

Short abstract
The relationship between regulatory complexity and gene spacing was examined in Caenorhabditis elegans and Drosophila melanogaster. Intergenic distance, and hence genome architecture, is shaped by regulatory information contained in noncoding DNA.
Abstract
Background
Factors affecting the organization and spacing of functionally unrelated genes in metazoan genomes are not well understood. Because of the vast size of a typical metazoan genome compared to known regulatory and protein-coding regions, functional DNA is generally considered to have a negligible impact on gene spacing and genome organization. In particular, it has been impossible to estimate the global impact, if any, of regulatory elements on genome architecture.
Results
To investigate this, we examined the relationship between regulatory complexity and gene spacing in Caenorhabditis elegans and Drosophila melanogaster. We found that gene density directly reflects local regulatory complexity, such that the amount of noncoding DNA between a gene and its nearest neighbors correlates positively with that gene's regulatory complexity. Genes with complex functions are flanked by significantly more noncoding DNA than genes with simple or housekeeping functions. Genes of low regulatory complexity are associated with approximately the same amount of noncoding DNA in D. melanogaster and C. elegans, while loci of high regulatory complexity are significantly larger in the more complex animal. Complex genes in C. elegans have larger 5' than 3' noncoding intervals, whereas those in D. melanogaster have roughly equivalent 5' and 3' noncoding intervals.
Conclusions
Intergenic distance, and hence genome architecture, is highly nonrandom. Rather, it is shaped by regulatory information contained in noncoding DNA. Our findings suggest that in compact genomes, the species-specific loss of nonfunctional DNA reveals a landscape of regulatory information by leaving a profile of functional DNA in its wake.
Background
Many basic issues regarding the organization of regulatory DNA remain unresolved. We do not know the portion of any genome comprising regulatory DNA. We do not understand the factors that govern the size, distance and orientation of regulatory elements relative to coding regions. Nor do we usually know the identity of the many transcription factors that bind any given element. For these reasons, it has been difficult to assess the impact of regulatory DNA on metazoan genome architecture.
Nevertheless, it is clear that metazoan genomes are not completely random assortments of genic and non-genic sequence. Genomes possess higher-order physical organization, including structural motifs such as centromeres and telomeres, reasonably distinct domains of heterochromatin and euchromatin [1], and less well-defined regions with biased base composition, such as isochores [2]. Various functional states have been correlated with these organizational groupings. GC-rich isochores, for instance, are relatively gene dense [3], and genes within these isochores tend to be more highly transcribed [4] than genes in less GC-rich regions of the genome.
Metazoan genomes also contain physical clusters of co-regulated genes. Highly conserved, tightly regulated clusters include the Hox genes, which specify anterior-posterior pattern in all bilaterians [5]. Other clusters that are more loosely arranged include human housekeeping genes [6-9], testis-specific genes in Drosophila melanogaster [10], and muscle-specific genes in Caenorhabditis elegans [11]. These observations suggest that the typical metazoan genome has more fine-scale architecture than is readily apparent. However, the vast majority of metazoan genes are not located in any known cluster and so it remains unclear whether or how these genes are organized. Furthermore, the majority of coexpressed clusters identified in D. melanogaster do not share common functional annotations, suggesting that the apparent coexpression of physically clustered genes may be the result of increased local accessibility of promoters in opened chromatin, rather than explicit regulatory similarity [12].
Despite sharing structural and organizational features, metazoan genomes vary in total size (C value) across several orders of magnitude [13]. Several explanations for this variation have been proposed. Noncoding, repetitive DNA elements, such as transposons, satellites and simple sequence repeats, can account for some fraction of genome size difference [14,15]. An extension of this model suggests that genome size is determined by the balance between insertions, such as rare bouts of invasion by self-replicating elements, and deletions of nonfunctional DNA from the genome [16-18]. Such mutational models of genome size can be contrasted to adaptive models, which suggest that selective constraints act on overall genome size, largely independent of any specific informational content of the DNA. For example, genome size and cell size are significantly correlated [19]. This correlation may influence the developmental rate and developmental complexity of an organism and thereby exert selective pressure on overall genome size [20].
While both mutational and adaptive models contribute to our understanding of metazoan genome size, neither addresses an important aspect of DNA function - the regulation of gene expression - and its possible effect on genome size and architecture. The effect of regulatory DNA on genome architecture has been ignored largely because of the difficulty of identifying regulatory elements and the general assumption that most intergenic DNA is nonfunctional. However, in lineages that have experienced high rates of DNA loss it is possible that the spatial requirements of regulatory DNA could shape intergenic distance and hence genome architecture. Here we examine how regulatory DNA influences gene distribution in two distantly related animals, D. melanogaster and C. elegans. We compare the regulatory complexity of a large sample of the genes from each animal with the spacing of these genes within each genome. We find a positive correlation between the inferred regulatory complexity of a gene and the distance from that gene to its nearest neighbor. We also find that while genes with common housekeeping functions occupy approximately the same amount of space in both D. melanogaster and C. elegans, genes that play a central role in development and pattern formation occupy significantly more space in D. melanogaster. Finally, it appears that C. elegans partitions its regulatory information upstream of the promoter, whereas no strong bias is apparent in D. melanogaster. We suggest that the interplay between the relatively high rate of nonfunctional DNA loss and selective pressure to maintain minimal spatial requirements for essential genetic regulatory information shapes genome architecture in these taxa.
Results
Genomes contain relatively few genes with highly complex expression patterns
Because we cannot directly measure regulatory complexity, we developed surrogate measurements for the regulatory complexity associated with individual genes. In many cases, complex expression patterns are composed of separable tissue-specific or spatially specific subpatterns, each of which is driven by a discrete cis-regulatory element (see for example [21-23]. Thus, genes expressed in a greater number of tissues and spatial domains tend to require a greater number of regulatory elements to drive this expression (see for example [24-28]). Accordingly, we use the complexity of a gene's expression pattern as a surrogate for its regulatory complexity.
In this study we measured complexity of expression pattern in two ways. First, we surveyed the curated literature-based resources of FlyBase and WormBase and generated an expression complexity index from each. FlyBase and WormBase contain information on expression pattern and mutant phenotype for every gene that has been studied in each animal. Our FlyBase index (FBx) counts domains of gene expression and tissues affected in mutant larvae, adults and embryos. FlyBase contains information on 1,879 of the 13,370 predicted genes in the euchromatic portion of the D. melanogaster genome, from which we generated FBx values. WormBase contains expression pattern entries for 1,125 genes of the 19,614 predicted genes in the C. elegans genome, from which we generated WormBase (WBx) values. Our second measure for complexity of expression pattern was obtained from the Berkeley Drosophila Genome Project (BDGP) in situ hybridization (ISH) project [29]. Using a random, nonredundant set of expressed sequence tags as probes, this project is systematically surveying gene expression during D. melanogaster embryogenesis. Annotation of the 1,728 genes surveyed (as of October 2003) was used to generate our BDGP index values (BDGPx).
These indices survey the complexity of gene expression patterns in approximately 14% (FBx) and approximately 13% (BDGPx) of D. melanogaster genes (3,156 unique genes, ~24% of the total predicted gene set), and approximately 6% of C. elegans genes (WBx). All three distributions contain many genes that have a low expression complexity value and far fewer genes that have a high expression complexity value (Figure 1). This result indicates that most of the genes in these genomes are deployed in a small number of tissues, whereas a small set of genes is used repeatedly in specific tissues at specific times. Therefore, most genes in these animals are likely to require a small number of cis-regulatory elements, whereas a much smaller group is likely to require large arrays of regulatory elements.
Figure 1.
