

Abstract
Intimal thickening is an early phase of atherosclerosis characterized by differentiation of plaque smooth muscle cells (SMCs) from a contractile to a synthetic phenotype. We used laser microdissection (LMD) plus real-time RT-PCR to quantify mRNAs for calponin-1 and smoothelin, markers of the contractile phenotype, and for serum response factor (SRF), a regulator of SMC differentiation, in intimal and medial SMCs of human coronary arteries with intimal thickening. RNA expression was also analyzed by ISH and protein expression was detected by IHC. LMD plus RT-PCR found similar levels of SRF mRNA in intimal and medial SMCs, while medial mRNA levels for calponin-1 and smoothelin were higher. ISH confirmed that smoothelin mRNA levels in media exceeded those in intima, whereas SRF mRNA levels were similar at both sites. For calponin-1 and smoothelin, protein levels mirrored respective mRNA levels. By contrast, more medial than intimal SRF protein was present. Our results indicate that intimal SMCs exhibit a largely synthetic phenotype, perhaps reflecting lower intimal levels of SRF protein; ISH and LMD plus real-time RT-PCR provide comparable results; as a valuable alternative to ISH, LMD plus RT-PCR allows parallel measurement of several transcripts; and tissue gene expression studies must measure both protein and mRNA levels.
Keywords: laser microdissection, real-time RT-PCR, smoothelin, calponin-1, serum response factor
In Western countries, atherosclerosis and its complications, myocardial infarction and stroke, are among the most common causes of death (WHO 1999). The process by which atherosclerosis develops is not fully understood and would benefit from an improved knowledge of levels of gene expression in arterial tissue. For this reason, much work has been devoted to the analysis of mRNA and/or protein expression in the arterial wall (see, e.g., Rolph et al. 2002). Up to now, ISH and IHC have mainly been used for gene expression analysis in tissues (see, e.g., Salame et al. 2000; Hilfiker et al. 2002; Kind 2002). However, these methods are not suitable for measurement of absolute levels of gene expression (Kind 2002). Quantification of ISH, for example, requires a time-consuming approach using radioactively labeled probes (Ky and Shughrue 2002), and measurement of protein abundance using IHC is demanding and semiquantitative at best (Burke et al. 2002).
In 1982, laser microdissection (LMD) was introduced. This method uses laser light to selectively isolate parts of a tissue, single cells, or even cellular components such as chromosomes (Burgemeister and Schütze 2002). Such dissected samples can be used to provide, e.g., total RNA that can then be used for absolute or relative quantitative analysis of mRNA expression using real-time RT-PCR or DNA microarrays (Lehmann et al. 2000; Alevizos et al. 2001; Rubin 2001; Burgemeister and Schütze 2002; Trogan et al. 2002).
Although technically demanding, LMD can be used to analyze single cells and homogeneous parts of a tissue (Burgemeister and Schütze 2002). New mRNA targets can be quickly measured using real-time RT-PCR. By contrast, ISH often requires time-consuming optimization when new mRNA targets are investigated. Furthermore, LMD allows fast and semiautomatic sample preparation, in contrast to the time-consuming procedure of ISH, for which automation is difficult (Kind 2002).
To identify the best means for quantifying gene expression in tissues, we compared the combination of LMD plus real-time RT-PCR to ISH in the analysis of gene (mRNA) expression in the smooth muscle cells (SMCs) of human coronary arteries with intimal thickening, a well-characterized early phase of atherosclerosis (Virmani et al. 2000). In general, for many gene products, a poor correlation between mRNA and protein levels has been found (Goodlett and Aebersold 2002). We therefore additionally used IHC to estimate gene expression at the protein level.
We chose smoothelin and calponin-1 for the present study because both of these gene products are well-established markers of the quiescent or contractile phenotype that characterizes SMCs of the arterial media (van der Loop et al. 1996; Sobue et al. 1999). In addition, we analyzed expression of serum response factor (SRF) mRNA and protein, a gene product known to regulate calponin-1 expression in vitro (Sobue et al. 1999; Miano et al. 2000).
Materials and Methods
Tissue Preparation
Human coronary arteries were obtained at the Papworth Hospital (Cambridge, UK) from hearts explanted during heart transplantation for advanced coronary heart disease as part of a tissue bank of human coronary arteries established by the MAFAPS consortium (Bellosta et al. 2002; Brinck et al. 2003). The explantation procedure was approved by the local Ethics Committee of Papworth Hospital. The arteries were cut into ~1 cm sections and snap-frozen in liquid nitrogen-cooled isopentane within minutes after explantation. Thereafter, coronary arteries were embedded in GSV tissue embedding medium (Slee Technik GmbH; Mainz, Germany) and stored at −80C until use. The grade of atherosclerosis of each sample was characterized and classified using histochemistry and immunohistology according to Virmani et al. (2000). Arteries showing intimal thickening and containing few or no macrophages in the intima or media were chosen to avoid falsification of results by contaminating cells. For this purpose, serial sections were stained with a monoclonal antibody (MAb) directed to the macrophage-specific marker CD68 (DAKO; Glostrup, Denmark) and with the MAb 25F9, which is specific for differentiated macrophages (BMA Biomedicals; Augst, Switzerland). For LMD, cryosections ~10 μm thick were fixed on polyethylene-naphthalene membrane-mounted slides with 70% (v/v) ethanol according to the manufacturer's instructions (PALM Technologies; Bernried, Germany). For ISH and IHC, cryosections ~6 μm thick were fixed on Superfros glass slides (Gerhard Menzel Glasbearbeitungswerk; Braunschweig, Germany).
