

Abstract
Protein kinase inhibitors have emerged as new drugs in various therapeutic areas, including leishmaniasis, an important parasitic disease. Members of the Leishmania casein kinase 1 (CK1) family represent promising therapeutic targets. Leishmania casein kinase 1 isoform 2 (CK1.2) has been identified as an exokinase capable of phosphorylating host proteins, thus exerting a potential immune-suppressive action on infected host cells. Moreover, its inhibition reduces promastigote growth. Despite these important properties, its requirement for intracellular infection and its chemical validation as a therapeutic target in the disease-relevant amastigote stage remain to be established. In this study, we used a multidisciplinary approach combining bioinformatics, biochemical, and pharmacological analyses with a macrophage infection assay to characterize and define Leishmania CK1.2 as a valid drug target. We show that recombinant and transgenic Leishmania CK1.2 (i) can phosphorylate CK1-specific substrates, (ii) is sensitive to temperature, and (iii) is susceptible to CK1-specific inhibitors. CK1.2 is constitutively expressed at both the promastigote insect stage and the vertebrate amastigote stage. We further demonstrated that reduction of CK1 activity by specific inhibitors, such as D4476, blocks promastigote growth, strongly compromises axenic amastigote viability, and decreases the number of intracellular Leishmania donovani and L. amazonensis amastigotes in infected macrophages. These results underline the potential role of CK1 kinases in intracellular survival. The identification of differences in structure and inhibition profiles compared to those of mammalian CK1 kinases opens new opportunities for Leishmania CK1.2 antileishmanial drug development. Our report provides the first chemical validation of Leishmania CK1 protein kinases, required for amastigote intracellular survival, as therapeutic targets.
INTRODUCTION
Leishmaniasis is an important human disease caused by protozoan parasites of the genus Leishmania. This parasite has two developmental stages, an extracellular promastigote form present in the insect vector and an intracellular amastigote form predominantly located inside phagolysosomes of macrophages in mammalian hosts. There are several types of the disease, cutaneous, mucocutaneous, and visceral leishmaniasis, with the last type being the most severe form, as it can affect vital organs (1). Several treatment options are available, and these either show severe side effects (pentavalent antimonials, miltefosine, amphotericin B [AMB]), are unaffordable (liposomal amphotericin B), or lead to parasite resistance (pentavalent antimonials) (2). There is thus an urgent need to discover new treatments.
Protein kinases represent one of the most important groups of drug targets currently in development and are the targets of several Food and Drug Administration (FDA)-approved drugs against cancer (3). Targeting Leishmania through its essential protein kinases is an attractive strategy, which can be broadly applied to diseases caused by unicellular parasites (4). Phosphorylation is indeed essential for almost all cellular processes, and as a result, inhibition of protein kinases should lead to interference with the regulation of key processes in metabolism, proliferation, or differentiation. In Leishmania, different protein kinases have already been identified as potential drug targets, such as mitogen-activated protein kinases (MAPKs) or cdc2-related kinase 3 (CRK3) (5–7). Members of another family of kinases have emerged as potential drug targets, casein kinase 1 (CK1), a family of multifunctional Ser/Thr protein kinases that are characterized by a highly conserved kinase domain and a specific C-terminal domain implicated in its regulation and localization (8). Although CK1 homologs in various protozoa share properties that are similar to those of higher eukaryotes, their functions have not yet been identified (9–14). There are six CK1 isoforms in Leishmania major, LmjF35.1000 (UniProtKB/Swiss-Prot accession number Q9NHE2; LmaCK1.1), LmjF35.1010 (UniProtKB/Swiss-Prot accession number Q9NHE1; LmaCK1.2), LmjF04.1210 (UniProtKB/Swiss-Prot accession number O96999), LmjF25.1580 (UniProtKB/Swiss-Prot accession number Q4Q9T3), LmjF27.1780 (GenBank accession number E9ADF5), and LmjF30.3470 (UniProtKB/Swiss-Prot accession number Q4Q6U1), for which the biochemical properties and the physiological functions are largely unknown.
