

Abstract
Nonnucleoside reverse transcriptase inhibitors (NNRTIs) are a mainstay of therapy for treating human immunodeficiency type 1 virus (HIV-1)-infected patients. MK-1439 is a novel NNRTI with a 50% inhibitory concentration (IC50) of 12, 9.7, and 9.7 nM against the wild type (WT) and K103N and Y181C reverse transcriptase (RT) mutants, respectively, in a biochemical assay. Selectivity and cytotoxicity studies confirmed that MK-1439 is a highly specific NNRTI with minimum off-target activities. In the presence of 50% normal human serum (NHS), MK-1439 showed excellent potency in suppressing the replication of WT virus, with a 95% effective concentration (EC95) of 20 nM, as well as K103N, Y181C, and K103N/Y181C mutant viruses with EC95 of 43, 27, and 55 nM, respectively. MK-1439 exhibited similar antiviral activities against 10 different HIV-1 subtype viruses (a total of 93 viruses). In addition, the susceptibility of a broader array of clinical NNRTI-associated mutant viruses (a total of 96 viruses) to MK-1439 and other benchmark NNRTIs was investigated. The results showed that the mutant profile of MK-1439 was superior overall to that of efavirenz (EFV) and comparable to that of etravirine (ETR) and rilpivirine (RPV). Furthermore, E138K, Y181C, and K101E mutant viruses that are associated with ETR and RPV were susceptible to MK-1439 with a fold change (FC) of <3. A two-drug in vitro combination study indicated that MK-1439 acts nonantagonistically in the antiviral activity with each of 18 FDA-licensed drugs for HIV infection. Taken together, these in vitro data suggest that MK-1439 possesses the desired properties for further development as a new antiviral agent.
INTRODUCTION
Human immunodeficiency type 1 virus (HIV-1) reverse transcriptase (RT) plays an essential role in the HIV-1 life cycle by converting a single-strand viral RNA into a double-stranded proviral DNA via its polymerase and RNase H activities (1). Therefore, inhibition of reverse transcriptase has been one of the primary therapeutic strategies for developing antiviral agents to suppress the replication of HIV-1 (2, 3). There are two classes of RT inhibitors: one is the nucleoside reverse transcriptase inhibitors (NRTIs), which are active-site inhibitors, such as AZT and 3TC, and the other is nonnucleoside reverse transcriptase inhibitors (NNRTIs), which are non-active-site competitive inhibitors, such as efavirenz (EFV), nevirapine (NVP), etravirine (ETR), and rilpivirine (RPV). The NNRTIs bind to a hydrophobic pocket in the p66 subunit of the p66/p51 heterodimer of RT at a distance of 10 Å from the polymerase active site (4). NNRTI binding causes conformational changes within p66 that reposition the active-site residues into an inactive conformation, resulting in inhibition of the chemical step of a polymerization reaction (5).
The current standard of treatment for HIV-1-infected patients is highly active antiretroviral therapy (HAART), which is typically composed of 3 or more drugs with complementary mechanisms of actions (6). Patients undergoing HAART have experienced profound and continuous viral suppression, in many cases with substantial immune system recovery and halt of progression to clinical disease (7). Consensus guidelines for the use of HAART in antiretroviral-naive subjects recommend the use of 2 NRTIs in combination with an NNRTI, a boosted protease inhibitor, or an integrase inhibitor (8, 9). Although NNRTIs are key components of effective combination regimens, like all antiretroviral agents, their effectiveness can be hampered by the emergence of resistance mutations in viruses. Moreover, a single mutation can lead to significant reductions in susceptibility, often to all available inhibitors within the same class (10, 11). Mutations identified in the viruses from patients who failed with an NNRTI-containing regimen mostly involve residues around the NNRTI binding pocket (NNRTIBP) that play important roles in inhibitor binding. As a result, viruses harboring substitutions in these residues often disrupt the crucial interactions between NNRTIs and RT, conferring resistance to the inhibitors. Among NNRTI-associated mutations, the K103N substitution represents the most prevalent mutant identified in patients who have failed with regimens containing earlier NNRTIs, such as EFV and NVP (present in 40 to 60% of NNRTI-associated mutant viruses) (12–16), and the mutant virus displays significantly reduced susceptibility to the NNRTIs (>20-fold to EFV and >50-fold to NVP) (17, 18). The prevalence of this mutation may be attributed to the frequent prescription of NVP and EFV as the first-line therapy, especially EFV, which is the NNRTI component in the single-tablet regimen Atripla. In addition, the K103N mutant maintains replication capacity similar to that of the wild type (WT), enabling the virus to continue replicating in the presence of high selective pressure (19, 20). Based on X-ray crystallography of RT/NNRTI complexes, the reduced inhibitory potency may be ascribed to the changes in hydrophobic and electrostatic interactions between the inhibitors and K103 or N103 in the NNRTIBP and formation of new hydrogen bond networks within NNRTIBP with the K103N substitution (21). NNRTIs with good activity against the K103N mutant would be expected to have favorable interactions with the mutant asparagine side chain, thereby compensating for resistance caused by stabilization of the mutant enzyme through a hydrogen bond network, including the interactions between N103 and Y188 side chains (22). Virus with Y181C substitution is the second most prevalent NNRTI-associated mutant, which is mostly detected in patients on NVP treatment (present in 15 to 25% of NNRTI-associated mutant viruses) (14–16). Furthermore, the Y181C mutant virus also accounts for the majority of virologic failures from patients who were on an ETR-containing regimen (23). The substitution is also one of the major mutations identified in virologic failures from the RPV-treated group in a phase III trial (24). The mutation at codon position 181 (Y181C mutant) has a minimal impact on the fitness of the mutant virus, allowing the mutant virus to replicate efficiently even in the presence of related NNRTIs (19, 20). Y181 plays an important role in the π-π interactions with some NNRTIs inside the NNRTIBP; thus, viruses with Y181C substitution in RT will be void of the crucial interactions with the NNRTIs and exhibit high degrees of resistance to NVP and delavirdine (DLV) (>100-fold) and moderate resistance to ETR and RPV (∼4-fold) (25–27).
Despite the success of HAART, drug resistance is generated in a proportion of treated patients and may be directly transmitted from them to treatment-naive individuals at the time of their primary infection. Transmitted drug resistance (TDR) mutations have been documented among treatment-naive HIV-1-infected patients with a prevalence ranging from 3% to 24%, depending on the cohort and geographic characteristics; the highest prevalence is noted in countries with long-established use of antiretrovirals (28). In the United States, it is estimated that approximately 1 in 6 newly infected patients harbors virus with TDR mutations. Given the prevalence of K103N and Y181C mutants in the patients treated with the NNRTI-containing regimen, not surprisingly, the mutants also are the two most prevalent NNRTI TDR mutants in newly infected patients, representing more than 84% of TDR mutations (29, 30). In addition, the TDR mutations were found to significantly increase the virologic failure rate under HAART, particularly K103N and Y181C TDR mutants (30–32). As a result, TDR mutations have emerged as a potential threat to the success of antiretroviral therapy.
Extensive efforts have been made to identify novel NNRTIs that are highly active against the prevalent NNRTI-resistant viruses (especially for viruses carrying K103N or Y181C substitutions) and suitable for a once-daily dosing with excellent safety and tolerability profiles. To this end, MK-1439 was identified as a novel NNRTI comprised of a pyridone core bearing an aryl ether and methyl-triazolone moieties (Fig. 1). It displays excellent activities against not only WT viruses but also a broader panel of NNRTI-resistant viruses, including viruses carrying K103N and/or Y181C substitutions. Given the large genetic diversity of HIV-1, antiviral activity of MK-1439 was determined against different viral subtypes (A, B, C, D, F, G, H, J, and K). The susceptibility of RPV-associated clinical mutant viruses to MK-1439 was tested to evaluate the resistance profile of MK-1439 against these mutants. In addition, a 2-drug in vitro combination study was performed to assess the potential for antagonism between the antiviral agents. Furthermore, specificity and cytotoxicity tests were also conducted to gain information on potential off-target activities of MK-1439.
FIG 1.
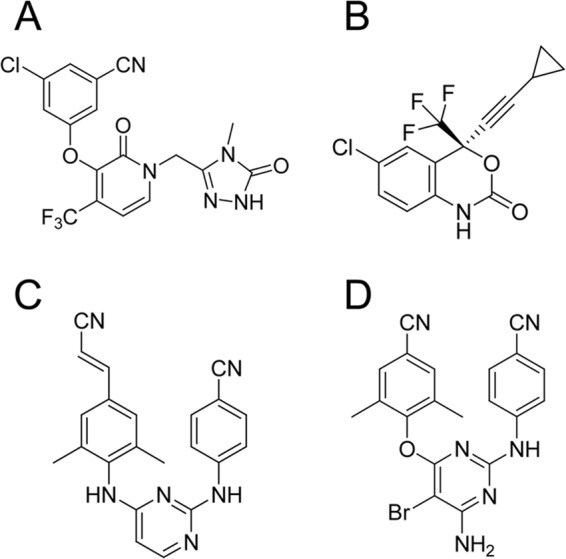
Structures of MK-1439 (A), EFV (B), RPV (C), and ETR (D).