Laser Microdissection
For LMD, the PALM (positioning and ablation with laser microbeams) Robot MicroBeam device (PALM Technologies) was used according to the manufacturer's instructions. To isolate total RNA from microdissected cells, we fixed arterial sections of 10 μm thickness with 70% (v/v) ethanol for 5 min and washed them three times with diethylpyrocarbonate (DEPC)-treated water for 5 min to remove embedding medium. Sections were finally washed with 70% (v/v), 90% (v/v), and absolute ethanol each time for 2 min and dried at room temperature. Next, endothelial cells and the elastica interna were removed by microdissection before dissection of SMCs from the media and intima of each section. SMCs were dissected from the arterial intima and media and the microdissected tissue was collected in a microcentrifuge cap. We localized the regions of tissue containing SMCs by staining serial sections with anti-myosin (smooth) MAbs specific for the smooth muscle myosin heavy chains SM-1 and SM-2 (clone HSM-V; Sigma, Darmstadt, Germany) using the protocol described below.
RNA Extraction
We isolated total RNA using the RNA Microprep kit (Stratagene; La Jolla, CA) according to the manufacturer's instructions. This kit allows isolation of total RNA from 10-50 ng of tissue, which corresponds to ~5 × 105 dissected cells. After lysis of ~5 × 105 dissected cells, the lysate was applied to the silica-based fiber matrix that is housed in the Stratagene's RNA Microprep spin cup to bind the RNA during DNase I treatment, washing, and removal steps. With an elution volume of 30 μl, the resulting RNA was sufficiently concentrated to allow synthesis of cDNA and RT-PCR.
RNA Quantification
RNA concentrations were measured using the RiboGreen RNA Quantitation kit (Molecular Probes; Eugene, OR) with a Fluorolite 1000 microplate reader (Dynatech Laboratories; Germantown, MD) and a commercial external RNA standard (Molecular Probes). The dye was diluted 2000-fold in 1 TE buffer (10 mM Tris-HCl, pH 7.5, 1 mM EDTA). The external RNA standard was diluted in 1 TE buffer to create a series of standards with known concentrations, and RNA samples of interest were also diluted in 1 TE buffer. A blank containing only the fluorescent dye and 1 TE buffer was also prepared. Equal volumes (100 μl) of dye in 1 TE buffer and 1 TE buffer containing the nucleic acids were mixed and added to individual flat-bottomed microplate wells. Standards and nucleic acid samples were prepared by pipetting and incubated for 5 min at room temperature while being protected from light. The microplate reader was set to scan fluorescence using filters with 485-nm excitation and 530-nm emission. The results were analyzed using the Fluorolite 1000 software (Dynatech Laboratories). A blank consisting of 1 TE buffer plus fluorescent dye was used for global background subtraction.
Reverse Transcription
We performed RT with 10-50 ng of total RNA in a total reaction volume of 30 μl, containing oligo(dT) primer (Invitrogen; Karlsruhe, Germany), the four deoxyribonucleotide triphosphates (2.5 mM each; MBI Fermentas, Heidelberg, Germany), 20 U of RNase inhibitor (Promega; Mannheim, Germany), 200 U of Superscript II reverse transcriptase (Invitrogen), and first-strand synthesis buffer. The RNA was incubated at 70C for 10 min to remove any secondary structures and was then immediately placed on ice before preparation of the reaction mixture. Synthesis of cDNA was performed at 42C for 1 hr with a final step at 75C for 10 min to inactivate the enzyme, as previously described (Lorkowski et al. 2001A, B). Aliquots of cDNA were stored at −20C or were immediately used for real-time RT-PCR. Controls without reverse transcriptase were performed using the same protocol to ensure absence of contaminating DNA.
Primer Design
Primers for real-time RT-PCR were designed using the Gen-Bank entries listed in Table 1 and the Primer Express v2.0.0 software (Applied Biosystems; Weiterstadt, Germany) using standard parameters with a few more stringent modifications: optimal melting temperature of primers 60C; maximal difference in melting temperature of primers 2C; GC content between 30 and 80%; no 3′-GC clamp; optimal primer length 20 bp; maximal base repeat 2; melting temperature of amplicon between 75C and 85C; amplicon size between 80 and 150 bp. Primers were from Invitrogen or MWG Biotech (Ebersberg, Germany) and the primer concentrations used in real-time RT-PCR were optimized according to the instructions given by Applied Biosystems using cDNA derived from total RNA isolated from cultured SMCs as described below.
Table 1.
