

Abstract
Recent evidence suggests that most influenza A virus gene segments can contribute to the pathogenicity of the virus. In this regard, the hemagglutinin (HA) subtype of the circulating strains has been closely surveyed, but the reassortment of internal gene segments is usually not monitored as a potential source of an increased pathogenicity. In this work, an oligonucleotide DNA microarray (PhyloFlu) designed to determine the phylogenetic origins of the eight segments of the influenza virus genome was constructed and validated. Clades were defined for each segment and also for the 16 HA and 9 neuraminidase (NA) subtypes. Viral genetic material was amplified by reverse transcription-PCR (RT-PCR) with primers specific to the conserved 5′ and 3′ ends of the influenza A virus genes, followed by PCR amplification with random primers and Cy3 labeling. The microarray unambiguously determined the clades for all eight influenza virus genes in 74% (28/38) of the samples. The microarray was validated with reference strains from different animal origins, as well as from human, swine, and avian viruses from field or clinical samples. In most cases, the phylogenetic clade of each segment defined its animal host of origin. The genomic fingerprint deduced by the combined information of the individual clades allowed for the determination of the time and place that strains with the same genomic pattern were previously reported. PhyloFlu is useful for characterizing and surveying the genetic diversity and variation of animal viruses circulating in different environmental niches and for obtaining a more detailed surveillance and follow up of reassortant events that can potentially modify virus pathogenicity.
INTRODUCTION
Influenza viruses are one of the most important pathogens of humans, causing seasonal epidemics and occasional pandemics. They are enveloped viruses with a genome composed of 8 segments of single-stranded RNA of negative polarity that encode at least 13 proteins (1). Based on the antigenicity of their hemagglutinin (HA) and neuraminidase (NA) proteins, influenza A viruses are classified into 16 HA subtypes and 9 NA subtypes (2). Recently, however, two new HA subtypes present in bats were described (3, 4).
The HA protein has been identified as a major determinant of virulence; it is responsible for receptor binding and virus entry into the cell, which explains at least in part the host range and tissue tropism of influenza viruses (5). In addition to HA, many other viral proteins have been shown to play a role in shaping the virulence of influenza A viruses, contributing to their adaptation to infection and transmission to new host species, ability to modulate the host immune response, and capacity to replicate efficiently at low temperatures (6–8). Thus, the ribonucleoprotein complex (polymerase acidic [PA], polymerase basic 1 [PB1], PB2, and nucleoprotein [NP]) plays an important role in interspecies transmission, and together with nonstructural 1 protein [NS1], which antagonizes the cellular response to interferon, it is a known determinant of host range restriction and pathogenicity. NA, which is important for virus release from the infected cell, and matrix 2 protein (M2), an ion channel needed for virus uncoating in the endosome, also participate in these events (6–8).
The virulence of influenza A viruses can be influenced by single point mutations, nonhomologous recombination, or by exchanges of complete genomic segments. All these mechanisms of genome evolution increase the genetic diversity of the viruses and have been linked to modifications of virus pathogenicity (6). Regarding gene reassortment, the exchange of HA and NA genes between human and animal strains was responsible for the emergence of the H2N2 and H3N2 viruses that caused the pandemics of 1957 and 1968, respectively, in the human population. Likewise, the influenza virus strain that caused the first pandemic of this century in 2009 to 2010 (A/H1N1pdm09) resulted from a genetic reassortment event between a Eurasian “avian-like” H1N1 virus and a swine triple reassortant strain (9). As a consequence of the global impact of these strains on human health, the appearances of new subtypes in viruses infecting human populations are closely monitored.
Influenza A virus infects many species of wild and domestic birds, as well as a large number of mammals, including humans. Despite the frequent occurrence of gene reassortment between influenza virus strains from the same or different animal hosts, and the growing evidence that links many of the virus segments to increased pathogenicity in certain viral genetic backgrounds (6), the reassortment of internal gene segments as a potential cause of the generation of viruses with increased pathogenicity has been neglected. Thus, for a more accurate assessment of new circulating virus strains, the reassortment of all eight viral genes should be monitored. In this work, we developed an oligonucleotide DNA microarray to determine the phylogenetic origin of each of the eight segments of the influenza A virus genome and to classify each viral gene into a clade. This microarray should also be useful for screening and characterizing the genomic fingerprint of influenza A virus strains circulating in wild or domesticated birds and swine. This assay was validated with human, swine, equine, and avian strains present in either field or clinical samples or that were grown in cell culture or egg embryos.
MATERIALS AND METHODS
Viruses.
Total RNA extracted from egg-grown reference influenza A viruses representing the 16 HA and the 9 NA subtypes was kindly provided by R. G. Webster (St. Jude Children's Research Hospital, Memphis, TN). Swine viruses were isolated from lung biopsy samples of pigs with respiratory symptoms from porcine farms in central Mexico and were adapted to grow in embryonated chicken eggs at the Veterinary Medicine School, Universidad Nacional Autónoma de Mexico (UNAM). Cloacal swabs positive for influenza virus were collected from migratory waterfowl in Sonora, Mexico. Human nasopharyngeal swabs of children with respiratory disease positive for influenza viruses were obtained from the School of Medicine, UNAM, which carried out an epidemiological study in Jalisco and Mexico City in 2010. Human influenza viruses adapted to grow in cell culture were obtained from the National Institute of Epidemiological Reference, the biosafety level 3 (BSL-3) laboratory of the Ministry of Health of the State of Veracruz, or the Institute of Biotechnology, UNAM, in Cuernavaca, Morelos, Mexico. Table 1 describes the influenza virus strains characterized in this work. The reference viral strains are listed in Table 2.
TABLE 1.
Influenza virus strains used in this worka
| Virus strain | Host | Collection yr | Collection siteb | Subtype |
|---|---|---|---|---|
| A/swine/Mexico/Ver29/2010 (H1N1) | Swine | 2010 | Veracruz | H1N1 |
| A/swine/Mexico/Ver31/2010 (H1N1) | Swine | 2010 | Veracruz | H1N1 |
| A/swine/Mexico/Qro32/2010 (H1N1) | Swine | 2010 | Querétaro | H1N1 |
| A/swine/Mexico/Ver37/2010 (H1N1) | Swine | 2010 | Veracruz | H1N1 |
| A/swine/Mexico/Mich40/2010 (H3N2) | Swine | 2010 | Michoacán | H3N2 |
| A/swine/Mexico/Mex50/2010 (H3N2) | Swine | 2010 | Edo. Mexico | H3N2 |
| A/swine/Mexico/Mex51/2010 (H3N2) | Swine | 2010 | Edo. Mexico | H3N2 |
| A/swine/Mexico/Mex52/2010 (H1N1) | Swine | 2010 | Edo. Mexico | H1N1 |
| A/Mexico City/IBT22/2009 (H1N1)pdm (pandemic) | Human | 2009 | México City | H1N1 |
| A/Mexico/DIF2662/2003 (H3N2) | Human | 2003 | México City | H3N2 |
| A/Mexico/DIF2664/2003 (H3N2) | Human | 2003 | México City | H3N2 |
| A/Mexico/Jal25216/2009 (H3N2) | Human | 2009 | Jalisco | H3N2 |
| A/Mexico/Ver1/2010 (H3N2) | Human | 2010 | Veracruz | H3N2 |
| A/Mexico/Mex33/2010 (H3N2) | Human | 2010 | México City | H3N2 |
| A/Mexico/Jal121/2011 (H3N2) | Human | 2011 | Jalisco | H3N2 |
| A/Mexico/Ver2/2009 (H1N1) (seasonal) | Human | 2009 | Veracruz | H1N1 |
| A/Eurasian teal/Mexico/Son701/2008 (H11N3) | Avian: Anas crecca | 2008 | Sonora | H11N3 |
| A/American widgeon/Mexico/Son769/2008 (H9N2) | Avian: Anas americana | 2008 | Sonora | H9N2 |
| A/northern shoveler/Mexico/Son797/2008 (H5N3) | Avian: Anas clypeata | 2008 | Sonora | H5N3 |
| A/Eurasian teal/Mexico/Son829/2009 (H6N5) | Avian: Anas crecca | 2009 | Sonora | H6N5 |
| A/Eurasian teal/Mexico/Son1132/2009 (H10N3) | Avian: Anas crecca | 2009 | Sonora | H10N3 |
Swine viruses were adapted to grow in chick embryos. Human A/H1N1 and A/H3N2 viruses were isolated in cell culture or characterized directly from nasopharyngeal swabs. Avian viruses were characterized directly from cloacal swab samples.
Edo. Mexico, Estado de Mexico.
TABLE 2.
