

Short abstract
This study reports an abundance of primate-specific segmental duplications at the breakpoints of syntenic blocks in the human genome. Segmental duplications are associated with syntenic rearrangements but do not necessarily cause them.
Abstract
Background
Chromosomal evolution is thought to occur through a random process of breakage and rearrangement that leads to karyotype differences and disruption of gene order. With the availability of both the human and mouse genomic sequences, detailed analysis of the sequence properties underlying these breakpoints is now possible.
Results
We report an abundance of primate-specific segmental duplications at the breakpoints of syntenic blocks in the human genome. Using conservative criteria, we find that 25% (122/461) of all breakpoints contain ≥ 10 kb of duplicated sequence. This association is highly significant (p < 0.0001) when compared to a simulated random-breakage model. The significance is robust under a variety of parameters, multiple sets of conserved synteny data, and for orthologous breakpoints between and within chromosomes. A comparison of mouse lineage-specific breakpoints since the divergence of rat and mouse showed a similar association with regions associated with segmental duplications in the primate genome.
Conclusion
These results indicate that segmental duplications are associated with syntenic rearrangements, even when pericentromeric and subtelomeric regions are excluded. However, segmental duplications are not necessarily the cause of the rearrangements. Rather, our analysis supports a nonrandom model of chromosomal evolution that implicates specific regions within the mammalian genome as having been predisposed to both recurrent small-scale duplication and large-scale evolutionary rearrangements.
Background
The random-breakage model has been the dominant paradigm of chromosomal evolution since the seminal work of Nadeau and Taylor [1]. At a gross level of resolution, comparative vertebrate mapping and sequencing efforts have, in general, upheld the apparent random nature of chromosomal rearrangements [2-4]. Recent detailed analyses comparing the nearly finished human and draft mouse genomes have, however, revealed an excess of small rearrangements and an extraordinary density of breakpoints within particular regions of the genome [3,5]. Many anecdotal reports have described apparent associations between segmental duplications and alterations in orientation and order between the human and mouse genomes [6-9]. Such regions of recurrent breakage suggest an alternative model of chromosomal evolution, termed 'fragile breakage' [5,10]. The molecular basis for such fragility is not understood.
In our studies of recent human segmental duplication, we have been impressed by the apparent correspondence between breakpoints in conserved synteny and blocks of segmental duplication (Figure 1a, which shows a graphic of chromosome 7). However, caution must be exercised in comparing regions of segmental duplication and breakpoints in synteny. It is well known that large expanses of genomic sequence near telomeres and pericentromeric regions of the human genome have emerged almost solely through segmental duplication events during primate evolution [11-13]. Such regions, therefore, would create artifacts if not properly excluded during global analyses. Using available genomic sequence data from human, mouse and rat, we sought to formally test the significance of this association by comparing the distribution of segmental duplications and conserved syntenic breakpoints where unique sequence was the basis for the assignment of syntenic breakpoints.
Figure 1.