MATERIALS AND METHODS
Full-length wild-type (WT) and two mutant RT proteins (K103N and Y181C) were expressed in Escherichia coli BL21(DE3) cells and purified as described previously (33). The t500 RNA template was made by IBA GmbH (Germany), and the biotinylated DNA primer was made by Integrated DNA Technology (IDT; Coralville, IA). The R8 virus was a kind gift from Christopher Aiken (Vanderbilt University, Nashville, TN). SupT1 cells were provided by the NIH AIDS Research and Reference Reagent Program. Ruthenylated dUTP (Ru-dUTP) was custom-made by Midland Certified Reagents Company (Midland, TX). Human peripheral blood mononuclear cells (PBMCs) were from Biological Specialty Corporation (Colma, PA). Fetal bovine serum (FBS) was purchased from HyClone (Logan, UT). Penicillin-streptomycin (1%) and alamarBlue cell viability reagent were bought from Invitrogen (Carlsbad, CA). Interleukin-2 (IL-2; 20 U/ml) was from Roche Applied Science (Indianapolis, IN). Gamma interferon (IFN-γ) and macrophage colony-stimulating factor (M-CSF; 50 ng/ml) were obtained from R&D Systems (Minneapolis, MN). Phytohemagglutinin P (PHA-P; 5 μg/ml) and phorbol myristate acetate (PMA) were from Sigma-Aldrich (St. Louis, MO). Clear-bottom black 96-well plates were purchased from Costar (Washington, DC). Human DNA polymerases α and ß were obtained from Chimerx (Milwaukee, WI), and human DNA polymerase γ was internally produced. Fish sperm DNA gapped with exonuclease III was from Affymetrix (Santa Clara, CA). Deoxynucleoside triphosphates (dNTPs) (1.6 μM each) were purchased from GE Healthcare (Buckinghamshire, United Kingdom). [33P]dATP (1 μCi) and an AlphaLISA kit, for p24 detection, and Wallac Microbeta and VICTOR luminometers were bought from PerkinElmer (Waltham, MA). Culture medium (RPMI 1640) and Dulbecco's modified Eagle medium-10% FBS (DMEM-FBS) were from Gibco (Carlsbad, CA). Versene-EDTA was ordered from BioWhittaker (Walkersville, MD). A Falcon microplate (384 wells) was purchased from BD (Franklin Lake, NJ). The integrase strand transfer inhibitor raltegravir (RAL) and benchmark NNRTIs were synthesized in-house. A Tecan Safire2 microplate reader was bought from Tecan US Inc. (Durham, NC). An electrochemiluminescence (ECL) detector Sector Imager S6000 and specific avidin standard plates were purchased from Meso Scale Discovery (Rockville, MD).
The following antiviral agents and cells employed for combination study were provided by Southern Research Institute (SRI, Birmingham, AL): the NRTIs lamivudine (3TC), abacavir (ABC), zidovudine (AZT), stavudine (d4T), zalcitabine (ddC), didanosine (ddI), emtricitabine (FTC), tenofovir DF (TDF), and ribavirin (RBV; hepatitis C drug); the NNRTIs DLV, EFV, NVP, ETR, and RPV; protease inhibitors (PIs) darunavir (DRV) and indinavir (IDV); the entry inhibitors (EI) maraviroc (MVC) and enfuvirtide (ENF; T20); CEM-SS cells; MAGI-CCR5 cells; CXCR4-tropic virus strain HIV-1IIIB; and CCR5-tropic virus strain HIV-1Ba-L.
HIV-1 reverse transcriptase biochemical assay.
The full-length WT and 2 mutant RT proteins were expressed in Escherichia coli BL21(DE3) cells and purified as described previously (33). The ECL RT biochemical assay was performed based on the protocol described previously (34). Briefly, the heterodimeric nucleic acid substrate used in the HIV-1 RT polymerase reactions was generated by annealing biotinylated DNA primer to a 500-nucleotide RNA template. The HIV-1 RT enzyme (final concentration of 50 pM) was combined with an inhibitor or dimethylsulfoxide (DMSO; 10% DMSO in the final reaction mixture) in assay buffer containing 62.5 mM Tris-HCl, pH 7.8, 1.25 mM dithiothreitol, 7.5 mM MgCl2, 100 mM KCl, 0.03% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate [CHAPS], 0.125 mM EGTA). This mixture was preincubated for 30 min at room temperature in microtiter plates. The polymerization reaction was initiated by the addition of template/primer substrate (final concentration, 16.6 nM), and dNTPs (final concentrations were 2 μM for dCTP, dGTP, and dATP and 66.6 nM for Ru-dUTP). After 90 min of incubation at 37°C, reactions were quenched by the addition of EDTA (25 mM). The resulting mixture was incubated for an additional 5 min at room temperature, followed by transferring the solution (50 μl) to a blocked avidin plate from Meso Scale Discovery (MSD). The mixtures were incubated at room temperature for 60 min prior to the quantification of the reaction product via an ECL 6000 imager instrument. EFV was used as a control compound in the assay, and all 50% inhibitory concentrations (IC50s) are relative values, as they are highly dependent on assay conditions.
Cytotoxicity of MK-1439 in resting and activated primary cells and proliferating cells. (i) Cell preparation.
Human PBMCs were isolated from anticoagulated blood by Ficoll gradient purification and washed 3 times in phosphate-buffered saline (PBS). PBMCs were incubated in RPMI 1640 medium supplemented with 10% FBS, 1% penicillin-streptomycin, and IL-2 (20 U/ml) for 72 h prior to compound treatment. Resting CD4+ T cells and peripheral blood monocytes were isolated by negative magnetic cell sorting. CD4+ T cells were incubated in RPMI 1640 medium supplemented with 10% heat-inactivated human serum and 1% penicillin-streptomycin, while monocytes were incubated in DMEM supplemented with 10% FBS, 2% heat-inactivated human serum, and 1% penicillin-streptomycin for 48 h prior to compound treatment. Macrophages were derived from peripheral blood monocytes cultured for 7 days in DMEM supplemented with 10% FBS, 1% penicillin-streptomycin, and M-CSF (50 ng/ml). The cells were maintained by replenishing with 50% fresh medium at least twice a week.
(ii) Cell activation.
Human PBMCs and CD4+ T cells were isolated as described above. The cells were resuspended in RPMI 1640 medium and transferred into T75 culture flasks at a density of 106 cells/ml in medium containing phytohemagglutinin P (PHA-P; 5 μg/ml) and incubated for 3 days at 37°C under 5% CO2. The PHA-treated PBMCs were collected and washed twice with RPMI 1640. The cells were then resuspended in RPMI 1640 containing IL-2 (20 U/ml) at a density of 106 cells/ml for MK-1439 cytotoxicity testing.
Monocytes and macrophages were prepared as described above. The cells were first primed with IFN-γ at 100 U/ml for 2 days. The media were removed, and the remaining cells were washed twice followed by the addition of media containing 20 nM phorbol myristate acetate (PMA). After incubation for 3 days, the cells were treated with MK-1439 for cytotoxicity testing.
Cytotoxicity test.
alamarBlue cell viability reagent was utilized for the cell cytotoxicity assay. The above-mentioned cell types were plated in clear-bottom black 96-well plates at predetermined densities (50,000 to 106 cells/well depending on the cell type) in an appropriate culture medium. A 12-point compound titration was performed with a starting concentration of 100 μM in DMSO, followed by 3-fold serial dilutions in culture medium. Serial dilutions of DMSO in parallel were included as negative controls. A previously identified cytotoxic compound was run as a positive control. After addition of the compounds to the cells, cells were incubated for approximately 72 h followed by addition of a 1/10 volume of alamarBlue cell viability reagent. Cells were incubated at 37°C for empirically determined time periods (2 to 6 h depending on cell types), and fluorescence intensity was recorded by a Tecan Safire2 microplate reader.
Human DNA polymerase assay.
MK-1439 was tested against purified human DNA polymerases α, ß, and γ at concentrations ranging from 1 to 100 μM. Buffer conditions for the DNA polymerase α were 50 mM Tris, pH 8.0, 10 mM MgCl2, 500 μg/ml bovine serum albumin (BSA; prepared and stored at 4°C until use), 2 mM dithiothreitol (DTT), 0.1 mM spermine (added fresh at time of assay), and 0.005 U/μl polymerase α. Buffer conditions for ß polymerase assays were 20 mM Tris, pH 7.5, 10 mM MgCl2,100 mM KCl, 200 μg/ml BSA (prepared and stored at 4°C until use), 2 mM β-mercaptoethanol (β-ME; added fresh at the time of assay), and 0.0025 U/μl polymerase ß. Buffer conditions for γ polymerase assays were 25 mM HEPES, pH 8.0 (adjusted up from pH 7.3), 10 mM MgCl2, 100 mM NaCl, 500 μg/ml BSA (prepared and stored at 4°C until use), 2.5 mM β-ME (added fresh at time of assay), and 0.00025 U/μl γ polymerase.