Characterization of influenza A reference strains by in silico and microarray hybridization assays
| Virus strain | Hybridization patterns by genea: |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PB2 |
PB1 |
PA |
HA |
NP |
NA |
M |
NS |
|||||||||
| MH | isA | MH | isA | MH | isA | MH | isA | MH | isA | MH | isA | MH | isA | MH | isA | |
| A/FortMonmouth/1/47 (H1N1) | 2 | 2 | 0 | 0 | 4 | 4 | H1-0 | H1-0 | 4 | 4 | N1-1 | N1-1 | NDb3 | 0 | 1 | 1 |
| A/New Jersey/1976 (H1N1) | 2 | 2 | 0 | 0 | 0 | 0c | H1-1 | H1-1 | 4 | 4 | N1-2 | N1-2 | ND | 0 | 1 | 1 |
| A/Singapore/1-MA12C/1957 (H2N2) | 2 | 2 | 3 | 3 | 4 | 4 | H2-0 | H2-0 | 4 | 4 | N2-0 | N2-0 | 0 | 0 | 1 | 1 |
| A/equine/Miami/1/1963 (H3N8) | 2 | 2 | 1 | 1 | 2 | 2 | H3-1 | H3-1 | 1 | 1 | N8-0 | N8-0 | 0 | 0 | 1 | 1 |
| A/duck/Czechoslovakia/1956 (H4N6) | 1 | 1 | 3 | 3 | 3 | 3 | H4-2 | H4-2 | 0 | 0 | N6-0 | N6-0 | ND | 0 | 1 | 1 |
| A/tern/South/Africa/1961 (H5N3) | 1 | 1 | 3 | 3 | 3 | 3 | H1-0 | H1-0 | 0 | 0 | N3-2 | N3-2 | 0 | 0 | 1 | 1 |
| A/shearwater/Australia/1/1972 (H6N5) | 1 | 1 | 3 | 3 | 3 | 3 | H6-0 | H6-0 | 0 | 0 | N5-1 | N5-1 | 0 | 0 | 1 | 1 |
| A/ruddy/turnstone/NJ/65/1985 (H7N3) | 2 | 2 | 3 | 3 | 3 | 3 | H7-0 | H7-0 | 0 | 0 | N3-0 | N3-0 | 0 | 0 | 0 | 0 |
| A/turkey/Ontario/6118/1968 (H8N4) | 2 | 2 | 0 | 0 | 3 | 3 | H8-0 | H8-0 | 0 | 0 | N4-0 | N4-0 | 0 | 0 | 1 | 1 |
| A/turkey/Wisconsin/1/1966 (H9N2) | 2 | 2 | 3 | 3 | 3 | 3 | H9-0 | H9-0 | 0 | 0 | N2-ND | N2-1 | 0 | 0 | 1 | 1 |
| A/chicken/Germany/n/1949 (H10N7) | 1 | 1 | 3 | 3 | 3 | 3 | H10-1 | H10-1 | 0 | 0 | N7-1 | N7-1 | 0 | 0 | 0 | 0 |
| A/duck/Memphis/546/1974 (H11N9) | 2 | 2 | 0 | 0 | 3 | 3 | H11-0 | H11-0 | 0 | 0 | N9-0 | N9-0 | 0 | 0 | 0 | 0 |
| A/duck/Alberta/60/1976 (H12N5) | 2 | 2 | 3 | 3 | 3 | 3 | H12-0 | H12-0 | 0 | 0 | N5-0 | N5-0 | 0 | 0 | 0 | 1 |
| A/gull/Maryland/704/1977 (H13N6) | 1 | 1 | 3 | 3 | 3 | 3 | H13-0 | H13-0 | 0 | 0 | N6-0 | N6-0 | 0 | 0 | 1 | 1 |
| A/mallard/Astrakhan/263/1982 (H14N5) | 1 | 1 | 3 | 3 | 3 | 3 | H14-0 | H14-0 | 0 | 0 | ND | ND | 0 | 0 | 1 | 1 |
| A//wedge-tailed shearwater/Australia/2576/1979 (H15N9) | 1 | 1 | 3 | 3 | 3 | 3 | H15-0 | H15-0 | 0 | 0 | N9-1 | N9-1 | 0 | 0 | 1 | 1 |
| A/shorebird/Delaware/168/06 (H16N3) | 1 | 1 | 3 | 3 | 3 | 3 | H16-2 | H16-2 | 0 | 0 | N3-1 | N3-1 | 0 | 0 | ND | 1 |
PB2, polymerase basic 2; PA, polymerase acidic; HA, hemagglutinin; NP, nucleoprotein; NA, neuraminidase; M, matrix; NS, nonstructural; MH, hybridization assay; isA, in silico assay.
ND, gene segments not detected or clades not defined.
This segment was originally assigned as clade 4 by the in silico assay, different from the clade 0 assigned by microarray analysis. Nucleotide sequencing of the segment revealed a divergence between the reported sequence used in the in silico assay (GenBank accession no. CY021962.1) and the real sequence of the hybridized sample. This finding highlights the reliability of the results generated with the microarray assay.
Probe design.
Three different sets of influenza A virus sequences were used in this study to design the probes. The sequences of the six internal segments and subtypes H1, H3, N1, and N2 of human, swine, and avian origin were those reported by Smith et al. (9). For all other HA and NA subtypes, a set of full-length sequences from all hosts reported prior to 2008 were downloaded from the NCBI Influenza Virus Resource (10). Finally, a set of influenza virus sequences was added from viruses reported prior to July 2010 from animal hosts other than swine, birds, or humans. All identical sequences were eliminated from the analysis. Only complete gene sequences were used, and the identical gene sequences were collapsed using the NCBI Influenza Virus Resource database algorithm, which collapses all identical sequences in the data set by representing them with the oldest sequence of the group, and the metadata of the collapsed sequences are not preserved.
A phylogenetic inference was carried out for each genomic segment, and in the case of HA and NA, for each subtype. The multiple sequence alignment of the nucleotide sequences was performed with the multiple sequence comparison by log-expectation (MUSCLE) algorithm (11), using the default parameters for nucleotide sequences. The sequences were translated and realigned using MUSCLE with the default parameters for protein sequences. The sequences were then converted back to nucleotides to perform the phylogenetic analysis. The phylogenetic trees were inferred with the PhyML algorithm (12) using the maximum likelihood method under the Hasegawa-Kishino-Yano model, with gamma-distributed rates among sites (HKY + G). Each monophyletic group in a given tree defined a clade for that particular gene segment. However, in three particular cases, two clades were defined within a monophyletic group. In these cases (N4, NP, and H1), independent probes were designed for each clade. The sequence identity of each clade was calculated using the cd-hit algorithm (13), targeting up to 90% of sequence identity within the groups. For the case of HA and NA genes, the clades were defined for every subtype. All phylogenetic trees and clades defined can be found on the PhyloFlu website (http://copernico.ibt.unam.mx/phyloflu).
For each defined clade, one sequence was chosen randomly, and this sequence was cut into fragments of 70 nucleotides (nt), with each 70 mer phased out by 5 bases. The generated oligonucleotides were classified by their capacity to recognize the sequences within their own clade while not recognizing the sequences from other clades. The selection of the 70 mers was based on the theoretical ΔG hybridization value calculated with the nearest neighbor method and the total aligned nucleotides. The oligonucleotides (14) that recognized all the sequences from their own clade with a ΔG of ≤−75 kcal/mol and had a match of ≥60 of 70 nucleotides, while at the same time not recognizing sequences from other clades with a ΔG of <−55 kcal/mol and that matched <55 of 70 nucleotides, were defined as specific. The oligonucleotides that did not meet the cutoff values were considered unspecific and were discarded from the analysis. Once a group of specific oligonucleotides was obtained, the minimum number of 70 mers that recognized all the sequences in their own clade at least three times and had the lowest theoretical ΔG hybridization value was chosen. The list of selected oligonucleotides can be found on the PhyloFlu website (see above).
In silico genomic patterns.
To identify genomic patterns from the hybridization data, in silico hybridization analyses were performed with complete genome sequences. A genomic pattern was defined as the collection of clades contributed by each segment of the influenza A virus genome. For the theoretical hybridization analyses, a database with all available human, swine, avian, equine, and canine influenza A virus genomes was created (see http://copernico.ibt.unam.mx/phyloflu). In silico hybridization was performed by aligning the designed specific oligonucleotides with the sequences of the influenza A virus genome database, using the BLASTALL 2.2.25 program (15). A positive match was scored if the hybridization consisted of ≥60 of 70 nucleotides paired and had a theoretical hybridization ΔG of <−70 kcal/mol. The database of the in silico hybridization genomic patterns was used to identify the genomic fingerprints obtained from the microarray assay.
Oligonucleotide synthesis and microarray printing.