Primers used in real-time RT-PCR
|
| ||||
|---|---|---|---|---|
| Primer | GenBank entry | Orientation | Sequence | Product size |
|
| ||||
| ARHA | NM_001664 | Forward | 5′-CAC GCC TCA GGC AAT TAG ACA-3′ | 140 bp |
| Reverse | 5′-TGG GAG GGA ACC TGG ATA CAG-3′ | |||
| Calponin-1 | NM_001299 | Forward | 5′-GAA CAT CGG CAA CTT CAT CA-3′ | 105 bp |
| Reverse | 5′-TGC ACC TGT GTA TGG TTG GT-3′ | |||
| GAPDH | NM_002046 | Forward | 5′-GTC AGT GGT GGA CCT GAC CT-3′ | 123 bp |
| Reverse | 5′-ACC TGG TGC TCA GTG TAG CC-3′ | |||
| ROCK1 | NM_005406 | Forward | 5′-CCA AAA TCA CAA AGG CCA TGA-3′ | 116 bp |
| Reverse | 5′-TGT GGC ACT TAA CAT GGC ATC T-3′ | |||
| Smoothelin | NM_134270 | Forward | 5′-TTG GAC AAG ATG CTG GAT CA-3′ | 95 bp |
| Reverse | 5′-CGC TGG TCT CTC TTC CTT TG-3′ | |||
| SRF | NM_003131 | Forward | 5′-CTT CCT CTC CTT CCC ACA AAA G-3′ | 120 bp |
| Reverse | 5′-TCA CCT CAA CTC ATC CCT GAC A-3′ | |||
|
| ||||
Real-time RT-PCR
We performed real-time RT-PCR using a 384-well ABI PRISM 7900 HT Sequence Detection System (Applied Biosystems) and the QuantiTect SYBR Green PCR kit (Qiagen; Hilden, Germany). Cycling parameters were as follows: initial denaturation at 95C for 10 min, followed by 40 cycles comprising 15 sec at 95C and 60 sec at 60C. The reaction included 2.25 μl of non-diluted cDNA (corresponds to 1.5 to 3.75 ng of total RNA) for samples, using the 2 × QuantiTect SYBR Green PCR Master Mix (Qiagen) and 200 nM of each forward and reverse primer. The forward and reverse primers used for real-time RT-PCR are shown in Table 1. All samples were assayed in duplicate and analyzed using the Sequence Detection System Software v2.0 (Applied Biosystems). After PCR amplification, we heated the samples from 60C to 95C to melt the PCR products so as to check their homogeneity. In addition, we checked the PCR products by gel electrophoresis on 2% (w/v) agarose gels using standard methods. Negative controls without reverse transcription were performed to confirm sufficient removal of contaminating DNA. In addition, controls containing all constituents but the template in the reaction mixture were performed to rule out contamination of reagents. We performed relative mRNA quantification using the standard curve method (user bulletin no. 2; Applied Biosystems 2001) using a dilution row of ten serial 1:5 dilutions of cDNA for calculating PCR efficiencies of each primer pair in all real-time RT-PCR experiments. The cDNA derived from total RNA isolated from vascular human SMCs was cultured to 80% confluence in medium supplemented with 5% (v/v) fetal calf serum (FCS), followed by 72 hr culture in medium supplemented with 1% (v/v) FCS. PCR efficiencies were taken into account for calculating fold changes of mRNA expression. Fold changes were normalized to the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
DNA Sequencing
To check the identity of amplicons, we sequenced all PCR products directly using the Big Dye Terminator Cycle Sequencing kit (Applied Biosystems) and the primers shown in Table 1 (Invitrogen) on an ABI Prism 3700 DNA sequencer or an ABI Prism 310 DNA sequencer (both Applied Biosystems). Reactions were performed using the Big Dye 2.0 Sequencing Reagent (Applied Biosystems) in a PTC-200 thermal cycler (MJ Research; Watertown, MA) using 300 ng DNA per 3200 bp × DNA length and the following temperature profile: 25 cycles of 96C for 10 sec, 50C for 5 sec, and 60C for 4 min, and a final hold at 4C. Products of the sequencing reaction were purified using the Sephadex system (Amersham Biosciences; Uppsala, Sweden) before electrophoretic separation and detection.
Immunohistochemical Staining
We fixed arterial cryosections mounted on glass slides with 4% (w/v) paraformaldehyde as previously published (Weitkamp et al. 1999). After washing with PBS, we treated the tissue with 0.05% (v/v) Tween in PBS. Sections were blocked for 30 min with 1% (w/v) BSA in PBS at room temperature and were then incubated with primary MAbs directed toward smoothelin (1:500; Chemicon International, Temecula, CA), calponin-1 (1:100; Sigma), SRF (1:100; Neo Markers, Fremont, CA), smooth muscle myosin heavy chains (1:400; SMC 1552, Sigma), von Willebrand factor (1:800; DAKO), CD68 (1:50; DAKO), and/or 25F9 (1:50; BMA Biomedicals) for 1 hr. We then washed the slides, incubated them for 1 hr with Cy2- or Cy3-labeled secondary antibodies (both 1:50; both from Jackson ImmunoResearch Laboratories, West Grove, PA) in 1% (w/v) BSA in 0.05% (v/v) Tween in PBS, and washed them again. Cell nuclei were stained with Hoechst Dye 33,258 (Hoechst; Frankfurt, Germany). To ensure specificity of the antibodies, we incubated blocked tissue sections with IgG1 control antibodies (DAKO) in 1% (w/v) BSA and 0.05% Tween in PBS (pH 7.3) instead of primary antibodies, using the same dilutions as used for the specific antibodies listed above. Stainings were visualized using an Axioplan 2 microscope (Zeiss; Jena, Germany), the Axiophot camera system (Zeiss), or a digital camera system (Color View; Soft Imaging System, Münster, Germany).
