellets were reconstituted in 25 μl digestion buffer (50 mM ammonium bicarbonate, 10% acetonitrile, and 2 mM dithiothreitol [pH 7.5]) and digested overnight at 37°C with 12 ng/μl sequencing grade modified trypsin (Roche Diagnostics GmbH, Mannheim, Germany).
High-resolution MALDI-FT-ICR mass spectrometry.
Solutions of viral peptides (1 μl) were diluted with a solution (4 μl) of matrix (5 mg/ml α-cyano-4-hydroxycinnaminic acid in 50% acetonitrile with 0.1% trifluoroacetic acid), and 1 μl was spotted onto a matrix-assisted laser desorption ionization–Fourier-transform ion cyclotron resonance (MALDI-FT-ICR) mass spectrometry sample plate (MTP AnchorChip 400/384 TF; Bruker Daltonics, Billerica, MA). MALDI-FT-ICR mass spectra were recorded with a 7T Bruker APEX-Qe instrument (Bruker Daltonics, Billerica, MA) in the positive-ion mode, as described previously (8–11). The mass spectra shown are representative of mass spectra obtained from three replicate experiments.
Mass spectral analysis.
Mass spectra were analyzed using Data Analysis v. 3.4 software (Bruker Daltonics, Billerica, MA) in conjunction with the FluTyper algorithm, using default parameters (17). A signature peptide was initially defined as any theoretical tryptic peptide M whose frequency of occurrence (Po) was greater than 0.95 for one type or subtype and 0 for all other types or subtypes for a given influenza protein (8). These were identified for type A H1N1 seasonal and pandemic and H3N2 and type B influenza viruses based on available translated gene sequences within the NCBI influenza resource database at the time of analysis for the hemagglutinin (HA), neuraminidase (NA), nucleoprotein (NP), and matrix 1 (M1) viral proteins. Since few signature peptides may be detected, the FluTyper algorithm uses a lower frequency of occurrence of 0.7, and indicator peptides are also used by the algorithm to assist with strain identification. An indicator peptide is defined in the same manner as a signature peptide except that it may occur with a frequency of occurrence of greater than 0.7 for one type and subtype and less than 0.1 for other types or subtypes. Period-specific signatures consider only influenza viruses in circulation since 2009 (2009 to 2012).
RESULTS
Human influenza infections are caused by type A influenza viruses of the H1N1 and H3N2 subtypes and type B influenza viruses (18). Type A H1N1 strains include both oseltamivir-resistant seasonal and 2009-originating pandemic strains that are in cocirculation throughout the globe (19). Human primary clinical specimens were obtained and grown in culture for four representative strains across these types and subtypes (Fig. 1). The mass spectrometric results were verified by performing at least three replicate analyses per specimen.
FIG 1.
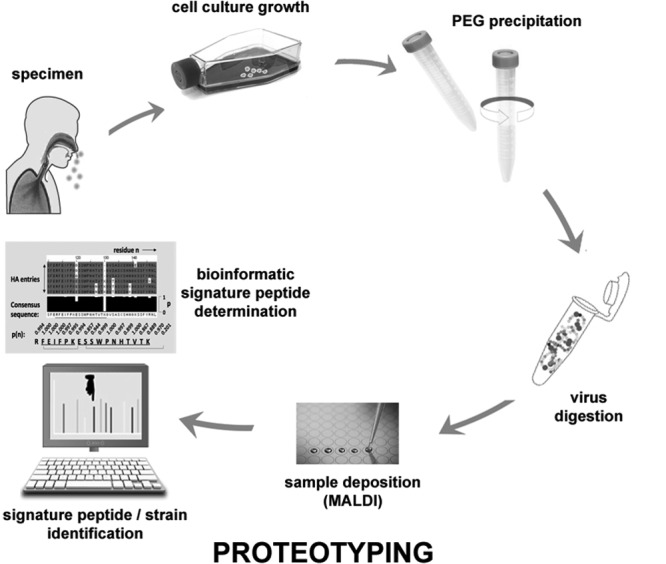
Overview of the proteotyping strategy for molecular characterization of influenza virus clinical specimens grown in cell culture. PEG, polyethylene glycol.
Viruses were harvested after 48 to 72 h of growth in MDCK cells. The average viral titers were measured by plaque assays to be 3.2 × 103 and 2.5 × 103 PFU/ml for seasonal and pandemic type A H1N1, respectively, 2.5 × 103 PFU/ml for type A H3N2, and 2.3 × 104 PFU/ml for type B virus strains, consistent with reported values using the same cell line (20).
An average of 5 ng of total virus, digested with trypsin, was deposited onto the MALDI plate for analysis by mass spectrometry. Since only 1/100 or less of the deposited sample is consumed during analysis, less than 50 pg of virus was consumed to produce the data shown in Fig. 2 to 6. Based upon the reported mass of an influenza virus particle of 5 femtograms or 5 × 10−16 g (21), this corresponds to ∼105 copies of the virus, a value comparable to that reported for the RT-PCR analysis of virus obtained from nasopharyngeal swabs (22).
FIG 2.

High-resolution MALDI-FT-ICR mass spectrum of a whole virus tryptic digest of the A/Puerto Rico/8/34/Mount Sinai (H1N1) reference influenza strain. All identified ions are listed in Table 1. Ions in bold indicate signature peptides.
FIG 6.

High-resolution MALDI-FT-ICR mass spectrum of a whole virus tryptic digest of a cell-cultured clinical specimen containing a type B virus strain. All identified ions are listed in Table 5. Ions in bold indicate signature peptides (*).
A bioinformatic analysis of all translated gene sequences for the hemagglutinin (HA), neuraminidase (NA), nucleoprotein (NP), and matrix (M1) viral proteins of influenza viruses reported in the NCBI Influenza Virus Resource Database (23) was conducted. This analysis was also performed with all strains isolated since January 2009. This period (2009 to 2012) was used to identify period-specific signature peptides, given that novel influenza antigenic variants have been shown to arise approximately every 3 years (24, 25). This analysis led to the identification of 74 period-specific signature peptides for seasonal type A H1N1, 65 peptides for 2009-originating pandemic type A H1N1, 66 peptides for type A H3N2, and 95 peptides for type B influenza viruses, for the HA, NA, NP, and M1 proteins, respectively (see Tables S2 to S5 in the supplemental material). These signature peptides were used to identify the influenza viruses within the primary clinical samples.
To establish that the proteotyping approach is viable for the analysis of cell-cultured strains (Fig. 1), it was first applied to a well-characterized reference strain, namely, A/Puerto Rico/8/34/Mount Sinai (H1N1). The analysis was aided by the availability of the complete genomic sequence for this strain (NCBI taxonomy identification no. 183764).