The oligonucleotides were obtained from Invitrogen-Illumina (San Diego, CA) and stored in 384-well plates at −70°C at a concentration of 40 mM. The microarray was printed on poly-l-lysine slides at the microarray core unit of the Institute of Cellular Physiology of the National University of Mexico (UNAM). Each probe was printed in quadruplicate in the microarray. A unique oligonucleotide of 70 nucleotides in length (spike70) that has no identity with sequences reported in public databases (16) was added (4 pmol) to each of the synthesized 70 mers and used as hybridization and spot location controls. Each microarray slide was processed prior to hybridization. First, the oligonucleotides were covalently attached to the glass by exposing them to 60 mJ UV light for 20 min. The slides were then immersed in a prewarmed 3× SSC-0.2% SDS solution (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) at 65°C for 5 min, washed twice with water for 30 s to remove the excess SDS, washed with 95% ethanol for 30 s, and dried by centrifugation for 5 min at 50 × g. Once dried, the slides were immersed in a solution containing 5.5 g succinic anhydride dissolved in 15 ml of 1 M sodium borate (pH 8.0) and 355 ml of 1-methyl-2-pyrrolidinone to block the exposed amino groups in the poly-l-lysine. Finally, the slides were washed twice with water for 1 min, once with 95% ethanol for 1 min, and dried again by centrifugation for 5 min at 50 × g.
Sample processing and hybridization.
Viral RNA from the reference samples was reverse transcribed using a random nonamer primer with a known tag (5′-CACTGGAGGATANNNNNNNNN-3′). Double-stranded DNA was synthesized by two rounds of Sequenase 2.0 (USB), and two steps of PCR amplification were then performed with a primer having a sequence complementary to the tag of the previous primer (5′-GTTCCCACTGGAGGATA-3′). In the second amplification step, a modified nucleotide (aminoallyl-dUTP) was added at a 7:3 ratio with dTTP, and the labeled PCR product was subsequently purified by column (Zymo Research). The aminoallyl-dUTP-labeled product was eluted in water and labeled with a Cy3 monofunctional dye (GE Healthcare Life Sciences) (16). Viral RNA from viruses grown in cell culture or chicken embryonated eggs or from viruses present in either human clinical samples or field avian specimens was extracted using the PureLink RNA minikit (Invitrogen) and stored at −70°C. To increase the amount of influenza A virus genetic material present in these samples, the complete viral genome was amplified by a one-step reverse transcription-PCR (RT-PCR) (Invitrogen) using the 5′ and 3′ conserved regions at the ends of each segment, as described previously (17). The PCR product was purified by a Zymo column, and two PCR amplification steps were performed as described above. The amplified DNA with incorporated aminoallyl-dUTP was then labeled with Cy3 and column purified. The complement of the spike70 probe was labeled with a Cy5 monofunctional dye (GE Healthcare Life Sciences) (16). Before hybridization, 2.5 ml (4 ng/ml) of the Cy5-labeled spike70 complement was added to each Cy3-labeled sample, and the samples were hybridized for 12 h in a Die Tech 5005 hybridization chamber submerged in a water bath at 65°C.
Data analysis.
The microarrays were scanned with a GenePix 4000B (Molecular Devices, Sunnyvale, CA) apparatus that generated a tagged image file format (TIFF) image for each channel. The images were analyzed with the GenePix 6.0.2 software included in the scanner. The output .gpr files containing the information collected by the scanner were analyzed with an R script (18). For each positive Cy3 fluorescent signal, we calculated the normalized intensity values (Ii) with the formula
where si is the spot's intensity median number of experiments; similarly, bi is the median of the background signals.
For a hybridization considered to be positive for a given spot, the fluorescent signal of the spot in the microarray was required to pass two cuts: first, the fluorescence intensity needed to be ≥5% of the maximum normalized intensity value of the experiment, and second, the raw fluorescence intensity value needed to be ≥5% of the maximum possible value of the scanner (65, 535); this second filter was set to avoid false positives in experiments in which the overall fluorescence was too low. Since each clade was defined by a different number of oligonucleotides, the probability value was weighted based on the number of oligonucleotides representing each clade, so that the clades with fewer oligonucleotides had a similar statistical power as those with more oligonucleotides per clade. To determine the true positive results, a hypergeometric distribution was used to distinguish the clades that were significantly overrepresented for each segment with a statistical significance, as follows:
in which P is the probability of observing k probes from a total of D for a given clade by chance, N is the total number of probes selected for the microarray, and n is the number of probes considered to be positives with the considerations previously mentioned.
RESULTS
Microarray design.
A summary of the process used to design the 70 mers included in the microarray is depicted in Fig. 1. A set of 1,492 complete gene sequences from influenza A viruses isolated from 18 animal hosts was downloaded from the NCBI Influenza Virus Resource (10). Additionally, 6,413 complete sequences from human, swine, and avian hosts (9) were included in the study. This work encompasses the sequence data present in the database before July 2010 (Table 3). For each influenza virus segment, a phylogenetic tree was inferred; in the particular cases of HA and NA, trees were constructed for every subtype of these gene segments. Each of the formed monophyletic groups formed is referred to in this work as a clade (see a few exceptions in Materials and Methods), and each clade was numbered (shown for the NA subtype 7 gene in Fig. 2). For every clade in the trees, a gene sequence was randomly selected to generate 70-mer probes. For large clades (those containing >100 sequences), probes were designed from 2 to 4 gene sequences, since these clades were composed of sequences with a lower identity among them. The minimum identities among the sequences within each group were calculated (Table 3). The criteria used to select the 70 mers were (i) a pairing ΔG of ≤−75 kcal/mol, (ii) a match of ≥60 nucleotides (nt) with influenza virus sequences within the clade, and (iii) a ΔG of >−55 kcal/mol with noninfluenza virus sequences in GenBank or with influenza virus sequences outside the clade. Every sequence present in the clade was recognized by at least three independent probes.
FIG 1.
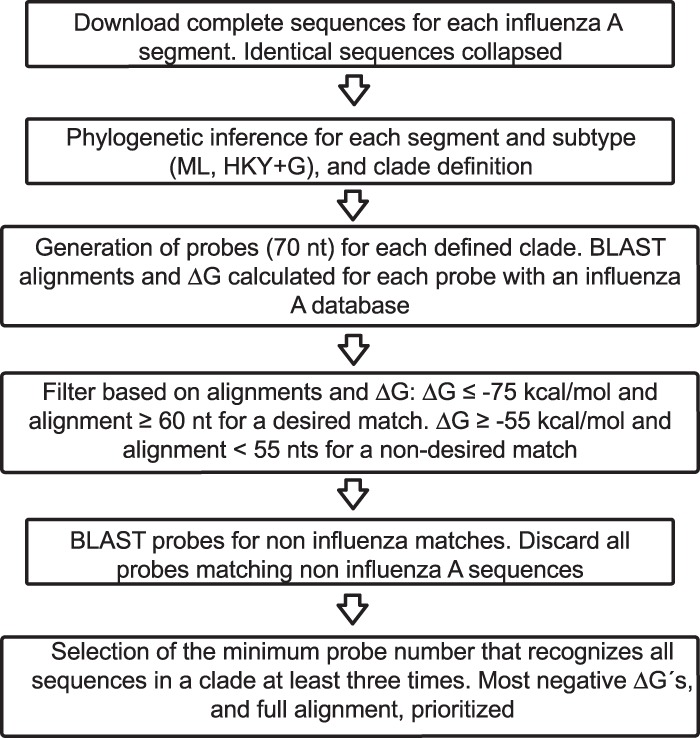
Diagrammatic description of the DNA microarray design. For details of the protocol, see Materials and Methods.
TABLE 3.
Gene sequences analyzed and probes designed for each of the eight segments and the HA and NA subtypes of influenza A virus genome
| Gene | No. of sequencesa | No. of cladesb | Min identity within the clades (%)c | No. of probes selectedd |
|---|---|---|---|---|
| PB2 | 926 | 3 | 84 | 54 |
| PB1 | 897 | 4 | 85 | 40 |
| PA | 910 | 5 | 84 | 57 |
| H1 | 206 | 4 | 85 | 21 |
| H2 | 41 | 2 | 85 | 10 |
| H3 | 379 | 5 | 86 | 24 |
| H4 | 18 | 4 | 87 | 14 |
| H5 | 189 | 2 | 87 | 11 |
| H6 | 45 | 3 | 87 | 12 |
| H7 | 77 | 4 | 89 | 15 |
| H8 | 5 | 1 | >90 | 4 |
| H9 | 61 | 3 | 87 | 18 |
| H10 | 8 | 2 | >90 | 7 |
| H11 | 9 | 2 | >90 | 6 |
| H12 | 8 | 2 | >90 | 6 |
| H13 | 3 | 2 | >90 | 6 |
| H14 | 2 | 1 | >90 | 4 |
| H15 | 7 | 1 | >90 | 4 |
| H16 | 8 | 4 | >90 | 12 |
| NP | 906 | 5 | 85 | 66 |
| N1 | 360 | 4 | 85 | 28 |
| N2 | 351 | 2 | 82 | 27 |
| N3 | 40 | 3 | 87 | 13 |
| N4 | 10 | 3 | 89 | 10 |
| N5 | 14 | 2 | >90 | 6 |
| N6 | 19 | 2 | 87 | 7 |
| N7 | 28 | 3 | 87 | 10 |
| N8 | 207 | 4 | 86 | 16 |
| N9 | 14 | 2 | >90 | 6 |
| M | 1,001 | 2 | 86 | 13 |
| NS | 1,146 | 2 | 81 | 17 |
Full-gene sequences available in GenBank before July 2010 or reported in reference 9.