In Situ Hybridization
The protocols for ISH were adapted from Plenz et al. (1997). Antisense and sense (control) RNA probes were synthesized from linearized pCRII plasmids (Invitrogen) containing either a 450-bp fragment of SRF cDNA or a 512-bp fragment of smoothelin cDNA. SP6 or T7 polymerase-mediated in vitro transcription was performed to label the probes with digoxigenin using the DIG RNA Labeling kit (Roche Diagnostics; Mannheim, Germany) and an rNTP mixture containing 3.5 mM digoxigenin-labeled rUTP, 6.5 mM rUTP, 10 mM rGTP, 10 mM rCTP, and 10 mM rATP. Cryostat sections 6 μm thick were collected from transplanted arteries and were mounted on RNase-free Superfrost microscope slides. Sections were washed with ice-cold 70% (v/v) ethanol for 5 min and then three times with DEPC-treated water for 5 min each time to avoid accumulation of probes in the embedding medium or in greasy tissue. Tissue sections were briefly dried at 50C and for half an hour at room temperature. The dried slides were processed as follows under RNase-free conditions (if not otherwise mentioned, all procedures were carried out at room temperature): tissues were fixed in 4% (w/v) paraformaldehyde (pH 7.4) for 10 min. After fixation, sections were washed three times in PBS (pH 7.4) for 5 min and permeabilized with 5 μg/ml proteinase K (MBI Fermentas) in 1 TE buffer for 7 min at 37C. Permeabilization was stopped by incubation with 2 mg/ml glycine in PBS for 30 sec. After equilibration in triethanolamine buffer (100 mM, pH 8.0), the slides were acetylated with freshly made 100 mM triethanolamine supplemented with 0.25% (v/v) acetic anhydride. Slides were maintained in this solution for 10 min to acetylate free amino groups and avoid unspecific binding of RNA probes with proteins. Tissue sections were then washed twice in twofold SSC for 5 min and dehydrated by washing in a graded series of ethanol solutions [50% (v/v), 70% (v/v), 90% (v/v)] and absolute ethanol at room temperature for 2 min each time. After dehydration, sections were prehybridized at 50C in a hybridization chamber saturated with 50% (v/v) formamide in DEPC-treated water using a hybridization buffer composed of 50% (v/v) deionized formamide (Roth; Karlsruhe, Germany), 2 mg/ml herring sperm DNA, 200 μg/ml yeast tRNA, 10 mM DTT, 1 mg/ml BSA, and twofold SSC (all from Sigma). Labeled RNA probes were heated at 80C for 3 min and quickly cooled on ice to stabilize the single-stranded probes and were diluted in preheated hybridization buffer (50C) to a final concentration of 0.3 μg/ml. Hybridization followed overnight with probes (sense or antisense) of smoothelin or SRF. Slides were incubated without coverslips in the saturated hybridization chamber at 50C as described above. After hybridization, the slides were washed twice with 2 × SSC and 0.2 × SSC at 50C for 15 min each time. After buffer changing to 0.05% (v/v) Tween in TBS (pH 8.0) at room temperature for 5 min, slides were blocked for 1 hr in blocking buffer [1% (w/v) BSA in 0.05% (v/v) Tween in TBS]. Incubation for 30 min with 1 μl/ml alkaline phosphatase-labeled anti-digoxigenin Fab fragments (Roche) in blocking buffer followed. After washing twice in 0.3% (v/v) Tween in TBS for 5 min, slides were preincubated in a Tris/MgCl2 buffer (100 mM Tris, 100 mM NaCl, 50 mM MgCl2, pH 9.5). Hybridization of probes was visualized by the alkaline phosphatase-catalyzed formation of a chromogenic complex using nitroblue tetrazolium-chloride (NBT) and 5-bromo-4-chloro-3-indolyl-phosphate toluidine sal (X-phosphate) from Biomol (Hamburg, Germany). For this, slides were incubated in a staining solution composed of 335 μg/ml NBT, 174 μg/ml X-phosphate, and 240 μg/ml levamisole in Tris/MgCl2 buffer. Staining was performed under microscopic control and was stopped with DEPC-treated water after 30 min to 2 hr.
Results
Quantification of Contaminating Cells
Serial arterial sections were microdissected and used to estimate the number of macrophages contaminating the SMCs. Macrophages were counted after staining the sections with an MAb directed to the macrophagespecific marker CD68 and with the MAb 25F9, which is specific for differentiated macrophages. This revealed that, on average, fewer than five macrophage cells per 1000 intimal SMCs were present in the sections used for microdissection, and no macrophages were detected in the media. Therefore, the contamination of dissected intimal SMCs with macrophages is likely to have had a negligible effect on our results.