A total of 93 peptides derived from the HA (17), NA (7), NP (42), and M1 (27) proteins were detected by mass spectrometry (Fig. 2), and these are grouped according to protein in Table 1. Not all are labeled in Fig. 2, due to space constraints, but those shown in bold are period-specific signature peptides based on all 1,934 characterized strains (see Table S1 in the supplemental material). Consistent with the data for all other strains, the tryptic peptides derived from the viral nucleoprotein and matrix M1 protein were found to predominate, in accordance with previous observations (12), due to the higher copy numbers of these proteins (3,000 and 1,000 molecules per virion, respectively, compared with 500 and 1,000 molecules per virion for hemagglutinin and neuraminidase proteins, respectively) (28). The successful characterization of the reference strain led to its subsequent application to the analysis of the clinical specimens obtained during the 2011 season, including representative cocirculating seasonal and pandemic type A H1N1, type A H3N2, and type B strains.
TABLE 1.
Peptides identified in a MALDI-FT-ICR mass spectrum (Fig. 2) of a whole-virus digest of the A/Puerto Rico/8/34/Mount Sinai (H1N1) reference strain
| [M+H]+
m/z mono.a |
Difference (ppm) | Viral protein | Residues | Sequence | |
|---|---|---|---|---|---|
| Theoretical | Experimental | ||||
| 545.3411 | 545.3418 | −1.23 | HA | 416–419 | LEKR |
| 659.3153 | 659.3166 | −1.97 | HA | 158–162 | SSFYR |
| 676.3816 | 676.383 | −2.04 | HA | 330–335 | MVTGLR |
| 780.4296 | 780.431 | −1.78 | HA | 131–136 | FEIFPK |
| 801.4331 | 801.4347 | −1.95 | HA | 236–242 | VRDQAGR |
| 896.5569 | 896.5585 | −1.74 | HA | 63–71 | GIAPLQLGK |
| 904.4675 | 904.4692 | −1.91 | HA | 419–425 | RMENLNK |
| 907.4889 | 907.49 | −1.19 | HA | 412–418 | EFNKLEK |
| 1,016.5781 | 1,016.5806 | −2.49 | HA | 163–170 | NLLWLTEK |
| 1,125.554 | 1,125.5559 | −1.66 | HA | 505–513 | YSEESKLNR |
| 1,137.736 | 1,137.7385 | −2.23 | HA | 61–71 | LKGIAPLQLGK |
| 1,187.6425 | 1,187.6452 | −2.31 | HA | 226–235 | FTPEIAERPK |
| 1,343.7436 | 1,343.7471 | −2.63 | HA | 225–235 | RFTPEIAERPK |
| 1,443.6367 | 1,443.6398 | −2.14 | HA | 475–486 | EIGNGCFEFYHK |
| 1,626.8314 | 1,626.8356 | −2.58 | HA | 402–415 | MNIQFTAVGKEFNK |
| 1,677.8852 | 1,677.8889 | −2.21 | HA | 163–176 | NLLWLTEKEGSYPK |
| 2,633.2232 | 2,633.225 | −0.69 | HA | 203–224 | EQQNIYQNENAYVSVVTSNYNR |
| 604.3055 | 604.3066 | −1.87 | NA | 88–92 | DNSIR |
| 629.3047 | 629.3053 | −0.89 | NA | 333–337 | GFSYR |
| 793.3957 | 793.3975 | −2.3 | NA | 136–141 | DRSPYR |
| 1,021.5219 | 1,021.5237 | −1.72 | NA | 338–346 | YGNGVWIGR |
| 1,409.7177 | 1,409.7187 | −0.68 | NA | 81–92 | GWAIYSKDNSIR |
| 2,149.9289 | 2,149.9327 | −1.75 | NA | 354–371 | HGFEMIWDPNGWTETDSK |
| 2,639.1989 | 2,639.2005 | −0.6 | NA | 354–375 | HGFEMIWDPNGWTETDSKFSVR |
| 574.2949 | 574.2961 | −2.07 | NP | 200–204 | GINDR |
| 622.3102 | 622.3119 | −2.79 | NP | 205–208 | NFWR |
| 631.3779 | 631.3788 | −1.41 | NP | 442–446 | TEIIR |
| 661.3997 | 661.3999 | −0.29 | NP | 356–361 | GKLSTR |
| 672.4408 | 672.4417 | −1.27 | NP | 262–267 | SALILR |
| 676.3088 | 676.3105 | −2.45 | NP | 157–162 | TGMDPR |
| 708.3833 | 708.3846 | −1.8 | NP | 385–389 | YWAIR |
| 731.4052 | 731.4063 | −1.52 | NP | 176–184 | SGAAGAAVK |
| 761.3946 | 761.396 | −1.81 | NP | 417–422 | NLPFDR |
| 763.4103 | 763.412 | −2.27 | NP | 92–98 | TGGPIYR |
| 831.4325 | 831.4336 | −1.36 | NP | 20–26 | QNATEIR |
| 850.3915 | 850.3922 | −0.79 | NP | 237–243 | AMMDQVR |
| 887.5063 | 887.507 | −0.79 | NP | 175–184 | RSGAAGAAVK |
| 891.5052 | 891.5077 | −2.77 | NP | 91–98 | KTGGPIYR |
| 919.5114 | 919.5134 | −2.2 | NP | 92–99 | TGGPIYRR |
| 951.5165 | 951.5179 | −1.51 | NP | 383–389 | SRYWAIR |
| 961.4452 | 961.4468 | −1.71 | NP | 392–400 | SGGNTNQQR |
| 978.5373 | 978.5399 | −2.69 | NP | 228–236 | GKFQTAAQK |
| 1,023.4999 | 1,023.4996 | 0.27 | NP | 462–470 | GVFELSDEK |
| 1,135.5397 | 1,135.5421 | −2.09 | NP | 205–213 | NFWRGENGR |
| 1,177.5867 | 1,177.5888 | −1.81 | NP | 200–208 | GINDRNFWR |
| 1,186.6796 | 1,186.6824 | −2.38 | NP | 56–65 | LIQNSLTIER |
| 1,191.623 | 1,191.6253 | −1.94 | NP | 185–195 | GVGTMVMELVR |
| 1,207.6179 | 1,207.6206 | −2.25 | NP | 185–195 | GVGTMoxVMELVR |