Monophyletic groups in each constructed phylogenetic tree.
Nucleotide sequence identity among the gene sequences comprising each clade.
Number of probes that recognize all sequences within a clade at least three times.
FIG 2.
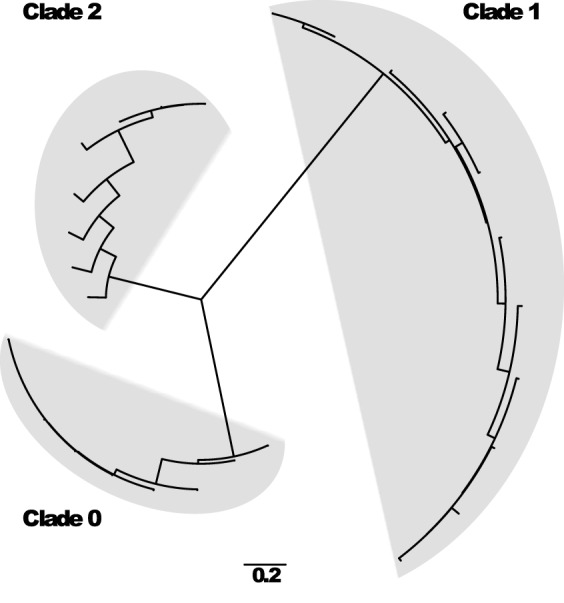
Phylogenetic tree of the subtype 7 neuraminidase gene. The tree was inferred by maximum likelihood under the HKY + G substitution model. The three clades defined with the methodology described are indicated. The minimum sequence identity within the clades is 89%.
During the analysis, an overrepresentation in the number of sequences of HA and NA subtypes of viruses that infect humans (H1, H3, N1, and N2) was found. In contrast, HA subtypes H4, H8, and H10 to H16 as well as NA subtypes N5, N6, N7, and N9 were underrepresented in the database (Table 3). For the internal genomic segments, we had a relatively similar number of sequences. A total of 544 specific probes were selected to identify the 8 segments of the influenza A virus genome distributed in 88 clades (Table 3). Each clade was defined by a different number of probes, with 4 being the lowest and 35 being the highest.
Phylogenetic origin of RNA segments and genomic fingerprint.
The microarray provides information about the phylogenetic origin of each of the eight segments of the influenza A virus genome. During the analysis, the influenza virus RNA segments were allocated to a clade within their corresponding phylogenetic tree (see an example in Fig. 3). Out of 88 defined clades, the origin of each segment was traced for 69 clades to viruses circulating in a particular animal host (Table 4); 54 clades were mapped to avian strains, 7 to equine strains, 5 to human strains, and 3 to swine strains. Eighteen more clades were traced to viruses that phylogenetically contain sequences circulating in 2 or 3 animal hosts, and 8 more clades could not be assigned to a particular host.
FIG 3.
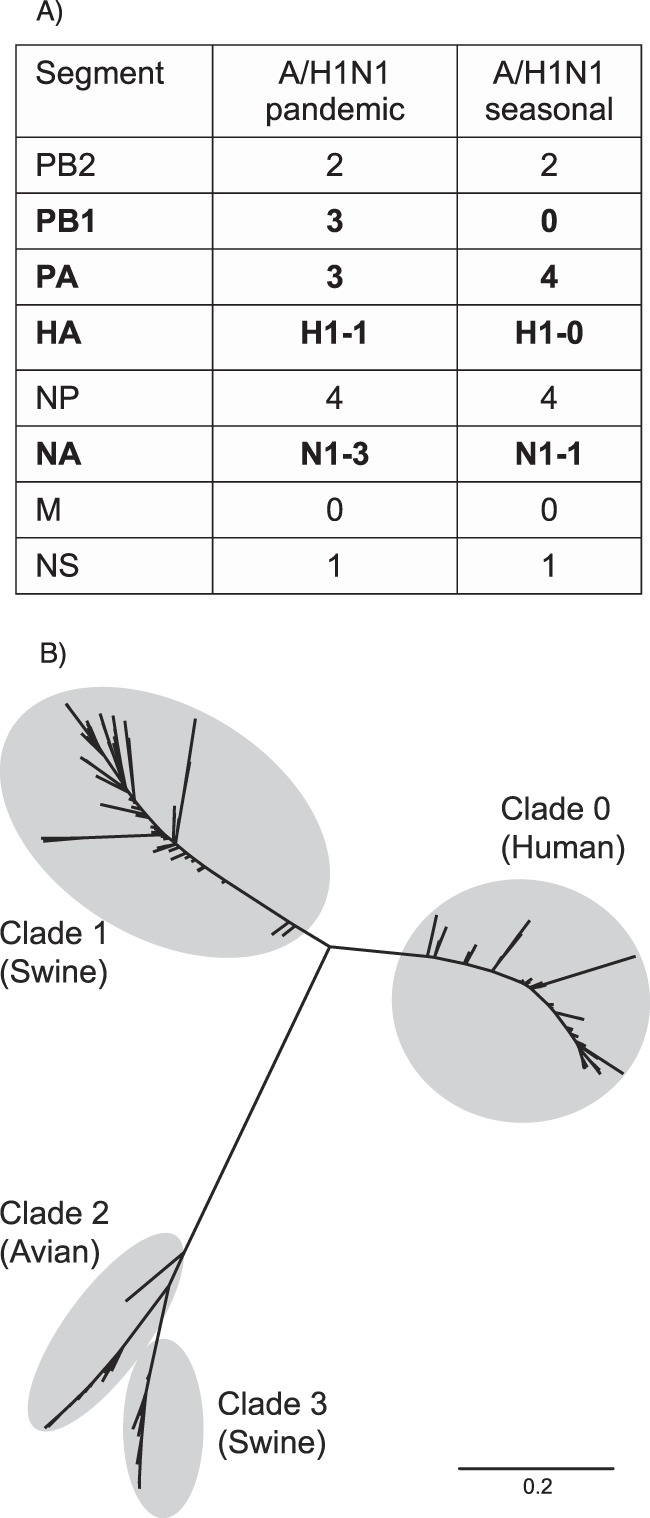
Clade characterization of H1N1 strains. (A) Clades determined by microarray hybridization for the gene segments of an A/H1N1pdm09 virus and a seasonal A/H1N1 virus. Gene segments that belong to different clades in the two strains are in bold. (B) Phylogenetic tree of the subtype 1 hemagglutinin gene showing the identified clades. The tree was inferred by maximum likelihood under the HKY + G substitution model. Classification of the H1 gene of the A/H1N1pdm09 virus within clade 1 indicates a swine origin, while the H1 from the A/H1N1 seasonal strain points to a human origin (clade 0).
TABLE 4.