Tissue Preparation and Establishment of Laser Microdissection
For LMD, the PALM Robot MicroBeam device was used. This system allows contact-free cell-specific sample isolation and makes use of a 337-nm nitrogen laser so that no damage to biological matter occurs due to absorption of laser light and warming of the sample is avoided. However, in a first step we tested the suitability of different protocols for preparing tissue samples for LMD-based real-time RT-PCR analysis. We compared the protocol for removing the embedding medium, as described in Materials and Methods, with protocols using acetone or xylol instead of ethanol. Tissue samples washed and fixed with acetone revealed less RNA than samples washed and fixed with ethanol. Xylol-washed and -fixed samples revealed similar yields and quality of RNA as obtained by the ethanolbased protocol, but samples washed and fixed with ethanol achieved the most reproducible yields of high-quality RNA. In our hands, quantification of RNA isolated from microdissected samples using the Ribo-Green RNA Quantitation kit was helpful for establishing the combined protocol of laser microdissection and real-time RT-PCR but was not mandatory for performing the quantification assays. Using no more than ten sections at once for LMD, isolation of cells from each section revealed comparable amounts of RNA such that the number of microdissected cells was sufficient for estimating the amount of RNA for each PCR.
Establishment of Real-time RT-PCR Assays
We established real-time RT-PCR assays for the mRNAs of RhoA GTPase (ARHA, member A of the Ras homologue gene family), Rho-associated coiled coil-containing protein kinase 1 (ROCK1), smoothelin, calponin-1, SRF, and the housekeeping gene GAPDH. Serial dilutions (1:5) of cDNA derived from total RNA isolated from human coronary SMCs that were cultured as described in Materials and Methods were used to determine the efficiency of each PCR primer pair according to the protocol published by Applied Biosystems (user bulletin no. 2). For each target, PCR efficiency was around 85% (data not shown). Therefore, PCR efficiencies were taken into account during calculation of fold changes in mRNA expression according to the protocol provided by Applied Biosystems. To check the identity of the amplicons, we sequenced all PCR products and compared the sequence obtained with the GenBank database. In all cases, sequence identity compared with the corresponding database entries for ARHA, ROCK1, calponin-1, smoothelin, SRF, and GAPDH was greater than 99%. Furthermore, we ensured that CT values of samples derived from coronary tissues were in the linear range of CT values of the diluted cDNA standard. We tested the suitability of GAPDH as a housekeeping gene by ensuring that the expression of GAPDH mRNA was constant in coronary SMCs cultured under various conditions. In all cases, SMCs were first cultured in medium containing 5% (v/v) FCS to 80% confluence and then maintained for 2 days at 1% (v/v) FCS. Cells were then cultured for 1 day in medium supplemented with 1% (v/v) FCS, in medium cultured with 10% (v/v) FCS, or under treatment with 10 ng/ml transforming growth factor-beta1 (TGFβ1) in medium containing 1% (v/v) FCS.
Contaminating DNA
Although RNA samples were treated with DNase I to remove contaminating DNA, we ensured that the samples were free of genomic DNA. Using RNA samples that were subjected to a reverse transcription according to the procedure described in Materials and Methods but lacking the reverse transcriptase, we found almost no contamination.
Real-time RT-PCR Results
Expression of ARHA, ROCK1, smoothelin, calponin-1, and SRF was analyzed at the RNA level in arteries derived from three explanted human hearts. Total RNA was isolated from SMCs dissected from the arterial intima and from the arterial media of three arteries. Differences in mRNA expression levels were calculated as fold changes and normalized to the expression of the housekeeping gene GAPDH. cDNA of three different samples derived from three individuals was synthesized and measured at least two times individually, and the analysis of each sample was performed in duplicate. Compared with the intima, the level of smoothelin mRNA was 4.3 ± 1.0-fold (mean ± SD; p<0.0005) greater in the media, while the level of calponin-1 mRNA was 2.9 ± 1.1-fold greater in the media compared with the intima (p<0.25). SRF mRNA levels did not differ between the intima and the media (Figure 1). As expected, quantification of ARHA and ROCK1 mRNA expression using real-time RT-PCR revealed no significant changes in mRNA expression of these two genes between media and intima (data not shown).
Figure 1.

Analysis of mRNA expression in arterial smooth muscle cells derived from the arterial media and intima using LMD followed by real-time RT-PCR. Glyceraldehyde-3-phosphate dehydrogenase was chosen as housekeeping gene for normalization. For serum response factor, no differences in expression of medial and intimal mRNA were seen (1.1 ± 0.3-fold; mean ± SD). The markers for contractile smooth muscle cells, smoothelin and calponin-1, show higher mRNA expression in the media than in the intima, for smoothelin 4.3 ± 1.0-fold and for calponin-1 2.9 ± 1.1-fold. Three different samples were measured at least two times individually and duplicates were performed for each analysis.
In Situ Hybridization
To compare our microdissection-based real-time RT-PCR results with an established method, we performed ISH for SRF and smoothelin mRNAs on cross-sections of arteries that had been used for the realtime RT-PCR. In accordance with our PCR results, ISH showed that SRF mRNA was expressed at similar levels in intima and media and that smoothelin mRNA expression was expressed at a higher level in the media (Figure 2). Because of the relatively small differences in smoothelin mRNA expression in the intima and the media, we found it difficult to estimate this difference in smoothelin mRNA expression using ISH.
Figure 2.
