

Abstract
Identification of a novel class of anti-Burkholderia compounds is key in addressing antimicrobial resistance to current therapies as well as naturally occurring resistance. The FabI enoyl-ACP reductase in Burkholderia is an underexploited target that presents an opportunity for development of a new class of inhibitors. A library of substituted diphenyl ethers was used to identify FabI1-specific inhibitors for assessment in Burkholderia pseudomallei ex vivo and murine efficacy models. Active FabI1 inhibitors were identified in a two-stage format consisting of percent inhibition screening and MIC determination by the broth microdilution method. Each compound was evaluated against the B. pseudomallei 1026b (efflux-proficient) and Bp400 (efflux-compromised) strains. In vitro screening identified candidate substituted diphenyl ethers that exhibited MICs of less than 1 μg/ml, and enzyme kinetic assays were used to assess potency and specificity against the FabI1 enzyme. These compounds demonstrated activity in a Burkholderia ex vivo efficacy model, and two demonstrated efficacy in an acute B. pseudomallei mouse infection model. This work establishes substituted diphenyl ethers as a suitable platform for development of novel anti-Burkholderia compounds that can be used for treatment of melioidosis.
INTRODUCTION
Burkholderia pseudomallei is a Gram-negative bacterium endemic to Southeast Asia and Northern Australia and is the causative agent of melioidosis. This disease is difficult to manage and often fatal in humans if not aggressively and appropriately treated (1–5). While there are current effective treatment options for Burkholderia infections, there has been increasing concern regarding drug-resistant and naturally resistant strains that render the current treatment regimen ineffective and negatively impact the ability to treat and manage the disease (2, 5–7). Also, mounting evidence indicates that B. pseudomallei is an emerging pathogen in other areas of the world, and demand for treatment of melioidosis will increase as a result (8–12). These reasons, along with the ability of Burkholderia to be easily spread via aerosol and ease of dissemination, provide the need to develop novel compounds that inhibit underexploited drug targets.
The enoyl-ACP reductase enzyme FabI1 (encoded on chromosome 1), involved in fatty acid elongation in the bacterial fatty acid biosynthesis type II (FASII) pathway, is an underexploited drug target in Burkholderia. Our own antibacterial discovery program has demonstrated that substituted diphenyl ethers inhibit the FabI enzyme of a variety of organisms, leading to in vivo efficacy (13, 14). The ability of this compound class to exhibit broad-spectrum activity and demonstrate efficacy in vivo makes it attractive for development of novel Burkholderia chemotherapeutics.
Using our library of substituted diphenyl ethers, our drug discovery efforts have been expanded to assess whether inhibition of the B. pseudomallei enoyl-ACP reductase FabI1 demonstrates efficacy in ex vivo and animal models of infection. To assess the activities of our library of substituted diphenyl ethers, we first identified lead inhibitors by whole bacterial screening against Burkholderia thailandensis strains E264 and Bt38 (E264 Δ[amrAB-oprA] Δ[bpeAB-oprB]) and followed up with MIC determination using B. pseudomallei strains 1026b (15) and Bp400 (1026b Δ[amrAB-oprA] Δ[bpeAB-oprB]) (16). We then assessed the most potent FabI1 inhibitors in both the Burkholderia ex vivo efficacy model and our rapid animal efficacy model. The use of selective FabI1 inhibitors, efflux mutant strains, and the ex vivo and animal models of efficacy allow us to directly assess whether or not targeted inhibition of FabI1 could be used as a suitable platform for novel anti-Burkholderia therapies.
MATERIALS AND METHODS
Kinetics and inhibition of FabI1.
FabI1 and octenoyl-coenzyme A (Oct-CoA) were prepared as described previously (17). Inhibition constants (Ki) were analyzed using the standard equation for uncompetitive inhibition. Initial velocities were determined at a fixed substrate concentration, and the data were fit to the following equation, using a value of Km (Oct-CoA) of 160 μM: vi/v0 = (Km + S)/(Km + S[1 + {I/Ki}]).
The parameters vi and v0 are the initial velocities in the presence and absence of inhibitor. The substrate concentration (S) was fixed at 30 μM, and the inhibitor concentration was varied from 0 to 8,000 nM. Data fitting was performed using the software program Grafit 4.0 (Erithacus Software Ltd.).