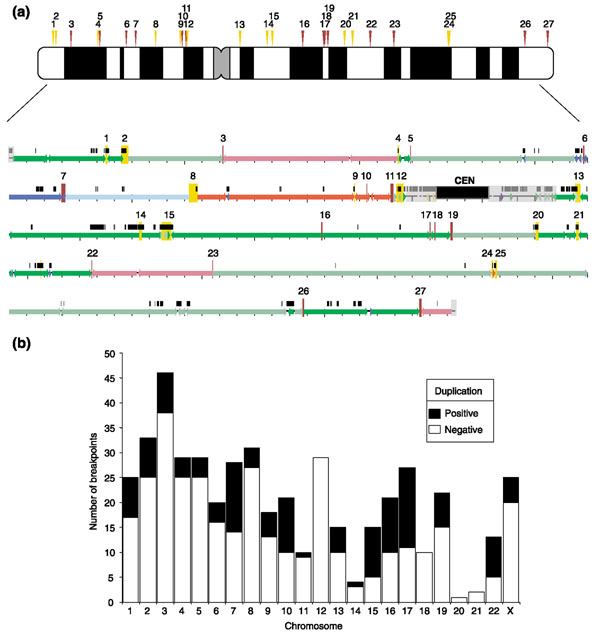
Association of duplications and syntenic breaks: human versus mouse. (a) Top: ideogram of human chromosome 7 showing the positions of breakpoints in mouse-human synteny. We assessed only breakpoints that represented a change in orientation or difference in chromosome compared with the mouse genome (arrowheads). These breakpoints are designated as duplication-positive and duplication-negative (yellow and brown, respectively). Bottom: human chromosome 7 with blocks of conserved synteny with mouse indicated by colored bars. Segmental duplications are indicated by black bars over the sequence. Regions of duplication abutting the centromere and telomeres were excluded (gray shading). Small tick marks under the colored line represent 1-Mb intervals and breakpoints in synteny are numbered as in the chromosome ideogram. Gaps in conserved synteny were excluded. Conserved syntenic regions less than 100 kb or more than 75% duplicated were ignored. This prevented very small duplicated blocks from interrupting orthologous conservation or inappropriately increasing the number of breaks within a duplicated region. Breakpoints were scored as duplication-positive if they contained ≥ 10 kb of duplicated sequence. By these criteria, 13 of the 27 breakpoints on chromosome 7 (yellow boxes/arrows) were associated with duplications. (b) A histogram of the chromosomal distribution of syntenic breakpoints and their duplication status.
Results
In this study we sought to determine the relationship between recent human segmental duplications and breakpoints in conserved synteny. Comparison of duplications and syntenic breakpoints is complicated by the fact that duplicated sequence can create potential non-orthologous assignments owing to the high degree of sequence identity to multiple locations within a genome. Alternatively, duplicated regions may lead to the inability to map a block of sequence to a particular orthologous locus, creating a de facto gap within the 'syntenic map'. To eliminate these potential problems, we applied a set of conservative criteria. First, we only considered breakpoints where orthologous sequence anchors had been unambiguously placed within unique sequence and the overall length of the conserved syntenic segment was ≥ 100 kb. A breakpoint was identified as either a change in orientation or in chromosomal location based on unique regions within the human genome. We ignored apparent gaps in conserved synteny in the human genome where flanking regions had the same chromosome and orientation assignment in the mouse.
In our analysis we considered only pairwise alignments (≥ 1 kb in length, ≥ 90% sequence identity) representing primate-specific segmental duplications within the human genome [14]. Segmental duplications are duplications of apparently normal genomic DNA that often contain genes or genic segments as well as common transposable elements. Using two independent methods of assessment [14,15], we have mapped the precise location of segmental duplications within the most recent human genome sequence assemblies (see Materials and methods). The working-draft nature of the mouse genome currently underestimates the content and location of recent duplications because of the effective collapse of whole-genome shotgun sequence [3]. As part of our analysis of conserved synteny, we excluded human pericentromeric and subtelomeric regions, where multiple megabases of recently acquired duplications have accumulated [14]. Comparative studies have shown that most of these regions have emerged as a consequence of primate-specific duplication events. Computational and phylogenetic analyses confirm that these regions have been populated by duplicative transposition of euchromatic sequences over the past 35 million years of evolution [16-19]. These regions are, therefore, derived specifically within the primate lineage and do not contain a sufficient number of unique sequence anchors to reliably establish orthologous relationships between rodents and humans [5,20,21]
We compared the distribution of human segmental duplication and of breakpoints in conserved human-mouse synteny (human NCBI build 31 and MSGC v.3). By count, 122/461 (26.5%) of the breakpoints contained one or more duplicated blocks of at least 10 kb in size (Table 1). By sequence content, breakpoint regions showed an eightfold enrichment for human segmental duplications (Table 1). To assess the significance of this association, we randomly reassigned breakpoints, without replacement, throughout the entire human genome. This procedure fixed the size and number of breakpoints, while allowing for their position to vary, effectively simulating a random-breakage model. The number of duplication-positive breakpoints was calculated for each replicate. On the basis of 10,000 replicates, the simulated count (maximum 50) never exceeded the observed count of 122, suggesting that this association is unlikely to have occurred by chance (p < 0.0001) (Figure 2).