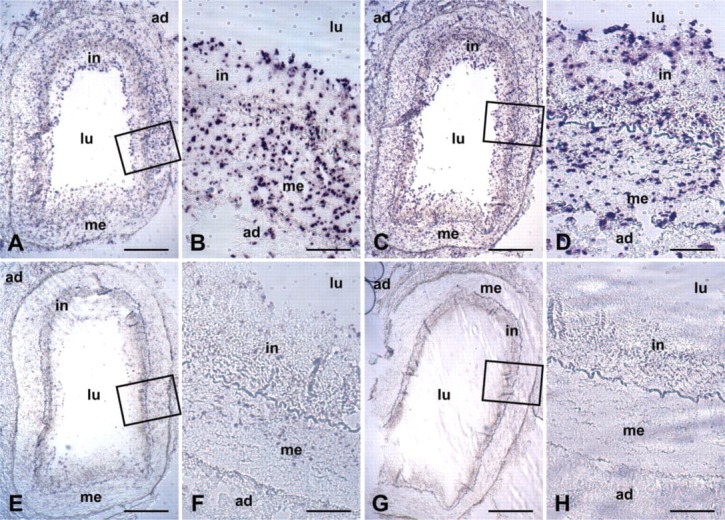
Analysis of mRNA expression of smoothelin and serum response factor in the arterial wall using ISH. Serum response factor mRNA is detected in intima and media at equal levels. Smoothelin mRNA is present mainly in the media and lower levels are detectable in the intima. (A, B) Smoothelin antisense probes; (C, D) serum response factor antisense probes; (E,F) smoothelin sense probes (negative controls); (G,H) serum response factor sense probes (negative controls). Rectangles in the overviews indicate magnified areas. ad, adventitia; in, intima; lu, arterial lumen; me, media. Bars: A, C, E, G = 500 μm; B, D, F, H = 100 μm.
Immunohistochemical Staining
We performed IHC staining for smoothelin, calponin-1, and SRF protein to compare differences in mRNA expression with changes in protein expression. Similar to our results at the mRNA level, more smoothelin and calponin-1 proteins were present in medial than in intimal SMCs. However, in contrast to the mRNA results, SRF protein was present at a significantly greater level in the media than in the intima. To check our results, we measured the occurrence of smooth muscle myosin heavy chains, well-established markers for differentiated SMCs, in the intima and media of our arteries using an antibody detecting the two isoforms SM-1 and SM-2. This confirmed that the distribution of SRF was similar to that of the smooth muscle myosin heavy chains (Figure 3).
Figure 3.
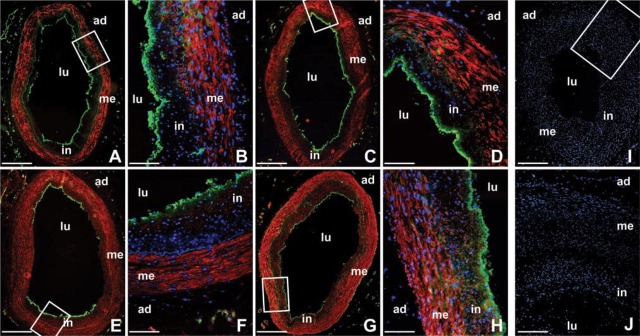
IHC staining of smoothelin, calponin-1, serum response factor, and myosin heavy chains SM-1 and SM-2 in coronary arteries. Abundance of the smoothelin and calponin-1 proteins is higher in medial smooth muscle cells of the arterial wall, in keeping with the results at the mRNA level (see Figure 1). However, expression of serum response factor protein also differs between the intima and the media, in contrast to the mRNA results (Figure 1). As expected, smooth muscle myosin heavy chains SM-1 and SM-2 were higher in the media than in the intima. (A, B) Smoothelin (red); (C, D) calponin-1 (red); (E, F) serum response factor (red); (G, H) smooth muscle myosin heavy chains SM-1 and SM-2 (red); (I, J) negative controls (mouse IgG1 was used instead of the primary antibody). Cell nuclei are shown in blue and endothelial cells are visualized in green by staining the von Willebrand factor (nuclei are not shown in the overviews). Rectangles in the overviews indicate magnified areas. ad, adventitia; in, intima; lu, arterial lumen; me, media. Bars: A, C, E, G, I = 500 μm; B, D, F, H = 100 μm; J = 200 μm.
Discussion
This study provides an interesting application of techniques for studying tissue expression of mRNA and protein, a task that is of particular importance in the postgenomic area.
Our results confirm the previously reported finding that SMCs of the thickened preatherosclerotic intima show a less-differentiated synthetic phenotype than in the media (Thyberg 1998) and suggest for the first time that this may be due to the lower amount of SRF protein present in the intima. As markers for the contractile phenotype, we chose smoothelin and calponin-1 (van der Loop et al. 1996; Sobue et al. 1999). Smoothelin is a constituent of the cytoskeleton and co-localizes with actin stress fibers (van der Loop 1997), while calponin-1, a calmodulin- and F-actin-binding troponin-like protein, forms an integral part of the actinlinked contractile machinery in SMCs (Miano et al. 2000). Smoothelin and calponin-1 are known to be expressed at higher levels in the quiescent contractile SMCs of the arterial media than in the active synthetic SMCs of the intima (van der Loop et al. 1996; Sobue et al. 1999), but up to now no information has been available on the relative expression of SRF. In vitro, however, SRF increases the expression of certain markers for SMC differentiation, including calponin-1 (Miano et al. 2000), and is therefore believed to regulate SMC differentiation at the level of gene transcription (Kim et al. 1997; Li et al. 1997; Madsen et al. 1997; Mack et al. 2000; Miano et al. 2000; Chai and Tarnawski 2002). Therfore, this triad of genes provided a useful model system in which to test methods for the in situ study of gene expression.