Progress curve analysis was performed as described previously to identify slow-onset inhibitors of FabI1 (13). Assays were performed by adding enzyme (5 nM) to solutions containing glycerol (8% [vol/vol]), bovine serum albumin (BSA) (0.1 mg/vol), dimethyl sulfoxide (DMSO) (2% [vol/vol]), Oct-CoA (300 μM), NADH (250 μM), NAD+ (200 μM), and inhibitor (50 nM). Reactions were monitored until the progress curve became linear, indicating that the steady state had been reached. In this protocol, the low enzyme concentration and high substrate concentration ensured that substrate depletion was minimized so that the progress curves were approximately linear over a period of 30 min in the absence of inhibitors.
Preincubation assays were performed to obtain the true inhibition constants and to determine the preference of slow-onset inhibitors for the different cofactor-bound forms of FabI1. Enzyme (10 nM) was preincubated in the presence of a fixed concentration of DMSO (2% [vol/vol]), NAD+ (20 to 400 μM), NADH (250 μM), and inhibitors (0 to 2000 nM) for 5 h at 4°C. After warming to 25°C, assays were initiated by the addition of Oct-CoA (30 μM). The inhibition constant was calculated as previously described (18).
Bacteria, screening, and MIC determination.
B. thailandensis E264 (efflux proficient) (19), B. thailandensis Bt38 (E264 Δ[amrAB-oprA] Δ[bpeAB-oprB]) (kindly provided by H. P. Schweizer, Colorado State University), B. pseudomallei 1026b (efflux proficient) (15), and B. pseudomallei Bp400 (1026b Δ[amrAB-oprA] Δ[bpeAB-oprB]) (16) were grown to an optical density at 600 nm (OD600) of ∼0.6, frozen at −80°C in 10% glycerol, and used as standard bacterial stocks for these studies. For each evaluation, bacteria were prepared fresh by growth from the standard stocks on Luria-Bertani (LB) agar, Miller (BD), grown at 37°C for 48 to 72 h. Bacteria recovered from the LB plates were inoculated in 50 ml LB broth. Broth cultures were then incubated for 24 h at 37°C passed 1:100, and incubated for an additional 6 h at 37°C. Bacteria were then diluted to a concentration of 1 × 104 CFU/ml in cation-adjusted Mueller-Hinton broth (caMHB) (BD, Franklin Lakes, NJ), and 50 μl of sample was added to each well of the test plate. Substituted diphenyl ethers were screened in a percent inhibition high-throughput fashion against B. thailandensis E264 and B. thailandensis Bt38 in a 96-well plate format to identify lead compounds. All compounds were diluted in caMHB to concentrations of 80 and 40 μg/ml in a 50-μl volume/well. MICs of candidate compounds were determined against B. pseudomallei 1026b and B. pseudomallei Bp400 by the broth microdilution method. For MIC determination, compounds were added to the 96-well plate starting at 512 μg/ml in the first column and serially diluted 1:2 to column 12 for a final concentration of 0.12 μg/ml caMHB. MIC plates were incubated at 37°C for 18 h, at which time 10 μl of alamarBlue (Invitrogen, Carlsbad, CA) was added to each well, and plates were incubated for an additional 4 h. Reduction of alamarBlue was determined by absorbance readings at wavelengths of 570 and 600 nm using a microplate reader (Biotek, Winooski, VT). Percent growth reduction was calculated from spectrophotometric readings over the drug concentration series. The MIC was determined as the lowest drug concentration to inhibit visible growth.
Cytotoxicity assay.
A Vero cell line (African green monkey kidney cells) was used to assess potential cytotoxicity, similar to a method described previously (20). Vero cells were maintained in Leibovitz's (L15) growth medium without phenol and supplemented with heat-inactivated newborn calf serum and penicillin-streptomycin. The cells were grown at 37°C and subcultured weekly. Cell suspensions containing 1.3 × 105 cells per ml and resazurin (21) were added to the 96-well plates. The compound was added, and 2-fold serial dilutions were completed with a 100-μl final volume in the wells. The plates were incubated for 72 h at 37°C in the dark. The plates were read at 570 nm and 600 nm in a plate reader, and the LD50 (lethal dose causing a 50% loss of cell viability) of the compound was calculated using standard procedures.
Burkholderia ex vivo model of efficacy.