Each 50-μl reaction mixture contained 0.05 mg/ml gapped fish sperm DNA and dNTPs (1.6 μM each with 3 μCi [33P]dATP). All reactions were initiated by the addition of the polymerases. After incubation for 30 min at 37°C (60 min for γ polymerase), each reaction was quenched with 50 μl EDTA (0.5 M) and samples were collected on filter disks. Disks were washed (6×) and dried before scintillation counting. MK-1439 was assayed in a concentration range from 1 to 100 μM. Inhibition for each reaction was calculated relative to the appropriate DMSO control. Aphidicolin was the positive control for α polymerase, and ddATP was the positive control for ß/γ polymerases.
HIV multiple-cycle replication assay.
MK-1439 was tested in HIV-1 multiple-cycle replication assays using genetically defined WT and laboratory mutant viruses (K103N, Y181C, and K103N/Y181C) as described previously (26, 27) Antiviral activity assays were performed using variants of a laboratory HIV-1 isolate, R8, and MT-4 human T-lymphoid cells in cell culture medium supplemented with 10% or 50% normal human serum (NHS). EFV was used as a control compound in the assay, and all EC50s are relative values as they are highly dependent on assay conditions.
HIV-1 single-cycle replication assay with viruses containing RT sequences obtained from patient isolates.
To assess how the potency of the compounds was affected by different HIV-1 subtypes and NNRTI-associated subtype B mutant viruses, Monogram Biosciences performed a PhenoScreen assay with MK-1439, EFV, ETR, and RPV against a group of isolates from different subtypes, as well as a broader panel of NNRTI-associated mutant viruses, using a variation of their clinical diagnostic assays as described previously (35).
Combination study.
The combination study was conducted by SRI in a 2-drug combination with MK-1439 and each of the 18 FDA-approved antiviral agents described above. The combination inhibitory effect of MK-1439 on replication of the HIV strains was analyzed using the Prichard and Shipman MacSynergy II three-dimensional model (36). RBV and d4T were used as the positive controls as an antagonism pair, because the antiviral activity of d4T is antagonized by RBV, as described previously (26).
For these studies, synergy is defined as drug combinations yielding synergy volumes greater than 50. Slightly synergistic activity and highly synergistic activity have been operationally defined as yielding synergy volumes of 50 to 100 and >100, respectively. Additive drug interactions have synergy volumes in the range from −50 to 50, while synergy volumes between −50 and −100 are considered slightly antagonistic and those less than −100 are highly antagonistic.
RESULTS
Biochemical inhibitory potency of MK-1439.
The potencies of MK-1439 against purified recombinant WT RT and RT with the 2 most prevalent amino acid substitutions (K103N or Y181C) were measured in biochemical assays. As shown in Table 1, the results showed that MK-1439 inhibited RNA-dependent DNA synthesis with IC50s of 12.2, 9.7, and 9.7 nM for WT, K103N, and Y181C RT viruses, respectively. These results suggest that K103N and Y181C substitutions in RT have no impact on the affinity of MK-1439 to the NNRTIBP. In the same assay, K103N RT showed approximately 10-fold reduced affinity to EFV, and Y181C RT displayed moderately reduced affinity to ETR and RPV, which is consistent with literature reports (18, 26, 27).
TABLE 1.
Inhibitory potency of MK-1439, EFV, and ETR against the WT and mutant viruses
| Compound | IC50a (nM) |
||
|---|---|---|---|
| WT | K103N | Y181C | |
| MK-1439 | 4.5 ± 0.8 (6) | 5.5 ± 0.8 (6) | 6.1 ± 1.3 (6) |
| EFV | 0.81 ± 0.27 (753) | 7.1 ± 3.2 (131) | 1.0 ± 0.49 (132) |
| ETR | 0.47 ± 0.15 (7) | 0.89 ± 0.21 (6) | 1.6 ± 0.4 (6) |
| RPV | 0.77 ± 0.30 (7) | 1.5 ± 0.5 (6) | 1.4 ± 0.4 (6) |
IC50s are means ± standard deviations. Numbers in parentheses indicate the number of times the assays were performed.
Cytotoxicity of MK-1439 with various cell types.
As HIV-1 is known to infect activated and resting primary cells as well as proliferating cells, it is important to assess the impact of antiviral agents on the viability of those cell types. Consequently, MK-1439 was tested for its effects on the viability of resting and activated CD4+ T cells, PBMCs, monocytes, and macrophages and in proliferating transformed cell lines, such as MT4, SupT1, and HL60 cell lines, with a previously identified cytotoxic compound as a control. MK-1439 did not display cytotoxicity in any cell type tested, whether proliferating, stationary, or activated, at concentrations of up to 100 μM.
Biochemical activity of MK-1439 against human DNA polymerases α, ß, and γ.
The potential for off-target activity of MK-1439 against mechanistically similar enzymes was evaluated by assessing its inhibitory activity against human DNA polymerases α, ß, and γ. The results showed that MK-1439 exhibited greater than 10,000-fold selectivity with respect to the cellular DNA polymerases α, ß, and γ with IC50s of >100 μM.
Off-target activity evaluation.
To assess its specificity, MK-1439 was evaluated against a large panel of enzymes, transporters, ion channels, and receptor assays at Ricerca Biosciences. In the screen with more than 110 protein targets, MK-1439 showed an IC50 of greater than 10 μM against all targets except 5-HT2ß, where an IC50 of 2.5 μM was noted in a ligand binding assay. However, the 5-HT2ß activity was not observed in a functional cell-based assay monitoring the accumulation of inositol-1-phosphate. Therefore, the binding activity does not appear to translate into a 5-HT2ß functional response.
Antiviral activity of MK-1439.
As mentioned above, K103N and Y181C mutant viruses account for much of the NNRTI-resistant mutations detected in patients who have failed with antiretroviral regimens containing earlier NNRTIs, such as EFV and NVP, and these mutations can sometimes be found in circulating HIV-1 that may infect people with the preexisting resistance mutation(s). Therefore, MK-1439 was evaluated for its potency against the WT and 2 mutant viruses in cell culture replication assays using a transformed human cell line and laboratory HIV-1 isolates. In addition to the conditions under 10% NHS, the inhibitory potency of MK-1439 was evaluated in serum concentrations of 50% NHS to assess the effect of serum concentration on the potency, since binding to serum proteins often can reduce the efficacy of antiviral agents. An MT-4 human T-lymphoid cell line infected with a laboratory HIV-1 isolate (R8) was employed to test the antiviral activity of MK-1439. The assay is a multiple-cycle replication assay in which infections are initiated at a low multiplicity of infection (MOI), and the virus is allowed to spread through cells in culture over a period of 3 days. The extent of replication is quantified by measuring the amount of HIV-1 capsid antigen, p24, in culture supernatants using a commercial AlphaScreen assay kit.
In the presence of 10% NHS, MK-1439 had an EC95 of 11.0 against WT virus and EC95 of 13.4, 16.4, and 30.5 against K103N, Y181C, and K103N/Y181C mutant viruses, respectively (Table 2). These results showed that MK-1439 had a less than 2-fold shift in potency against the 2 most prevalent NNRTI-resistant viruses (K103N and Y181C mutants) and less than a 3-fold shift against the K103N/Y181C double mutant. Under the same conditions, both ETR and RPV did not show a shift in potency for the K103N mutant virus from that of the WT virus. However, ETR displayed more than 5- and 11-fold shifts in potency in inhibiting the replication of Y181C and K103N/Y181C mutant viruses, respectively, compared to the WT virus. RPV exhibited a smaller potency shift of 2- and 6-fold against Y181C and K103N/Y181C mutant viruses, respectively. K103N and K103N/Y181C mutant viruses conferred greater than 40- and 50-fold reductions in susceptibility to EFV, and EFV had a less than 2-fold shift in potency with the Y181C mutant virus.
TABLE 2.