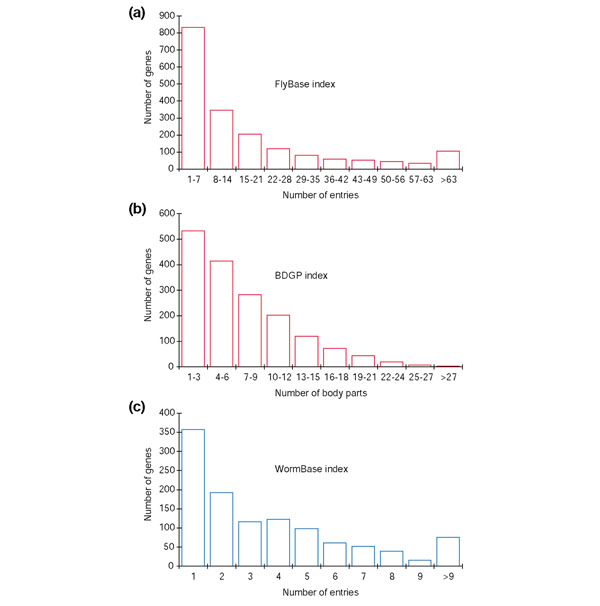
Genes of low regulatory complexity are common and genes of high regulatory complexity are rare in D. melanogaster and C. elegans. Distribution of genes with respect to complexity of expression in (a) FlyBase index (FBx), (b) BDGP in situ hybridization index (BDGPx), and (c) WormBase index (WBx). In all three cases, the distributions are heavily weighted toward genes expressed in a small number of locations and show relatively few genes deployed in a large number of tissues.
Regulatory complexity and gene spacing
To accommodate a large number of separate regulatory elements, organisms could employ two basic approaches. They could increase the density of regulatory elements - that is, increase the informational content, but maintain overall size of a regulatory region (as in viruses). Alternatively, they could add elements by expanding the physical size of a regulatory region - that is, maintain the density of information, and increase the space occupied by that regulatory information. If a regulatory element requires a minimal threshold of physical space, then genes with a complex expression pattern that require more regulatory elements will also require more physical space in the genome to contain those elements. Therefore, we determined whether there is a correlation between regulatory complexity (as estimated by our expression complexity indices) and the amount of noncoding DNA flanking each gene.
We determined intergenic distance for all genes in the euchromatic portions of the D. melanogaster and C. elegans genomes (intergenic distance is defined as the sum of upstream and downstream distance to the nearest neighboring genes; see Materials and methods for details) and compared this distance to each gene's expression index value. For each of the three expression indices we divided index values into bins containing roughly 10% of the genes in each sample and plotted the mean intergenic distance for each bin (division of the data into precise 10% bins was constrained by integral data values; see Materials and methods for details). We found that intergenic distance is positively correlated with expression diversity (FBx, Pearson r = 0.23, least-squares linear regression r2 = 0.05, p < 0.0001; BDGPx, r = 0.13, r2 = 0.02, p < 0.0001; WBx, r = 0.19, r2 = 0.04, p < 0.0001). More intergenic DNA flanks bins of genes inferred to have greater regulatory complexity than bins inferred to have low regulatory complexity (Tukey-Kramer HSD, α < 0.05; see Figure 2 and Materials and methods). This is true in both D. melanogaster and C. elegans, regardless of the index used to estimate regulatory complexity (literature-derived or in-situ derived).
Figure 2.
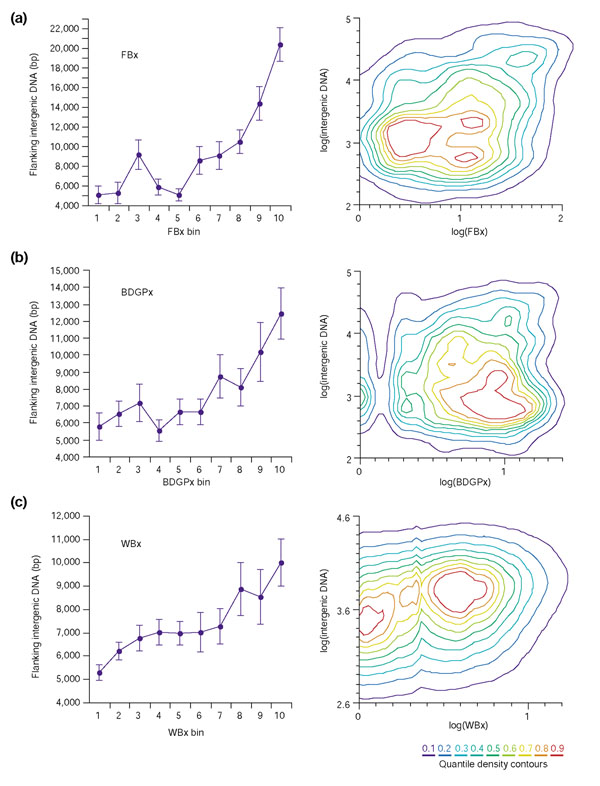
Intergenic DNA increases with regulatory complexity in D. melanogaster and C. elegans. Expression indices were divided into bins, each containing approximately 10% of the entries in an index. Mean amount of intergenic DNA for each bin (± standard error) was plotted for all three expression indices (left): (a) FBx; (b) BDGPx; (c) WBx. The average amount of intergenic DNA flanking the genes in bins of greater regulatory complexity is significantly greater than that of bins of lower regulatory complexity in all three indices (Tukey-Kramer HSD, α = 0.05). In the nonparametric bivariate density plots of intergenic DNA versus index value (right), each contour represents a boundary including 10% of the data. The innermost red contour includes 10% of the data points and excludes the other 90%. The outermost purple contour includes 90% of the data points, whereas 10% fall outside this boundary.
Measurement of intergenic distance does not account for the possibility of regulatory information contained within the boundaries of a gene itself (for example, 5' and 3' untranslated regions and introns). However, transcriptional regulatory elements do occur in these regions (see for example [30,31]). In addition, regulatory elements can lie within or beyond adjacent genes (see for example [32]). Therefore, we established an alternative means of measuring the footprint of a gene that would take these scenarios into account. We generated sliding windows spanning many genes along each D. melanogaster chromosome and graphed the size of each window (in base pairs) relative to position on the chromosome. Of the window sizes tested (ranging from 5 to 50 genes), an 11-gene window was judged to provide the best resolution of peaks from background variation (Figure 3 and data not shown). This window measures the size of the immediate neighborhood of the central gene in an 11-gene interval (1 central gene and 5 genes on either side), providing a broader view of the arrangement of nearby genes and potential regulatory regions. Each chromosome contains regions of high gene density, where 11 genes are tightly packed with little intervening DNA, and peaks of low gene density, where 11 genes and their associated intergenic DNA are widely spaced (for a typical example see Figure 3). Low gene density indicates that one or more genes within a window have a large amount of associated noncoding DNA. By our model, peaks of low gene density, which contain more intergenic DNA, should be more likely to contain genes of high regulatory complexity. To test this prediction on the X chromosome, we identified all genes within peaks greater than a visually selected cutoff of 250 kb. We then assessed the expression complexity of genes in these large windows using our expression indices. Although most genes in the D. melanogaster genome are unknown with respect to expression pattern and as a result do not have index values, peaks greater than 250 kb in size contain significantly more genes of high expression complexity than the average 11-gene window on the X chromosome (Figure 3; Welch ANOVA, p < 0.008; Wilcoxon two-sample test, p < 0.03). Thus, we observe a significant correlation between gene spacing and regulatory complexity using three independent measures of expression complexity, two independent measures of locus size, and in two very different animals.
Figure 3.