Infected RAW 264.7 mouse macrophages (American Type Tissue Collection, Manassas, VA) were used to assess intracellular efficacies of our compounds. RAW cells were cultured in complete medium that consisted of minimal essential medium (Invitrogen), 10% fetal bovine serum (HyClone, Logan, UT), 2 mM l-glutamine (Invitrogen), 1× nonessential amino acids (Invitrogen), and 0.075% sodium bicarbonate (EMD Science, Gibbstown, NJ). Bacteria were added to 2.5 × 105 RAW cells per well in a 24-well tissue culture plate at a multiplicity of infection of 5 CFU per cell in a 0.5 ml medium. The plates were then centrifuged at 2,400 × g for 2 min and placed at 37°C/5% CO2 to incubate for 1 h. Supernatant was then removed; plates were washed once with 2 ml phosphate-buffered saline (PBS) and 1 ml of either 0.35 mg/ml kanamycin (Sigma-Aldrich, St. Louis, MO) for wild-type strains or 0.1 mg/ml kanamycin (Sigma-Aldrich) for double efflux mutant strains in complete medium. Plates were then incubated for 1 h and washed 2 times with 2 ml PBS.
Selected compounds were then added to each well at either 10, 1, or 0.1 μg/ml in complete medium in triplicate. No treatment, 1 μg/ml doxycycline (Sigma-Aldrich), and 10 μg/ml ceftazidime were included as controls. Plates were incubated for 18 h, and cells were observed for signs of infection. Cells were washed 3 times with 2 ml PBS, and 1 ml sterile double-distilled water (ddH2O) was added to each well. Each well was thoroughly mixed/scraped to ensure complete cell lysis. Lysates were then serially diluted 1:10, inoculum was plated from the neat, 10−1, 10−2, 10−3 and 10−4 dilutions onto LB agar plates, and plates were incubated for 48 h at 37°C. Colonies from each plate was counted, and the log10 CFU/ml lysate value was calculated.
Acute B. pseudomallei mouse model of infection for evaluation of efficacy.
Five- to six-week-old BALB/c female mice (Charles River Laboratories, Wilmington, MA) were challenged by intranasal infection with 5,000 CFU/mouse B. pseudomallei Bp400. Animals were anesthetized with a mixture of 100 mg/kg of body weight ketamine and 10 mg/kg xylazine delivered intraperitoneally. The bacteria were diluted to the appropriate concentration in PBS to achieve an inoculum concentration of 2.5 × 105 CFU/ml. The inoculum was then delivered in a 20-μl volume dropwise in alternating nostrils. Ceftazidime was formulated for injection in a 0.1% BSA solution in PBS (pH 7.4) at a concentration of 40 mg/ml. PT52 and PT68 were formulated for injection in a lipid-based delivery system of a combined lipid and surfactant mixture consisting of 40% Captex 200 (Abitec Corp., Janesville, WI)–40% Solutol HS15 (BASF, Mount Olive, NJ)–20% Capmul MCM (Abitec Corp., Janesville, WI). The mixture was gently heated at 50°C to homogenize components. The resulting homogenous isotropic mixture was then diluted with PBS to yield a fine dispersion at a concentration of 40 mg/ml. Two hundred milligrams per kilogram ceftazidime, PT52, or PT68 was delivered intraperitoneally, with treatment beginning at the time of infection and repeated doses delivered every 12 h. The number of viable bacteria in lung and spleen tissue was determined at 60 h postexposure by plating serial 10-fold dilutions of homogenates onto LB agar and incubating for 48 h at 37°C. The bacterial burden was assessed using a one-way analysis of variance (ANOVA) followed by Dunnett's multiple-comparison test, with significance determined by a P value of <0.05. Outliers were determined by Grubbs' test.
RESULTS
Substituted diphenyl ethers are potent inhibitors of Burkholderia.
A library of diphenyl ethers was screened for potency in an established high-throughput percent growth inhibition assay and confirmed using the broth microdilution method. The substituted diphenyl ethers were tested against the efflux mutant strain B. thailandensis Bt38 and efflux-proficient strain B. thailandensis E264 to assess each candidate as a pump substrate and to determine inhibitory activity against whole bacteria. Thirty-two compounds out of the library tested exhibited greater than 50% growth reduction and 22 showed greater than 90% growth reduction at 20 μg/ml against B. thailandensis Bt38. Only PT52 inhibited more than 50% growth of B. thailandensis E264 at 40 μg/ml. PT51, PT52, PT55, and PT68 were further investigated due to findings of previous structure activity relationship (SAR) studies (17) and to evaluate the effect of A-ring substitutions on efficacy (20).