Association of phylogenetic influenza virus gene clades with host species
| Probe name | Determined animal source(s)a |
|---|---|
| PhyloFlu_S1_PB2_C0 | Equine |
| PhyloFlu_S1_PB2_C1 | AVIAN/human, swineb |
| PhyloFlu_S1_PB2_C2 | NAc |
| PhyloFlu_S2_PB1_C0 | HUMAN/swine |
| PhyloFlu_S2_PB1_C1 | EQUINE/canine |
| PhyloFlu_S2_PB1_C2 | Equine |
| PhyloFlu_S2_PB1_C3 | NA |
| PhyloFlu_S3_PA_C0 | Swine |
| PhyloFlu_S3_PA_C1 | Equine |
| PhyloFlu_S3_PA_C2 | EQUINE/canine |
| PhyloFlu_S3_PA_C3 | AVIAN/human |
| PhyloFlu_S3_PA_C4 | Human |
| PhyloFlu_S4_H1_C0 | Human |
| PhyloFlu_S4_H1_C1 | SWINE/human |
| PhyloFlu_S4_H1_C2 | Avian |
| PhyloFlu_S4_H1_C3 | Swine |
| PhyloFlu_S4_H2_C0 | NA |
| PhyloFlu_S4_H2_C1 | Avian |
| PhyloFlu_S4_H3_C0 | HUMAN/swine |
| PhyloFlu_S4_H3_C1 | Equine |
| PhyloFlu_S4_H3_C2 | Avian |
| PhyloFlu_S4_H3_C3 | Avian |
| PhyloFlu_S4_H3_C4 | Avian |
| PhyloFlu_S4_H4_C0 | Avian |
| PhyloFlu_S4_H4_C1 | Avian |
| PhyloFlu_S4_H4_C2 | Avian |
| PhyloFlu_S4_H4_C3 | Avian |
| PhyloFlu_S4_H5_C0 | AVIAN/human |
| PhyloFlu_S4_H5_C1 | Avian |
| PhyloFlu_S4_H6_C0 | Avian |
| PhyloFlu_S4_H6_C1 | Avian |
| PhyloFlu_S4_H6_C2 | Avian |
| PhyloFlu_S4_H7_C0 | Avian |
| PhyloFlu_S4_H7_C1 | Equine |
| PhyloFlu_S4_H7_C2 | Avian |
| PhyloFlu_S4_H7_C3 | Avian |
| PhyloFlu_S4_H8_C0 | Avian |
| PhyloFlu_S4_H9_C0 | Avian |
| PhyloFlu_S4_H9_C1 | Avian |
| PhyloFlu_S4_H9_C2 | Avian |
| PhyloFlu_S4_H10_C0 | Avian |
| PhyloFlu_S4_H10_C1 | Avian |
| PhyloFlu_S4_H11_C0 | Avian |
| PhyloFlu_S4_H11_C1 | Avian |
| PhyloFlu_S4_H12_C0 | Avian |
| PhyloFlu_S4_H12_C1 | Avian |
| PhyloFlu_S4_H13_C0 | Avian |
| PhyloFlu_S4_H13_C1 | AVIAN/otherd |
| PhyloFlu_S4_H14_C0 | Avian |
| PhyloFlu_S4_H15_C0 | Avian |
| PhyloFlu_S4_H16_C0 | Avian |
| PhyloFlu_S4_H16_C1 | Avian |
| PhyloFlu_S4_H16_C2 | Avian |
| PhyloFlu_S4_H16_C3 | Avian |
| PhyloFlu_S5_NP_C0 | Avian |
| PhyloFlu_S5_NP_C1 | NA |
| PhyloFlu_S5_NP_C2 | Equine |
| PhyloFlu_S5_NP_C4 | Human |
| PhyloFlu_S5_NP_C5 | Avian |
| PhyloFlu_S6_N1_C0 | Avian |
| PhyloFlu_S6_N1_C1 | Human |
| PhyloFlu_S6_N1_C2 | Swine |
| PhyloFlu_S6_N1_C3 | NA |
| PhyloFlu_S6_N2_C0 | Human |
| PhyloFlu_S6_N2_C1 | Avian |
| PhyloFlu_S6_N3_C0 | Avian |
| PhyloFlu_S6_N3_C1 | Avian |
| PhyloFlu_S6_N3_C2 | Avian |
| PhyloFlu_S6_N4_C0 | Avian |
| PhyloFlu_S6_N4_C1 | Avian |
| PhyloFlu_S6_N4_C2 | AVIAN/other |
| PhyloFlu_S6_N5_C0 | Avian |
| PhyloFlu_S6_N5_C1 | Avian |
| PhyloFlu_S6_N6_C0 | Avian |
| PhyloFlu_S6_N6_C1 | Avian |
| PhyloFlu_S6_N7_C0 | Avian |
| PhyloFlu_S6_N7_C1 | Avian |
| PhyloFlu_S6_N7_C2 | EQUINE/swine/other |
| PhyloFlu_S6_N8_C0 | NA |
| PhyloFlu_S6_N8_C1 | Avian |
| PhyloFlu_S6_N8_C2 | Avian |
| PhyloFlu_S6_N8_C3 | Avian |
| PhyloFlu_S6_N9_C0 | Avian |
| PhyloFlu_S6_N9_C1 | Avian |
| PhyloFlu_S7_M_C0 | NA |
| PhyloFlu_S7_M_C1 | Equine |
| PhyloFlu_S8_NS_C0 | Avian |
| PhyloFlu_S8_NS_C1 | NA |
An animal source was defined when ≥90% of the sequences of the clade were from a single host. Sources written in uppercase letters indicate the host represents at least 75% of all sequences identified; source in lowercase letters beside it represent the other significant host(s).
More than one animal source was defined.
NA, not assigned.
Other is defined as any other mammal from which influenza A virus has been collected; see the full list at http://www.ncbi.nlm.nih.gov/genomes/FLU/Database/nph-select.cgi.
The information obtained from the individual RNA segments was used to define the genomic fingerprint of the strain, i.e., the genomic pattern of the virus determined by the combined phylogenetic origin of all eight RNA segments (Fig. 3). This genomic pattern helps to trace the origins of the strains (see below), i.e., when and where strains with the same genomic fingerprint were reported, or if a strain represents a new, unreported genetic pattern for an influenza virus.
Microarray validation with reference viruses.
To validate the microarray, a set of 17 reference viruses was initially used. These strains represent 16 HA and 9 NA influenza A virus subtypes isolated from a variety of hosts, including human, horse, and domestic and wild birds. In silico hybridizations with all reference strains were first conducted to compare the theoretical performance of the microarray with the experimental results (see below). The sequences of the individual gene segments of each strain available at the NCBI nucleotide database were downloaded and aligned with the 70 mers included in the microarray. A result was scored as positive if the gene segment correctly paired with ≥60 of 70 nucleotides, and if the theoretical hybridization had a ΔG value of ≤−65 kcal/mol. For the microarray hybridization assay, the total RNA extracted from the influenza virus-infected cells was used for random RT-PCR amplification and Cy3 labeling.
The results generated in silico were compared with those obtained by microarray hybridization. In all cases, but one, the in silico results matched the hybridization experimental data (Table 2). The divergent result was resolved by nucleotide sequencing of the relevant region of the PA gene of the reference strain (see Table 2). The obtained sequence sequence revealed a divergence between the reported sequence used in the in silico assay (GenBank accession no. CY021962.1) and the actual sequence of the hybridized sample. This finding highlights the reliability of the results generated with the microarray assay.
The microarray correctly identified the HA and NA subtypes of all tested strains and also detected and classified the 6 internal segments of most strains, with the exception of segments 7 and 8 in some cases (Table 2). The lower detection efficiency of the small genomic RNA segments seems to be the result of a limited amount of viral RNA, since when the whole genome of the virus was amplified with specific primers (see Materials and Methods), this problem was less common. Of interest, among the viruses isolated from humans, the in silico assay detected four in which a reassortment had occurred. One of them (isolate A/Iowa/CEID23/05) shared its genotype with triple reassortant (H1N1) viruses that emerged in U.S. swine in the late 1990s following the emergence of related human/swine/avian triple reassortant H3N2 and H1N2 subtypes in American pigs (19). The other three reassortant viruses identified by the microarray were the product of a genetic reassortment between human seasonal H1N1 and swine triple reassortant virus strains; the HA and NA genes of strains A/Saskatchewan/5350/2009, A/Saskatchewan/5351/2009, and A/Saskatchewan/5131/2009 were derived from the human H1N1 virus, while the other six genes came from the triple reassortant strain (20).
Swine influenza A viruses.
A set of 8 viruses collected from Mexican farm pigs, adapted to grow in chicken embryos, was analyzed by PhyloFlu hybridization. In this case, as with the human and avian viruses described in the following sections, before random PCR amplification and microarray hybridization, the influenza virus genomes were amplified by RT-PCR using primers that specifically recognize the conserved 5′ and 3′ terminal regions of the eight viral segments.
Two different genomic patterns were found in the samples analyzed. The first pattern, represented by viruses Ver29, Ver31, Qro32, Ver37, and Mex52, had an H1N1 subtype specificity, while the genetic fingerprint of viruses Mich40, Mex50, and Mex51 belonged to subtype H3N2 (Table 5). The genomic patterns of strains Mich40 and Mex51 were only partially determined (6 and 7 genes classified, respectively), but the partial fingerprint was the same as that of virus Mex50. The genomic fingerprints determined by the microarray matched the genomic patterns of triple reassortant H1N1 and H3N2 viruses in our database. The swine H1N1 viruses from Mexico shared a genomic pattern with H1N1 triple reassortants reported between 1995 and 2009 in the United States (Table 6) (21, 22). Similarly, the H3N2 swine viruses had the same fingerprint as the H3N2 triple reassortant strains detected first in 1990 and then reported continuously until 2010 (23, 24) (Table 6). To our knowledge, this is the first description of triple reassortant viruses circulating in pigs in Mexico. Of interest, none of the swine isolates analyzed had the genomic pattern of the A/H1N1pdm09 virus.