| 1,221.6262 | 1,221.6283 | −1.75 | NP | 437–446 | TSDMRTEIIR |
| 1,223.6207 | 1,223.6224 | −1.41 | NP | 66–75 | MVLSAFDERR |
| 1,239.6156 | 1,239.6178 | −1.79 | NP | 66–75 | MoxVLSAFDERR |
| 1,323.6223 | 1,323.6252 | −2.16 | NP | 163–174 | MCSLMQGSTLPR |
| 1,334.6745 | 1,334.6748 | −0.2 | NP | 294–305 | py-EGYSLVGIDPFR |
| 1,339.6172 | 1,339.6175 | −0.2 | NP | 163–174 | MoxCSLMQGSTLPR |
| 1,352.685 | 1,352.6878 | −2.04 | NP | 294–305 | EGYSLVGIDPFR |
| 1,420.7688 | 1,420.772 | −2.27 | NP | 107–117 | ELILYDKEEIR |
| 1,482.7011 | 1,482.704 | −1.95 | NP | 423–436 | TTIMAAFNGNTEGR |
| 1,739.7845 | 1,739.7869 | −1.37 | NP | 447–461 | MMESARPEDVSFQGR |
| 1,755.7794 | 1,755.7838 | −2.49 | NP | 447–461 | MoxMESARPEDVSFQGR |
| 2,065.9943 | 2,066.0005 | −2.99 | NP | 244–261 | ESRNPGNAEFEDLTFLAR |
| 2,104.9472 | 2,104.946 | 0.58 | NP | 423–441 | SCLPACVYGPAVASGYDFER |
| 2,156.9519 | 2,156.9565 | −2.15 | NP | Sep-26 | SYEQMETDGERQNATEIR |
| 2,225.0773 | 2,225.0786 | −0.57 | NP | 417–436 | NLPFDRTTIMAAFNGNTEGR |
| 2,355.0808 | 2,355.086 | −2.2 | NP | 362–382 | GVQIASNENMoxETMESSTLELR |
| 3,245.4493 | 3,245.4584 | −2.81 | NP | 122–150 | QANNGDDATAGLTHMMIWHSNLNDATYQR |
| 3,438.6139 | 3,438.6104 | 1.01 | NP | 274–305 | SCLPACVYGPAVASGYDFEREGYSLVGIDPFR |
| 399.2105 | 399.2104 | 0.13 | M1 | 161–163 | SHR |
| 473.2836 | 473.2842 | −1.23 | M1 | 73–76 | GLQR |
| 555.2639 | 555.2647 | −1.36 | M1 | 175–178 | HENR |
| 687.379 | 687.3803 | −1.93 | M1 | 22–27 | AEIAQR |
| 846.3967 | 846.3984 | −2.05 | M1 | 211–217 | py-QMVQAMR |
| 849.4075 | 849.4089 | −1.63 | M1 | 244–250 | MGVQMQR |
| 863.4232 | 863.4251 | −2.24 | M1 | 211–217 | QMVQAMR |
| 865.4024 | 865.4036 | −1.37 | M1 | 244–250 | MoxGVQMQR |
| 879.4181 | 879.4204 | −2.66 | M1 | 211–217 | QMoxVQAMR |
| 902.4736 | 902.4747 | −1.22 | M1 | 106–113 | EITFHGAK |
| 921.5079 | 921.5101 | −2.34 | M1 | 179–187 | MVLASTTAK |
| 1,005.5086 | 1,005.5105 | −1.86 | M1 | 243–250 | RMGVQMQR |
| 1,125.6996 | 1,125.7013 | −1.53 | M1 | 48–57 | TRPILSPLTK |
| 1,256.6673 | 1,256.6692 | −1.48 | M1 | 164–174 | py-QMVTTTNPLIR |
| 1,272.6622 | 1,272.6647 | −1.93 | M1 | 164–174 | py-QMoxVTTTNPLIR |
| 1,273.6938 | 1,273.697 | −2.48 | M1 | 164–174 | QMVTTTNPLIR |
| 1,289.6887 | 1,289.6882 | 0.42 | M1 | 164–174 | QMoxVTTTNPLIR |
| 1,490.7565 | 1,490.76 | −2.35 | M1 | 36–47 | NTDLEVLMEWLK |
| 1,604.8396 | 1,604.8424 | −1.72 | M1 | 231–243 | NDLLENLQAYQKR |
| 1,635.911 | 1,635.9138 | −1.69 | M1 | 58–72 | GILGFVFTLTVPSER |
| 1,669.8808 | 1,669.8829 | −1.26 | M1 | 161–174 | SHRQMoxVTTTNPLIR |
| 1,809.9394 | 1,809.9342 | 2.87 | M1 | 164–178 | QMVTTTNPLIRHENR |
| 2,003.9358 | 2,003.9314 | 2.17 | M1 | 78–95 | RFVQNALNGNGDPNNMDK |
| 2,219.084 | 2,219.0865 | −1.11 | M1 | 114–134 | EISLSYSAGALASCMGLIYNR |
| 2,350.2005 | 2,350.2068 | −2.69 | M1 | 28–47 | LEDVFAGKNTDLEVLMEWLK |
| 2,685.3848 | 2,685.3847 | 0.03 | M1 | 218–242 | TIGTHPSSSAGLKNDLLENLQAYQK |
| 2,737.2748 | 2,737.2733 | 0.55 | M1 | 135–160 | MGAVTTEVAFGLVCATCEQIADSQHR |
“mono” indicates monoisotopic values.
A high-resolution MALDI-FT-ICR mass spectrum of a whole virus digest obtained from a type A H1N1 seasonal specimen is shown Fig. 3 and summarized in Table 2. In total, 36 peptides were identified by the FluTyper algorithm, to identify the strain as a type A strain of the H1N1 subtype with 100% certainty. The detection of one seasonal signature peptide, consisting of HA residues 164 to 171 (m/z 944.5560), and three indicator peptides, consisting of HA residues 331 to 336 (m/z 676.3814) and NP residues 462 to 470 (m/z 1,051.5049) and 185 to 195 (m/z 1,187.6804) (all shown in bold in Fig. 3), enabled the strain to be confidently identified as being of the type A H1N1 subtype. The detection of a total of 18 period-specific signature peptides also enabled the strain to be differentiated from type A H1N1 pandemic strains cocirculating during this period (see shaded entries in Table S2 in the supplemental material).
FIG 3.

High-resolution MALDI-FT-ICR mass spectrum of a whole virus tryptic digest of a cell-cultured clinical specimen containing a seasonal type A H1N1 virus strain. All identified ions are listed in Table 2. Ions in bold indicate signature (*) and indicator (^) peptides.
TABLE 2.