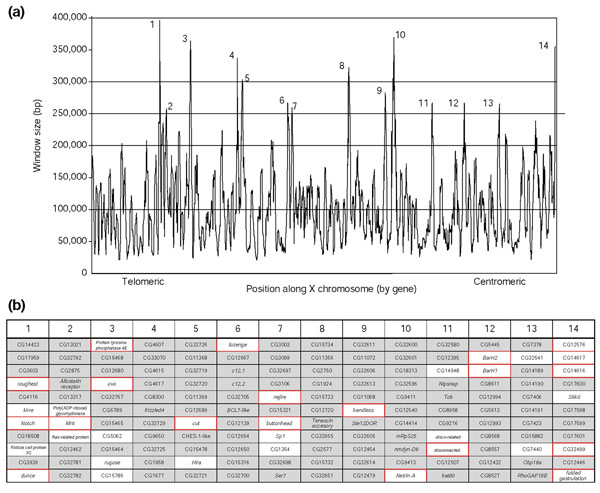
Regions of low gene density contain significantly more genes of high regulatory complexity. (a) Window size (in base pairs) of an 11-gene sliding window across the X chromosome versus position along the chromosome. The horizontal line at 250,000 bp indicates the cutoff above which a window was designated as low density. A total of 53 windows larger than 250,000 bp were identified on the X chromosome. These windows overlap to generate 14 independent peaks, numbered 1 through 14. Normalized FBx and BDGPx scores for each gene were calculated by dividing the raw index score by the maximum score for that index. The normalized scores of all low-density windows were compared to the scores of all 11-gene windows on the chromosome. The expression complexity score for low gene density windows was significantly greater than the average score for all possible windows on the X chromosome (Welch ANOVA, p < 0.008; Wilcoxon two-sample test, p < 0.03). (b) The 11 genes flanking the highest point of each numbered peak on the X chromosome. Genes boxed in red fall in the top 20% of expression complexity by FBx or the top 24% by BDGPx. Genes in unshaded boxes have expression data available, but do not fall in the upper range of the FBx or BDGP indices. Genes that are shaded, which represent the majority of genes in these windows, have no expression data available. This panel indicates only genes in the highest central peak. However, all genes within windows exceeding 250,000 bp in size were used for the statistical analysis described above.
Functional classification and gene spacing
Much study of the evolution of development has focused on a relatively small subset of genes that govern multiple developmental processes [33-35]. These genes typically encode transcription factors and signaling molecules, rather than metabolic enzymes or structural components of the cell. The repeated utilization of genes in these developmentally important classes predicts that these genes should require greater numbers of regulatory elements and larger stretches of intergenic DNA than genes with primarily housekeeping functions.
To test this prediction we used functional categories based on Gene Ontology (GO) [36] and additional literature-derived functional groupings to investigate the correlation between gene spacing and functional classification. Because GO annotations for D. melanogaster and C. elegans use different categorizations, they are not directly comparable. Therefore, we selected GO categories of interest from D. melanogaster and used BLAST to determine the best match for each fly protein in the C. elegans proteome. The GO categories used were: pattern specification (GO:0007389), embryonic development (GO:0009790), specific RNA polymerase II transcription factors (GO:0003704), receptor activity (GO:0004872), cell differentiation (GO:0030154), metabolism (GO:0008152), structural constituents of the ribosome (GO:0003735), and general RNA polymerase II transcription factors (GO:0016251). Some genes (for example, caudal, Notch, twist, and others) are members of more than one selected GO category; however, we accounted for this in our analysis (see below and Materials and methods). In addition to the GO categories, we generated a list of housekeeping genes (HK set) by combining three lists of human housekeeping genes [6-8] and using BLAST to identify the best single match for these genes in the D. melanogaster and C. elegans proteomes. Finally, we analyzed genes present in single copy in C. elegans, D. melanogaster and the yeast Saccharomyces cerevisiae, (CDY set) [37], which are likely to represent genes with primarily housekeeping functions [38].
In both C. elegans and D. melanogaster, 'simple' gene groups with primarily ubiquitous or 'housekeeping' functions (CDY, general transcription factors, ribosomal constituents, metabolism and HK sets) are flanked by an average of 4-5 kb of intergenic DNA. In contrast, 'complex' groups with more diverse roles (embryonic development, pattern specification, and specific TFs) average 8-11 kb of intergenic DNA in C. elegans and 17-25 kb in D. melanogaster (Figure 4). Two groups, receptor activity and cell differentiation genes, were more variable between the two species, suggesting possible differences in the biological roles of these groups in the two organisms.
Figure 4.
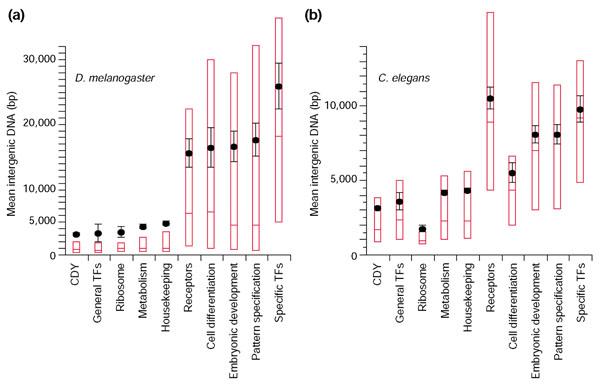
Functionally complex genes have more intergenic DNA than functionally simple genes. A comparison of intergenic distances among genes of different GO groups. The mean and median amounts of flanking intergenic DNA are shown for various functional categories of genes in (a) D. melanogaster and (b) C. elegans (black points and bars indicate mean value ± standard error; red bars indicate median values, red boxes enclose 25th-75th percentiles). Genes with low regulatory complexity are represented by the CDY, general RNA polymerase II (PolII) transcription factors, ribosomal components, metabolism, and housekeeping gene sets. Genes of high regulatory complexity are represented by receptor activity, cell differentiation, genes involved in embryonic development, genes involved in pattern specification, and specific RNA PolII transcription factors. All sets of low regulatory complexity have significantly less flanking intergenic DNA than all sets of high regulatory complexity regardless of species (Tukey-Kramer HSD, α = 1 × 10-4).
We next pooled all genes in the five simple groups and all genes in the three complex groups to generate nonredundant gene sets. For these sets, we assessed the contribution of 5' and 3' noncoding regions to the total intergenic distance (Figure 5a). In both the C. elegans and D. melanogaster simple gene sets, 5' and 3' noncoding regions each contribute approximately 2 kb of DNA to the total intergenic distance. For the complex gene sets, total intergenic DNA is partitioned nearly equally between upstream and downstream sequences in D. melanogaster, whereas upstream DNA is significantly larger than downstream DNA in C. elegans (Figure 5a, Wilcoxon two sample test, p < 0.0001). These results suggest that C. elegans cis-regulatory elements largely occupy space upstream of the regulated gene, consistent with analysis of several C. elegans enhancers [39]. In contrast, D. melanogaster appears equally likely to distribute regulatory information upstream or downstream of the gene, consistent with observations of extensive 3' regulatory regions in D. melanogaster [40-42]. It is important to note that while the amount of intergenic DNA flanking groups of simple genes is not significantly different between animals (Figure 5a), genes that have complex functions in D. melanogaster are flanked by significantly more intergenic DNA than their C. elegans counterparts (Tukey-Kramer HSD, α = 1e-4; Wilcoxon two sample test, p < 0.001; see Materials and methods).
Figure 5.