MIC values were determined for the efflux-compromised B. pseudomallei Bp400 mutant strain and efflux-proficient strain B. pseudomallei 1026b. PT52, PT55, and PT68 had MIC values of less than 1 μg/ml against B. pseudomallei Bp400 (Table 1). Evaluation of PT51, PT52, PT55, and PT68 against the efflux-proficient strain B. pseudomallei 1026b resulted in increased MIC values due to drug efflux (Table 1). While the majority of substituted diphenyl ethers demonstrated potency against Burkholderia, they were also readily effluxed.
TABLE 1.
In vitro activities of FabI inhibitors against B. pseudomalleia
| FabI inhibitor | MIC (mg/liter) for strain |
Vero cell cytotoxicity, LD50 (mg/liter) | |
|---|---|---|---|
| B. pseudomallei 1026b | B. pseudomallei Bp400 | ||
| PT51 | 2 | 2 | 190.2 |
| PT52 | 1 | 0.125 | 89.8 |
| PT55 | 4 | 0.5 | 164.9 |
| PT68 | 4 | 0.5 | 152.9 |
| Doxycycline | 0.5 | 0.125 | NDb |
| Ceftazidime | 2 | 1 | ND |
MICs of FabI1 inhibitors against wild-type and efflux-compromised B. pseudomallei strains. MICs were determined using the broth microdilution method.
ND, not determined in this study.
The cytotoxicity of each compound was evaluated using a standard Vero cell assay (Table 1). Each diphenyl ether compound exhibited LD50 values 100-fold greater than the MIC, which is consistent with our previous reports for compounds in this drug series (20). There was no toxicity observed in the ex vivo RAW 264.7 macrophage assay for the clinical drugs ceftazidime and doxycycline, which is consistent with published reports (22).
Diphenyl ethers with whole-bacterium activity have potency against FabI1 enoyl-ACP reductase enzyme.
Based on the analysis of MIC values, PT51, PT52, PT55, and PT68 were selected as lead compounds, and their binding affinities and modes of inhibition against FabI1 were determined (Table 2). Compounds PT51 and PT55 were rapid reversible inhibitors and displayed moderate binding affinity to FabI1, with 50% inhibitory concentrations (IC50s) of 2,699 ± 287 nM and 695 ± 132 nM.
TABLE 2.
Inhibition of B. pseudomallei FabI1 by substituted diphenyl ethersa
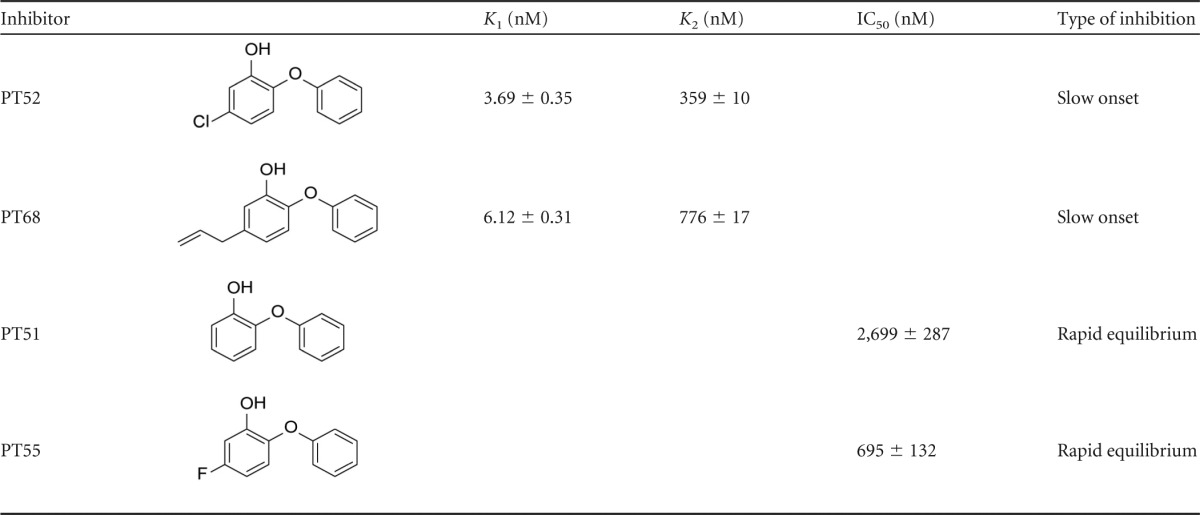
Structure-activity relationship studies performed show that A-ring substitutions are critical in determining the type of inhibition and binding affinity for both E-NAD+ (K1) and E-NADH (K2).