Peptides identified in a MALDI-FT-ICR mass spectrum (Fig. 3) of a whole-virus digest of a cell-cultured clinical specimen containing a type A H1N1 seasonal influenza straina
| [M+H]+
m/z mono. |
Difference (ppm) | Viral protein | Residues | Sequence | Po | Signature or indicator | Period-specific signature | Bayes | |
|---|---|---|---|---|---|---|---|---|---|
| Theoretical | Experimental | ||||||||
| 622.3096 | 622.3106 | −1.60 | NP | 205–208 | NFWR | 1.00 | S | S | |
| 676.3811 | 676.3814 | −0.51 | HA | 331–336 | MVTGLR | 0.95 | S | S | |
| 687.3784 | 687.3783 | 0.17 | M1 | 22–27 | AEIAQR | 0.96 | S | S | |
| 708.3828 | 708.3835 | −1.04 | NP | 385–389 | YWAIR | 1.00 | S | S | |
| 780.4291 | 780.4288 | 0.32 | HA | 131–136 | FEIFPK | 0.99 | S | ||
| 827.4410 | 827.4413 | −0.31 | HA | 270–276 | YAFALSR | 0.96 | S | ||
| 846.3961 | 846.3953 | 0.89 | M1 | 211–217 | py-QMVQAMR | 0.97 | S | ||
| 849.4070 | 849.4067 | 0.30 | M1 | 244–250 | MGVQMQR | 0.97 | S | ||
| 850.3910 | 850.3913 | −0.39 | NP | 237–243 | AMMDQVR | 0.97 | S | S | |
| 863.4226 | 863.4223 | 0.35 | M1 | 211–217 | QMVQAMR | 0.97 | S | ||
| 865.4019 | 865.4021 | −0.26 | M1 | 244–250 | MoxGVQMQR | 0.97 | S | ||
| 878.4618 | 878.4608 | 1.15 | M1 | 28–35 | LEDVFAGK | 0.91 | S | S | |
| 879.4175 | 879.4176 | −0.09 | M1 | 211–217 | QMoxVQAMR | 0.97 | |||
| 881.3968 | 881.3966 | 0.21 | M1 | 244–250 | MoxGVQMoxQR | 0.97 | S | ||
| 902.4730 | 902.4728 | 0.27 | M1 | 106–113 | EITFHGAK | 0.98 | S | S | |
| 944.5564 | 944.5560 | 0.40 | HA | 164–171 | NLLWLTGK | 0.77 | S | S | |
| 951.5159 | 951.5153 | 0.63 | NP | 383–389 | SRYWAIR | 0.83 | S | ||
| 961.4446 | 961.4446 | 0.00 | NP | 392–400 | SGGNTNQQR | 0.89 | S | S | |
| 1,021.5214 | 1,021.5201 | 1.25 | NA | 366–374 | YGNGVWIGR | 0.85 | S | S | |
| 1,051.5055 | 1,051.5049 | 0.54 | NP | 462–470 | GVFELSDER | 0.72 | S | S | |
| 1,067.5190 | 1,067.5198 | −0.74 | NP | 66–74 | MVLSAFDER | 0.99 | S | ||
| 1,096.5456 | 1,096.5433 | 2.06 | HA | 403–412 | MNTQFTAVGK | 0.98 | S | ||
| 1,125.6990 | 1,125.6988 | 0.20 | M1 | 48–57 | TRPILSPLTK | 1.00 | S | S | |
| 1,186.6790 | 1,186.6787 | 0.26 | NP | 56–65 | LIQNSLTIER | 0.91 | S | ||
| 1,187.6817 | 1,187.6804 | 1.06 | NP | 185–195 | GVGTMVLELIR | 0.74 | S | ||
| 1,203.6766 | 1,203.6762 | 0.31 | NP | 185–195 | GVGTM*VLELIR | 0.74 | |||
| 1,223.6201 | 1,223.6176 | 2.06 | NP | 66–75 | MVLSAFDERR | 0.95 | S | ||
| 1,239.6150 | 1,239.6135 | 1.24 | NP | 66–75 | MoxVLSAFDERR | 0.95 | S | ||
| 1,256.6667 | 1,256.6655 | 0.98 | M1 | 164–174 | py-QMVTTTNPLIR | 0.89 | |||
| 1,272.6617 | 1,272.6584 | 2.55 | M1 | 164–174 | py-QMoxVTTTNPLIR | 0.89 | |||
| 1,273.6933 | 1,273.6916 | 1.32 | M1 | 164–174 | QMVTTTNPLIR | 0.89 | |||
| 1,275.6112 | 1,275.6117 | −0.39 | NP | 39–48 | FYIQMCTELK | 0.95 | S | ||
| 1,289.6882 | 1,289.6853 | 2.25 | M1 | 164–174 | QMoxVTTTNPLIR | 0.89 | |||
| 1,462.7246 | 1,462.7207 | 2.70 | M1 | 36–47 | NTDLEALMEWLK | 0.94 | S | ||
| 1,478.7196 | 1,478.7170 | 1.73 | M1 | 36–47 | NTDLEALM*EWLK | 0.94 | |||
| 1,635.9104 | 1,635.9055 | 3.00 | M1 | 58–72 | GILGFVFTLTVPSER | 0.94 | S | ||
S, signature peptide; I, indicator peptide; “mono.” indicates monoisotopic values.
A high-resolution MALDI-FT-ICR mass spectrum of a whole virus digest obtained from a type A H1N1 pandemic strain is shown in Fig. 4 and summarized in Table 3. In total, 36 peptides belonging exclusively to strains of the type A H1N1 subtype were identified by the FluTyper algorithm. The detection of two signature peptides, consisting of HA residues 343 to 348 (m/z 630.3944) and 279 to 285 (m/z 887.4091), and two indicator peptides, consisting of NP residues 191 to 201 (m/z 1,159.6498) and HA residues 236 to 245 (m/z 1,198.7303) (shown in bold in Fig. 4), enabled the strain to be differentiated from cocirculating seasonal strains. When strains circulating since 2009 were considered, 6 additional period-specific signature peptides were detected (shown underlined in Fig. 4; also see shaded entries in Table S3 in the supplemental material).
FIG 4.

High-resolution MALDI-FT-ICR mass spectrum of a whole virus tryptic digest of a cell-cultured clinical specimen containing a pandemic type A H1N1 virus strain. All identified ions are listed in Table 3. Ions in bold indicate signature (*) and indicator (^) peptides.
TABLE 3.
Peptides identified in a MALDI-FT-ICR mass spectrum (Fig. 4) of a whole-virus tryptic digest of a cell-cultured clinical specimen containing a pandemic type A H1N1 virus straina
| [M+H]+
m/z mono. |
Difference (ppm) | Viral protein | Residues | Sequence | Po | Signature or indicator | Period-specific signature | Bayes | |
|---|---|---|---|---|---|---|---|---|---|
| Theoretical | Experimental | ||||||||
| 555.2634 | 555.2646 | −2.19 | M1 | 175–178 | HENR | 0.99 | S | ||
| 630.3933 | 630.3944 | −1.69 | HA | 343–348 | LATGLR | 0.99 | S | S | |
| 775.4097 | 775.4113 | −2.05 | NP | 423–428 | NLPFER | 0.99 | S | S | |
| 849.4070 | 849.4079 | −1.11 | M1 | 244–250 | MGVQMQR | 0.98 | S | S | |
| 850.3910 | 850.3917 | −0.86 | NP | 243–249 | AMMDQVR | 0.98 | S | ||
| 855.3964 | 855.3979 | −1.76 | M1 | 211–217 | py-QMVHAMR | 0.96 | S | ||
| 859.4381 | 859.4389 | −0.98 | HA | 246–252 | VRDQEGR | 0.76 | S | ||
| 865.4019 | 865.4027 | −0.96 | M1 | 244–250 | MoxGVQMQR | 0.98 | S | ||
| 871.3913 | 871.3896 | 1.96 | M1 | 211–217 | py-QMoxVHAMR | 0.96 | S | ||
| 872.4229 | 872.4237 | −0.87 | M1 | 211–217 | QMVHAMR | 0.96 | S | S | |
| 885.5557 | 885.5564 | −0.85 | HA | 173–179 | NLIWLVK | 0.99 | S | ||
| 887.4080 | 887.4091 | −1.25 | HA | 279–285 | YAFAMER | 0.97 | S | S | S |