TABLE 5.
Microarray hybridization of swine influenza A viruses
| Virus strain | No. of segments classified | Phylogenetic clades by genea: |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| PB2 | PB1 | PA | HA | NP | NA | M | NS | ||
| A/swine/Mexico/Ver29/2010 (H1N1) | 8 | 2 | 3 | 3 | H1-1 | 4 | N1-2 | 0 | 1 |
| A/swine/Mexico/Ver31/2010 (H1N1) | 8 | 2 | 3 | 3 | H1-1 | 4 | N1-NDb | 0 | 1 |
| A/swine/Mexico/Ver37/2010 (H1N1) | 8 | 2 | 3 | 3 | H1-1 | 4 | N1-2 | 0 | 1 |
| A/swine/Mexico/Qro32/2010 (H1N1) | 8 | 2 | 3 | 3 | H1-1 | 4 | N1-2 | 0 | 1 |
| A/swine/Mexico/Mex52/2010 (H1N1) | 8 | 2 | 3 | 3 | H1-1 | 4 | N1-2 | 0 | 1 |
| A/swine/Mexico/Mex50/2010 (H3N2) | 8 | 2 | 3 | 3 | H3-0 | 4 | N2-0 | 0 | 1 |
| A/swine/Mexico/Mex51/2010 (H3N2) | 7 | 2 | 3 | 3 | H3-0 | 4 | N2-0 | ND | 1 |
| A/swine/Mexico/Mich40/2010 (H3N2) | 6 | 2 | 3 | 3 | H3-0 | 4 | N2-0 | ND | ND |
Phylogenetic clades assigned to each influenza A virus strain by microarray hybridization.
ND, gene segments not detected or clades not defined.
TABLE 6.
Most frequent genomic patterns reported for swine influenza A viruses
| Virus straina | Reported yrb |
In silico genomic pattern by genec: |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| PB2 | PB1 | PA | HA | NP | NA | M | NS | ||
| A/swine/Minnesota/SG239/2007 (H1N2) | 2007-2010 | 2 | 3 | 3 | H1-0 | 4 | N2-0 | 0 | 1 |
| A/swine/Wisconsin/1/1957 (H1N1) | 1957, 1961, 1966-2009 | 2 | 0 | 0 | H1-1 | 4 | N1-2 | 0 | 1 |
| A/swine/Iowa/H03G1/2003 (H1N1)d | 1995-2009 | 2 | 3 | 3 | H1-1 | 4 | N1-2 | 0 | 1 |
| A/swine/Minnesota/074A/2009 (H1N1) | 2009-2010 | 2 | 3 | 3 | H1-1 | 4 | N1-3 | 0 | 1 |
| A/swine/Ehime/1/1980 (H1N2) | 1980, 1996, 1999-2006 | 2 | 0 | 0 | H1-1 | 4 | N2-0 | 0 | 1 |
| A/swine/France/WVL4/1985 (H1N1) | 1985-2009 | 1 | 3 | 3 | H1-3 | 0 | N1-3 | 0 | 1 |
| A/swine/Iowa/H02AS8/2002 (H3N2) | 1990-2010 | 2 | 3 | 3 | H3-0 | 4 | N2-0 | 0 | 1 |
| A/swine/Colorado/1/1977 (H3N2) | 1977, 1981, 1997-2006 | 2 | 3 | 4 | H3-0 | 4 | N2-0 | 0 | 1 |
| A/swine/Fujian/F1/2001 (H5N1) | 2001-2007 | 1 | 3 | 3 | H5-0 | 0 | N1-3 | 0 | 1 |
Representative strains for the particular genomic pattern shown.
Years when strains with the same genomic pattern were reported in the NCBI Influenza Virus Resource.
Genomic pattern determined for the different virus strains based on in silico assays.
Viruses with the same genomic fingerprint found in the tested samples from Mexico are shown in bold type.
Human virus strains.
Eight human influenza A viruses collected in Mexico between 2003 and 2009 from patients with acute respiratory infections were characterized with the microarray. The amplification was carried out either directly from nasopharyngeal swabs or from virus-infected cultured cells. Three genomic patterns were identified in these viruses, corresponding to seasonal A/H3N2 (six viruses), seasonal A/H1N1 (1 virus), and A/H1N1pdm09 (1 virus) viral strains (Table 7). The microarray clearly detected the different HA and NA virus subtypes of H1N1 and H3N2 viruses. Importantly, it also detected four clade differences between the genomic patterns of the seasonal and pandemic H1N1 virus strains, including the HA and NA genes, and two internal segments encoding proteins PB1 and PA (Table 7). A comparison of the genomic patterns of these influenza viruses with our database showed that all H3N2 strains share their genomic fingerprint with virus strain A/Hong Kong/1-10-MA21-1/1968 (H3N2), a strain from Hong Kong that caused the third pandemic of 20th century in 1968 and has circulated since then to this day. The pattern of the seasonal A/H1N1 virus was the same as that of the influenza virus strain first reported in 1933 (A/Wilson-Smith/33 [H1N1]) that circulated from 1933 to 1957 and from 1977 to 2009; the genomic fingerprint of the A/H1N1pdm09 virus is shared with the A/human/California/07/2009 (H1N1) virus strain (Table 8).
TABLE 7.
Microarray hybridization of human influenza A viruses
| Virus straina | No. of segments classified | Phylogenetic cladesb |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| PB2 | PB1 | PA | HA | NP | NA | M | NS | ||
| A/Mexico/DIF2662/2003 (H3N2) | 8 | 2 | 3 | 4 | H3-0 | 4 | N2-0 | 0 | 1 |
| A/Mexico/DIF2664/2003 (H3N2) | 7 | 2 | 3 | 4 | H3-0 | 4 | N2-0 | 0 | NDc |
| A/Mexico/Jal25216/2009 (H3N2) | 8 | 2 | 3 | 4 | H3-0 | 4 | N2-0 | 0 | 1 |
| A/Mexico/Ver1/2010 (H3N2) | 8 | 2 | 3 | 4 | H3-0 | 4 | N2-0 | 0 | 1 |
| A/Mexico/Mex33/2010 (H3N2) | 8 | 2 | 3 | 4 | H3-0 | 4 | N2-0 | 0 | 1 |
| A/Mexico/Jal121/2011 (H3N2) | 8 | 2 | 3 | 4 | H3-0 | 4 | N2-0 | 0 | 1 |
| A/Mexico City/IBT22/2009 (H1N1)pdm | 8 | 2 | 3 | 3 | H1-1 | 4 | N1-3 | 0 | 1 |
| A/Mexico/Ver2/2009 (H1N1) (seasonal) | 8 | 2 | 0 | 4 | H1-0 | 4 | N1-1 | 0 | 1 |
The A/H3N2 viruses were analyzed directly from clinic samples. The A/H1N1 strains were are adapted to grow in cell culture.
Phylogenetic clades assigned to each influenza A virus strain by microarray hybridization.
ND, not detected.
TABLE 8.
Most frequent genomic patterns reported for human influenza A viruses
| Virus straina | Reported yrb |
In silico genomic patternc |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| PB2 | PB1 | PA | HA | NP | NA | M | NS | ||
| A/Wilson-Smith/33 (H1N1)d | 1933-1957, 1977-2009 | 2 | 0 | 4 | H1-0 | 4 | N1-1 | 0 | 1 |
| A/human/California/07/2009 (H1N1) | 2009-2013 | 2 | 3 | 3 | H1-1 | 4 | N1-3 | 0 | 1 |
| A/Singapore/1/1957 (H2N2) | 1957-1968 | 2 | 3 | 4 | H2-0 | 4 | N2-0 | 0 | 1 |
| A/Hong Kong/1-10-MA21-1/1968 (H3N2) | 1968-2013 | 2 | 3 | 4 | H3--0 | 4 | N2-0 | 0 | 1 |
| A/Hong Kong/481/97 (H5N1) | 1997, 2003-2008, 2011 | 1 | 3 | 3 | H5-0 | 0 | N1-3 | 0 | 1 |
Representative strains for the particular genomic pattern shown.
Years when strains with the same genomic pattern were reported in the NCBI Influenza Virus Resource.
Genomic pattern determined for the different virus strains based on in silico assays.
Viruses with the same genomic fingerprint found in the tested samples from Mexico are shown in bold type.
Avian influenza virus field isolates.