The addition of a fluoro substituent to the A ring (PT51 versus PT55) resulted in only a 4-fold improvement in binding affinity. However, replacement of the fluoro substituent with a larger functional group, such as a chlorine (PT52) or a propenyl group (PT68), dramatically enhanced binding affinity to the FabI1-NAD+ binary complex, with K1 values of 3.69 ± 0.35 nM or 6.12 ± 0.31 nM, respectively. Progress curve analysis also indicated that the compounds PT52 and PT68 were time-dependent inhibitors, displaying a slow-onset inhibition mechanism against FabI1. It is not unusual for some inhibitors to exhibit this kinetic behavior, since triclosan and selected diphenyl ethers were shown to be slow-onset inhibitors of FabI1 (17). Preincubation experiments allowed us to evaluate the dependence of enzyme inhibition on NADH or NAD+, which provides the true thermodynamic affinity of the inhibitor for E-NAD+ (K1) and E-NADH (K2). For both PT52 and PT68, the dependence of Ki′ (apparent inhibition constant) on [NAD+] was best described by the equation that includes the inhibitor binding to both the E-NAD+ and E-NADH forms of FabI1, although based on the K1 and K2 values of these two compounds, there is a 100-fold preference for E-NAD+.
Substituted diphenyl ethers demonstrate efficacy in a Burkholderia ex vivo model and in the Burkholderia rapid animal model.
PT51, PT52, PT55, and PT68 were further evaluated in a Burkholderia ex vivo model of efficacy based on their whole bacterial activities. Evaluation of activity in the ex vivo model was conducted against B. pseudomallei 1026b at 10 μg/ml and at multiple concentrations against B. pseudomallei Bp400 (Fig. 1A and B). PT51, PT52, PT55, and PT68 all inhibited the growth of the efflux pump mutant B. pseudomallei Bp400 by >90% at 10 μg/ml, and growth of B. pseudomallei 1026b was not inhibited by PT51, PT52, PT55, and PT68.
FIG 1.
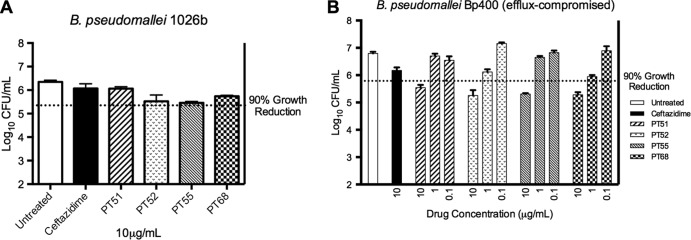
RAW 264.6 macrophages were infected with B. pseudomallei at a multiplicity of infection (MOI) of 5 and treated overnight with either ceftazidime or select substituted diphenyl ethers. B. pseudomallei 1026b was treated with 10 μg/ml compounds for 18 h, macrophages were lysed, and lysate was plated on solid medium for bacterial enumeration (A). B. pseudomallei Bp400 was treated at the same concentration with ceftazidime and at 10, 1, and 0.1 mg/ml with substituted diphenyl ethers to observe concentration-dependent inhibition. Again macrophages were lysed, and lysate was plated onto solid medium for bacterial enumeration (B).
To assess whether PT51, PT52, PT55, and PT68 demonstrated inhibition in a concentration-dependent manner, they were further evaluated for activities against B. pseudomallei Bp400 in the ex vivo model. Each of the compounds inhibited growth of B. pseudomallei Bp400 in a dose-dependent fashion (Fig. 1B), and the precise 90% growth reduction values for each compound in the ex vivo model were determined (Table 3). Importantly, PT51, PT52, PT55, and PT68 had MIC values in the ex vivo model comparable to those determined in artificial medium.
TABLE 3.