Table 1.
Duplications and syntenic breakpoints in the human genome compared with mouse
| Total analyzed | Duplication-positive counts* | Duplication-positive bases | |||||||
| Number | bp | %† | Number | % | p-value‡ | bp | % | p-value‡ | |
| Duplicated blocks | 1,643 | 72,151,679 | 2.6 | NA | NA | NA | NA | NA | NA |
| Break points | 461 | 83,985,399 | 3.1 | 122 | 26.5 | < 0.0001 | 20,030,062 | 23.8 | < 0.0001 |
| Between chromosomes | 163 | 42,496,016 | 1.5 | 43 | 26.4 | < 0.0001 | 7,265,961 | 17.1 | < 0.0001 |
| Different orientations | 298 | 41,489,383 | 1.5 | 79 | 26.5 | < 0.0001 | 12,555,425 | 30.3 | < 0.0001 |
*A positive breakpoint contained ≥ 10 kb duplicated sequence (continuous sequence).†Fraction of genome size (2,745,533,264 bp, which excludes highly duplicated pericentromeric and subtelomeric regions).‡p < 0.0001, simulated value never exceeded observed in 10,000 replicates. NA, not applicable.
Figure 2.
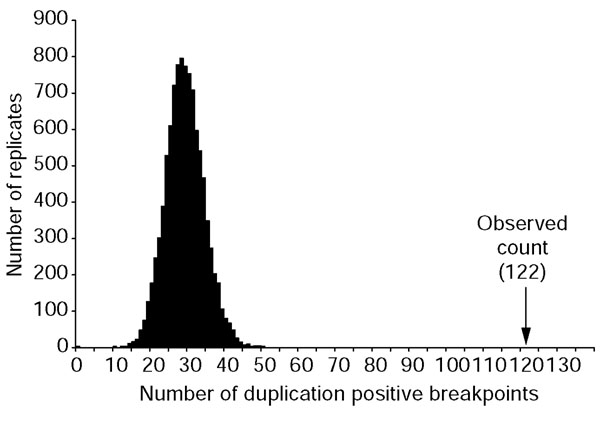
Simulation of random chromosomal breakage. We performed simulation studies to determine the significance of the observed association of segmental duplications with breakpoints by randomly reassigning breakpoints to positions within the assayed genomic sequence (see Materials and methods). Not once in 10,000 replicates did the simulated count exceed the observed number of duplication-positive breakpoints.
In addition, size thresholds for conserved syntenic regions (200 kb, 500 kb, 1,000 kb) and duplication thresholds (10 kb, 20 kb, 50 kb), were also examined. All parameter combinations showed a highly significant association with human segmental duplications (p < 0.001) (see Additional data file 1). Conserved syntenic breakpoints both within chromosomes and between chromosomes showed an association (Figure 1b and Table 1). We also analyzed two other datasets of mouse-human conserved synteny: the published mouse draft [3] and a further refinement of the Pevzner-Tesler analyses [5]. Both sets showed a significant association between segmental duplication and orthologous breakpoints, suggesting that methodological differences are not responsible for these observations (see Additional data files 2 and 3). It should be noted that if we do not apply these stringent criteria for assignment of orthologous syntenic blocks and duplicated breakpoints, the association rises to 555/1,070 breakpoints (51%).
To examine the potential causal relationship between duplications and breakpoints in synteny requires determination of the relative timing and therefore the order of these events. On the basis of the high degree of sequence identity and estimates of neutral sequence divergence among primates [11,22,23], the duplications are primate specific, having occurred within the past 35 million years of evolution. Studies of neutral mutation differences (single base-pair events, indels and rearrangements) between human and mouse have suggested an increased rate within the rodent lineage [3]. The nearly completed draft sequence of the rat genome provides an additional rodent species for comparison, allowing us to identify breakpoints that are shared between mouse and rat. We compared the human and rat genomes for equivalents to the 439 human-mouse syntenic breakpoints (Table 2). Mouse-human breakpoints absent in the human-rat comparison suggest rearrangements specific to the mouse lineage (mouse-specific breakpoints). Breakpoints supported by human-rat comparisons suggest rearrangements that occurred either within the human/primate lineage or the common rat-mouse rodent lineage (shared mouse-rat breakpoints). Thus, if a causal relationship exists, there should be no association of primate-specific duplications and mouse-specific breakpoints, as they have occurred in two separate lineages. However, direct causality is not supported as no significant difference (p = 0.4626, chi-squared 1 df = 0.5397) was observed in the prevalence of associated duplications between mouse-specific and shared mouse-rat breakpoints.