Antiviral activity of MK-1439, EFV, and ETR against WT and mutant viruses in the presence of 10% and 50% NHS
| Compound and supplement | EC95a (nM) |
|||
|---|---|---|---|---|
| WT | K103N | Y181C | K103N/Y181C | |
| 10% NHS | ||||
| MK-1439 | 11.0 ± 2.6 (11) | 13.4 ± 3.3 (11) | 16.4 ± 3.2 (9) | 30.5 ± 3.9 (8) |
| EFV | 5.0 ± 3.2 (203) | 247 ± 83 (152) | 8.6 ± 4.5 (45) | 297 ± 129 (41) |
| ETR | 4.4 ± 2.1 (29) | 5.3 ± 3.9 (28) | 23.5 ± 12 (11) | 52.5 ± 23.9 (11) |
| RPV | 2.0 ± 1.0 (8) | 1.7 ± 1.0 (5) | 4.3 ± 3.1 (6) | 12.9 ± 9.9 (6) |
| 50% NHS | ||||
| MK-1439 | 20 ± 6.7 (9) | 42 ± 7.8 (7) | 27 ± 14 (5) | 55 ± 14 (7) |
| EFV | 41 ± 24 (193) | 1427 ± 53 (22) | 80 ± 34 (25) | 2943 ± 903 (12) |
| ETR | 38 ± 22 (42) | 36 ± 9.8 (24) | 263 ± 191 (19) | 653 ± 216 (22) |
| RPV | 37 ± 16 (11) | 48 ± 18 (7) | 120 ± 26 (6) | 407 ± 153 (6) |
EC95 are means ± standard deviation. Numbers in parentheses indicate the number of times the assays were performed.
In the presence of 50% serum, MK-1439 displayed an EC95 of 20.3, 42.5, 27.4, and 55.1 nM against WT virus and virus carrying K103N, Y181C, and K103N/Y181C substitutions, respectively. As expected, all 4 NNRTIs showed that fold changes (FCs) in potency with mutant viruses relative to WT were similar to what was observed in the presence of 10% serum.
With respect to the effect of serum concentration on the potency of NNRTIs, MK-1439 had a 2- to 3-fold shift in potency against the WT and the 3 variants when the serum level increased from 10% to 50%. In contrast, serum content exhibited a greater impact on the potency of EFV and ETR, with a 6- to 10-fold and a 7- to 12-fold increase in EC95, respectively, as the serum concentration was increased from 10% to 50%. Furthermore, RPV displayed the biggest shift in potency (18- to 31-fold) among the 4 NNRTIs when the serum was increased from 10% to 50%. Consistent with the results in 10% serum, viruses containing the K103N mutant also displayed significant resistance to EFV in the presence of 50% serum. As shown in Table 2, K103N and K103N/Y181C mutant viruses conferred 35- and 70-fold resistance to EFV, respectively, compared to WT virus. As a result, MK-1439 was 34- and 54-fold more potent than EFV against K103N and K103N/Y181C mutants, respectively, and was 2- to 3-fold more potent than EFV against WT virus and Y181C mutant virus under the same conditions. Compared to ETR and RPV, MK-1439 displayed approximately 2-fold better potency against the WT virus and had a comparable potency against the K103N mutant. However, MK-1439 was 9- and 4-fold more potent than ETR and RPV, respectively, in suppressing the replication of Y181C mutant viruses. The K103N/Y181C mutant virus was 10- and 6-fold less susceptible to ETR and RPV, respectively, than to MK-1439. Therefore, MK-1439 exhibited better potency overall against WT and the most prevalent NNRTI-associated mutant viruses than EFV, ETR, and RPV in the presence of a high percentage of serum.
Susceptibility of different HIV-1 subtypes to NNRTIs.
The antiviral activity of MK-1439 and other NNRTIs was also evaluated against 10 different subtypes of viruses. Several isolates from each subtype were selected for testing (a total of 93 viruses). As shown in Table 3, no viral subtype displayed a greater than 1.5-fold shift in potency with MK-1439 compared to that of the control virus (EC50 of 4.2 nM), a drug-sensitive reference strain (CNDO) with PR and RT sequence derived from laboratory HIV strain NL4-3 (35). Other NNRTIs tested in this study also did not show an obvious shift in potency with different HIV-1 subtypes. Among the 10 subtype viruses, subtype H virus appeared to be hypersensitive to the NNRTIs with FCs from 0.17 to 0.3, which was reported previously (26).
TABLE 3.
Fold change of inhibitory potency of NNRTIs tested with different HIV-1 subtypes
| Subtype (n) | Fold change in inhibitory potency |
|||
|---|---|---|---|---|
| EFV | ETR | RPV | MK-1439 | |
| A (5) | 0.81 ± 0.34 | 0.76 ± 0.36 | 0.84 ± 0.45 | 0.84 ± 0.33 |
| A1 (13) | 0.70 ± 0.16 | 0.73 ± 0.36 | 0.73 ± 0.32 | 0.68 ± 0.19 |
| AE (5) | 0.75 ± 0.27 | 072 ± 0.28 | 0.71 ± 0.27 | 0.79 ± 0.44 |
| AG (18) | 0.75 ± 0.21 | 0.66 ± 0.15 | 0.68 ± 0.17 | 0.92 ± 0.40 |
| B (7) | 1.02 ± 0.52 | 0.79 ± 0.22 | 0.75 ± 0.20 | 0.99 ± 0.42 |
| BF (4) | 1.02 ± 0.39 | 1.02 ± 0.37 | 0.92 ± 0.32 | 1.44 ± 0.73 |
| C (22) | 0.89 ± 0.27 | 0.77 ± 0.19 | 0.72 ± 0.19 | 1.07 ± 0.36 |
| D (9) | 0.91 ± 0.16 | 0.74 ± 0.12 | 0.71 ± 0.15 | 0.94 ± 0.29 |
| G (8) | 0.74 ± 0.16 | 0.62 ± 0.15 | 0.61 ± 0.15 | 0.93 ± 0.27 |
| H (2) | 0.29 ± 0.01 | 0.18 ± 0.08 | 0.17 ± 0.07 | 0.30 ± 0.01 |
A drug-sensitive strain was employed as the control to calculate the fold changes, and fold changes are means ± standard deviations.
Susceptibility of NNRTI-associated mutant viruses to MK-1439.
HIV-1 variants selected for resistance to NNRTIs either in cell culture or in the clinic harbor amino acid substitutions in the NNRTIBP, such as K103N, Y181C, V106A, G190A, P236L, E138K, and K101E (37). To evaluate the impact of various known NNRTI-resistant mutants on the potency of MK-1439, the inhibitor was tested against wild-type HIV-1 and a panel of mutant viruses that are commonly associated with currently marketed NNRTIs (EFV, NVP, ETR, and RPV) in a single-cycle replication assay.
As shown in Fig. 2, of the 23 resistant mutant viruses tested, only 2 mutant viruses (carrying Y188L and V106A/G190A/F227L substitutions) displayed more than a 50-fold shift in potency against MK-1439. On the other hand, 9 mutant viruses showed fold changes of more than 50 with EFV. For instance, double mutant K103N/G190A virus conferred greater than 250-fold resistance versus EFV but showed no reduced susceptibility to MK-1439. The other example is the G190S mutant, which conferred more than 150-fold resistance to EFV, yet the mutant virus was more susceptible to MK-1439, with an FC of 9. In this assay format, the V106A mutant virus conferred more than 40-fold reduced susceptibility to MK-1439 but was susceptible to EFV, ETR, and RPV. However, in the multiple-cycle assay, the V106A mutant showed 11-fold reduced susceptibility to MK-1439 (data not shown). The discrepancy may be due to the different backgrounds of viruses and assay platforms used in the respective assays, as described in Materials and Methods. The overall mutant profile of MK-1439 is superior to that of EFV and similar to those of ETR and RPV, except the virus carries V106A/G190A/F227L substitutions.
FIG 2.

Susceptibility of mutant viruses containing NNRTI-associated mutants to NNRTIs. (A) MK-1439; (B) EFV; (C) RPV; (D) ETR. An asterisk indicates that the EC50 was not reached at highest concentration tested. CNDO is a drug-sensitive reference standard that is used to determine the fold changes in drug susceptibility of patient samples. The EC50 were 1, 0.9, 0.6, and 2.4 nM for EFV, RPV, ETR, and MK-1439, respectively, against CNDO. MDRC4 is a multidrug-resistant virus control that is used to evaluate and monitor assay performance. The fold change was defined by the ratio of EC50 between the mutant virus and CNDO. The EC50 was obtained from a single compound titration (n = 1).
To further assess the mutant profile of MK-1439 compared to other NNRTIs, an extended panel of NNRTI-associated clinical isolates (a total of 96 mutant viruses) was employed in the study, and the antiviral activity of the NNRTIs against these mutant viruses was performed by Monogram Biosciences under the same assay format (see Table S1 in the supplemental material).