We were able to show that LMD plus real-time RT-PCR correlates well with both standard IHC and ISH. Moreover, LMD plus RT-PCR proved superior to ISH in detecting small differences in mRNA expression. Our results also demonstrate the need to measure both mRNA and protein abundance to investigate the functional relevance of gene expression in tissues and to indicate that regulation of SRF activity may occur, e.g., at the level of synthesis or degradation of SRF protein and not just at the level of gene expression or mRNA abundance.
In summary, therefore, our data show that the combination of LMD and real-time RT-PCR is a valuable method to reproducibly quantify RNA expression and that this technique is suitable for analyzing gene expression in arteries. In addition, quantitative data derived from real-time RT-PCR are consistent with qualitative data derived from ISH. These methods for the analysis of cell-specific gene expression are likely to be a useful tool in analyzing gene expression in a wide range of healthy and diseased tissues.
Acknowledgments
This work forms part of a project entitled “Macrophage Function and Stability of the Atherosclerotic Plaque” (MA-FAPS, QLG2-CT-1999-01007) supported by the European Union as part of the Fifth Framework Program (Bellosta et al. 2002).
We wish to express our appreciation to Mr Andrew Ritchie, Dr Andrew Exley, and Dr Martin Goddard at Papworth Hospital NHS Trust, Cambridge, UK, for providing arterial tissue samples, and to Dr Gabriele Plenz for help on in situ hybridization. We are also grateful to Monika Greive, Silke Kummer, and Sabine Sczcuka for excellent technical assistance. This work forms part of the doctoral thesis of K.S., and we are thankful to Prof Friedrich Spener for supervising this thesis.
Literature Cited
- Alevizos I, Mahadevappa M, Zhang X, Ohyama H, Kohno Y, Posner M, Gallagher G, et al. (2001) Oral cancer in vivo gene expression profiling assisted by laser capture microdissection and microarray analysis. Oncogene 20:6196–6204 [DOI] [PubMed] [Google Scholar]
- Applied Biosystems (2001) Relative quantitation of gene expression: ABI PRISM 7700 Sequence Detection System. User bulletin #2: Rev. B. Weiterstadt, Germany, Applied Biosystems; [Google Scholar]
- Bellosta S, Bernini F, Chinetti G, Cignarella A, Cullen P, von Eckardstein A, Exley A, et al. (2002) Macrophage function and stability of the atherosclerotic plaque: progress report of a European project. Nutr Metab Cardiovasc Dis 12:3–11 [PubMed] [Google Scholar]
- Brinck H, Cullen P, Exley A, Goddard MJ, Kummer S, Lorkowski S, Perrey S, et al. (2003) Internet based image database for atherosclerosis research. Proc 17th Int Congr Exhibition Computer Assisted Radiology and Surgery (CARS) 2003. Oxford, Elsevier, 1301 [Google Scholar]
- Burgemeister R, Schütze K. (2002) Tissue microdissection techniques. In Lorkowski S, Cullen P, eds. Analysing Gene Expression. A Handbook of Methods: Possibilities and Pitfalls. Weinheim, Wiley-VCH, 120–132 [Google Scholar]
- Burke AP, Tracy RP, Kolodgie F, Malcom GT, Zieske A, Kutys R, Pestaner J, et al. (2002) Elevated C-reactive protein values and atherosclerosis in sudden coronary death. Circulation 105:2019–2023 [DOI] [PubMed] [Google Scholar]
- Chai J, Tarnawski AS. (2002) Serum response factor: discovery, biochemistry, biological roles and implications for tissue injury healing. J Physiol Pharmacol 53:147–157 [PubMed] [Google Scholar]
- Goodlett D, Aebersold R. (2002) Correlation of mRNA and protein expression. In Lorkowski S, Cullen P, eds. Analysing Gene Expression. A Handbook of Methods: Possibilities and Pitfalls. Weinheim, Wiley-VCH, 58–63 [Google Scholar]
- Hilfiker A, Hilfiker-Kleiner D, Fuchs M, Kaminski K, Lichtenberg A, Rothkotter HJ, Schieffer B, et al. (2002) Expression of CYR61, an angiogenic immediate early gene, in arteriosclerosis and its regulation by angiotensin II. Circulation 106:254–260 [DOI] [PubMed] [Google Scholar]
- Kim S, Ip HS, Lu MM, Clendenin C, Parmacek MS. (1997) A serum response factor-dependent transcriptional regulatory program identifies distinct smooth muscle cell sublineages. Mol Cell Biol 17:2266–2278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kind CN. (2002) In situ hybridisation, immunocytochemistry and immunohistochemistry. In Lorkowski S, Cullen P, eds. Analysing Gene Expression. A Handbook of Methods: Possibilities and Pitfalls. Weinheim, Wiley-VCH, 704–716 [Google Scholar]