Five avian field cloacal samples collected from migratory birds in Sonora, Mexico, from 2007 to 2009, were characterized with the microarray. Three complete and two partial genomic patterns were identified in these strains, each of them being unique, even for those viruses from which only a partial pattern was determined. The fingerprints differed in some internal genes and in their HA (H5, H6, H9, H10, and H11) and NA (N2, N3, N5) subtypes (Table 9). Compared to the genomic pattern database, the genetic fingerprints of these viruses were found to match patterns previously reported for viruses infecting waterfowl in California, Washington, Wisconsin, Ohio, and Alberta, Canada, from 1983 to 2007 (Table 10) (25). In mid-2012 and early 2013, two outbreaks of avian influenza virus A/H7N3 affected Mexican poultry farms. This strain was not found among the influenza viruses characterized from these migratory birds.
TABLE 9.
Microarray hybridization of avian influenza A viruses
| Virus strain | No. of segments classified | Phylogenetic clades by genea: |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| PB2 | PB1 | PA | HA | NP | NA | M | NS | ||
| A/Eurasian teal/Mexico/Son701/2008 (H11N3) | 8 | 2 | 3 | 3 | H11-(0/1)c | 0 | N3-0 | 0 | 1 |
| A/American widgeon/Mexico/Son769/2008 (H9N2) | 8 | 2 | 3 | 3 | H9-(0/1) | 0 | N2-1 | 0 | 1 |
| A/northern shoveler/Mexico/Son797/2008 (H5N3) | 6 | NDb | 3 | 3 | H5-1 | 0 | ND | 0 | 1 |
| A/Eurasian teal/Mexico/Son829/2009 (H6N5) | 8 | 2 | 3 | 3 | H6-0 | 0 | N5-0 | 0 | 1 |
| A/Eurasian teal/Mexico/Son1132/2009 (H10N3) | 6 | 2 | 3 | 3 | ND | 0 | ND | 0 | 0 |
Phylogenetic clades assigned to each influenza A virus strain by microarray hybridization.
ND, not detected.
In parentheses are clades that could not be resolved.
TABLE 10.
Genomic patterns of avian influenza A viruses
| Virus straina | Reported yrb |
In silico genomic pattern by genec: |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| PB2 | PB1 | PA | HA | NP | NA | M | NS | ||
| A/avian/mallard-duck/Alberta/797/1983 (H11N3) | 1983, 1986, 2006 | 2 | 3 | 3 | H11-0 | 0 | N3-0 | 0 | 1 |
| A/avian/mallard/Alberta/199/1992 (H6N5) | 1992, 2003, 2007 | 2 | 3 | 3 | H6-0 | 0 | N5-0 | 0 | 1 |
| A/chicken/Taiwan/A703-1/2008 (H5N2) | 2008 | 1 | 3 | 3 | H5-1 | 0 | N2-1 | 0 | 1 |
| A/turkey/CO/118899/1972 (H5N2) | 1972-2007 | 2 | 3 | 3 | H5-1 | 0 | N2-1 | 0 | 1 |
| A/avian/turkey/Wisconsin/1/1966 (H9N2) | 1966 | 2 | 3 | 3 | H9-0 | 0 | N2-1 | 0 | 1 |
| A/avian/mallard/Alberta/11/1991 (H9N2) | 1991, 2007 | 2 | 3 | 3 | H9-1 | 0 | N2-1 | 0 | 1 |
Representative strains for the particular genomic pattern shown.
Years when strains with the same genomic pattern were reported in the NCBI Influenza Virus Resource.
Genomic pattern determined for the different virus strains based on in silico assays. The patterns shown are only those related to the genomic fingerprints of the avian strains characterized in this work (shown in Table 5).
DISCUSSION
Several studies have reported the use of DNA microarrays for the detection or classification of influenza viruses (26–31). These microarrays were designed to detect specific influenza A virus strains (29, 30) or to subtype and pathotype the HA of avian influenza viruses (30) but not to determine both the HA and NA subtypes. One of those reports detected the virus at the species, hemagglutinin (HA) subtype, and gene segment levels (28). In this work, we developed and validated PhyloFlu, a new method designed to identify and classify the eight gene segments of the influenza A virus genome based on a DNA microarray and phylogenetic analysis. Each segment, including genes encoding each of the 16 HA and 9 NA subtypes, was classified into a clade that identified its phylogenetic origin. In most cases, the phylogenetic information allowed us to trace the origin of the segment to a virus infecting a particular animal host.
Even though DNA microarray analysis, such as the one reported in this work, and next-generation sequencing methods are both powerful tools for detecting influenza virus gene rearrangements, microarrays have the advantage of being faster and more cost-effective than high-throughput sequencing (32). Although these methodologies are being rapidly improved, especially in the case of massive sequencing, and the costs are being reduced, microarray currently seems to be the method of choice for the initial characterization of the genomic patterns of influenza-positive samples, while next-generation sequencing can be used as a complementary approach to characterize in more detail those influenza virus strains of particular interest.
The PhyloFlu microarray unambiguously determined 16 HA and 9 NA subtypes of a collection of virus reference strains, and it also performed well in the characterization of viruses present in different kinds of samples, including cultured cells, chicken embryo amniotic fluid, human nasopharyngeal swabs, and cloacal swab specimens. Most of the eight RNA segments were detected and classified into different clades when random primers were used for direct RT-PCR amplification of the total RNA extracted from the sample; however, the results were more consistent if the influenza virus genome was previously amplified with primers specific for the conserved 5′ and 3′ ends of the gene segments before random amplification and microarray hybridization. Using this last method, we were able to identify and classify the eight gene segments in 28 of 38 (74%) of the tested influenza-positive samples, and the phylogenetic origins of at least six segments were traced in all the samples evaluated. In samples in which the amount of genetic material was limited, the genomic pattern was only partially determined. In most cases, for those segments that were classified, a clear signal-to-background difference was observed in the fluorescence intensity values of the positive spots in the microarray (Fig. 4).
FIG 4.

Microarray hybridization specificity. Shown are the signal and background fluorescence intensities for the hybridization spots defining the different clades (numbers on top of bars) of each gene segment. The names of the strains used to illustrate the hybridization pattern for the reference (A), swine (B), human (C), and avian (D) influenza viruses are indicated. The clade assigned for each segment of the four strains is shown. Gray bars, signal; black bars, background.
In addition to identifying the phylogenetic origin of each individual segment of a virus in a single assay, the combination of the phylogenetic information for the segments provides a genomic fingerprint of the strain. This fingerprint can then be used to investigate whether strains with the same genomic pattern have circulated previously in human, swine, or bird populations, and when and where they were reported. The potential of the microarray to detect reassortment events was shown by its ability to disclose the gene reassortments that originated the influenza virus A/H1N1pdm09 strain. In this case, a change of clade was clearly detected in four segments of the pandemic 2009 strain compared to seasonal A/H1N1 viruses. Of note, the HA and NA subtypes of the reassortant A/H1N1pdm09 virus were the same as those of the A/H1N1 seasonal strains circulating at that time, but the subtype genes of these strains were readily differentiated into different clades by the microarray. The assay also efficiently detected the circulation of triple reassortant viruses in swine, as well as a variety of HA and NA subtypes in migratory birds from northern Mexico that had not been previously reported.
The assay should be useful for screening and characterizing environmental influenza virus niches, such as wild migratory and nonmigratory birds, poultry, swine farms, and backyard pigs, to determine the genetic pool of the circulating strains. PhyloFlu should also be useful for maintaining an epidemiological surveillance of influenza viruses in humans to detect reassortment events that imply a change in clade for any of the eight viral genes, even if the HA and NA subtypes remain the same. This type of surveillance should allow us to assess more carefully the genetic diversity and variation of circulating viruses and investigate potential implications for changes in virus pathogenicity.
ACKNOWLEDGMENTS
This work was partially supported by grants S0008-126823 and 2008-87691 from CONACyT and grant IN213410 from DGAPA, UNAM. L.F.P. is a recipient of a scholarship from CONACyT.