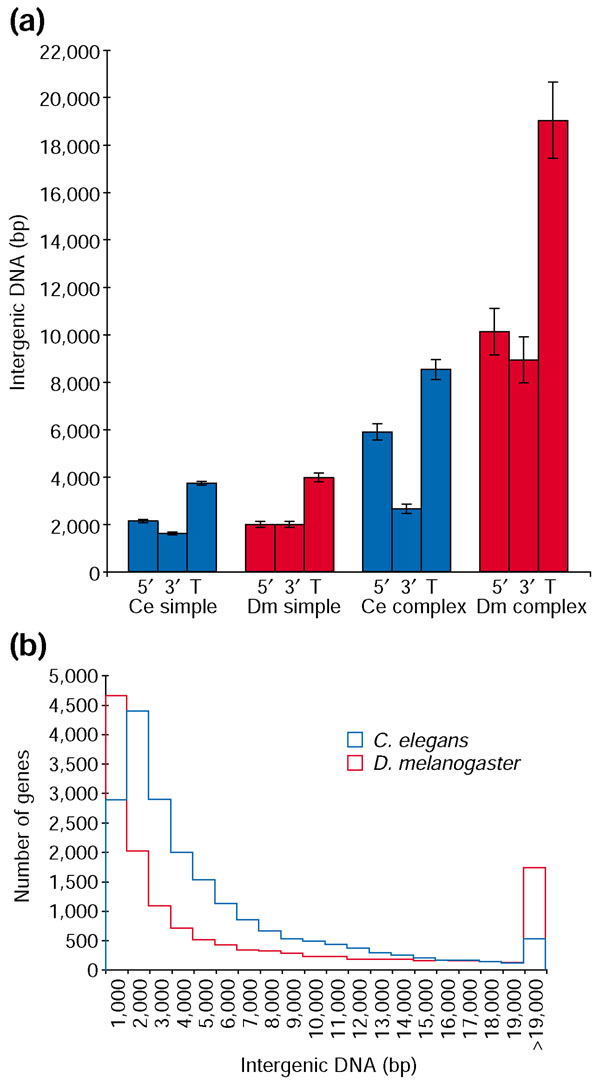
Complex genes have more intergenic DNA in D. melanogaster than in C. elegans. (a) Mean 5' flanking DNA (5'), 3' flanking DNA (3'), and total intergenic DNA (T; all ± standard error) is shown for nonredundant groups of simple genes (CDY, general RNA PolII transcription factors, ribosomal components, metabolism, and housekeeping) and complex genes (embryonic development, pattern specification, and specific RNA PolII transcription factors) in C. elegans (blue) and D. melanogaster (red). C. elegans complex genes have significantly more 5' flanking DNA than 3' flanking DNA (Wilcoxon two-sample test, p < 0.0001). The C. elegans complex group is flanked by significantly less DNA than the D. melanogaster complex group (Tukey-Kramer HSD, α = 1 × 10-4). (b) Distribution of intergenic DNA for all genes in C. elegans (blue) and D. melanogaster (red). In general, genes in C. elegans are more evenly spaced than in D. melanogaster. The largest class of genes in D. melanogaster has less than 1,000 bp of intergenic DNA separating neighboring genes, whereas the largest class in C. elegans has 1,000-2,000 bp. Thus, D. melanogaster does not have a euchromatic genome that is generally expanded with respect to C. elegans, even though it has many more genes with greater than 19,000 bp of flanking intergenic DNA.
Approximately 15% of C. elegans genes are predicted to be located in co-regulated operons [43]. Intergenic distance between genes within operons is likely to underestimate the size of DNA used to regulate these genes and this underestimate could contribute to the observed difference in complex gene spacing between C. elegans and D. melanogaster, which does not organize genes into operons. We determined that approximately 12% of genes in the complex groups and approximately 37% of genes in the simple groups are predicted to be organized into operons in C. elegans (data not shown). Removing these genes from their respective datasets had no effect on the observed difference between D. melanogaster and C. elegans gene groups (Tukey-Kramer HSD, α = 1 × 10-4).
We were also concerned that general euchromatic genome expansion in D. melanogaster or euchromatic genome compaction in C. elegans could account for the difference in amount of intergenic DNA associated with complex genes. To assess this possibility, we analyzed the distribution of intergenic DNA measurements for all genes in both animals (Figure 5b). The D. melanogaster genome, which has approximately 55 Mb of intergenic DNA, has more genes with large amounts of intergenic DNA than does the C. elegans genome, which has approximately 47 Mb of intergenic DNA (estimated using upstream and downstream intergenic distances as calculated in this study). However, this difference in intergenic spacing is not uniformly distributed, as D. melanogaster shows both more regions of dense gene spacing and highly dispersed gene spacing than C. elegans, whose genes are more evenly distributed (Figure 5b). Thus, the larger intergenic regions seen in D. melanogaster genes of complex function is not consistent with a general genome-wide expansion in flies or compaction in worms.
Finally, we examined individual genes of complex function to examine how the difference observed at the group level would be reflected at the level of individual genes. From the CDY set and KOG (euKaryotic clusters of Orthologous Genes [44]) we identified orthologous pairs of genes or gene families in D. melanogaster and C. elegans. We then selected genes known or expected to be developmentally important in D. melanogaster, and confirmed their orthologous relationships with C. elegans genes using the KOGnitor comparison tool. These candidate groups yielded 29 relatively clear single-copy orthologs and many orthologous gene families. For a representative group of 49 D. melanogaster genes and their C. elegans counterparts (including all 29 single-copy orthologs identified and 5 gene families, Figure 6a), the mean intergenic interval is 27,928 bp in D. melanogaster and 7,670 bp in C. elegans, thoroughly consistent with the trend observed at the group level (Figure 4a). In addition, many of the D. melanogaster genes are located in gene-sparse regions of the genome and have larger introns (Figure 6b), suggesting that they have even more space available for potential regulatory elements than indicated by the larger flanking regions alone.
Figure 6.
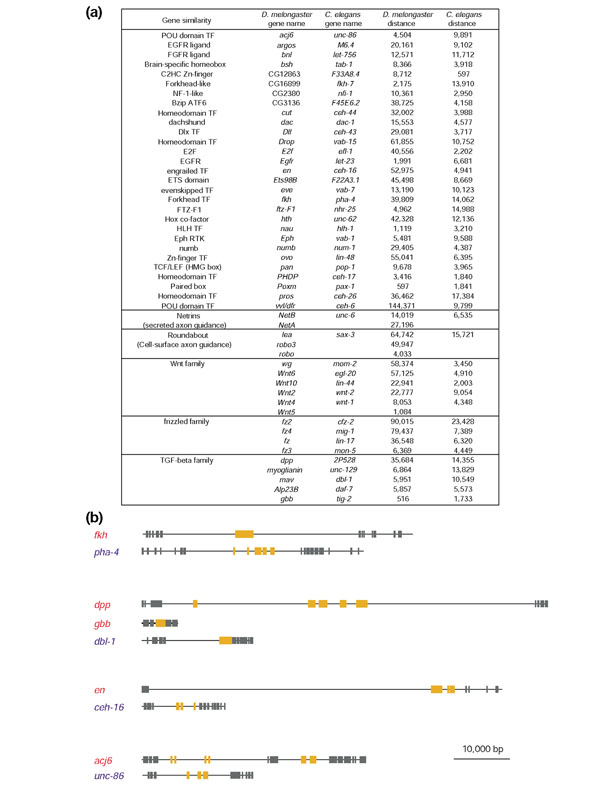
Developmentally important genes in D. melanogaster have larger intergenic intervals than their C. elegans counterparts. (a) Forty-nine developmentally important genes from D. melanogaster and their C. elegans counterparts. Genes in the top section represent orthologs, defined by KOG. Subsequent sections represent gene families. Listing of genes in different species on the same line within gene families does not imply that they are orthologous. The mean intergenic size for the D. melanogaster genes is 27,928 bp. Then mean intergenic size for the C. elegans genes is 7,670 bp. (b) Genomic regions of four representative gene sets in D. melanogaster (red) and C. elegans (blue). Orange boxes designate exons of the indicated genes. Gray boxes designate exons of neighboring genes. Note that genomic intervals are typically larger in D. melanogaster than in C. elegans, often owing to both larger flanking noncoding regions and larger introns. The total euchromatic genome of D. melanogaster is estimated at 117 Mb and the euchromatic genome of C. elegans is estimated at 100 Mb. The overall gene distribution within the genome is denser in flies than worms, suggesting that the larger regions of noncoding DNA associated with these representative complex genes are specifically allocated to these loci.
Discussion
We have examined the relationship between the regulatory complexity of a gene and the spacing of that gene with respect to its neighbors in D. melanogaster and C. elegans. We show that in each animal developmentally important genes expected to possess high levels of regulatory information occupy more space in the genome than other gene classes. This regulatory information may comprise enhancer elements with well-defined binding sites for transcription factors, insulator elements, which contribute to the precise expression pattern of a gene by preventing cross-talk between enhancers [45], and other known and unknown regulatory motifs. In addition, developmentally important genes in D. melanogaster have more space for regulatory information than the corresponding C. elegans genes, and C. elegans tends to apportion its noncoding DNA upstream of the gene whereas D. melanogaster shows no significant bias. These results show that regulatory information shapes genome architecture and provide support at the genomic level for a model in which the expansion of regulatory information facilitates increased morphological complexity in metazoa.
Reliability of expression indices
Because direct measurement of regulatory complexity for all genes in the D. melanogaster and C. elegans genomes is not possible, we used several surrogate measures of regulatory complexity. These surrogates necessarily introduce uncertainty into our assessment of regulatory complexity, and here we attempt to assess the effect of these uncertainties on our conclusions.