According to recent proteomic analyses, the most studied CK1 in Leishmania, CK1.2, is also the most abundant isoform (13, 15–17). This kinase can be shed in the extracellular environment, where it can directly phosphorylate extracellular substrates, such as the C3a polypeptide of the human complement system, or can be exported into infected host cells by exosomal release from intracellular parasites, with potential immunosuppressive effects through the phosphorylation of cellular host proteins, including, for example, human IFNAR1, a receptor essential for alpha/beta interferon (IFN-α/β) signaling (15, 17–19). These data suggest that Leishmania CK1.2 might be a suitable target to identify efficient antiparasitic inhibitors. This kinase displays an unusual ATP binding pocket substantially different from that of mammalian CK1, despite their very similar kinase domains (20), and its inhibition by trisubstituted pyrrole blocks promastigote growth (13). However, many aspects of this protein remain unknown, such as its implication in intracellular amastigote survival, as the only available data on the impact of CK1 inhibition on parasite survival were obtained on cultured promastigotes. Yet, proteomics and phosphoproteomics studies revealed important differences in stage-specific protein expression and protein phosphorylation between promastigotes and amastigotes (21–23). As a consequence, pharmacological data obtained for CK1.2 in promastigotes cannot be directly extrapolated to axenic amastigotes or intracellular parasites.
Here we used D4476 {4-[4-(2,3-dihydro-1,4-benzodioxin-6-yl)-5-(2-pyridinyl)-1H-imidazol-2-yl]benzamide}, an established CK1 inhibitor, to investigate the effects of casein kinase 1 inhibition on axenic and intracellular amastigote survival. We show that D4476 inhibits recombinant as well as transgenic CK1.2 purified from parasite extracts. At the 50% effective concentration (EC50) dose, D4476 has only a cytostatic effect on promastigotes but a cytotoxic effect on axenic amastigotes. Finally, we show that D4476 can significantly decrease the parasite burden of infected macrophages. Altogether, these findings validate CK1.2 as an important novel therapeutic target.
MATERIALS AND METHODS
Leishmania donovani culture and axenic amastigote differentiation.
Infectious L. donovani strain 1S2D (MHOM/SD/62/1S-CL2D) was obtained from Henry Murray, Weill Cornell Medical College, New York, NY, and maintained in infected hamsters. Axenic L. donovani 1S2D, clone LdB, was obtained from Steve Beverley, Washington University School of Medicine, St. Louis, MO, and cultured as described previously (22, 24, 25).
Parasite growth inhibition assay.
L. donovani promastigotes and axenic amastigotes (2 × 106 cells/ml) in their respective media were distributed in 96-well plates (125 μl/well). An equal volume of medium containing inhibitor at the indicated concentrations (in dimethyl sulfoxide [DMSO] at a final concentration of 1%) was added. After 24 h of incubation in the dark at 26°C (promastigotes) or 37°C (amastigotes), 25 μl of resazurin solution at 0.001% was added and the plates were incubated as described above for an additional 24 h. The plates were read (excitation λ, 544 nm; emission λ, 590 nm) using a fluorescent microplate reader (Safas Xenius XML) (26).
Hamster infection and isolation of infectious parasites.
Anesthetized hamsters were inoculated by intracardiac injection of 5 × 107 infectious promastigotes obtained from infected hamster spleens. After up to 4 months of infection, the infected hamsters were euthanized with CO2, and spleens were collected, homogenized in RPMI 1640 supplemented with antibiotics, and cleared by centrifugation at 130 × g for 5 min at room temperature (RT). Saponin (1 mg/ml) was added under gentle agitation, and parasites were harvested 5 min later by centrifugation at 1,800 × g for 10 min at room temperature. After a second treatment with saponin followed by two washing steps, remaining host cell contaminants were removed by Percoll centrifugation. Parasites were resuspended in 3 ml of 45% Percoll and layered on top of a cushion of 2 ml 90% Percoll in a 15-ml Falcon tube. After 30 min centrifugation at 3,500 × g and 15°C, parasites were recovered from the interface of the gradient and washed three times. These purified parasites were differentiated into promastigotes to be used for infection of peritoneal exudate macrophages (PEMs).
Macrophage infection and assessment of intracellular parasite survival. (i) L. donovani infection assay.