Table 2.
Comparison of human-mouse and human-rat breakpoints versus segmental duplication content
| Duplicated | Unduplicated | Total | |||
| Number | % | Number | % | Number | |
| Mouse-specific | 29 | 29.9 | 68 | 70.1 | 97 |
| Shared breakpoint | 110 | 34.2 | 212 | 65.8 | 322 |
| Undetermined* | 12 | 28.6 | 30 | 71.4 | 42 |
Mouse-human breakpoints were classified as either shared with rat or specific to the mouse assembly. No significant difference in segmental duplication content was found when shared and mouse-specific breakpoints were compared. Chi-squared test (χ2 = 0.5397, p = 0.4626). *Forty-two human-mouse breakpoints showed no evidence of conserved synteny within 1 Mb of the breakpoint when the human and rat genomes were compared.
Discussion
Several recent comparative mapping studies in a wide variety of closely related eukaryotic organisms have shown a relationship between large-scale chromosomal rearrangement and repetitive DNA. The nature of the repetitive DNA within these breakpoint regions varies significantly, from clusters of rRNA and tRNA genes to various transposable elements [24-26]. Between human and mouse, an association with segmental duplications and repetitive DNA has been previously suggested although never rigorously tested [6,27]. Recent published reports of three out of seven different conserved syntenic breakpoints that distinguish the human and great-ape karyotype uncovered segmental duplications precisely at the site of these breakpoints [28-32]. Interestingly, a few of these primate segmental duplications also function as breakpoints of recurrent chromosomal structural rearrangements associated with disease and polymorphism within the human population [11,32].
In a very recent study, Armengol and colleagues suggested an enrichment of segmental duplications near sites of evolutionary rearrangement [33]. They reported that 53% of all evolutionary rearrangement breakpoints between human and mouse associate with segmental duplications, as compared to 18% expected in a random assignment of breaks. This number is significantly higher than our estimate and is likely to be due to methodological differences between the two studies. For example, we specifically excluded highly duplicated pericentromeric and subtelomeric regions because of their dynamic evolution within the primate lineages and the difficulties associated with assigning 'true' orthologous relationships. The Armengol study did not make this distinction. Second, Armengol and colleagues considered shorter segments of conserved synteny (down to 20 kb in size) that fell within larger blocks of synteny. In our study, we required large tracts of unique sequence (> 100 kb) to establish conserved synteny, purposefully excluding short regions which might provide false associations due to genomic duplications and deletions since the divergence of mouse and humans. Using conservative criteria, we find that 25% (122/461) of all breakpoints contain ≥ 10 kb of duplicated sequence.
Both of these studies considered the location of primate-specific segmental duplications only from the perspective of the human genome sequence assembly. While it is tempting to speculate that nonhomologous recombination of blocks of duplicated DNA might have a direct role in mediating rearrangements [33], the temporal order of these events and therefore the cause-consequence relationship has not been previously investigated. In the case of mouse-human comparisons, it seems unlikely that the segmental duplications are the direct cause of the rearrangement. On the basis of levels of sequence divergence, the segmental duplications considered in this analysis emerged over the past 35 million years of primate evolution [11]. In contrast, the conserved synteny breaks have occurred in both human and mouse lineages since their separation 75 million years ago. Also, the association is just as strong when only mouse-specific syntenic breakpoints are considered (Table 2). It is therefore unlikely that segmental duplications are driving chromosomal rearrangements through nonhomologous recombination, as no correlation between primate-specific duplications and mouse-lineage-specific syntenic rearrangements would be expected. Rather, our analysis supports a nonrandom model of chromosomal evolution that implicates a predominance of recurrent small-scale duplication and large-scale evolutionary rearrangements within specific 'fragile' regions of the mammalian genome. Understanding the nature and pattern of segmental duplications within mammalian genomes will be pivotal in revealing the molecular basis of chromosomal evolution among these species.