All three indices will tend to underestimate the true complexity of a gene's full expression pattern simply because the expression of very few genes has been surveyed in all tissues throughout the life cycle of any animal. For instance, the BDGPx only considers embryonic expression. Furthermore, little information is available on environmentally responsive gene expression, as most investigation has focused on developmental profiles of expression under standardized conditions. However, the systematic underestimation of regulatory complexity due to limited sampling across environmental conditions or developmental stages applies to all genes, not preferentially to genes expressed in either a simple or complex pattern, and therefore should not significantly bias our conclusions.
Our two literature-derived indices (FBx and WBx) suffer from ascertainment bias. Genes involved in multiple developmental processes or genes that have large genomic footprints are more readily identified in genetic screens and are more likely to elicit sustained investigation. This situation has led to a relative over-representation of developmentally important genes in the literature-based indices and a probable overestimation of regulatory complexity for genes with very high FBx or WBx values. By combining genes with the highest index values into a single group, the binning of individual index values reduces the effect of overestimating regulatory complexity. In addition, GO groups and the in situ hybridization index (BDGPx) are immune to this sampling issue because they consider either functional classification or a completely random gene set, respectively, and each clearly shows the same trend as the literature-derived indices.
Curation of the data in all three indices may also introduce uncertainty into our results. For instance, the BDGP in situ project annotates gene expression maintained over multiple developmental stages in a single organ as multiple distinct entries [29]. Similarly, housekeeping genes, whose expression may be driven by only one cis-regulatory element, are found in many tissues, and so the BDGPx will tend to overestimate the regulatory complexity of these genes. However, the BDGP project only annotates genes with some degree of tissue specificity, omitting ubiquitously expressed genes [29]. A simple gene whose regulatory complexity has been overestimated would introduce a smaller value for intergenic distance into the high regulatory complexity group. Therefore, overestimation of regulatory complexity for simple genes should dilute, rather than enhance, the positive correlation between regulatory complexity and intergenic distance. Manually collapsing tissue annotations across developmental stages improved the correlation between intergenic DNA size and the BDGPx (data not shown), but we report the unmodified BDGP data here to avoid investigator-derived bias in our estimates of regulatory complexity. Moreover, the GO-derived groups are not subject to the same systematic biases as the other indices but show the same overall result.
While it is generally accepted that complex gene expression requires complex regulatory control, we must consider the degree to which expression complexity is a legitimate proxy for regulatory complexity. The expression of particular genes in distinct morphological fields, tissues and organs is consistently controlled by physically and functionally discrete cis-regulatory elements (reviewed in [33-35]). Conversely, gene expression in populations of cells with shared identity is often controlled by a single regulatory element (see for example [46-48]). Thus, genes that have a complex expression pattern tend to use a greater number of cis-regulatory elements than genes expressed in a single tissue, location or cell type. This trend clearly supports the use of expression complexity as a surrogate for regulatory complexity. However, even genes that have a simple expression pattern occasionally use multiple cis-regulatory elements (see for example [49]), and an apparently complex expression pattern will sometimes be driven by a relatively simple control element (see for example [50,51]). As a relative measure, therefore, complexity of expression pattern should faithfully approximate regulatory complexity for a group of genes, but will not reliably predict the absolute number of cis-regulatory elements used by any individual gene.
Regulatory DNA and genome architecture
The distribution of regulatory information among genes in the genomes of D. melanogaster and C. elegans is not uniform. All three expression indices indicate that most genes are expressed in simple or limited domains whereas relatively few genes are expressed in a wide variety of specific tissues (Figure 1). This observation is consistent with known principles of animal development. A relatively small set of genes, primarily transcription factors and signaling molecules, play a disproportionate role in the development of metazoans (reviewed in [33-35]). These genes are used repeatedly during development to generate the basic body plan and specify organ identity. Once this morphological ground plan is established, a larger suite of tissue-specific genes is deployed during terminal differentiation. Accordingly, transcription factors and signaling molecules consistently have high values in our expression indices (Figure 4 and data not shown) while genes of low regulatory complexity comprise the bulk of the genome.
We show here how these relatively few genes of high regulatory complexity have accommodated their need for increased amounts of regulatory information. An increase in regulatory information will require either an increase in information density or an increase in the space allocated to storing that information. If the size of intergenic DNA in metazoan genomes were essentially unconstrained, an increase in the space devoted to information storage would escape notice in the background fluctuation of intergenic distance and would have no discernable effect on the distribution of genes within the genome. DNA with little informational content would predominate, and even genes that require a large number of regulatory elements would have more than enough intergenic DNA to accommodate those elements without apparent expansion. If, however, functional regulatory DNA represents a significant portion of the intergenic DNA in a genome, then there should be a direct correlation between regulatory information content and quantity of intergenic DNA [52]. That is, genes with many regulatory elements will require more space, and this space will have a significant impact on the local arrangement of genes. Indeed, we find that genes predicted to have more regulatory elements occupy significantly more space than do their simple neighbors. The fact that we can see this relationship suggests that the genomes of C. elegans and D. melanogaster possess a high ratio of functional regulatory DNA to nonfunctional noncoding DNA.
It is interesting to note that evidence suggesting regulatory DNA in C. elegans is most often positioned upstream of a gene's promoter [39] is strongly supported by our analysis of the relative size of 5' and 3' noncoding intervals for the complex gene sets. No such bias in the distribution of noncoding DNA is apparent in D. melanogaster, suggesting that these two animals may have different constraints on the location of regulatory information relative to the promoter of a gene.
Evolution of genome architecture
How does this architecture arise? The net difference between the rate of DNA deletion and insertion appears to determine the direction of genome expansion or compaction in many organisms [16,17]. Both the D. melanogaster and C. elegans lineages have unusually high rates of DNA deletion, leading to compact genomes [53-55]. For instance, the rate of DNA loss is 40 times higher in the approximately 180 Mb D. melanogaster genome than in the approximately 1,980 Mb genome of Hawaiian crickets [17], and is 60 times faster in Drosophila than in mammals [56]. When the DNA-deletion rate is significantly greater than the rate of DNA insertion, deletion will predominate in reducing genome size and sculpting genome architecture. As deletions become more and more likely to remove functional DNA, selection against further deletion should tend to stabilize the minimum size of intergenic regions, and the underlying architecture of the genome will emerge.
Our work suggests that high rates of DNA loss may sculpt the spacing of genes toward minimum functional requirements for regulatory DNA. Such functional constraints in noncoding DNA are known to affect distributions of insertions and/or deletions (indels). For example, constraints imposed by intronic splicing requirements influence the pattern of deletion and insertion observed in D. melanogaster introns [57]. Comparison of noncoding regions of different Drosophila species indicates that conserved noncoding sequences are often found in small blocks, with conserved spacing between the blocks [58,59]. This suggests that spacing constraints also act in intergenic regions, potentially to preserve spacing between specific transcription factor binding sites or other regulatory elements, or more generally to provide sufficient physical space to insulate regulatory elements from one another. In addition, interference selection, lowered recombination due to segregation of weakly selected mutations, was suggested to account for a correlation between intergenic distance and coding region length [60]. A proposed alternative, that longer genes are functionally more complex and therefore require larger noncoding regions [60], now finds support in our observed correlation between intergenic distance and regulatory complexity. Interference selection may itself contribute to the evolution of complex regulatory regions: minimum spacers, favored in the reduction of recombination interference, may be required for recombination of complex modular regulatory elements.
Other compact genomes, such as that of the teleost fish Fugu rubripes, are also likely to be the product of greater rates of DNA loss and are expected to show the relationship between regulatory complexity and intergenic distance demonstrated here. Even in the large human genome, there is evidence that some regions have experienced compaction where gene density is increased. Dense gene clustering implies a relative lack of local regulatory complexity and predicts that the clustered genes should have relatively simple expression patterns. This prediction is indeed supported by the presence of tissue-specific and housekeeping gene clusters and regions of high gene density in the human genome [4,8,9,61]. Thus, the emergence of some regions of high gene density and clusters may reflect deletion acting to reveal local regulatory complexity, rather than the organization of the genome into chromatin domains or multigene transcriptional groups. In addition, the association between gene spacing and regulatory complexity could be exploited in the analysis of novel genes and genomes. Based on our results, the relative regulatory complexity of a 'novel' gene might be inferred on the basis of the architecture of its local genomic neighborhood.