PEMs from female C57BL/6 mice were seeded onto coverslips and incubated on the following day in RPMI–0.7% bovine serum albumin for 2 h at 37°C with mouse serum-opsonized parasites at a multiplicity of infection of 10. Free parasites were removed by washing, and the plates were further incubated at 37°C in a CO2 incubator. After 48 h, 1% DMSO (control), 50 μM D4476 in 1% DMSO, or 1 μM amphotericin B in 1% DMSO was added to the medium. Intracellular parasites were counted in paraformaldehyde (PFA)-fixed cells, after 24 h or 48 h of contact with the drugs, by nuclear staining with Hoechst 33342 (0.5 μg/ml) using a Leica DMI6000B microscope (magnification, ×100). Two independent experiments were performed in triplicate, and 300 macrophages were analyzed per coverslip. Results were expressed as mean values ± standard deviations.
(ii) L. amazonensis infection assay.
A biologically relevant, high-content cell-based assay was used to determine the antileishmanial activity of D4476, as described by Aulner et al. (27). Briefly, 6-day-old BALB/c bone marrow-derived macrophages were seeded in 384-well plates and infected with lesion-derived, DsRed2-transgenic L. amazonensis amastigotes (MPRO/BR/1972/M1841). Twenty hours later, D4476 was added at 10 and 1 μM final concentrations (in 1% DMSO). Live macrophage cultures were then analyzed using a spinning-disk confocal microscope (Opera QEHS; PerkinElmer Technologies). About 10,000 macrophages were analyzed per well, which allows at least 40,000 cells to be analyzed per group in quadruplicate experiments. To measure antileishmanial activity and toxicity on host macrophages for the compound D4476, the robust version of the strictly standardized mean difference (SSMD*) was calculated for chosen variables; SSMD* represents the difference between the result of D4476 treatment and the result for the negative DMSO reference phenotype (28).
Ethics statement.
All animals were housed in an A3 animal facility in compliance with the guidelines of the A3 animal facilities at the Institut Pasteur, approved by the Comité d'Ethique pour l'Expérimentation Animale (CETEA; approval number 89), as referenced by the Ministry of Research of France. Animal housing conditions and the protocols used in the work described herein were approved by the Direction des Transports et de la Protection du Public, Sous-Direction de la Protection Sanitaire et de l'Environnement, Police Sanitaire des Animaux, under approval numbers B75-15-27 and B75-15-28, in accordance with the Ethics Charter of Animal Experimentation, which includes appropriate procedures to minimize pain and animal suffering. G.F.S. and E.P. are authorized to perform experiments on vertebrate animals (licenses B75-1159 and 75-1265, respectively, issued by the Direction Départementale de la Protection des Populations de Paris) and are responsible for all the experiments conducted personally or under their supervision, as governed by the laws and regulations relating to the protection of animals.
Bioinformatics.
Multiple-sequence alignments (MSA) were computed using the PSI-Coffee mode of the T-Coffee program (29), which uses homology extension against Non Redundant (version 8.91). The resulting alignments were visualized using the Jalview Java alignment editor (30, 31). To determine the conservation of Leishmania protein kinases across Leishmania species, homologs of all Pfam Leishmania major protein kinases were identified by BLAST analysis against a subset containing only eukaryotic proteins, as released by UniProt in September 2011 (32, 33). Only hits aligned to the L. major proteins with an E value lower than 10−10 were used for the analysis. L. braziliensis, L. infantum, and L. mexicana were included to evaluate the average similarity inside the genus.
Cell death analysis.
Parasites were diluted in phosphate-buffered saline (PBS) and incubated with 4 μg/μl propidium iodide (Sigma-Aldrich). Cells were analyzed with a FACSCalibur flow cytometer (excitation λ, 488 nm; emission λ, 617 nm; Beckman Coulter). Analysis was performed using the Watson pragmatic model of FlowJo (version 7.6) software (Tree Star, Inc., San Carlos, CA).
Protein extraction.
Parasites were harvested from logarithmic-phase cultures by centrifugation at 1,500 × g for 10 min. Cells were washed in PBS without CaCl2 and MgCl2 and resuspended in lysis buffer containing 50 mM Tris HCl, pH 8, 150 mM NaCl, 1% Triton X-100, 100 μM MgCl2, 100 U/ml Benzonase, and protease inhibitor cocktail (Complete EDTA-free tablets; Roche Applied Science). Samples were incubated on ice for 30 min and vortexed every 10 min, followed by 5 min sonication (10 s on/20 s off). The lysates were centrifuged for 15 min at 12,000 × g to eliminate cell debris. Supernatants were either used immediately or stored at −80°C.