Materials and methods
To examine the association between duplication and orthologous breakpoints, we initially compared the published mouse (MGSCv3) and human (Nov 2002 build31) sequence assemblies. Syntenic anchoring regions were built from BLASTZ mouse-human DNA alignments [34]. High-scoring alignments (≥ 900; calculated as 3 × matches - mismatches - gaps) were used to define well-conserved syntenic anchor regions (100 kb regions showing ≥ 10% of the sequence aligned with a sum alignment score of ≥ 10,000). These anchor regions were extended if adjacent 100-kb sliding windows matched the mouse chromosome and orientation with a sum score of ≥ 7,000. These extended regions were then joined together if they agreed in orientation and were within 500 kb of each other in the human genome and within 4 Mb of each other in the mouse. These conservative criteria restricted mouse-human synteny comparisons to either large-scale orientation changes or translocations between chromosomes. For this study, as a further safeguard against mouse misassembly, gaps between these syntenic segments were joined if the syntenic segments between two flanking regions agreed in terms of assigned mouse chromosome and orientation. In addition to this updated mouse-human synteny map, we also considered two earlier published versions of conserved synteny [3,5].
Segmental duplications were detected as pairwise alignments within the human genome (≥ 90% and ≥ 1 kb) as previously described and verified by assembly-independent methods [15,35]. Pairwise alignments were collapsed into a nonredundant set on the basis of genome coordinates, essentially assigning each base in the genome as duplicated or not. Both sets of data are available as part of the University of California, Santa Cruz genome browser data [36].
Duplications and syntenic regions were displayed using the graphic viewer Parasight [37] for each human chromosome. This analysis excluded the Y chromosome, which was not sequenced in the mouse. Our goal was to study breakpoints between the species that were based on the alignment of unique sequence. Genomic sequence from each centromere and telomere to the first conserved synteny that showed essentially no duplication was excluded from the analysis. These areas represent highly duplicated pericentromeric and subtelomeric regions where assignment of human-mouse orthology is problematic. The syntenic breakpoints and duplications within the remaining genomic regions were then analyzed for association using a series of Perl scripts. Conserved syntenic blocks less than 100 kb in length and/or composed of ≥ 75% duplicated bases were deleted to eliminate breakpoints created as a consequence of duplicative transposition. Breakpoint regions were defined as the gaps between syntenic blocks that represented a difference in mouse chromosome assignment or orientation of unique sequence. Gaps within conserved synteny were not counted as breakpoints (although shown in Figure 1a). Breakpoint regions were scored as duplication positive if the duplication content exceeded 10 kb (Table 1; 122/461 breakpoints).
To assess the significance of the duplication-breakpoint association, computer simulations of a random-breakage model reassigned the observed breakpoints to random positions within the genome. This was done without replacement, and the positions of breakpoints were limited in that they could only be placed as close together as the minimum length of the syntenic regions assayed (100 kb). For each replicate, the number of duplication-positive breakpoints was calculated as well as the number of duplicated bases within the breakpoints (see Additional data file 1). It is important to note that our assessment is conservative in its approach. A similar analysis, removing size constraints and including pericentromeric and subtelomeric regions, shows that up to half of all breakpoints (555/1,070 = 51%) are associated with segmental duplications.