Ex vivo activities of FabI inhibitors against B. pseudomallei Bp400a
| FabI inhibitor | 90% growth reduction (mg/liter) |
|---|---|
| PT51 | 6.1 ± 2.6 |
| PT52 | 2.5 ± 2.5 |
| PT55 | 6 ± 4.3 |
| PT68 | 2.0 ± 2.0 |
| Ceftazidime | >10 |
| Doxycycline | <1 |
Compounds were further evaluated for activityagainst B. pseudomallei Bp400 ex vivo. The drug concentrationthat inhibits 90% growth was calculated from linear regression analysis of dataobtained at 10, 1, and 0.1 μg/ml.
PT52 and PT68 were tested in the acute B. pseudomallei animal model of infection to determine efficacy. These compounds were chosen due to their slow-onset inhibitor characteristics observed in enzyme kinetic assays, in addition to inhibitory activity in the ex vivo efficacy model. Mice were challenged intranasally with 5,000 CFU B. pseudomallei Bp400. The bacterial burden was assessed at 60 h postinfection, and treated groups were compared to the untreated control group. PT52-treated mice exhibited a 0.6-log10 CFU/ml reduction (P < 0.05) in the lung and a 1.1-log10 CFU/ml reduction (P < 0.01) in the spleen (Fig. 2A and B). Mice treated with PT68 showed reduction only in the spleen at 1.3 log10 CFU/ml (Fig. 2B). Mice treated with 200 mg/kg ceftazidime were included as a positive control and were determined to have a bacterial burden at the level of detection in both lung and spleen tissue. These studies substantiated that these substituted diphenyl ethers can act by subterfuge and gain access to the efflux-deficient bacteria under host conditions and inhibit growth through inhibition of FabI1 (23).
FIG 2.
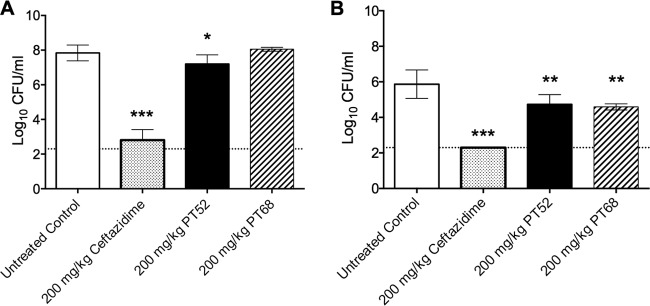
Bacterial burden in mouse lung (A) or spleen (B) at 60 h postinfection. The mean of each group was plotted, with error bars indicating ± standard deviations and a dashed line indicating the level of detection. Significance was determined by one-way ANOVA and a P value < 0.05. These data are representative of two independent experiments.
DISCUSSION
Discovery of new drug targets and chemotherapeutics is key in managing emerging pathogens, such as B. pseudomallei. B. pseudomallei can be particularly challenging to treat due to intrinsic and emerging antimicrobial resistance and the ability to efflux antibiotics (24). The enoyl-ACP reductase enzymes, particularly FabI in the FASII pathway, are underexploited new targets for the development of anti-Burkholderia drugs. Our compounds were developed around the diphenyl ether pharmacophore and in previous studies have exhibited whole-bacterium activity and in vivo efficacy against Francisella tularensis Schu4 (13, 14).
In this study, we have shown this same class of compounds to be efficacious for in vitro and ex vivo activity against efflux-compromised B. pseudomallei Bp400. Unfortunately, the majority of compounds in this class are efflux pump substrates, and while they are effluxed similarly to existing clinically used drugs for treatment of melioidosis, for instance, doxycycline, modifications to existing compounds will be made to address this issue or current compounds will need to be formulated to deliver the higher dose requirement that exceeds the higher MIC value.
Importantly, PT52 and PT68 were shown to significantly reduce the bacterial burden in the spleen, and only PT52 significantly reduced the bacterial burden in the lung compared to results for untreated mice. This observation, along with the ability of these compounds to display a low nanomolar binding affinity for FabI1 in vitro, suggests that the use of substituted diphenyl ethers to inhibit FabI1 enoyl-ACP reductase in Burkholderia can be exploited to control infection. Steady-state kinetic analysis showed that PT52 and PT68 were promising inhibitors due to favorable optimization of thermodynamic parameters, specifically inhibitor binding to the FabI1-NAD+ complex, represented by K1. This final stable ground state was coupled with a slow-onset inhibitor mechanism, which was lacking in compounds PT51 and PT55, which displayed higher K1 values. The loss of a slow-onset inhibitor mechanism of PT51 and PT55 against FabI1 may arise from lacking an A-ring substituent and from the smaller van der Waals radius of fluorine than of chlorine (PT52), respectively. Also, it has been demonstrated that A-ring substituents play a key role in hydrophobic interaction between diphenyl ether inhibitors and FabI from F. tularensis (13), which leads to slow-onset inhibition and high binding affinity in this system.