- Ky B, Shughrue P. (2002) Methods to enhance signal using isotopic in situ hybridization. J Histochem Cytochem 50:1031–1037 [DOI] [PubMed] [Google Scholar]
- Lehmann U, Bock O, Glockner S, Kreipe H. (2000) Quantitative molecular analysis of laser-microdissected paraffin-embedded human tissues. Pathobiology 68:202–208 [DOI] [PubMed] [Google Scholar]
- Li L, Liu Z, Mercer B, Overbeek P, Olson EN. (1997) Evidence for serum response factor-mediated regulatory networks governing SM22alpha transcription in smooth, skeletal, and cardiac muscle cells. Dev Biol 187:311–321 [DOI] [PubMed] [Google Scholar]
- Lorkowski S, Kratz M, Wenner C, Schmidt R, Weitkamp B, Fobker M, Reinhardt J, et al. (2001a) Expression of the ATP-binding cassette transporter gene ABCG1 (ABC8) in Tangier disease. Biochem Biophys Res Commun 283:821–830 [DOI] [PubMed] [Google Scholar]
- Lorkowski S, Rust S, Engel T, Jung E, Tegelkamp K, Galinski EA, Assmann G, et al. (2001b) Genomic sequence and structure of the human ABCG1 (ABC8) gene. Biochem Biophys Res Commun 280:121–131 [DOI] [PubMed] [Google Scholar]
- Mack CP, Thompson MM, Lawrenz-Smith S, Owens GK. (2000) Smooth muscle alpha-actin CArG elements coordinate formation of a smooth muscle cell-selective, serum response factor-containing activation complex. Circ Res 86:221–232 [DOI] [PubMed] [Google Scholar]
- Madsen CS, Hershey JC, Hautmann MB, White SL, Owens GK. (1997) Expression of the smooth muscle myosin heavy chain gene is regulated by a negative-acting GC-rich element located between two positive-acting serum response factor-binding elements. J Biol Chem 272:6332–6340 [DOI] [PubMed] [Google Scholar]
- Miano JM, Carlson MJ, Spencer JA, Misra RP. (2000) Serum response factor-dependent regulation of the smooth muscle calponin gene. J Biol Chem 275:9814–9822 [DOI] [PubMed] [Google Scholar]
- Plenz G, Koenig C, Severs NJ, Robenek H. (1997) Smooth muscle cells express granulocyte-macrophage colony-stimulating factor in the undiseased and atherosclerotic human coronary artery. Arterioscler Thromb Vasc Biol 17:2489–2499 [DOI] [PubMed] [Google Scholar]
- Rolph M, Zimmer S, Bottazzi B, Garlanda C, Mantovani A, Hansson G. (2002) Production of the long pentraxin PTX3 in advanced atherosclerotic plaques. Arterioscler Thromb Vasc Biol 22:10–14 [DOI] [PubMed] [Google Scholar]
- Rubin MA. (2001) Use of laser capture microdissection, cDNA microarrays, and tissue microarrays in advancing our understanding of prostate cancer. J Pathol 195:80–86 [DOI] [PubMed] [Google Scholar]
- Salame MY, Samani NJ, Masood I, deBono DP. (2000) Expression of the plasminogen activator system in the human vascular wall. Atherosclerosis 152:19–28 [DOI] [PubMed] [Google Scholar]
- Sobue K, Hayashi K, Nishida W. (1999) Expressional regulation of smooth muscle cell-specific genes in association with phenotypic modulation. Mol Cell Biochem 190:105–118 [PubMed] [Google Scholar]
- Thyberg J. (1998) Phenotypic modulation of smooth muscle cells during formation of neointimal thickenings following vascular injury. Histol Histopathol 13:871–891 [DOI] [PubMed] [Google Scholar]
- Trogan E, Choudhury R, Dansky H, Rong J, Breslow J, Fisher E. (2002) Laser capture microdissection analysis of gene expression in macrophages from atherosclerotic lesions of apolipoprotein E-deficient mice. Proc Natl Acad Sci USA 99:2234–2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Loop FT. (1997) Differentiation of smooth muscle cells in human blood vessels as identified by smoothelin, a novel marker for the contractile phenotype. Arterioscl Thromb Vasc Biol 17:665–671 [DOI] [PubMed] [Google Scholar]
- van der Loop FT, Schaart G, Timmer ED, Ramaekers FC, van Eys GJ. (1996) Smoothelin, a novel cytoskeletal protein specific for smooth muscle cells. J Cell Biol 134:401–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. (2000) Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol 20:1262–1275 [DOI] [PubMed] [Google Scholar]
- Weitkamp B, Cullen P, Plenz G, Robenek H, Rauterberg J. (1999) Human macrophages synthesize type VIII collagen in vitro and in the atherosclerotic plaque. FASEB J 13:1445–1457 [DOI] [PubMed] [Google Scholar]
- World Health Organization (1999) The World Health Report 1999: Making a Difference. Geneva, World Health Organization, 13–15 [Google Scholar]