| 902.4730 | 902.4732 | −0.17 | M1 | 106–113 | EITFHGAK | 0.97 | S | ||
| 903.4029 | 903.4024 | 0.56 | HA | 279–285 | YAFAMoxER | 0.97 | S | ||
| 905.4662 | 905.4653 | 0.98 | NP | 106–112 | IDGKWMR | 0.76 | |||
| 921.5074 | 921.5068 | 0.63 | M1 | 179–187 | MVLASTTAK | 0.98 | |||
| 1,125.6990 | 1,125.6993 | −0.25 | M1 | 48–57 | TRPILSPLTK | 0.96 | S | ||
| 1,159.6504 | 1,159.6498 | 0.48 | NP | 191–201 | GVGTIAMELIR | 0.95 | I | S | S |
| 1,160.7262 | 1,160.7264 | −0.14 | HA | 70–80 | LRGVAPLHLGK | 0.91 | S | ||
| 1,175.6453 | 1,175.6447 | 0.49 | NP | 191–201 | GVGTIAMoxELIR | 0.95 | S | ||
| 1,177.5861 | 1,177.5867 | −0.50 | NP | 206–214 | GINDRNFWR | 0.94 | S | ||
| 1,198.7307 | 1,198.7303 | 0.30 | HA | 236–245 | FKPEIAIRPK | 0.95 | I | S | S |
| 1,223.6201 | 1,223.6192 | 0.75 | NP | 72–81 | MVLSAFDERR | 0.98 | S | ||
| 1,245.6620 | 1,245.6620 | −0.01 | M1 | 164–174 | QMATTTNPLIR | 0.94 | S | ||
| 1,261.6569 | 1,261.6584 | −1.19 | M1 | 164–174 | QMoxATTTNPLIR | 0.94 | |||
| 1,323.6218 | 1,323.6222 | −0.32 | NP | 169–180 | MCSLMQGSTLPR | 1.00 | S | ||
| 1,326.8256 | 1,326.8248 | 0.62 | HA | 235–245 | KFKPEIAIRPK | 0.83 | S | ||
| 1,420.7682 | 1,420.7689 | −0.48 | NP | 113–123 | ELILYDKEEIR | 0.94 | |||
| 1,443.6362 | 1,443.6380 | −1.28 | HA | 488–499 | EIGNGCFEFYHK | 0.97 | |||
| 1,462.7246 | 1,462.7211 | 2.42 | M1 | 36–47 | NTDLEALMEWLK | 0.94 | |||
| 1,635.9105 | 1,635.9067 | 2.31 | M1 | 58–72 | GILGFVFTLTVPSER | 0.95 | |||
| 1,671.8701 | 1,671.8711 | −0.62 | NP | 253–267 | NPGNAEIEDLIFLAR | 0.95 | S | S | |
| 1,675.8762 | 1,675.8760 | 0.13 | NP | 407–422 | ASAGQISVQPTFSVQR | 0.96 | |||
| 1,725.7935 | 1,725.7873 | 3.57 | NP | 453–467 | MMESAKPEDLSFQGR | 0.92 | |||
| 1,781.9075 | 1,781.9032 | 2.43 | M1 | 164–178 | QMATTTNPLIRHENR | 0.90 | |||
| 2,044.0458 | 2,044.0417 | 2.00 | NP | 250–267 | ESRNPGNAEIEDLIFLAR | 0.92 | |||
| 2,320.1085 | 2,320.1116 | −1.32 | NP | 368–388 | GVQIASNENVETMDSNTLELR | 0.83 | S | S | |
S, signature peptide; I, indicator peptide; “mono.” indicates monoisotopic values.
A high-resolution MALDI-FT-ICR mass spectrum of a whole virus digest obtained from a type A H3N2 strain is shown in Fig. 5 and summarized in Table 4. In total, 23 subtype-specific peptides were identified by the FluTyper algorithm. The detection of a signature peptide, consisting of HA residues 317 to 324 (m/z 880.4353), and two indicator peptides, consisting of NP residues 389 to 395 (m/z 852.4377) and 453 to 465 (m/z 1,510.7010), for the type A H3N2 subtype enabled confident identification of this virus. When only strains circulating since 2009 were considered, 6 period-specific signature peptides were detected (see shaded entries in Table S4 in the supplemental material). The lower number of period-specific signature peptides for this subtype can be attributed to the observation that human type A H3N2 viruses appear to evolve faster than strains of the type A H1N1 subtype (24, 25). A novel antigenic variant of the type A H3N2 virus arises every 3 to 8 years, necessitating an update of the recommended influenza vaccine strain (27).
FIG 5.

High-resolution MALDI-FT-ICR mass spectrum of a whole virus tryptic digest of a cell-cultured clinical specimen containing a type A H3N2 virus strain. All identified ions are listed in Table 4. Ions in bold indicated signature (*) and indicator (^) peptides.
TABLE 4.
Peptides identified in a MALDI-FT-ICR mass spectrum (Fig. 5) of a whole-virus digest of a cell-cultured clinical specimen containing a type A H3N2 seasonal influenza straina
| [M+H]+
m/z mono. |
Difference (ppm) | Viral protein | Residues | Sequence | Po | Signature or indicator | Period-specific signature | Bayes | |
|---|---|---|---|---|---|---|---|---|---|
| Theoretical | Experimental | ||||||||
| 846.3961 | 846.3971 | −1.23 | M1 | 211–217 | py-QMVQAMR | 0.91 | S | ||
| 849.4070 | 849.4086 | −1.94 | M1 | 244–250 | MGVQMQR | 0.97 | S | ||
| 852.4363 | 852.4377 | −1.70 | NP | 389–395 | SGYWAIR | 0.72 | I | S | S |
| 863.4226 | 863.4246 | −2.31 | M1 | 211–217 | QMVQAMR | 0.91 | |||
| 880.4345 | 880.4353 | −0.86 | HA | 317–324 | ITYGACPR | 0.92 | S | S | |
| 964.4887 | 964.4900 | −1.34 | HA | 100–106 | WDLFVER | 0.97 | S | ||
| 1,177.5861 | 1,177.5864 | −0.24 | NP | 206–214 | GINDRNFWR | 0.93 | S | ||
| 1,191.6150 | 1,191.6162 | −0.98 | NP | 443–452 | TSDMRAEIIR | 0.74 | S | ||
| 1,219.6537 | 1,219.6535 | 0.15 | NP | 185–195 | GIGTMVMELIR | 0.91 | S | ||
| 1,223.6201 | 1,223.6193 | 0.67 | NP | 72–81 | MVLSAFDERR | 0.98 | S | ||
| 1,235.6486 | 1,235.6493 | −0.53 | NP | 191–201 | GIGTMoxVMELIR | 0.84 | S | ||
| 1,323.6218 | 1,323.6224 | −0.47 | NP | 169–180 | MCSLMQGSTLPR | 1.00 | |||
| 1,406.7526 | 1,406.7523 | 0.19 | NP | 113–123 | ELVLYDKEEIR | 0.92 | |||
| 1,455.6897 | 1,455.6883 | 0.96 | NP | 423–436 | STIMAAFTGNTEGR | 0.90 | S | ||
| 1,510.7029 | 1,510.7010 | 1.23 | NP | 453–465 | MMEGAKPEEVSFR | 0.79 | I | ||
| 1,620.8275 | 1,620.8299 | −1.49 | NP | 236–249 | FQTAAQRAMVDQVR | 0.64 | |||
| 1,635.9105 | 1,635.9085 | 1.20 | M1 | 58–72 | GILGFVFTLTVPSER | 0.97 | |||
| 1,663.8398 | 1,663.8398 | 0.03 | NP | 401–416 | ASAGQTSVQPTFSVQR | 0.96 | S | ||
| 1,671.8701 | 1,671.8700 | 0.04 | NP | 253–267 | NPGNAEIEDLIFLAR | 0.81 | S | ||
| 2,044.0458 | 2,044.0431 | 1.32 | NP | 250–267 | ESRNPGNAEIEDLIFLAR | 0.78 | S | ||
| 2,266.0437 | 2,266.0423 | 0.62 | NP | 362–382 | GVQIASNENMDNMGSSTLELR | 0.95 | S | ||
| 2,266.0438 | 2,266.0423 | 0.67 | NP | 368–388 | GVQIASNENMDNMGSSTLELR | 0.63 | S | ||
| 2,413.0428 | 2,413.0457 | −1.19 | M1 | 188–210 | AMoxEQMAGSSEQAAEAMEIASQAR | 0.72 | |||
S, signature peptide; I, indicator peptide; “mono.” indicates monoisotopic values.