Expression and purification of recombinant LmaCK1.2-His6.
The open reading frame of the Leishmania major CK1.2 homolog LmjF.35.1010 (termed LmaCK1.2) was synthesized by GeneCust and cloned into pBAD-thio-Topo (Invitrogen). pBAD-thio-LmaCK1.2 was also subjected to site-directed mutagenesis to generate a pBAD-thio-LmaCK1.2-K40A mutant using a QuikChange kit (Stratagene) and primers 5′-AGTTGCTATTgcACTGGAACAG-3′ and 5′-CCTGTTCCAGTgcAATAGCAACT-3′, where the lowercase nucleotides represent the mutated nucleotides. After induction, cells were harvested and resuspended in lysis buffer (60 mM β-glycerophosphate, 1 mM sodium vanadate, 1 mM sodium fluoride, 1 mM disodium phenylphosphate, 150 mM sodium chloride, 10 mM imidazole) supplemented with protease inhibitor cocktail (Complete EDTA-free tablets; Roche Applied Science). After sonication, Triton X-100 (final concentration, 0.1%) was added and the sample was incubated for 30 min at 4°C (shaking) and centrifuged for 30 min at 4°C. The proteins from the supernatant were purified on Co-nitrilotriacetic acid agarose (Pierce) and eluted in a PBS elution buffer at pH 7.5 (60 mM β-glycerophosphate, 1 mM sodium vanadate, 1 mM sodium fluoride, 1 mM disodium phenylphosphate) containing 300 mM imidazole. The eluate was supplemented with 15% glycerol and stored at −80°C.
Protein kinase assay.
Both recombinant LmaCK1.2 and parasite lysates were assayed in buffer C at pH 7.0 (60 mM β-glycerophosphate, 30 mM p-nitrophenyl phosphate, 25 mM MOPS [morpholinepropanesulfonic acid], 5 mM EGTA, 15 mM MgCl2, 1 mM dithiothreitol, 0.1 mM sodium vanadate) with 15 μM [γ-33P]ATP or [γ-32P]ATP in a 30-μl final volume. We used the following substrates: 27 μM CK-specific peptide substrate CK-S (RRKHAAIGpSAYSITA; synthesized by Proteogenix [Oberhausbergen, France]), 36 μg dephosphorylated casein, 16 μg histone H1, or 6 μg myelin basic protein (MBP). After 30 min of incubation at 30°C, the reaction was stopped by loading 25 μl of the reaction mixture onto Whatman P81 phosphocellulose paper. The filters were washed in 1% phosphoric acid and then assessed in a scintillation counter. Alternatively, for protein substrates, the reaction was stopped by adding an equal volume of 2× electrophoresis loading buffer to the 20-μl reaction mix. Incorporated 33P or 32P was monitored by SDS-PAGE and autoradiography. Native mammalian CK1 isoforms (essentially a mixture of CK1δ and CK1ε) were extracted from porcine brain and purified by affinity chromatography on an immobilized axin fragment (34). The kinase activities were expressed as a percentage of the maximal activity, i.e., the activity in the absence of inhibitors. Controls were performed with appropriate dilutions of DMSO.
ATP depletion and competition.
Two milligrams of amastigote total protein extract were dialyzed overnight at 4°C in 1 liter of dialysis solution (1× PBS, 1 mM EDTA, 1 mM dithiothreitol) with a Slide-A-Lyzer dialysis cassette (Pierce) to eliminate free ATP. Extracts were split in half; one half was mock treated (assay samples), whereas 10 mM MgCl2 and 250 μM D4476 were added to the other half (competition samples). Both samples were incubated for 30 min at 37°C and centrifuged at 800 rpm. ATP binding proteins from the assay and the competition samples were enriched by loading onto an ATP column, according to the manufacturer's instructions (ATP AffiPur kit I; Jena Bioscience), and eluted with an excess of free ATP. Finally, the eluted samples were concentrated using Amicon Ultra centrifugal filters to a final volume of approximately 80 μl.
For both the assay and the competition, 5 μl of the flowthrough and 20 μl of the eluate were separated on a Novex NuPAGE 4 to 12% bis-Tris gel (Life Technologies). The gel was stained with SYPRO Ruby (Life Technologies) according to the manufacturer's instructions and revealed using a Typhoon scanner. Alternatively, proteins separated by SDS-PAGE were transferred onto a polyvinylidene difluoride membrane and probed with SY3535 antibody. Signals were revealed by use of Super Signal ECL chemiluminescent substrate (Pierce).