Conclusions
Because of the vast size of animal genomes compared to the small, relatively discrete functional elements within them, regulatory DNA has been presumed to exert little, if any, global effect on metazoan genome organization. Here we have shown that spatial requirements for regulatory DNA shape the density of genes in the genomes of D. melanogaster and C. elegans. Further, we propose that small DNA deletions, constrained by functional blocks of DNA, are the primary mechanism for sculpting genome architecture. Repeated bouts of insertion and deletion may actively shape gene distribution - globally in organisms with compact genomes, and locally in organisms with expanded genomes.
Materials and methods
Datasets
The D. melanogaster genome annotations version 3.1 [62] were obtained from the BDGP. Only genes in the euchromatic portion of the genome were used for analysis. C. elegans genomic data were obtained from WormBase genome freeze WS100 [63,64].
Expression data for D. melanogaster were obtained from two independent sources. First, we determined the number of 'Expression and Phenotype' tags for all D. melanogaster genes listed in FlyBase [65]. Second, we measured embryonic expression complexity by counting the 'body parts' listed in the BDGP in situ hybridization database [66] (accessed 10 October 2003). This project uses a controlled vocabulary to annotate the expression of each gene during embryogenesis [29]. C. elegans expression data was obtained through AQL (Acedb Query Language) queries of WormBase for all genes that possessed 'Expr_pattern' entries.
The housekeeping (HK) gene set was generated by combining three lists of proposed human housekeeping genes [6-8]. This nonredundant list was compared by BLAST [67] to the D. melanogaster and C. elegans genomes. We retained only the best hit in each genome that exceeded an E-value of 1 × 10-20. The CDY (C. elegans, D. melanogaster, and yeast) dataset is derived from single-copy genes shared by Saccharomyces, Drosophila and Caenorhabditis [37]. We infer that these genes will largely have shared basal functions and few cell-type-specific functions [38]. Gene lists and sequences were retrieved by EnsMart from the Ensembl Genome Browser [68]. Because the C. elegans genome annotation employs different GO terms from that of Drosophila, we placed C. elegans genes into corresponding GO categories by BLAST of the D. melanogaster GO gene sets against the C. elegans proteome.
Spacing analysis
We wrote several PERL programs (available upon request) to parse C. elegans and D. melanogaster genomic data and calculate intergenic distances. For most genes, we defined upstream distance as the distance between the start of a gene's first exon and the boundary of the closest upstream neighboring exon (irrespective of DNA strand). We defined downstream distance as the distance between the end of a gene's last exon and the boundary of the closest downstream neighboring exon. Total intergenic distance was defined as the sum of the upstream and downstream distances. However, both genomes contained examples of genes with overlapping or interdigitated exons. In cases where exons overlapped with one another, intergenic distance was defined as zero. In cases where an exon was located within the intron of another gene, the intergenic distance was calculated from the boundary of the exon of interest to the nearest intron/exon boundary.
Data analysis and statistics
Data management and analysis were performed using a combination of PERL programs, Microsoft Excel and JMP 3.0 (SAS Institute).
Composition of individual indices and bins. FlyBase index (1,879 genes): Bin 1, genes with an index value of 1, corresponding to 1 'Expression and Phenotype' entry in FlyBase, N = 108 entries; Bin 2, two entries, N = 227; Bin 3, three entries, N = 172; Bin 4, four to five entries, N = 184; Bin 5, six to eight, N = 206; Bin 6, 9-13, N = 235; Bin 7, 14-18, N = 184; Bin 8, 19-29, N = 187; Bin 9, 30-49, N = 193; Bin 10, 50-336, N = 183.
BDGP index (1,698 genes): Bin1, one body part listed, N = 163; Bin 2, two body parts, N = 184; Bin 3, three body parts, N = 172; Bin 4, four body parts, N = 159; Bin 5, five body parts, N = 145; Bin 6, six to seven body parts, N = 201; Bin 7, eight to nine body parts, N = 180; Bin 8, 10-13, N = 144; Bin 9, 12-14, N = 142; Bin 10, 15-42, N = 208.
WormBase index (1,130 genes): Bin 1, one 'Expr_pattern' entry, N = 357; Bin 2, two entries, N = 192; Bin 3, three entries, N = 116; Bin 4, four entiries, N = 123; Bin 5, five entries, N = 98; Bin 6, six entries, N = 61; Bin 7, seven entries, N = 52; Bin 8, eight entries, N = 39; Bin 9, 9-11, N = 43; Bin 10, 12-27, N = 49.
Comparison of all pairs of bins in each index was performed using Tukey-Kramer HSD. As the size of intergenic DNA in each bin approximates a log-normal distribution (Figure 4, and data not shown) we compared both raw and log-transformed measurements. In all cases bins of higher inferred complexity tended to have higher average measures of intergenic DNA than bins of lower inferred complexity (Tukey-Kramer HSD, α = 0.05).
Composition of functional groups: CDY, Ce N = 1,237, Dm N = 1,250; general transcription factors, Ce N = 43, Dm N = 43; HK, Ce N = 540, Dm N = 609; pattern specification, Ce N = 73, Dm N = 73; embryonic development, Ce N = 88, Dm N = 88; specific transcription factors, Ce N = 45, Dm N = 45; metabolism, Ce N = 881, Dm N = 881; cell differentiation, Ce N = 46, Dm N = 46; receptor activity, Ce N = 106, Dm N = 106; ribosome constituents, Ce N = 93, Dm N = 93. The mean size of the intergenic DNA associated with each group suggested that the simple gene groups are not significantly different between species, but that both simple groups are smaller than both complex groups and that the C. elegans complex group is smaller than the D. melanogaster complex group (Tukey-Kramer HSD, α < 1e-4). This interpretation was confirmed by independent inspection of the intergenic DNA size distributions for each group. Complex groups had many more genes with large intergenic regions than simple groups did. Comparison between the C. elegans complex group and the D. melanogaster complex group was complicated by the observation that the D. melanogaster group contained both more genes with smaller than average intergenic regions and many more genes with much larger than average intergenic measures. We divided both raw and log-transformed measures from D. melanogaster and C. elegans into halves containing the largest and smallest 50% of genes. The largest 50% of complex genes in D. melanogaster is flanked by significantly more DNA than the largest 50% of C. elegans complex genes (Wilcoxon two-sample test, p < 0.001).
Additional data files
An Excel file containing the primary data used for the three expression indices, the D. melanogaster X chromosome, and the GO groups, is included (Additional data file 1).
Supplementary Material
An Excel file containing the primary data used for the three expression indices, the D. melanogaster X chromosome, and the GO groups
Acknowledgments
Acknowledgements
We thank Dan Lautenschleger for help with PERL scripts, and Barry Williams, John Yoder and ShengQiang Shu for their input and assistance. C.E.N. is supported by NRSA#5 F32 HD41314-02. B.M.H. is supported by NRSA #F32GM65737-02. S.B.C. is an Investigator of the Howard Hughes Medical Institute.