A high-resolution MALDI-FT-ICR mass spectrum of a whole virus digest obtained from an influenza type B strain is shown in Fig. 6 and summarized in Table 5. In total, 25 peptides belonging to type B influenza viruses were identified by the FluTyper algorithm, 13 of which were signature peptides for all type B viruses and 19 of which were period-specific signature peptides (see shaded entries in Table S5 in the supplemental material). The higher number of signature peptides (13) in this case is associated with the slower rates of evolution evident in type B influenza strains.
TABLE 5.
Peptides identified in a MALDI-FT-ICR mass spectrum (Fig. 6) of a whole virus digest of a cell-cultured clinical specimen containing a type B influenza straina
| [M+H]+
m/z mono. |
Difference (ppm) | Viral protein | Residues | Sequence | Po | Signature or indicator | Period-specific signature | Bayes | |
|---|---|---|---|---|---|---|---|---|---|
| Theoretical | Experimental | ||||||||
| 731.4199 | 731.4209 | −1.42 | HA | 352–357 | YRPPAK | 0.96 | S | S | S |
| 771.4723 | 771.4733 | −1.31 | M1 | 95–101 | GLILAER | 1.00 | S | ||
| 778.3665 | 778.3663 | 0.20 | HA | 28–294 | VWCASGR | 0.97 | S | S | S |
| 816.4363 | 816.4370 | −0.90 | HA | 54–60 | SHFANLK | 0.67 | S | ||
| 836.4989 | 836.4987 | 0.20 | HA | 121–127 | py-QLPNLLR | 0.95 | S | ||
| 853.5254 | 853.5258 | −0.48 | HA | 121–127 | QLPNLLR | 0.99 | S | S | S |
| 939.4893 | 939.4893 | 0.00 | HA | 273–280 | TGTITYQR | 0.99 | S | ||
| 996.5513 | 996.5509 | 0.39 | NP | 163–170 | TIYFSPIR | 0.93 | S | S | |
| 1,043.5844 | 1,043.5834 | 0.91 | NP | 246–256 | GGGTLVAEAIR | 1.00 | S | ||
| 1,057.6000 | 1,057.5995 | 0.45 | NP | 295–304 | ALVDQVIGSR | 1.00 | S | ||
| 1,100.5922 | 1,100.5910 | 1.06 | HA | 332–341 | AIGNCPIWVK | 0.97 | S | ||
| 1,102.6870 | 1,102.6866 | 0.37 | NP | 332–341 | VVLPISIYAK | 1.00 | S | S | S |
| 1,149.5874 | 1,149.5863 | 0.96 | NP | 439–447 | LQFWAPMTR | 1.00 | S | S | S |
| 1,165.5823 | 1,165.5806 | 1.00 | NP | 439–447 | LQFWAPMoxTR | 1.44 | |||
| 1,209.6051 | 1,209.6047 | 0.35 | NP | 540–549 | py-QTIPNFFFGR | 0.96 | S | ||
| 1,226.6317 | 1,226.6301 | 1.32 | NP | 540–549 | QTIPNFFFGR | 0.96 | S | S | S |
| 1,245.6685 | 1,245.6658 | 2.13 | HA | 413–423 | NLNSLSELEVK | 0.97 | S | ||
| 1,259.7140 | 1,259.7091 | 3.88 | NP | 320–331 | SMVVVRPSVASK | 1.00 | S | ||
| 1,264.6757 | 1,264.6744 | 1.00 | NP | 106–116 | NLIQNAHAVER | 0.95 | S | ||
| 1,280.6270 | 1,280.6278 | −0.66 | HA | 321–331 | SKPYYTGEHAK | 0.96 | S | S | S |
| 1,359.6436 | 1,359.6412 | 1.73 | NP | 86–96 | LGEFYNQMMVK | 1.00 | S | S | S |
| 1,418.8002 | 1,418.7981 | 1.46 | NP | 399–411 | VLSALTGTEFKPR | 0.96 | S | S | S |
| 1,441.8009 | 1,441.7976 | 2.29 | M1 | 201–213 | LAEELQSNIGVLR | 1.00 | S | S | S |
| 1,465.7209 | 1,465.7184 | 1.74 | M1 | 36–47 | EFDLDSALEWIK | 1.00 | S | ||
| 1,581.7941 | 1,581.7903 | 2.38 | HA | 300–314 | GSLPLIGEADCLHEK | 0.97 | S | S | S |
| 1,610.8748 | 1,610.8706 | 2.58 | NP | 305–319 | NPGIADIEDLTLLAR | 1.00 | S | S | S |
S, signature peptide; “mono.” indicates monoisotopic values.
The presence of an additional signature peptide at m/z 939.4893 in Fig. 6, consisting of HA residues 273 to 280, unequivocally establishes that the type B virus belongs to the Victoria lineage. Signature peptides to establish the correct lineage of type B human influenza viruses using the proteotyping approach have been reported previously by this laboratory (11). Since the 1970s, influenza B viruses have diverged into two antigenically distinct virus lineages, called the Yamagata and Victoria lineages (29). Viruses of the two lineages cocirculate, and predicting which lineage will predominate is challenging (30). In the past decade in the United States, the annual seasonal influenza vaccine contained a type B virus of the incorrect lineage, with respect to that in circulation, in five of nine seasons. It thus provided less-effective protection against circulating strains of the virus.
DISCUSSION
Effective vaccines rely on a close match between the current vaccine composition and the antigenicity of circulating strains. Present surveillance approaches employ a hemagglutinin inhibition assay that measures the antigenic distance between a tested isolate and a known reference virus, but it provides no molecular detail (31). It also requires reference antisera that need to be replaced annually during surveillance to keep pace with antigenic drift (32). Viruses that react poorly or not at all with the reference antisera are considered to represent antigenic variants or novel subtypes that warrant further molecular characterization by RT-PCR (31).
As circulating strains of influenza continue to evolve and to diverge, such diagnostic assays continue to fail as a consequence of sequence variations in primer and probe targets (33). As an illustration, PCR approaches for the diagnosis of type A H1N1 influenza were found to be unsuitable for pandemic strains due to sequence differences (34). Within months of the 2009 pandemic, rare mutations in the matrix gene were found to invalidate or to limit the suitability of standardized RT-PCR assays developed especially for pandemic strains (35, 36). These constraints can also limit methods that employ mass spectrometry to detect amplified products (37).