To compare the mutant profiles of the NNRTIs, here we define a viral variant (or clinical isolate) as resistant to a specific NNRTI when the FC in potency is greater than 10 compared to the control virus. Based on this criterion, among the 96 viruses, 62 (65%), 16 (17%), 15 (16%), and 18 (19%) viruses were resistant to EFV, MK-1439, ETR, and RPV, respectively (Fig. 3). A majority of the mutant viruses that displayed reduced susceptibility to MK-1439 (>10-fold shift in EC50) also showed resistance to EFV (15/16; 94%) and RPV (12/16; 75%), whereas less than 50% (44%; 7/16) of MK-1439-resistant viruses were found to have reduced susceptibility to ETR (data not shown). In addition, more than 30% of the mutant viruses exhibited greater than 100-fold resistance to EFV (Fig. 3B). On the other hand, the majority of the mutant viruses (>60%) displayed an FC of ≤5 when tested with MK-1439 and the other two NNRTIs (Fig. 3A, C, and D).
FIG 3.

Susceptibility of a panel of 96 clinically relevant NNRTI-associated mutant viruses to NNRTIs. (A) MK-1439; (B) EFV; (C) RPV; (D) ETR.
Susceptibility of clinical isolates that are resistant to EFV, ETR, and RPV (FC >10) to MK-1439 was also analyzed. As shown in Fig. 4, more than 60% (38/62) of EFV-resistant viruses were susceptible to MK-1439, with a ≤5-fold shift in EC50 (Fig. 4A). A majority (12/18; 67%) of RPV-resistant viruses also conferred reduced susceptibility to MK-1439, with an FC of >10. Among 15 mutant viruses that showed a greater than 10-fold shift in EC50 with ETR, 8 of them exhibited a <10-fold shift in potency with MK-1439. Therefore, MK-1439 not only shows superior mutant profile to EFV but also is highly active against the majority of EFV-resistant viruses.
FIG 4.

Susceptibility of EFV, ETR, and RPV Resistant Viruses to MK-1439. (A) MK-1439- versus EFV-resistant viruses; (B) MK-1439- versus RPV-resistant viruses; (C) MK-1439- versus ETR-resistant viruses.
Susceptibility of RPV-associated mutant viruses to MK-1439.
In the ECHO and THRIVE phase III trials of RPV, the most common NNRTI resistance-associated mutants emerging at failure in the RPV-treated group were E138K (45%) followed by K101E (13%), H221Y (10%), and Y181C (8%) (38). In these studies, most of the E138K substitutions occurred together with NRTI-resistant mutants, particularly the M184I/V substitution, with the majority in a combination of E138K/M184I (47%) substitutions. In addition, K101E mutant virus was also identified as one of the frequently selected viruses upon ETR treatment. To evaluate the susceptibility of these mutant viruses to MK-1439, site-directed mutagenesis was employed to generate E138K, K101E, M184I, and M184V substitutions as single mutants as well as double mutants with E138K/M184I or E138K/M184V and K101E/M184I or K101E/M184V substitutions. The level of resistance was evaluated in a single-cycle replication assay. As shown in Fig. 5, the E138K mutant displayed an ∼4-fold reduction in susceptibility to RPV and ETR and less than 3-fold reduction in susceptibility to MK-1439. M181I and M184V single mutants, as expected, did not show a reduction in susceptibility to any of the 3 NNRTIs, as they are NRTI-associated mutant viruses. The level of susceptibility of the K101E mutant to ETR, RPV, and MK-1439 is similar to the level of susceptibility displayed by E138K mutant virus. However, K101E mutant virus displays a higher level of resistance to EFV than the E138K mutant (FC of 9.2 and 1.9, respectively). The double mutant E138K/M181I virus maintained levels of susceptibility to the NNRTIs similar to those of the single E138K mutant. In contrast, the double mutant E138K/M184V virus conferred a higher level of susceptibility to the NNRTIs (FC of ≤2) than E138K and E138K/M184I mutants. The same phenomenon was also observed in the mutants carrying the K101E substitution in combination with the M181I or M184V substitution. These results are consistent with reports from the literature that the single and double mutant viruses did not show significant resistance to RPV (39–42).
FIG 5.

Susceptibility of RPV-associated mutant viruses to NNRTIs. Black bar, EFV; gray bar, RPV; cross-hatched bar, ETR; open bar, MK-1439. The experiments were repeated at least 3 times. Fold changes are in means ± standard deviations.
Two-drug combination study.
The necessity of combination therapy for the treatment of HIV-1 infection often presents challenges owing to potential antagonistic interactions among different classes of antiviral agents. In particular, the potential effect of 1 component in the combination therapy may have an effect on the antiviral activity of another component(s), which could be synergistic, additive, or antagonistic. Therefore, the anti-HIV-1 activity of MK-1439 was evaluated in 2-drug combination studies with each of 18 FDA-approved HIV-1 antiviral agents. The cytotoxicity of each 2-drug combination was also evaluated in parallel with the antiviral activities. The positive antagonism control of d4T in combination with RBV was tested in parallel with each of the assays. Analysis of drug interactions for each of the 2-drug combinations was performed using a MacSynergy II (Prichard and Shipman), a three-dimensional model for statistical evaluation of combination assays (36). As a result, based on a 3-dimensional model, a merely additive effect will result in a horizontal plane at 0% inhibition (plane of additivity) when the calculated individual antiviral activity was subtracted from the total antiviral activity determined in the 2-drug combination study. A synergistic or antagonistic effect, on the other hand, will render a surface above or below the plane of additivity, respectively. Four representative 3-dimensional plots are presented in Fig. 6. Figure 6A, B, C, and D represent the combination study of MK-1439 with FTC (a nucleoside inhibitor), TDF (a nucleotide inhibitor), RAL (an integrase inhibitor), and DRV (a PI), respectively. All results show that the percent inhibition after subtraction was on or close to the plane of additivity, which is indicative of an additive effect based on criteria described in Materials and Methods. In contrast, the positive antagonism control for the combination study (d4T in combination with RBV) exhibited a highly antagonistic interaction in all experiments (mean antagonism volume of −298 μM2% in CEM-SS cells and −412 μM2% in MAGI-CCR5 cells). The results are summarized in Table 4. No antagonistic interactions were observed within the concentration ranges examined for antiviral efficacy between MK-1439 and each of the 18 FDA-approved antiretroviral drugs used in this study. A slightly synergistic interaction was observed between MK-1439 and RAL (average synergy volume of 60.8 nM2%; slight synergy observed in 2/3 experiments), suggesting a possible beneficial interaction between these 2 drugs. Therefore, the data suggest a lack of antagonism of the antiviral effects between MK-1439 and each of the 18 FDA-approved antiretroviral drugs evaluated in this study, and it should not be problematic with the use of MK-1439 in combination with other antiviral agents in clinical settings.
FIG 6.

Mean three-dimensional plot of MK-1439 and other antiviral agents in the two-drug combination study. (A) MK-1439 and FTC; (B) MK-1439 and TDF; (C) MK-1439 and RAL; (D) MK-1439 and DRV.
TABLE 4.
Antiviral efficacy of MK-1439 in combination with 18 FDA-approved antiretroviral compounds in CEM-SS or MAGI-CCR5 cells (95% confidence values)
| Compounda | Mean synergy/antagonism volumeb (n = 3) | Antiviral effects |
|---|---|---|
| NRTI | ||
| Lamivudine (3TC) | 32.4/−0.15 nM2% | Nonantagonistic |
| Abacavir (ABC) | 43.9/−2.13 nMµM% | Nonantagonistic |
| Zidovudine (AZT) | 20.6/−7.06 nM2% | Nonantagonistic |
| Stavudine (d4T) | 43.7/−3.90 nMµM% | Nonantagonistic |
| Zalcitabine (ddC) | 15.1/−3.43 nM2% | Nonantagonistic |
| Didanosine (ddI) | 11.4/−0.70 nMµM% | Nonantagonistic |
| Emtricitabine (FTC) | 19.6/0 nM2% | Nonantagonistic |
| Tenofovir DF (TDF) | 26.7/−6.46 nM2% | Nonantagonistic |
| NNRTI | ||
| Delavirdine (DLV) | 18.6/−11.4 nM2% | Nonantagonistic |
| Efavirenz (EFV) | 4.81/−4.05 nM2% | Nonantagonistic |
| Etravirine (ETR) | 36.6/−11.4 nM2% | Nonantagonistic |
| Nevirapine (NVP) | 3.79/−3.04 nM2% | Nonantagonistic |
| Rilpivirine (RPV) | 6.78/−3.33 nM2% | Nonantagonistic |
| PI | ||
| Darunavir (DRV) | 40.9/−4.54 nM2% | Nonantagonistic |
| Indinavir (IDV) | 5.13/−5.93 nM2% | Nonantagonistic |
| EI | ||
| Maraviroc (MVC) | 35.7/−0 nM2% | Nonantagonistic |
| Enfuvirtide (ENF) | 14.4/−20.4 nM2% | Nonantagonistic |
| InI | ||
| Raltegravir (RAL) | 60.8/−7.27 nM2% | Nonantagonistic |
| Positive control | ||
| Stavudine/ribavirin | ||
| CEM-SS cells | 1.50/−298 µM2% | Highly antagonistic |
| MAGI-CCR5 cells | 27.2/−412 µM2% | Highly antagonistic |
The antiviral efficacy of MK-1439 in combination with maraviroc was performed in MAGI-CCR5 cells. All other evaluations were performed in CEM-SS cells.