References
- Grewal SI, Moazed D. Heterochromatin and epigenetic control of gene expression. Science. 2003;301:798–802. doi: 10.1126/science.1086887. [DOI] [PubMed] [Google Scholar]
- Bernardi G. The human genome: organization and evolutionary history. Annu Rev Genet. 1995;29:445–476. doi: 10.1146/annurev.ge.29.120195.002305. [DOI] [PubMed] [Google Scholar]
- Mouchiroud D, D'Onofrio G, Aissani B, Macaya G, Gautier C, Bernardi G. The distribution of genes in the human genome. Gene. 1991;100:181–187. doi: 10.1016/0378-1119(91)90364-H. [DOI] [PubMed] [Google Scholar]
- D'Onofrio G. Expression patterns and gene distribution in the human genome. Gene. 2002;300:155–160. doi: 10.1016/S0378-1119(02)01048-X. [DOI] [PubMed] [Google Scholar]
- Gellon G, McGinnis W. Shaping animal body plans in development and evolution by modulation of Hox expression patterns. BioEssays. 1998;20:116–125. doi: 10.1002/(SICI)1521-1878(199802)20:2<116::AID-BIES4>3.3.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Warrington JA, Nair A, Mahadevappa M, Tsyganskaya M. Comparison of human adult and fetal expression and identification of 535 housekeeping/maintenance genes. Physiol Genomics. 2000;2:143–147. doi: 10.1152/physiolgenomics.2000.2.3.143. [DOI] [PubMed] [Google Scholar]
- Hsiao LL, Dangond F, Yoshida T, Hong R, Jensen RV, Misra J, Dillon W, Lee KF, Clark KE, Haverty P, et al. A compendium of gene expression in normal human tissues. Physiol Genomics. 2001;7:97–104. doi: 10.1152/physiolgenomics.00040.2001. [DOI] [PubMed] [Google Scholar]
- Eisenberg E, Levanon EY. Human housekeeping genes are compact. Trends Genet. 2003;19:362–365. doi: 10.1016/S0168-9525(03)00140-9. [DOI] [PubMed] [Google Scholar]
- Lercher MJ, Urrutia AO, Hurst LD. Clustering of housekeeping genes provides a unified model of gene order in the human genome. Nat Genet. 2002;31:180–183. doi: 10.1038/ng887. [DOI] [PubMed] [Google Scholar]
- Boutanaev AM, Kalmykova AI, Shevelyov YY, Nurminsky DI. Large clusters of co-expressed genes in the Drosophila genome. Nature. 2002;420:666–669. doi: 10.1038/nature01216. [DOI] [PubMed] [Google Scholar]
- Roy PJ, Stuart JM, Lund J, Kim SK. Chromosomal clustering of muscle-expressed genes in Caenorhabditis elegans. Nature. 2002;418:975–979. doi: 10.1038/nature01012. [DOI] [PubMed] [Google Scholar]
- Spellman PT, Rubin GM. Evidence for large domains of similarly expressed genes in the Drosophila genome. J Biol. 2002;1:5. doi: 10.1186/1475-4924-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalier-Smith T. The Evolution of Genome Size. New York: John Wiley and Sons; 1985. [Google Scholar]
- Ohno S. So much "junk" DNA in our genome. In: Smith HH, editor. In Evolution of Genetic Systems. New York: Gordon and Breach; 1972. pp. 366–370. [Google Scholar]
- Kidwell MG. Transposable elements and the evolution of genome size in eukaryotes. Genetica. 2002;115:49–63. doi: 10.1023/A:1016072014259. [DOI] [PubMed] [Google Scholar]
- Lozovskaya ER, Nurminsky DI, Petrov DA, Hartl DL. Genome size as a mutation-selection-drift process. Genes Genet Syst. 1999;74:201–207. doi: 10.1266/ggs.74.201. [DOI] [PubMed] [Google Scholar]
- Petrov DA, Sangster TA, Johnston JS, Hartl DL, Shaw KL. Evidence for DNA loss as a determinant of genome size. Science. 2000;287:1060–1062. doi: 10.1126/science.287.5455.1060. [DOI] [PubMed] [Google Scholar]
- Petrov DA. Mutational equilibrium model of genome size evolution. Theor Popul Biol. 2002;61:531–544. doi: 10.1006/tpbi.2002.1605. [DOI] [PubMed] [Google Scholar]
- Gregory TR. The bigger the C-value, the larger the cell: genome size and red blood cell size in vertebrates. Blood Cells Mol Dis. 2001;27:830–843. doi: 10.1006/bcmd.2001.0457. [DOI] [PubMed] [Google Scholar]
- Gregory TR. Genome size and developmental complexity. Genetica. 2002;115:131–146. doi: 10.1023/A:1016032400147. [DOI] [PubMed] [Google Scholar]
- Baumeister R, Liu Y, Ruvkun G. Lineage-specific regulators couple cell lineage asymmetry to the transcription of the Caenorhabditis elegans POU gene unc-86 during neurogenesis. Genes Dev. 1996;10:1395–1410. doi: 10.1101/gad.10.11.1395. [DOI] [PubMed] [Google Scholar]
- Schwartz RJ, Olson EN. Building the heart piece by piece: modularity of cis-elements regulating Nkx2-5 transcription. Development. 1999;126:4187–4192. doi: 10.1242/dev.126.19.4187. [DOI] [PubMed] [Google Scholar]
- Fu W, Duan H, Frei E, Noll M. shaven and sparkling are mutations in separate enhancers of the Drosophila Pax2 homolog. Development. 1998;125:2943–2950. doi: 10.1242/dev.125.15.2943. [DOI] [PubMed] [Google Scholar]
- Goto T, Macdonald P, Maniatis T. Early and late periodic patterns of even skipped expression are controlled by distinct regulatory elements that respond to different spatial cues. Cell. 1989;57:413–422. doi: 10.1016/0092-8674(89)90916-1. [DOI] [PubMed] [Google Scholar]
- Boll W, Noll M. The Drosophila Pox neuro gene: control of male courtship behavior and fertility as revealed by a complete dissection of all enhancers. Development. 2002;129:5667–5681. doi: 10.1242/dev.00157. [DOI] [PubMed] [Google Scholar]
- Kim J, Sebring A, Esch JJ, Kraus ME, Vorwerk K, Magee J, Carroll SB. Integration of positional signals and regulation of wing formation and identity by Drosophila vestigial gene. Nature. 1996;382:133–138. doi: 10.1038/382133a0. [DOI] [PubMed] [Google Scholar]
- DiLeone RJ, Russell LB, Kingsley DM. An extensive 3' regulatory region controls expression of Bmp5 in specific anatomical structures of the mouse embryo. Genetics. 1998;148:401–408. doi: 10.1093/genetics/148.1.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Jan LY, Jan YN. Transcriptional regulation of atonal during development of the Drosophila peripheral nervous system. Development. 1998;125:3731–3740. doi: 10.1242/dev.125.18.3731. [DOI] [PubMed] [Google Scholar]
- Tomancak P, Beaton A, Weiszmann R, Kwan E, Shu S, Lewis SE, Richards S, Ashburner M, Hartenstein V, Celniker SE, et al. Systematic determination of patterns of gene expression during Drosophila embryogenesis. Genome Biol. 2002;3:research0088.1–0088.14. doi: 10.1186/gb-2002-3-12-research0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun VC, Stathopoulos A, Levine M. Promoter-proximal tethering elements regulate enhancer-promoter specificity in the Drosophila Antennapedia complex. Proc Natl Acad Sci USA. 2002;99:9243–9247. doi: 10.1073/pnas.142291299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuh CH, Bolouri H, Davidson EH. Cis-regulatory logic in the endo16 gene: switching from a specification to a differentiation mode of control. Development. 2001;128:617–629. doi: 10.1242/dev.128.5.617. [DOI] [PubMed] [Google Scholar]
- Lettice LA, Horikoshi T, Heaney SJ, van Baren MJ, van der Linde HC, Breedveld GJ, Joosse M, Akarsu N, Oostra BA, Endo N, et al. Disruption of a long-range cis-acting regulator for Shh causes preaxial polydactyly. Proc Natl Acad Sci USA. 2002;99:7548–7553. doi: 10.1073/pnas.112212199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhart J, Kirschner M. Cells Embryos and Evolution. Malden, MA: Blackwell Science; 1997. [Google Scholar]
- Carroll SB, Grenier JK, Weatherbee SD. From DNA to Diversity: Molecular Genetics and the Evolution of Animal Design. Malden, MA: Blackwell Science; 2001. [Google Scholar]