To determine the robustness of the association, a variety of syntenic size thresholds (100 kb, 200 kb, 500 kb, 1,000 kb) and duplication-positive thresholds (10 kb, 20 kb, 50 kb) were assessed (see Additional data file 1). Because such an association may be due to methodological considerations regarding the initial ascertainment of conserved syntenic blocks, we examined two other datasets. The first, published with the initial mouse draft (human NCBI build30 versus MGSC version 2), measured conserved syntenic regions with a minimum size of 300 kb [3]. The second utilized the same Pattern Hunter genomic alignment anchors but incorporated a refined algorithm [5] and measured syntenic regions greater than 1 Mb. Both sets showed significant (p < 0.0001) enrichment of duplications with breakpoints (see additional data files 2 and 3).
To determine the timing of mouse-human syntenic breakpoints we examined rat-human conserved synteny (rat v.2.1), using the same parameters described for human/mouse. For each mouse-human breakpoint, we examined conserved synteny between human and rat. If the region was not interrupted between human and rat genomes, then the breakpoint was assigned as mouse-specific. If the breakpoint was shared, then the mouse-human breakpoint was assigned as common to the mouse and rat. If no conserved synteny relationship could be identified within 500 kb on either side of the mouse-human breakpoint, the breakpoint was classified as 'undetermined' and excluded from further analysis. This allowed a subset of rearrangements to be generally classified into two different parts of the human-mouse-rat phylogeny. The frequency of duplication-positive and negative breakpoints for the two categories was compared using the chi-squared test (1 df).
Additional data files
Additional data available with the online version of this paper include: Additional data file 1, which shows the calculation of the number of duplication-positive breakpoints and the number of duplicated bases within the breakpoints; and Additional data files 2 and 3, which show the results of analysis of the published mouse draft genome [3] and a further refinement of the Pevzner-Tesler analyses [5].
Supplementary Material
The calculation of the number of duplication-positive breakpoints and the number of duplicated bases within the breakpoints
The results of analysis of the published mouse draft genome
A further refinement of the Pevzner-Tesler analyses
Acknowledgments
Acknowledgements
We thank D. Locke and J. Nadeau for helpful comments regarding the manuscript. This work was supported, in part, by NIH grants GM58815 and HG002385 and US Department of Energy grant ER62862 to E.E.E., a NIH Career Development Program in Genomic Epidemiology of Cancer (CA094816) to J.A.B., NHGRI grant 1P41HG02371 to D.H., the W.M. Keck Foundation and the Charles B. Wang Foundation.
References
- Nadeau JH, Taylor BA. Lengths of chromosomal segments conserved since divergence of man and mouse. Proc Natl Acad Sci USA. 1984;81:814–818. doi: 10.1073/pnas.81.3.814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- Aparicio S, Chapman J, Stupka E, Putnam N, Chia JM, Dehal P, Christoffels A, Rash S, Hoon S, Smit A, et al. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science. 2002;297:1301–1310. doi: 10.1126/science.1072104. [DOI] [PubMed] [Google Scholar]
- Pevzner P, Tesler G. Genome rearrangements in mammalian evolution: lessons from human and mouse genomes. Genome Res. 2003;13:37–45. doi: 10.1101/gr.757503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valero MC, de Luis O, Cruces J, Perez Jurado LA. Fine-scale comparative mapping of the human 7q11.23 region and the orthologous region on mouse chromosome 5G: the low-copy repeats that flank the Williams-Beuren syndrome deletion arose at breakpoint sites of an evolutionary inversion(s). Genomics. 2000;69:1–13. doi: 10.1006/geno.2000.6312. [DOI] [PubMed] [Google Scholar]