The antiviral synergy plot (95%) datasets from multiple experiments (n = 3) are combined, and arithmetic means are calculated for each drug-drug concentration. The positive and negative values are individually summed to give mean volumes for synergistic and antagonistic interactions, respectively.
DISCUSSION
NNRTIs are a cornerstone of HAART; however, a lower barrier to the development of resistant viruses of this class of inhibitors renders a broad spectrum of prevalent NNRTI-associated mutant viruses that would limit the options for the treatment of HIV-1-infected patients. In addition, EFV is associated with significant central nervous system (CNS) effects, and RPV is not approved to treat high-viral-load patients (>100,000 copies/ml). Consequently, new NNRTIs that are able to address these issues are intensively sought after in the field.
MK-1439 is a novel NNRTI which shows a lower level of potency shift as the serum concentration increases from 10% to 50% than other NNRTIs tested in this study. This is not unexpected, because MK-1439 has plasma protein binding of 76% compared to EFV, ETR, and RPV, which all have plasma protein binding greater than 99.5%. In the presence of 50% serum, MK-1439 showed more than 30-fold improved potency compared to that of EFV against K103N mutant virus and more than 9- and 4-fold better potency than ETR and RPV, respectively, against Y181C mutant virus. Therefore, it seems likely that, in the presence of 100% serum, the difference in potency between MK-1439 and the other NNRTIs is even greater against the respective mutant viruses, which would make MK-1439 the most potent NNRTI against K103N and Y181C mutant viruses to date in cell-based assays. As mentioned above, K103N and Y181C mutant viruses are not only the most prevalent NNRTI-associated mutants identified in patients who have failed on NNRTI containing regimen but also the most prevalent transmitted NNRTI-associated mutants in treatment-naive patients. Therefore, both mutant viruses present a significant threat to naive patients receiving an NNRTI-containing regimen, as transmitted mutant viruses increase the virologic failure rate of HAART (30–32). Furthermore, the Y181C mutant was frequently associated with patients who have failed with ETR- or RPV-containing regimens in clinical settings (23, 24). Given the association of the K103N mutant with the earlier NNRTIs (EFV and NVP) and the association of the Y181C mutant with the newer NNRTIs (ETR and RPV), it is highly desirable that new NNRTIs display excellent potency against both mutants. The resistance profile of MK-1439 suggests it is capable of fulfilling this medical need. More importantly, a long-term toxicity study in preclinical species suggested that MK-1439 exhibits a much better safety and tolerability profile than EFV and RPV in clinical settings (data not shown).
Although single Y188L mutant viruses exhibited a significant reduction in the susceptibility to MK-1439 (FC of >100), the mutant was not selected by MK-1439 during in vitro resistance selection (data not shown). Virus carrying the Y188L substitution also showed greater than 100-fold resistance to EFV, but it was only observed in 5% of the patients who have failed in treatments with an EFV-containing regimen. The replication capacity of Y188L mutant virus reportedly is comparable to that of K103N mutant virus (43). The lack of a prevalent Y188L mutant may have been due to the fact that two base changes are required to generate the Y188L mutant, while the K103N mutant only needs a single base change (44, 45) The Y188L mutant is derived from an intermediate mutant, either Y188H or Y188F substitution. Both mutants are highly susceptible to NNRTIs; thus, the opportunity to further develop into Y188L mutant may be limited (45).
Based on X-ray structures of RT, K101 (in subunit p66) and E138 (in subunit p51) are located at the top of the NNRTIBP and form a salt bridge via a water molecule, representing one of the entrances for NNRTIs (40, 46). Interactions between the 2 residues may play an important role in controlling the NNRTIBP in an open or closed conformation of this entrance. As a result, substitution of either residue (E138K or K101E) prevents the formation of such a salt bridge and keeps the entrance in an open form. The fact that viruses carrying E138K (45%) and K101E (13%) substitutions are the 2 most frequent mutants associated with the RPV-containing regimen suggests that RPV enters the NNRTIBP via this entrance. This hypothesis is supported by the finding that the E138K mutant enhanced the dissociation of RPV from the NNRTIBP, which is in an open-form conformation due to the replacement of glutamic acid with lysine (46). In addition, the fact that K101E/M184I or K101E/M184V mutants showed a phenomenon similar to that of the E138K/M184I or E138K/M184V mutants with respect to the susceptibility to NNRTIs also provides evidence that the salt bridge plays an important role in binding of RPV and ETR to the NNRTIBP. The K101E/M184I or K101E/M184V mutant is not as prevalent as the I or E138K/M184V mutant in patients who have failed with the RPV-containing regimen. This may be ascribed to the K101E mutant not being able to restore the fitness of M181I or V virus to the level that the E138K mutant did. E138K substitution is the most frequently identified mutant in the resistance selection study with RPV, but the mutant was not selected by MK-1439 in the study (data not shown). These results suggest that both inhibitors enter the NNRTIBP through different channels. Further investigation is required to test this hypothesis.
Although the E138K mutant virus confers only a low level of reduction in susceptibility to RPV (∼4-fold), it is the most prevalent mutant identified in patients with virologic failure while on an RPV-containing regimen. This may be due to the low trough level of RPV at 24 h (C24h with 25 mg dose, ∼260 nM) relative to the EC95 of RPV (37 nM) in the presence of 50% serum (C24h/EC95, ∼7). Given the high protein binding of RPV (99.6%), it is anticipated that the EC95 will be further increased in the presence of 100% serum; hence, the ratio of trough level to EC95 (in 100% serum) will be further reduced (<7-fold). Furthermore, in a clinical setting, it is not uncommon to see high variation in the pharmacokinetic profile of drug candidates among HIV-1 patients; thus, some patients may have trough levels significantly lower than 260 nM. As a result, even mutant viruses with a low level of resistance would be able to circumvent the suboptimal trough level. On the other hand, given that MK-1439 has a 2-fold better potency than RPV, coupled with its lower protein binding (76%) and higher trough levels (267 nM at 25 mg and 1,650 nM at 200 mg in Ph1b) (47) relative to the mutant EC95 values (20 nM), the survival of the E138K or K101E mutant virus becomes greatly diminished under such high drug pressures.
In summary, MK-1439 may represent the most effective NNRTI against the two most prevalent NNRTI-associated mutant viruses (with K103N and Y181C substitutions) compared to the NNRTIs approved by the FDA in the presence of 100% serum in a cell-based assay. When tested against a broad panel of common NNRTI mutant viruses, MK-1439 displayed a mutant profile superior to that of EFV and similar to those of ETR and RPV. Furthermore, given the high trough levels of MK-1439 observed in the clinical setting, the chance for E138K and K101E mutant viruses surviving is relatively low due to the low FCs with MK-1439. MK-1439 showed similar potency against different HIV subtypes and did not show any antagonistic effects with other antiviral agents tested in the combination study. In addition, MK-1439 is a highly specific RT inhibitor with a selectivity factor of >1,000 (based on in vitro activities) versus a broad panel of enzymes, transporters, ion channels, and receptors. Taking these results together, the virologic profile of MK-1439 supports its further development as a new NNRTI for the treatment of HIV-1 infection.
Supplementary Material
Footnotes
Published ahead of print 30 December 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02403-13.