By using the diphenyl ethers as tool compounds, we were able to show that inhibition of B. pseudomallei FabI1 results in efficacy in vitro and in the animal model of infection. Further optimization of this class or similar classes of FabI inhibitors could open up the possibility for the development of a novel compound that could be used to treat melioidosis.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Rocky Mountain Regional Center of Excellence funded by NIH, NIAID grant AI065357 (to R.A.S., P.J.T., and H.P.S.).
Footnotes
Published ahead of print 30 December 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02296-13.
REFERENCES
- 1.Cheng AC, Currie BJ. 2005. Melioidosis: epidemiology, pathophysiology, and management. Clin. Microbiol. Rev. 18:383–416. 10.1128/CMR.18.2.383-416.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wiersinga WJ, van der Poll T, White NJ, Day NP, Peacock SJ. 2006. Melioidosis: insights into the pathogenicity of Burkholderia pseudomallei. Nat. Rev. Microbiol. 4:272–282. 10.1038/nrmicro1385 [DOI] [PubMed] [Google Scholar]
- 3.Lazar Adler NR, Govan B, Cullinane M, Harper M, Adler B, Boyce JD. 2009. The molecular and cellular basis of pathogenesis in melioidosis: how does Burkholderia pseudomallei cause disease? FEMS Microbiol. Rev. 33:1079–1099. 10.1111/j.1574-6976.2009.00189.x [DOI] [PubMed] [Google Scholar]
- 4.Peacock SJ. 2006. Melioidosis. Curr. Opin. Infect. Dis. 19:421–428. 10.1097/01.qco.0000244046.31135.b3 [DOI] [PubMed] [Google Scholar]
- 5.Sarovich DS, Price EP, Limmathurotsakul D, Cook JM, Von Schulze AT, Wolken SR, Keim P, Peacock SJ, Pearson T. 2012. Development of ceftazidime resistance in an acute Burkholderia pseudomallei infection. Infect. Drug Resist. 5:129–132. 10.2147/IDR.S35529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Inglis TJ, Rolim DB, Rodriguez JL. 2006. Clinical guideline for diagnosis and management of melioidosis. Rev. Inst. Med. Trop. Sao Paulo 48:1–4. 10.1590/S0036-46652006000100001 [DOI] [PubMed] [Google Scholar]
- 7.Peacock SJ, Schweizer HP, Dance DA, Smith TL, Gee JE, Wuthiekanun V, DeShazer D, Steinmetz I, Tan P, Currie BJ. 2008. Management of accidental laboratory exposure to Burkholderia pseudomallei and B. mallei. Emerg. Infect. Dis. 14:e2. 10.3201/eid1407.071501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Currie BJ, Dance DA, Cheng AC. 2008. The global distribution of Burkholderia pseudomallei and melioidosis: an update. Trans. R. Soc. Trop. Med. Hyg. 102(Suppl 1):S1–S4. 10.1016/S0035-9203(08)70002-6 [DOI] [PubMed] [Google Scholar]
- 9.Kanungo R, Padhan P, Bhattacharya S, Srimannarayana J, Jayanthi S, Swaminathan RP. 2002. Melioidosis—a report from Pondicherry, South India. J. Assoc. Physicians India 50:1438–1439 [PubMed] [Google Scholar]
- 10.Rolim DB, Vilar DC, Sousa AQ, Miralles IS, de Oliveira DC, Harnett G, O'Reilly L, Howard K, Sampson I, Inglis TJ. 2005. Melioidosis, northeastern Brazil. Emerg. Infect. Dis. 11:1458–1460. 10.3201/eid1109.050493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rolim DB, Rocha MF, Brilhante RS, Cordeiro RA, Leitao NP, Jr, Inglis TJ, Sidrim JJ. 2009. Environmental isolates of Burkholderia pseudomallei in Ceara State, northeastern Brazil. Appl. Environ. Microbiol. 75:1215–1218. 10.1128/AEM.01953-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mukhopadhyay C, Kaestli M, Vandana KE, Sushma K, Mayo M, Richardson L, Tuanyok A, Keim P, Godoy D, Spratt BG, Currie BJ. 2011. Molecular characterization of clinical Burkholderia pseudomallei isolates from India. Am. J. Trop. Med. Hyg. 85:121–123. 