- Davidson EH. Genomic Regulatory Systems: Development and Evolution. San Diego, CA: Academic Press; 2001. [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panopoulou G, Hennig S, Groth D, Krause A, Poustka AJ, Herwig R, Vingron M, Lehrach H. New evidence for genome-wide duplications at the origin of vertebrates using an amphioxus gene set and completed animal genomes. Genome Res. 2003;13:1056–1066. doi: 10.1101/gr.874803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin GM, Yandell MD, Wortman JR, Gabor Miklos GL, Nelson CR, Hariharan IK, Fortini ME, Li PW, Apweiler R, Fleischmann W, et al. Comparative genomics of the eukaryotes. Science. 2000;287:2204–2215. doi: 10.1126/science.287.5461.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGhee JD, Krause MW. Transcription factors and transcriptional regulation. In: Riddle DL, Blumenthal T, Meyer BJ, Priess JR, editor. In C elegans II. Plainview, NY: Cold Spring Harbor Laboratory Press;; 1997. pp. 147–184. [PubMed] [Google Scholar]
- Blackman RK, Sanicola M, Raftery LA, Gillevet T, Gelbart WM. An extensive 3' cis-regulatory region directs the imaginal disk expression of decapentaplegic, a member of the TGF-beta family in Drosophila. Development. 1991;111:657–666. doi: 10.1242/dev.111.3.657. [DOI] [PubMed] [Google Scholar]
- Masucci JD, Miltenberger RJ, Hoffmann FM. Pattern-specific expression of the Drosophila decapentaplegic gene in imaginal disks is regulated by 3' cis-regulatory elements. Genes Dev. 1990;4:2011–2023. doi: 10.1101/gad.4.11.2011. [DOI] [PubMed] [Google Scholar]
- Sackerson C, Fujioka M, Goto T. The even-skipped locus is contained in a 16-kb chromatin domain. Dev Biol. 1999;211:39–52. doi: 10.1006/dbio.1999.9301. [DOI] [PubMed] [Google Scholar]
- Blumenthal T, Evans D, Link CD, Guffanti A, Lawson D, Thierry-Mieg J, Thierry-Mieg D, Chiu WL, Duke K, Kiraly M, Kim SK. A global analysis of Caenorhabditis elegans operons. Nature. 2002;417:851–854. doi: 10.1038/nature00831. [DOI] [PubMed] [Google Scholar]
- Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell AC, West AG, Felsenfeld G. Insulators and boundaries: versatile regulatory elements in the eukaryotic genome. Science. 2001;291:447–450. doi: 10.1126/science.291.5503.447. [DOI] [PubMed] [Google Scholar]
- Halfon MS, Carmena A, Gisselbrecht S, Sackerson CM, Jimenez F, Baylies MK, Michelson AM. Ras pathway specificity is determined by the integration of multiple signal-activated and tissue-restricted transcription factors. Cell. 2000;103:63–74. doi: 10.1016/s0092-8674(00)00105-7. [DOI] [PubMed] [Google Scholar]
- Xu C, Kauffmann RC, Zhang J, Kladny S, Carthew RW. Overlapping activators and repressors delimit transcriptional response to receptor tyrosine kinase signals in the Drosophila eye. Cell. 2000;103:87–97. doi: 10.1016/s0092-8674(00)00107-0. [DOI] [PubMed] [Google Scholar]
- Flores GV, Duan H, Yan H, Nagaraj R, Fu W, Zou Y, Noll M, Banerjee U. Combinatorial signaling in the specification of unique cell fates. Cell. 2000;103:75–85. doi: 10.1016/s0092-8674(00)00106-9. [DOI] [PubMed] [Google Scholar]
- Kuchenthal CA, Chen W, Okkema PG. Multiple enhancers contribute to expression of the NK-2 homeobox gene ceh-22 in C. elegans pharyngeal muscle. Genesis. 2001;31:156–166. doi: 10.1002/gene.10018. [DOI] [PubMed] [Google Scholar]
- Hiromi Y, Gehring WJ. Regulation and function of the Drosophila segmentation gene fushi tarazu. Cell. 1987;50:963–974. doi: 10.1016/0092-8674(87)90523-x. [DOI] [PubMed] [Google Scholar]
- Arnone MI, Martin EL, Davidson EH. Cis-regulation downstream of cell type specification: a single compact element controls the complex expression of the CyIIa gene in sea urchin embryos. Development. 1998;125:1381–1395. doi: 10.1242/dev.125.8.1381. [DOI] [PubMed] [Google Scholar]
- Comeron JM. What controls the length of noncoding DNA? Curr Opin Genet Dev. 2001;11:652–659. doi: 10.1016/S0959-437X(00)00249-5. [DOI] [PubMed] [Google Scholar]
- Petrov DA, Hartl DL. High rate of DNA loss in the Drosophila melanogaster and Drosophila virilis species groups. Mol Biol Evol. 1998;15:293–302. doi: 10.1093/oxfordjournals.molbev.a025926. [DOI] [PubMed] [Google Scholar]
- Petrov DA, Hartl DL. Trash DNA is what gets thrown away: high rate of DNA loss in Drosophila. Gene. 1997;205:279–289. doi: 10.1016/S0378-1119(97)00516-7. [DOI] [PubMed] [Google Scholar]
- Robertson HM. The large srh family of chemoreceptor genes in Caenorhabditis nematodes reveals processes of genome evolution involving large duplications and deletions and intron gains and losses. Genome Res. 2000;10:192–203. doi: 10.1101/gr.10.2.192. [DOI] [PubMed] [Google Scholar]
- Hartl DL. Molecular melodies in high and low C. Nat Rev Genet. 2000;1:145–149. doi: 10.1038/35038580. [DOI] [PubMed] [Google Scholar]
- Ptak SE, Petrov DA. How intron splicing affects the deletion and insertion profile in Drosophila melanogaster. Genetics. 2002;162:1233–1244. doi: 10.1093/genetics/162.3.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman CM, Kreitman M. Analysis of conserved noncoding DNA in Drosophila reveals similar constraints in intergenic and intronic sequences. Genome Res. 2001;11:1335–1345. doi: 10.1101/gr.178701. [DOI] [PubMed] [Google Scholar]
- Bergman CM, Pfeiffer BD, Rincon-Limas DE, Hoskins RA, Gnirke A, Mungall CJ, Wang AM, Kronmiller B, Pacleb J, Park S, et al. Assessing the impact of comparative genomic sequence data on the functional annotation of the Drosophila genome. Genome Biol. 2002;3:research0086.1–0086.20. doi: 10.1186/gb-2002-3-12-research0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comeron JM, Kreitman M. Population, evolutionary and genomic consequences of interference selection. Genetics. 2002;161:389–410. doi: 10.1093/genetics/161.1.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versteeg R, van Schaik BD, van Batenburg MF, Roos M, Monajemi R, Caron H, Bussemaker HJ, van Kampen AH. The human transcriptome map reveals extremes in gene density, intron length, GC content, and repeat pattern for domains of highly and weakly expressed genes. Genome Res. 2003;13:1998–2004. doi: 10.1101/gr.1649303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra S, Crosby M, Mungall C, Matthews B, Campbell K, Hradecky P, Huang Y, Kaminker J, Millburn G, Prochnik S, et al. Annotation of the Drosophila melanogaster euchromatic genome: a systematic review. Genome Biol. 2002;3:research0083.1–0083.22. doi: 10.1186/gb-2002-3-12-research0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris TW, Lee R, Schwarz E, Bradnam K, Lawson D, Chen W, Blasier D, Kenny E, Cunningham F, Kishore R, et al. WormBase: a cross-species database for comparative genomics. Nucleic Acids Res. 2003;31:133–137. doi: 10.1093/nar/gkg053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WormBase http://www.wormbase.org
- FlyBase http://flybase.bio.indiana.edu
- BDGP in situ homepage http://www.fruitfly.org/cgi-bin/ex/insitu.pl
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1006/jmbi.1990.9999. [DOI] [PubMed] [Google Scholar]
- Clamp M, Andrews D, Barker D, Bevan P, Cameron G, Chen Y, Clark L, Cox T, Cuff J, Curwen V, et al. Ensembl 2002: accommodating comparative genomics. Nucleic Acids Res. 2003;31:38–42. doi: 10.1093/nar/gkg083. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
An Excel file containing the primary data used for the three expression indices, the D. melanogaster X chromosome, and the GO groups