- Bi W, Yan J, Stankiewicz P, Park SS, Walz K, Boerkoel CF, Potocki L, Shaffer LG, Devriendt K, Nowaczyk MJ, et al. Genes in a refined Smith-Magenis syndrome critical deletion interval on chromosome 17p11.2 and the syntenic region of the mouse. Genome Res. 2002;12:713–728. doi: 10.1101/gr.73702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimelli G, Pujana MA, Patricelli MG, Russo S, Giardino D, Larizza L, Cheung J, Armengol L, Schinzel A, Estivill X, Zuffardi O. Genomic inversions of human chromosome 15q11-q13 in mothers of Angelman syndrome patients with class II (BP2/3) deletions. Hum Mol Genet. 2003;12:849–858. doi: 10.1093/hmg/ddg101. [DOI] [PubMed] [Google Scholar]
- DeSilva U, Elnitski L, Idol JR, Doyle JL, Gan W, Thomas JW, Schwartz S, Dietrich NL, Beckstrom-Sternberg SM, McDowell JC, et al. Generation and comparative analysis of approximately 3.3 Mb of mouse genomic sequence orthologous to the region of human chromosome 7q11.23 implicated in Williams syndrome. Genome Res. 2002;12:3–15. doi: 10.1101/gr.214802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pevzner P, Tesler G. Human and mouse genomic sequences reveal extensive breakpoint reuse in mammalian evolution. Proc Natl Acad Sci USA. 2003;100:7672–7677. doi: 10.1073/pnas.1330369100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samonte RV, Eichler EE. Segmental duplications and the evolution of the primate genome. Nat Rev Genet. 2002;3:65–72. doi: 10.1038/nrg705. [DOI] [PubMed] [Google Scholar]
- Horvath J, Schwartz S, Eichler E. The mosaic structure of human pericentromeric DNA: A strategy for characterizing complex regions of the human genome. Genome Res. 2000;10:839–852. doi: 10.1101/gr.10.6.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Spalluto C, McMurray A, Hearn T, Crosier M, Viggiano L, Miolla V, Archidiacono N, Rocchi M, Scott C, et al. Genomic sequence and transcriptional profile of the boundary between pericentromeric satellites and genes on human chromosome arm 10q. Hum Mol Genet. 2000;9:2029–2042. doi: 10.1093/hmg/9.13.2029. [DOI] [PubMed] [Google Scholar]
- Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, Adams MD, Myers EW, Li PW, Eichler EE. Recent segmental duplications in the human genome. Science. 2002;297:1003–1007. doi: 10.1126/science.1072047. [DOI] [PubMed] [Google Scholar]
- Bailey JA, Yavor AM, Massa HF, Trask BJ, Eichler EE. Segmental duplications: organization and impact within the current human genome project assembly. Genome Res. 2001;11:1005–1017. doi: 10.1101/gr.GR-1871R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman T, Trask BJ. Complex evolution of 7E olfactory receptor genes in segmental duplications. Genome Res. 2003;13:781–793. doi: 10.1101/gr.769003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy J, Hearn T, Crosier M, Mudge J, Viggiano L, Koczan D, Thiesen HJ, Bailey JA, Horvath JE, Eichler EE, et al. Genomic sequence and transcriptional profile of the boundary between pericentromeric satellites and genes on human chromosome arm 10p. Genome Res. 2003;13:159–172. doi: 10.1101/gr.644503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath JE, Gulden CL, Bailey JA, Yohn C, McPherson JD, Prescott A, Roe BA, De Jong PJ, Ventura M, Misceo D, et al. Using a pericentromeric interspersed repeat to recapitulate the phylogeny and expansion of human centromeric segmental duplications. Mol Biol Evol. 2003;20:1463–1479. doi: 10.1093/molbev/msg158. [DOI] [PubMed] [Google Scholar]
- Locke DP, Jaing Z, Pertz LM, Misceo D, Archidiacono N, Eichler EE. Molecular evolution of the human chromosome 15 pericentromeric region. Cytogenet Genome Res. 2004. [DOI] [PubMed]
- Eichler EE, Sankoff D. Structural dynamics of eukaryotic chromosome evolution. Science. 2003;301:793–797. doi: 10.1126/science.1086132. [DOI] [PubMed] [Google Scholar]
- Kent WJ, Baertsch R, Hinrichs A, Miller W, Haussler D. Evolution's cauldron: duplication, deletion, and rearrangement in the mouse and human genomes. Proc Natl Acad Sci USA. 2003;100:11484–11489. doi: 10.1073/pnas.1932072100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaessmann H, Heissig F, von Haeseler A, Paabo S. DNA sequence variation in a non-coding region of low recombination on the human X chromosome. Nat Genet. 1999;22:78–81. doi: 10.1038/8785. [DOI] [PubMed] [Google Scholar]