10.4269/ajtmh.2011.11-0166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu H, England K, am Ende C, Truglio JJ, Luckner S, Reddy BG, Marlenee NL, Knudson SE, Knudson DL, Bowen RA, Kisker C, Slayden RA, Tonge PJ. 2009. Slow-onset inhibition of the FabI enoyl reductase from Francisella tularensis: residence time and in vivo activity. ACS Chem. Biol. 4:221–231. 10.1021/cb800306y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.England K, am Ende C, Lu H, Sullivan TJ, Marlenee NL, Bowen RA, Knudson SE, Knudson DL, Tonge PJ, Slayden RA. 2009. Substituted diphenyl ethers as a broad-spectrum platform for the development of chemotherapeutics for the treatment of tularaemia. J. Antimicrob. Chemother. 64:1052–1061. 10.1093/jac/dkp307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeShazer D, Brett PJ, Carlyon R, Woods DE. 1997. Mutagenesis of Burkholderia pseudomallei with Tn5-OT182: isolation of motility mutants and molecular characterization of the flagellin structural gene. J. Bacteriol. 179:2116–2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mima T, Schweizer HP. 2010. The BpeAB-OprB efflux pump of Burkholderia pseudomallei 1026b does not play a role in quorum sensing, virulence factor production, or extrusion of aminoglycosides but is a broad-spectrum drug efflux system. Antimicrob. Agents Chemother. 54:3113–3120. 10.1128/AAC.01803-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu N, Cummings JE, England K, Slayden RA, Tonge PJ. 2011. Mechanism and inhibition of the FabI enoyl-ACP reductase from Burkholderia pseudomallei. J. Antimicrob. Chemother. 66:564–573. 10.1093/jac/dkq509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sivaraman S, Zwahlen J, Bell AF, Hedstrom L, Tonge PJ. 2003. Structure-activity studies of the inhibition of FabI, the enoyl reductase from Escherichia coli, by triclosan: kinetic analysis of mutant FabIs. Biochemistry 42:4406–4413. 10.1021/bi0300229 [DOI] [PubMed] [Google Scholar]
- 19.Brett PJ, DeShazer D, Woods DE. 1998. Burkholderia thailandensis sp. nov., a Burkholderia pseudomallei-like species. Int. J. Syst. Bacteriol. 48(Part 1):317–320. 10.1099/00207713-48-1-317 [DOI] [PubMed] [Google Scholar]
- 20.Boyne ME, Sullivan TJ, am Ende CW, Lu H, Gruppo V, Heaslip D, Amin AG, Chatterjee D, Lenaerts A, Tonge PJ, Slayden RA. 2007. Targeting fatty acid biosynthesis for the development of novel chemotherapeutics against Mycobacterium tuberculosis: evaluation of A-ring-modified diphenyl ethers as high-affinity InhA inhibitors. Antimicrob. Agents Chemother. 51:3562–3567. 10.1128/AAC.00383-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O'Brien J, Wilson I, Orton T, Pognan F. 2000. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 267:5421–5426. 10.1046/j.1432-1327.2000.01606.x [DOI] [PubMed] [Google Scholar]
- 22.Judy BM, Whitlock GC, Torres AG, Estes DM. 2009. Comparison of the in vitro and in vivo susceptibilities of Burkholderia mallei to ceftazidime and levofloxacin. BMC Microbiol. 9:88. 10.1186/1471-2180-9-88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cummings JE, Kingry LC, Rholl DA, Schweizer HP, Tonge PJ, Slayden RA. 25 November 2013. The Burkholderia pseudomallei enoyl-ACP reductase FabI1 is essential for in vivo growth and is the target of a novel chemotherapeutic with efficacy. Antimicrob. Agents Chemother. 10.1128/AAC.00176-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schweizer HP. 2012. Mechanisms of antibiotic resistance in Burkholderia pseudomallei: implications for treatment of melioidosis. Fut. Microbiol. 7:1389–1399. 10.2217/fmb.12.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.