REFERENCES
- 1.Gotte M, Li XG, Wainberg MA. 1999. HIV-1 reverse transcription: a brief overview focused on structure-function relationships among molecules involved in initiation of the reaction. Arch. Biochem. Biophys. 365:199–210. 10.1006/abbi.1999.1209 [DOI] [PubMed] [Google Scholar]
- 2.Castro HC, Loureiro NIV, Pujol-Luz M, Souza AMT, Albuquerque MG, Santos DO, Cabral LM, Frugulhetti IC, Rodrigues CR. 2006. HIV-1 reverse transcriptase: a therapeutical target in the spotlight. Curr. Med. Chem. 13:313–324. 10.2174/092986706775476089 [DOI] [PubMed] [Google Scholar]
- 3.Moore JP, Stevenson M. 2000. New targets for inhibitors of HIV-1 replication. Nat. Rev. Mol. Cell Biol. 1:40–49. 10.1038/35036060 [DOI] [PubMed] [Google Scholar]
- 4.Hopkins AL, Ren J, Esnouf RM, Willcox BE, Jones EY, Ross C, Miyasaka T, Walker RT, Tanaka H, Stammers DK, Stuart DI. 1996. Complexes of HIV-1 reverse transcriptase with inhibitors of the HEPT series reveal conformational changes relevant to the design of potent non-nucleoside inhibitors. J. Med. Chem. 39:1589–1600. 10.1021/jm960056x [DOI] [PubMed] [Google Scholar]
- 5.Ambrose Z, Julias JG, Boyer PL, Kewalramani VN, Hughes SH. 2006. The level of reverse transcriptase (RT) in human immunodeficiency virus type 1 particles affects susceptibility to nonnucleoside RT inhibitors but not to lamivudine. J. Virol. 80:2578–2581. 10.1128/JVI.80.5.2578-2581.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palella FJ, Delaney KM, Moorman AC, Loveless MO, Fuhrer J, Satten GA, Aschman DJ, Holmberg SD. 1998. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. N. Engl. J. Med. 338:853–860. 10.1056/NEJM199803263381301 [DOI] [PubMed] [Google Scholar]
- 7.Schneider MF, Gange SJ, Williams CM, Anastos K, Greenblatt RM, Kingsley L, Detels R, Munoz A. 2005. Patterns of the hazard of death after AIDS through the evolution of antiretroviral therapy: 1984–2004. AIDS 19:2009–2018. 10.1097/01.aids.0000189864.90053.22 [DOI] [PubMed] [Google Scholar]
- 8.Gazzard B, Bernard AJ, Boffito M, Churchill D, Edwards S, Fisher N, Geretti AM, Johnson M, Leen C, Peters B, Pozniak A, Ross J, Walsh J, Wilkins E, Youle M, Writing Committee British HIV Association 2006. British HIV Association (BHIVA) guidelines for the treatment of HIV-infected adults with antiretroviral therapy (2006). HIV Med. 7:487–503. 10.1111/j.1468-1293.2006.00424.x [DOI] [PubMed] [Google Scholar]
- 9.Hammer SM, Saag MS, Schechter M, Montaner JS, Schooley RT, Jacobsen DM, Thompson MA, Carpenter CC, Fischl MA, Gazzard BG, Gatell JM, Hirsch MS, Katzenstein DA, Richman DD, Vella S, Yeni PG, Volberding PA, International AIDS Society–USA Panel 2006. Treatment for adult HIV infection: 2006 recommendations of the International AIDS Society–USA panel. JAMA 296:827–843. 10.1001/jama.296.7.827 [DOI] [PubMed] [Google Scholar]
- 10.Bacheler L, Jeffrey S, Hanna G, D'Aquila R, Wallace L, Logue K, Cordova B, Hertogs K, Larder B, Buckery R, Baker D, Gallagher K, Scarnati H, Tritch R, Rizzo C. 2001. Genotypic correlates of phenotypic resistance to efavirenz in virus isolates from patients failing nonnucleoside reverse transcriptase inhibitor therapy. J. Virol. 75:4999–5008. 10.1128/JVI.75.11.4999-5008.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Delaugerre C, Rohban R, Simon A, Mouroux M, Tricot C, Agher R, Huraux JM, Katlama C, Calvez V. 2001. Resistance profile and cross-resistance of HIV-1 among patients failing a non-nucleoside reverse transcriptase inhibitor-containing regimen. J. Med. Virol. 65:445–448. 10.1002/jmv.2055 [DOI] [PubMed] [Google Scholar]
- 12.Johnson LB, Saravolatz LD. 2009. Etravirine, a next-generation nonnucleoside reverse-transcriptase inhibitor. Clin. Infect. Dis. 48:1123–1128. 10.1086/597469 [DOI] [PubMed] [Google Scholar]
- 13.Xu H, Quan Y, Brenner BG, Bar-Magen T, Oliveira M, Schader SM, Wainberg MA. 2009. Human immunodeficiency virus type 1 recombinant reverse transcriptase enzymes containing the G190A and Y181C resistance mutations remain sensitive to etravirine. Antimicrob. Agents Chemother. 53:4667–4672. 10.1128/AAC.00800-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barth RE, van der Loeff MF, Schuurman R, Hoepelman AI, Wensing AM. 2010. Virological follow-up of adult patients in antiretroviral treatment programmes in sub-Saharan Africa: a systematic review. Lancet Infect. Dis. 10:155–166. 10.1016/S1473-3099(09)70328-7 [DOI] [PubMed] [Google Scholar]
- 15.Shahriar R, Rhee SY, Liu TF, Fessel WJ, Scarsella A, Towner W, Holmes SP, Zolopa AR, Shafer RW. 2009. Nonpolymorphic human immunodeficiency virus type 1 protease and reverse transcriptase treatment-selected mutations. Antimicrob. Agents Chemother. 53:4869–4878. 10.1128/AAC.00592-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tambuyzer L, Azijn H, Rimsky LT, Vingerhoets J, Lecocq P, Kraus G, Picchio G, de Bethune MP. 2009. Compilation and prevalence of mutations associated with resistance to non-nucleoside reverse transcriptase inhibitors. Antivir. Ther. 14:103–109 [PubMed] [Google Scholar]
- 17.Johnson VA, Brun-Vezinet F, Clotet B, Gunthard HF, Kuritzkes DR, Pillay D, Schapiro JM, Richman DD. 2007. Update of the drug resistance mutations in HIV-1: 2007. Top. HIV Med. 15:119–125 [PubMed] [Google Scholar]
- 18.Nunberg JH, Schleif WA, Boots EJ, O'Brien JA, Quintero JC, Hoffman JM, Emini EA, Goldman ME. 1991. Viral resistance to human immunodeficiency virus type 1-specific pyridinone reverse transcriptase inhibitors. J. Virol. 65:4887–4892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nicastri E, Sarmati L, d'Ettorre G, Palmisano L, Parisi SG, Uccella I, Rianda A, Concia E, Vullo V, Vella S, Andreoni M. 2003. Replication capacity, biological phenotype, and drug resistance of HIV strains isolated from patients failing antiretroviral therapy. J. Med. Virol. 69:1–6. 10.1002/jmv.10269 [DOI] [PubMed] [Google Scholar]
- 20.Wang J, Bambara RA, Demeter LM, Dykes C. 2010. Reduced fitness in cell culture of HIV-1 with nonnucleoside reverse transcriptase inhibitor-resistant mutations correlates with relative levels of reverse transcriptase content and RNase H activity in virions. J. Virol. 84:9377–9389. 10.1128/JVI.00618-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lindberg J, Sigurdsson S, Lowgren S, Andersson HO, Sahlberg C, Noreen R, Fridborg K, Zhang H, Unge T. 2002. Structural basis for the inhibitory efficacy of efavirenz (DMP-266), MSC194 and PNU142721 towards the HIV-1 RT K103N mutant. Eur. J. Biochem. 269:1670–1677. 10.1046/j.1432-1327.2002.02811.x [DOI] [PubMed] [Google Scholar]
- 22.Hsiou Y, Ding J, Das K, Clark AD, Jr, Boyer PL, Lewi P, Janssen PA, Kleim JP, Rosner M, Hughes SH, Arnold E. 2001. The Lys103Asn mutation of HIV-1 RT: a novel mechanism of drug resistance. J. Mol. Biol. 309:437–445. 10.1006/jmbi.2001.4648 [DOI] [PubMed] [Google Scholar]
- 23.Alcaro S, Alteri C, Artese A, Ceccherini-Silberstein F, Costa G, Ortuso F, Bertoli A, Forbici F, Santoro MM, Parrotta L, Flandre P, Masquelier B, Descamps D, Calvez V, Marcelin AG, Perno CF, Sing T, Svicher V. 2011. Docking analysis and resistance evaluation of clinically relevant mutations associated with the HIV-1 non-nucleoside reverse transcriptase inhibitors nevirapine, efavirenz and etravirine. ChemMedChem 6:2203–2213. 10.1002/cmdc.201100362 [DOI] [PubMed] [Google Scholar]
- 24.Molina JM, Cahn P, Grinsztejn B, Lazzarin A, Mills A, Saag M, Supparatpinyo K, Walmsley S, Crauwels H, Rimsky LT, Vanveggel S, Boven K, ECHO Study Group 2011. Rilpivirine versus efavirenz with tenofovir and emtricitabine in treatment-naive adults infected with HIV-1 (ECHO): a phase 3 randomised double-blind active-controlled trial. Lancet 378:238–246. 10.1016/S0140-6736(11)60936-7 [DOI] [PubMed] [Google Scholar]