- Chen FC, Li WH. Genomic divergences between humans and other hominoids and the effective population size of the common ancestor of humans and chimpanzees. Am J Hum Genet. 2001;68:444–456. doi: 10.1086/318206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coghlan A, Wolfe KH. Fourfold faster rate of genome rearrangement in nematodes than in Drosophila. Genome Res. 2002;12:857–867. doi: 10.1101/gr.172702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlton JM, Angiuoli SV, Suh BB, Kooij TW, Pertea M, Silva JC, Ermolaeva MD, Allen JE, Selengut JD, Koo HL, et al. Genome sequence and comparative analysis of the model rodent malaria parasite Plasmodium yoelii yoelii. Nature. 2002;419:512–519. doi: 10.1038/nature01099. [DOI] [PubMed] [Google Scholar]
- Kellis M, Patterson N, Endrizzi M, Birren B, Lander ES. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature. 2003;423:241–254. doi: 10.1038/nature01644. [DOI] [PubMed] [Google Scholar]
- Dehal P, Predki P, Olsen AS, Kobayashi A, Folta P, Lucas S, Land M, Terry A, Ecale Zhou CL, Rash S, et al. Human chromosome 19 and related regions in mouse: conservative and lineage specific evolution. Science. 2001;293:104–111. doi: 10.1126/science.1060310. [DOI] [PubMed] [Google Scholar]
- Eder V, Mario V, Ianigro M, Teti M, Rocchi M, Archidiacono N. Chromosome 6 phylogeny in primates and centromere repositioning. Mol Biol Evol. 2003;20:1506–1512. doi: 10.1093/molbev/msg165. [DOI] [PubMed] [Google Scholar]
- Kellis M, Patterson N, Endrizzi M, Birren B, Lander ES. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature. 2003;423:241–254. doi: 10.1038/nature01644. [DOI] [PubMed] [Google Scholar]
- Nickerson E, Gibbs RA, Nelson DL. Sequence analysis of the breakpoints of a pericentric inversion distinguishing the human and chimpanzee chromosomes 12. Am J Hum Genet. 1999;65:A291. [Google Scholar]
- Locke DP, Archidiacono N, Misceo D, Cardone MF, Dechamps S, Roe BA, Rocchi M, Eichler EE. Refinement of a chimpanzee pericentric inversion breakpoiint to a segmental duplication cluster. Genome Biol. 2003;4:R50. doi: 10.1186/gb-2003-4-8-r50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankiewicz P, Park SS, Inoue K, Lupski JR. The evolutionary chromosome translocation 4;19 in Gorilla gorilla is associated with microduplication of the chromosome fragment syntenic to sequences surrounding the human proximal CMT1A-REP. Genome Res. 2001;11:1205–1210. doi: 10.1101/gr.181101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armengol L, Pujana MA, Cheung J, Scherer SW, Estivill X. Enrichment of segmental duplications in regions of breaks of synteny between the human and mouse genomes suggest their involvement in evolutionary rearrangements. Hum Mol Genet. 2003;12:2201–2208. doi: 10.1093/hmg/ddg223. [DOI] [PubMed] [Google Scholar]
- Schwartz S, Kent WJ, Smit A, Zhang Z, Baertsch R, Hardison RC, Haussler D, Miller W. Human-mouse alignments with BLASTZ. Genome Res. 2003;13:103–107. doi: 10.1101/gr.809403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey JA, Yavor AM, Viggiano L, Misceo D, Horvath JE, Archidiacono N, Schwartz S, Rocchi M, Eichler EE. Human-specific duplication and mosaic transcripts: the recent paralogous structure of chromosome 22. Am J Hum Genet. 2002;70:83–100. doi: 10.1086/338458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- University of California Santa Cruz genome browser http://genome.ucsc.edu
- Parasight http://humanparalogy.cwru.edu/parasight
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The calculation of the number of duplication-positive breakpoints and the number of duplicated bases within the breakpoints
The results of analysis of the published mouse draft genome
A further refinement of the Pevzner-Tesler analyses