Sequencing the full influenza viral genome involves amplification of all 8 gene segments and requires around 30 type- and subtype-specific PCR primers (5). Recently, a pyrosequencing methodology has been described for subtyping of type A human influenza viruses and identification of reassortants (38). However, this approach also depends on the use of a universal set of primers to amplify and to subtype each of the six internal genes and crucially does not target the HA and NA genes.
These observations demonstrate the need to develop new molecular methods for the surveillance of influenza virus. Our results, obtained for representative primary clinical specimens grown in culture, demonstrate the applicability of the proteotyping approach and the confident typing and subtyping of strains. Influenza viruses in common circulation were all correctly identified, even when viral protein coverage ranged from 10 to 35% in these spectra.
The design of the MALDI target plate makes it possible for mass spectra to be acquired from hundreds of digested virus samples within a few minutes. The time required for proteolytic digestion can be minimized by using microwave irradiation (39), and many samples can be processed in parallel. Although a high-resolution mass spectrometer comes at a considerable cost ($500,000 to $1,000,000), it is not inconsistent with the costs required for multiple PCR sequencers and associated equipment. The proteotyping approach also has broad applicability to other respiratory viruses that threaten human and animal health, as recently demonstrated by its application to the typing of parainfluenza viruses (26).
The identification and characterization at the molecular level of influenza type A and B viruses in human primary clinical specimens using a new proteotyping approach provide a convenient, rapid, and reliable means with which to meet the requirements for clinical patient specimens. The specimens contain representative strains of all currently widely circulating types and subtypes of the virus that pose a threat to human health. Importantly, the approach is not encumbered by problems associated with viral evolution that affect PCR assays, and multiple proteins (associated with multiple genes) are screened simultaneously. Signature peptides are detected in mass spectra to enable direct surveillance, with the potential for immediate diagnosis without the need for chemical or biochemical treatments or the use of specific reagents. Signature peptides for other, less common subtypes of human and animal viruses have also been detected and can be characterized in the same manner (40, 41). Another advantage of this approach is that it is open to the detection of novel virus strains that can escape detection by RT-PCR-based approaches, and it is also capable of detecting and differentiating other respiratory pathogens (26).
Supplementary Material
ACKNOWLEDGMENTS
Funding for this work was provided by the Australian Research Council (grant DP110101702 awarded to Kevin M. Downard).
We thank Dominic Dwyer and Janette Taylor at Westmead Hospital (Sydney, Australia), who provided the primary clinical specimens.
Footnotes
Published ahead of print 13 November 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JCM.02315-13.
REFERENCES
- 1.Dwyer DE, Smith DW, Catton MG, Barr IG. 2006. Laboratory diagnosis of human seasonal and pandemic influenza virus infection. Med. J. Aust. 185(Suppl):S48–S53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Butler D. 2012. Flu surveillance lacking. Nature 483:520–522. 10.1038/483520a [DOI] [PubMed] [Google Scholar]
- 3.Girard MP, Tam JS, Assossou OM, Kieny MP. 2010. The A (H1N1) influenza virus pandemic: a review. Vaccine 28:4895–4902. 10.1016/j.vaccine.2010.05.031 [DOI] [PubMed] [Google Scholar]
- 4.Smith GJ, Vijaykrishna D, Bahl J, Lycett SJ, Worobey M, Pybus OG, Ma SK, Cheung CL, Raghwani J, Bhatt S, Peiris JS, Guan Y, Rambaut A. 2009. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 459:1122–1125. 10.1038/nature08182 [DOI] [PubMed] [Google Scholar]
- 5.Layne SP. 2006. Human influenza surveillance: the demand to expand. Emerg. Infect. Dis. 12:562–568. 10.3201/eid1204.051198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schweiger B, Zadow I, Heckler R, Timm H, Pauli G. 2000. Application of a fluorogenic PCR assay for typing and subtyping of influenza viruses in respiratory samples. J. Clin. Microbiol. 38:1552–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spackman E, Senne DA, Myers TJ, Bulaga LL, Garber LP, Perdue ML, Lohman K, Daum LT, Suarez DL. 2002. Development of a real-time reverse transcriptase PCR assay for type A influenza virus and the avian H5 and H7 hemagglutinin subtypes. J. Clin. Microbiol. 40:3256–3260. 10.1128/JCM.40.9.3256-3260.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwahn AB, Wong JWH, Downard KM. 2009. Subtyping of the influenza virus by high resolution mass spectrometry. Anal. Chem. 81:3500–3506. 10.1021/ac900026f [DOI] [PubMed] [Google Scholar]
- 9.Schwahn AB, Wong JWH, Downard KM. 2009. Signature peptides of influenza nucleoprotein for the typing and subtyping of the virus by high resolution mass spectrometry. Analyst 134:2253–2261. 10.1039/b912234f [DOI] [PubMed] [Google Scholar]
- 10.Schwahn AB, Wong JWH, Downard KM. 2010. Rapid differentiation of seasonal and pandemic H1N1 influenza through proteotyping of viral neuraminidase with mass spectrometry. Anal. Chem. 82:4584–4590. 10.1021/ac100594j [DOI] [PubMed] [Google Scholar]
- 11.Schwahn AB, Downard KM. 2011. Proteotyping to establish the lineage of type A H1N1 and type B human influenza virus. J. Virol. Methods 171:117–122. 10.1016/j.jviromet.2010.10.011 [DOI] [PubMed] [Google Scholar]
- 12.Lun ATL, Wong JWH, Downard KM. 2012. FluShuffle and FluResort: new algorithms to identify reassorted strains of the influenza virus by mass spectrometry. BMC Bioinformatics 13:208. 10.1186/1471-2105-13-208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Szretter KJ, Balish AL, Katz JM. 2006. Influenza: propagation, quantification, and storage. Curr. Protoc. Microbiol. Dec Unit 15G.1. 10.1002/0471729256.mc15g01s3 [DOI] [PubMed] [Google Scholar]
- 14.Heyward JT, Klimas RA, Stapp MD, Obijeski JF. 1977. The rapid concentration and purification of influenza virus from allantoic fluid. Arch. Virol. 55:107–119 [DOI] [PubMed] [Google Scholar]
- 15.Polson A. 1974. Purification and aggregation of influenza virus by precipitation with polyethylene glycol. Prep. Biochem. 4:435–456 [DOI] [PubMed] [Google Scholar]
- 16.Jeong EK, Bae JE, Kim IS. 2010. Inactivation of influenza A virus H1N1 by disinfection process. Am. J. Infect. Control 38:354–360. 10.1016/j.ajic.2010.03.003 [DOI] [PubMed] [Google Scholar]