Footnotes
Published ahead of print 18 December 2013
REFERENCES
- 1.Bouvier NM, Palese P. 2008. The biology of influenza viruses. Vaccine 26:D49–D53. 10.1016/j.vaccine.2008.07.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Centers for Disease Control and Prevention 2013. Influenza type A viruses and subtypes. CDC, Atlanta, GA: http://www.cdc.gov/flu/avianflu/influenza-a-virus-subtypes.htm [Google Scholar]
- 3.Tong S, Li Y, Rivailler P, Conrardy C, Castillo DA, Chen LM, Recuenco S, Ellison JA, Davis CT, York IA, Turmelle AS, Moran D, Rogers S, Shi M, Tao Y, Weil MR, Tang K, Rowe LA, Sammons S, Xu X, Frace M, Lindblade KA, Cox NJ, Anderson LJ, Rupprecht CE, Donis RO. 2012. A distinct lineage of influenza A virus from bats. Proc. Natl. Acad. Sci. U. S. A. 109:4269–4274. 10.1073/pnas.1116200109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tong S, Zhu X, Li Y, Shi M, Zhang J, Bourgeois M, Yang H, Chen X, Recuenco S, Gomez J, Chen LM, Johnson A, Tao Y, Dreyfus C, Yu W, McBride R, Carney PJ, Gilbert AT, Chang J, Guo Z, Davis CT, Paulson JC, Stevens J, Rupprecht CE, Holmes EC, Wilson IA, Donis RO. 2013. New World bats harbor diverse influenza A viruses. PLoS Pathog. 9:e1003657. 10.1371/journal.ppat.1003657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wright P, Neumann G, Kawaoka Y. 2013. Orthomyxoviruses, p 1186–1234 In Knipe D, Howley P, (ed) Fields virology, 6th ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 6.Huang CH, Chen CJ, Yen CT, Yu CP, Huang PN, Kuo RL, Lin SJ, Chang CK, Shih SR. 2013. Caspase-1 deficient mice are more susceptible to influenza A virus infection with PA variation. J. Infect. Dis. 10.1093/infdis/jit381 [DOI] [PubMed] [Google Scholar]
- 7.Zhang Y, Zhang Q, Kong H, Jiang Y, Gao Y, Deng G, Shi J, Tian G, Liu L, Liu J, Guan Y, Bu Z, Chen H. 2013. H5N1 hybrid viruses bearing 2009/H1N1 virus genes transmit in guinea pigs by respiratory droplet. Science 340:1459–1463. 10.1126/science.1229455 [DOI] [PubMed] [Google Scholar]
- 8.Pappas C, Aguilar PV, Basler CF, Solórzano A, Zeng H, Perrone LA, Palese P, García-Sastre A, Katz JM, Tumpey TM. 2008. Single gene reassortants identify a critical role for PB1, HA, and NA in the high virulence of the 1918 pandemic influenza virus. Proc. Natl. Acad. Sci. U. S. A. 105:3064–3069. 10.1073/pnas.0711815105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith GJD, Vijaykrishna D, Bahl J, Lycett SJ, Worobey M, Pybus OG, Ma SK, Cheung CL, Raghwani J, Bhatt S, Peiris JS, Guan Y, Rambaut A. 2009. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 459:1122–1125. 10.1038/nature08182 [DOI] [PubMed] [Google Scholar]
- 10.Bao Y, Bolotov P, Dernovoy D, Kiryutin B, Zaslavsky L, Tatusova T, Ostell J, Lipman D. 2008. The influenza virus resource at the National Center for Biotechnology Information. J. Virol. 82:596–601. 10.1128/JVI.02005-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797. 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59:307–321. 10.1093/sysbio/syq010 [DOI] [PubMed] [Google Scholar]
- 13.Li W, Godzik A. 2006. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22:1658–1659. 10.1093/bioinformatics/btl158 [DOI] [PubMed] [Google Scholar]
- 14.Weckx S, Carlon E, DeVuyst L, Van Hummelen P. 2007. Thermodynamic behavior of short oligonucleotides in microarray hybridizations can be described using Gibbs free energy in a nearest-neighbor model. J. Phys. Chem. B. 111:13583–13590. 10.1021/jp075197x [DOI] [PubMed] [Google Scholar]
- 15.Mount DW. 2007. Using the Basic Local Alignment Search Tool (BLAST). Cold Spring Harb. Protoc. 2007:pdb.top17. 10.1101/pdb.top17 [DOI] [PubMed] [Google Scholar]
- 16.Urisman A, Fischer KF, Chiu CY, Kistler AL, Beck S, Wang D, DeRisi JL. 2005. E-Predict: a computational strategy for species identification based on observed DNA microarray hybridization patterns. Genome Biol. 6:R78. 10.1186/gb-2005-6-9-r78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou B, Donnelly ME, Scholes DT, St. George K, Hatta M, Kawaoka Y, Wentworth DE. 2009. Single-reaction genomic amplification accelerates sequencing and vaccine production for classical and Swine origin human influenza A viruses. J. Virol. 83:10309–10313. 10.1128/JVI.01109-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.R Development Core Team 2012. R: a language and environment for statistical computing: reference index. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org [Google Scholar]
- 19.Gray GC, McCarthy T, Capuano AW, Setterquist SF, Olsen CW, Alavanja MC, Lynch CF. 2007. Swine workers and swine influenza virus infections. Emerg. Infect. Dis. 13:1871–1878. 10.3201/eid1312.061323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bastien N, Antonishyn NA, Brandt K, Wong CE, Chokani K, Vegh N, Horsman GB, Tyler S, Graham MR, Plummer FA, Levett PN, Li Y. 2010. Human infection with a triple-reassortant swine influenza A(H1N1) virus containing the hemagglutinin and neuraminidase genes of seasonal influenza virus. J. Infect. Dis. 201:1178–1182. 10.1086/651507 [DOI] [PubMed] [Google Scholar]
- 21.Barman S, Krylov PS, Fabrizio TP, Franks J, Turner JC, Seiler P, Wang D, Rehg JE, Erickson GA, Gramer M, Webster RG, Webby RJ. 2012. Pathogenicity and transmissibility of North American triple reassortant swine influenza A viruses in ferrets. PLoS Pathog. 8:e1002791. 10.1371/journal.ppat.1002791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Terebuh P, Olsen CW, Wright J, Klimov A, Karasin A, Todd K, Zhou H, Hall H, Xu X, Kniffen T, Madsen D, Garten R, Bridges CB. 2010. Transmission of influenza A viruses between pigs and people, Iowa, 2002–2004. Influenza Other Respir. Viruses. 4:387–396. 10.1111/j.1750-2659.2010.00175.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Webby RJ, Swenson SL, Krauss SL, Gerrish PJ, Goyal SM, Webster RG. 2000. Evolution of swine H3N2 influenza viruses in the United States. J. Virol. 74:8243–8251. 10.1128/JVI.74.18.8243-8251.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumar SR, Deflube L, Biswas M, Shobana R, Elankumaran S. 2011. Genetic characterization of swine influenza viruses (H3N2) isolated from Minnesota in 2006–2007. Virus Genes 43:161–176. 10.1007/s11262-011-0618-4 [DOI] [PubMed] [Google Scholar]
- 25.Obenauer JC, Denson J, Mehta PK, Su X, Mukatira S, Finkelstein DB, Xu X, Wang J, Ma J, Fan Y, Rakestraw KM, Webster RG, Hoffmann E, Krauss S, Zheng J, Zhang Z, Naeve CW. 2006. Large-scale sequence analysis of avian influenza isolates. Science 311:1576–1580. 10.1126/science.1121586 [DOI] [PubMed] [Google Scholar]
- 26.Li J, Chen S, Evans DH. 2001. Typing and subtyping influenza virus using DNA microarrays and multiplex reverse transcriptase PCR. J. Clin. Microbiol. 39:696–704. 10.1128/JCM.39.2.696-704.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang D, Coscoy L, Zylberberg M, Avila PC, Boushey HA, Ganem D, DeRisi JL. 2002. Microarray-based detection and genotyping of viral pathogens. Proc. Natl. Acad. Sci. U. S. A. 99:15687–15692. 10.1073/pnas.242579699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sengupta S, Onodera K, Lai A, Melcher U. 2003. Molecular detection and identification of influenza viruses by oligonucleotide microarray hybridization. J. Clin. Microbiol. 41:4542–4550. 10.1128/JCM.41.10.4542-4550.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Townsend MB, Dawson ED, Mehlmann M, Smagala JA, Dankbar DM, Moore CL, Smith CB, Cox NJ, Kuchta RD, Rowlen KL. 2006. Experimental evaluation of the FluChip diagnostic microarray for influenza virus surveillance. J. Clin. Microbiol. 44:2863–2871. 10.1128/JCM.00134-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gall A, Hoffmann B, Harder T, Grund C, Höper D, Beer M. 2009. Design and validation of a microarray for detection, hemagglutinin subtyping, and pathotyping of avian influenza viruses. J. Clin. Microbiol. 47:327–334. 10.1128/JCM.01330-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gardner SN, Jaing CJ, McLoughlin KS, Slezak TR. 2010. A microbial detection array (MDA) for viral and bacterial detection. BMC Genomics 11:668. 10.1186/1471-2164-11-668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tang P, Chiu C. 2010. Metagenomics for the discovery of novel human viruses. Future Microbiol. 5:177–189. 10.2217/fmb.09.120 [DOI] [PubMed] [Google Scholar]