- 25.Vingerhoets J, Azijn H, Fransen E, De Baere I, Smeulders L, Jochmans D, Andries K, Pauwels R, de Bethune MP. 2005. TMC125 displays a high genetic barrier to the development of resistance: evidence from in vitro selection experiments. J. Virol. 79:12773–12782. 10.1128/JVI.79.20.12773-12782.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lai MT, Munshi V, Touch S, Tynebor RM, Tucker TJ, McKenna PM, Williams TM, DiStefano DJ, Hazuda DJ, Miller MD. 2009. Antiviral activity of MK-4965, a novel nonnucleoside reverse transcriptase inhibitor. Antimicrob. Agents Chemother. 53:2424–2431. 10.1128/AAC.01559-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu M, Felock PJ, Munshi V, Hrin RC, Wang YJ, Yan Y, Munshi S, McGaughey GB, Gomez R, Anthony NJ, Williams TM, Grobler JA, Hazuda DJ, McKenna PM, Miller MD, Lai MT. 2012. Antiviral activity and in vitro mutation development pathways of MK-6186, a novel nonnucleoside reverse transcriptase inhibitor. Antimicrob. Agents Chemother. 56:3324–3335. 10.1128/AAC.00102-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taniguchi T, Nurutdinova D, Grubb JR, Onen NF, Shacham E, Donovan M, Overton ET. 2012. Transmitted drug-resistant HIV type 1 remains prevalent and impacts virologic outcomes despite genotype-guided antiretroviral therapy. AIDS Res. Hum. Retrovir. 28:259–264. 10.1089/aid.2011.0022 [DOI] [PubMed] [Google Scholar]
- 29.Bennett DE, Camacho RJ, Otelea D, Kuritzkes DR, Fleury H, Kiuchi M, Heneine W, Kantor R, Jordan MR, Schapiro JM, Vandamme AM, Sandstrom P, Boucher CA, van de Vijver D, Rhee SY, Liu TF, Pillay D, Shafer RW. 2009. Drug resistance mutations for surveillance of transmitted HIV-1 drug-resistance: 2009 update. PLoS One 4:e4724. 10.1371/journal.pone.0004724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li JZ, Paredes R, Ribaudo HJ, Svarovskaia ES, Metzner KJ, Kozal MJ, Hullsiek KH, Balduin M, Jakobsen MR, Geretti AM, Thiebaut R, Ostergaard L, Masquelier B, Johnson JA, Miller MD, Kuritzkes DR. 2011. Low-frequency HIV-1 drug resistance mutations and risk of NNRTI-based antiretroviral treatment failure: a systematic review and pooled analysis. JAMA 305:1327–1335. 10.1001/jama.2011.375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Geretti AM, Fox ZV, Booth CL, Smith CJ, Phillips AN, Johnson M, Li JF, Heneine W, Johnson JA. 2009. Low-frequency K103N strengthens the impact of transmitted drug resistance on virologic responses to first-line efavirenz or nevirapine-based highly active antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 52:569–573. 10.1097/QAI.0b013e3181ba11e8 [DOI] [PubMed] [Google Scholar]
- 32.Paredes R, Lalama CM, Ribaudo HJ, Schackman BR, Shikuma C, Giguel F, Meyer WA, Johnson VA, III, Fiscus SA, D'Aquila RT, Gulick RM, Kuritzkes DR, AIDS Clinical Trials Group (ACTG) A5095 Study Team 2010. Pre-existing minority drug-resistant HIV-1 variants, adherence, and risk of antiretroviral treatment failure. J. Infect. Dis. 201:662–671. 10.1086/650543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shaw-Reid CA, Munshi V, Graham P, Wolfe A, Witmer M, Danzeisen R, Olsen DB, Carroll SS, Embrey M, Wai JS, Miller MD, Cole JL, Hazuda DJ. 2003. Inhibition of HIV-1 ribonuclease H by a novel diketo acid, 4-[5-(benzoylamino)thien-2-yl]-2,4-dioxobutanoic acid. J. Biol. Chem. 278:2777–2780. 10.1074/jbc.C200621200 [DOI] [PubMed] [Google Scholar]
- 34.Munshi V, Lu M, Felock P, Barnard RJ, Hazuda DJ, Miller MD, Lai MT. 2008. Monitoring the development of non-nucleoside reverse transcriptase inhibitor-associated resistant HIV-1 using an electrochemiluminescence-based reverse transcriptase polymerase assay. Anal. Biochem. 374:121–132. 10.1016/j.ab.2007.10.004 [DOI] [PubMed] [Google Scholar]
- 35.Petropoulos CJ, Parkin NT, Limoli KL, Lie YS, Wrin T, Huang W, Tian H, Smith D, Winslow GA, Capon DJ, Whitcomb JM. 2000. A novel phenotypic drug susceptibility assay for human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 44:920–928. 10.1128/AAC.44.4.920-928.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Prichard MN, Shipman C., Jr 1990. A three-dimensional model to analyze drug-drug interactions. Antiviral Res. 14:181–205. 10.1016/0166-3542(90)90001-N [DOI] [PubMed] [Google Scholar]
- 37.Das K, Lewi PJ, Hughes SH, Arnold E. 2005. Crystallography and the design of anti-AIDS drugs: conformational flexibility and positional adaptability are important in the design of non-nucleoside HIV-1 reverse transcriptase inhibitors. Prog. Biophys. Mol. Biol. 88:209–231. 10.1016/j.pbiomolbio.2004.07.001 [DOI] [PubMed] [Google Scholar]
- 38.Rimsky L, Vingerhoets J, Van E, Eron VJ, Clotet B, Hoogstoel A, Boven K, Picchio G. 2012. Genotypic and phenotypic characterization of HIV-1 isolates obtained from patients on rilpivirine therapy experiencing virologic failure in the phase 3 ECHO and THRIVE studies: 48-week analysis. J. Acquir. Immune. Defic. Syndr. 59:39–46. 10.1097/QAI.0b013e31823df4da [DOI] [PubMed] [Google Scholar]
- 39.Hu Z, Kuritzkes DR. 2011. Interaction of reverse transcriptase (RT) mutations conferring resistance to lamivudine and etravirine: effects on fitness and RT activity of human immunodeficiency virus type 1. J. Virol. 85:11309–11314. 10.1128/JVI.05578-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kulkarni R, Babaoglu K, Lansdon EB, Rimsky L, Van E, Picchio VG, Svarovskaia E, Miller MD, White KL. 2012. The HIV-1 reverse transcriptase M184I mutation enhances the E138K-associated resistance to rilpivirine and decreases viral fitness. J. Acquir. Immune Defic. Syndr. 59:47–54. 10.1097/QAI.0b013e31823aca74 [DOI] [PubMed] [Google Scholar]
- 41.Xu HT, Asahchop EL, Oliveira M, Quashie PK, Quan Y, Brenner BG, Wainberg MA. 2011. Compensation by the E138K mutation in HIV-1 reverse transcriptase for deficits in viral replication capacity and enzyme processivity associated with the M184I/V mutations. J. Virol. 85:11300–11308. 10.1128/JVI.05584-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu HT, Colby-Germinario SP, Asahchop EL, Oliveira M, McCallum M, Schader SM, Han Y, Quan Y, Sarafianos SG, Wainberg MA. 2013. Effect of mutations at position E138 in HIV-1 reverse transcriptase and their interactions with the M184I mutation on defining patterns of resistance to nonnucleoside reverse transcriptase inhibitors rilpivirine and etravirine. Antimicrob. Agents Chemother. 57:3100–3109. 10.1128/AAC.00348-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang W, Wrin T, Gamarnik AV. 2002. RT mutations that confer NNRTI resistance may also impair replication capacity. Antivir. Ther. 7:S79 [Google Scholar]
- 44.Lai MT, Lu M, Felock PJ, Hrin RC, Wang YJ, Yan Y, Munshi S, McGaughey GB, Tynebor RM, Tucker TJ, Williams TM, Grobler JA, Hazuda DJ, McKenna PM, Miller MD. 2010. Distinct mutation pathways of non-subtype B HIV-1 during in vitro resistance selection with nonnucleoside reverse transcriptase inhibitors. Antimicrob. Agents Chemother. 54:4812–4824. 10.1128/AAC.00829-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Soriano V, de Mendoza C. 2002. Genetic mechanisms of resistance to NRTI and NNRTI. HIV. Clin. Trials 3:237–248. 10.1310/06DD-UN4D-9AW2-RLBY [DOI] [PubMed] [Google Scholar]
- 46.Singh K, Marchand B, Rai DK, Sharma B, Michailidis E, Ryan EM, Matzek KB, Leslie MD, Hagedorn AN, Li Z, Norden PR, Hachiya A, Parniak MA, Xu HT, Wainberg MA, Sarafianos SG. 2012. Biochemical mechanism of HIV-1 resistance to rilpivirine. J. Biol. Chem. 287:38110–38123. 10.1074/jbc.M112.398180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anderson MS, Gilmartin J, Robberechts M, De Lepeleire I, Tetteh E, Guo Y, Schürmann D, Wagner F, Wagner JA, Butterton JR. 2013. Safety and antiviral activity of MK-1439, a novel non-nucleoside reverse transcriptase inhibitor (NNRTI), in treatment-naïve HIV-infected patients, abstr 100 Abstr. 20th Conf. Retrovir. Opportun. Infect., Atlanta, GA [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.