- 17.Wong JWH, Schwahn AB, Downard KM. 2010. FluTyper: an algorithm for automated typing and subtyping of the influenza virus from high resolution mass spectral data. BMC Bioinformatics 11:266. 10.1186/1471-2105-11-266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klimov AI, Garten R, Russell C, Barr IG, Besselaar TG, Daniels R, Engelhardt OG, Grohmann G, Itamura S, Kelso A, McCauley J, Odagiri T, Smith D, Tashiro M, Xu X, Webby R, Wang D, Ye Z, Yuelong S, Zhang W, Cox N. 2012. WHO recommendations for the viruses to be used in the 2012 Southern Hemisphere influenza vaccine: epidemiology, antigenic and genetic characteristics of influenza A (H1N1) pdm09, A (H3N2) and B influenza viruses collected from February to September 2011. Vaccine 30:6461–6471. 10.1016/j.vaccine.2012.07.089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Janies DA, Voronkin IO, Studer J, Hardman J, Alexandrov BB, Treseder TW, Valson C. 2010. Selection for resistance to oseltamivir in seasonal and pandemic H1N1 influenza and widespread co-circulation of the lineages. Int. J. Health Geogr. 9:13. 10.1186/1476-072X-9-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hatakeyama S, Sakai-Tagawa Y, Kiso M, Goto H, Kawakami C, Mitamura K, Sugaya N, Suzuki Y, Kawaoka Y. 2005. Enhanced expression of an alpha-2,6-linked sialic acid on MDCK cells improves isolation of human influenza viruses and evaluation of their sensitivity to a neuraminidase inhibitor. J. Clin. Microbiol. 43:4139–4146. 10.1128/JCM.43.8.4139-4146.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vollmer F, Arnold S, Keng D. 2008. Single virus detection from the reactive shift of a whispering-gallery mode. Proc. Natl. Acad. Sci. U. S. A. 105:20701–20704. 10.1073/pnas.0808988106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boivin G, Coulombe Z, Wat C. 2003. Quantification of the influenza virus load by real-time polymerase chain reaction in nasopharyngeal swabs of patients treated with oseltamivir. J. Infect. Dis. 188:578–580. 10.1086/377046 [DOI] [PubMed] [Google Scholar]
- 23.Bao Y, Bolotov P, Dernovoy D, Kiryutin B, Zaslavsky L, Tatusova T, Ostell J, Lipman D. 2008. The influenza virus resource at the National Center for Biotechnology Information. J. Virol. 82:596–601. 10.1128/JVI.02005-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferguson NM, Galvani AP, Bush RM. 2003. Ecological and immunological determinants of influenza evolution. Nature 422:428–433. 10.1038/nature01509 [DOI] [PubMed] [Google Scholar]
- 25.Rambaut A, Pybus OG, Nelson MI, Viboud C, Taubenberger JK, Holmes EC. 2008. The genomic and epidemiological dynamics of human influenza A virus. Nature 453:615–619. 10.1038/nature06945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nguyen AP, Downard KM. 2013. Proteotyping of the parainfluenza virus with high-resolution mass spectrometry. Anal. Chem. 85:1097–1105. 10.1021/ac302962u [DOI] [PubMed] [Google Scholar]
- 27.Smith DJ, Lapedes AS, de Jong JC, Bestebroer TM, Rimmelzwaan GF, Osterhaus ADME, Fouchier RAM. 2004. Mapping the antigenic and genetic evolution of influenza virus. Science 305:371–376. 10.1126/science.1097211 [DOI] [PubMed] [Google Scholar]
- 28.Lamb RA, Krug RM. 1996. Orthomyxoviridae: the viruses and their replication. In Fields BN. (ed), Fields virology, 3rd ed. Lippincott-Raven, New York, NY [Google Scholar]
- 29.Rota PA, Wallis TR, Harmon MW, Rota JS, Kendal AP, Nerome K. 1990. Cocirculation of two distinct evolutionary lineages of influenza type B virus since 1983. Virology 175:59–68. 10.1016/0042-6822(90)90186-U [DOI] [PubMed] [Google Scholar]
- 30.Reed C, Meltzer MI, Finelli L, Fiore A. 2012. Public health impact of including two lineages of influenza B in a quadrivalent seasonal influenza vaccine. Vaccine 30:1993–1998. 10.1016/j.vaccine.2011.12.098 [DOI] [PubMed] [Google Scholar]
- 31.WHO 2011. Global Influenza Surveillance Network: manual for the laboratory diagnosis and virological surveillance of influenza. World Health Organization, Geneva, Switzerland [Google Scholar]
- 32.Katz JM, Hancock K, Xu X. 2011. Serologic assays for influenza surveillance, diagnosis and vaccine evaluation. Expert Rev. Anti Infect. Ther. 9:669–683. 10.1586/eri.11.51 [DOI] [PubMed] [Google Scholar]
- 33.Gray MC, Su WY, Hal SJ. 2012. Improving influenza virus detection. Expert Opin. Med. Diagn. 6:75–87. 10.1517/17530059.2012.642860 [DOI] [PubMed] [Google Scholar]
- 34.Lalle E, Bordi L, Castilletti C, Meschi S, Selleri M, Carletti F, Lapa D, Travaglini D, Ippolito G, Capobianchi MR, Di Caro A. 2010. Design and clinical application of a molecular method for detection and typing of the influenza A/H1N1pdm virus. J. Virol. Methods 163:486–488. 10.1016/j.jviromet.2009.10.004 [DOI] [PubMed] [Google Scholar]
- 35.Zheng X, Todd KM, Yen-Lieberman B, Kaul K, Mangold K, Shulman ST. 2010. Unique finding of a 2009 H1N1 influenza virus-positive clinical sample suggests matrix gene sequence variation. J. Clin. Microbiol. 48:665–666. 10.1128/JCM.02318-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dhiman N, Espy MJ, Irish C, Wright P, Smith TF, Pritt BS. 2010. Mutability in the matrix gene of novel influenza A H1N1 virus detected using a FRET probe-based real-time reverse transcriptase PCR assay. J. Clin. Microbiol. 48:677–679. 10.1128/JCM.02225-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sampath R, Hall TA, Massire C, Li F, Blyn LB, Eshoo MW, Hofstadler SA, Ecker DJ. 2007. Rapid identification of emerging infectious agents using PCR and electrospray ionization mass spectrometry. Ann. N. Y. Acad. Sci. 1102:109–120. 10.1196/annals.1408.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deng YM, Caldwell N, Barr IG. 2011. Rapid detection and subtyping of human influenza A viruses and reassortants by pyrosequencing. PLoS One 6:e23400. 10.1371/journal.pone.0023400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pramanik NB, Mirza UA, Ning YH, Liu YH, Bartner PL, Weber PC, Bose AK. 2002. Microwave-enhanced enzyme reaction for protein mapping by mass spectrometry: a new approach to protein digestion in minutes. Protein Sci. 11:2676–2687. 10.1110/ps.0213702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ha JW, Downard KM. 2011. Evolution of H5N1 influenza virus through proteotyping of hemagglutinin with high resolution mass spectrometry. Analyst 136:3259–3267. 10.1039/c1an15354d [DOI] [PubMed] [Google Scholar]
- 41.Nguyen AP, Downard KM. 2013. Subtyping of influenza neuraminidase using mass spectrometry. Analyst 138:1787–1793. 10.1039/c3an00086a [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.