Antibodies.
Peptide IPYKEGKSLTGTARYC, corresponding to a part of the kinase domain, was used to raise an anti-LmCK1.2 antibody in rabbit (SY3535). The antibody was purified by peptide affinity chromatography. Peptides and antibodies were generated and purified by Eurogentec. The affinity-purified antipeptide polyclonal antiserum was used at a 1/500 dilution to identify LmCK1.2 in L. donovani protein extracts. We used a polyclonal anti-human CK1 antibody at a 1/500 dilution (Cell Signaling Technology) and a mouse anti-V5 antibody at a 1/1,000 dilution (Invitrogen).
Compounds.
The following inhibitors used in this study were purchased from Sigma: D4476 (CAS registry number 301836-43-1), IC261 {1,3-dihydro-3-[(2,4,6-trimethoxyphenyl)methylene]-2H-indol-2-one; CAS registry number 186611-52-9}, and CKI-7 [N-(2-aminoethyl)-5-chloroisoquinoline-8-sulfonamide dihydrochloride; CAS registry number 1177141-67-1].
RESULTS
LmjF.35.1010 is a highly conserved casein kinase I homolog.
LmjF35.1010 (LmaCK1.2) is 353 amino acids long and has a predicted molecular mass of 39.75 kDa. We found LmaCK1.2 to be more closely related to trypanosomatid CK1 (80% identity with Trypanosoma cruzi Tc00.1047053508541.240 and up to 81% identity with T. brucei CK1 Tb927.5.800; data not shown) than to the other Leishmania CK1 isoforms (32 to 67% identity; Table 1). LmaCK1.2 is also characterized by its high degree of conservation among Leishmania species (L. major, L. infantum, L. donovani, L. mexicana, and L. braziliensis), showing more than 99% identity (Fig. 1A), which is particularly high, given that, on average, orthologous Leishmania kinases display only 88% identity (data not shown). LmjF35.1010 lacks only one amino acid compared to the L. infantum (LinJ35.1030) or L. donovani (LdBPK-351030.1) ortholog and shows three and four amino acid changes compared to the L. mexicana (LmxM34.1010) and L. braziliensis (LbrM34.1000) orthologs, respectively. All these differences are restricted to the last 30 C-terminal amino acids (Fig. 1A). This level of conservation among Leishmania species does not apply to all CK1 isoforms but is unique to LmaCK1.2, which, in fact, is the most conserved kinase in Leishmania (Fig. 1B, horizontal axis). This genomic stability of LmaCK1.2 is not due to its chromosomal location, as the adjacent CK1 homolog (LmaCK1.1, which is 67% identical to LmaCK1.2) does not show the same degree of sequence preservation (data not shown). This finding indicates that there is a very strong purifying selection against mutations within LmaCK1.2, probably pointing to very important physiological functions of this CK1 family member. This high level of conservation is common to other protozoan parasites. For example, Plasmodium falciparum CK1 (PfCK1) is more than 99% homologous to the CK1 orthologs of other Plasmodium species, such as P. berghei CK1 (PbCK1), P. knowlesi CK1 (PkCK1), P. chabaudi CK1 (PcCK1), P. reichenowi CK1 (PrCK1), and P. yoelii (PyCK1), with no more than two amino acid changes (data not shown). The conservation was also high when considering human CK1. LmaCK1.2 was 59% identical to CK1δ and CK1ε and 58% identical to CK1α. The percent identity increased up to 71% and 66%, respectively, when restricting the comparison to the catalytic domain. These data are significantly higher than the average percent identity between human and Leishmania kinases (37%; Fig. 1B). When comparing the Leishmania kinome with the human kinome, LmaCK1.2 is the kinase with the highest similarity to its human ortholog (Fig. 1B, vertical axis). Even though Leishmania CK1.2 is highly conserved among eukaryote CK1 (Fig. 1C), its sequence displays some characteristics specific to trypanosomatids, such as a GGA insertion between domains III and IV in the kinase domain (Fig. 1C, bar) (11) or the presence of six amino acid changes identical in all trypanosomatids (Fig. 1C, dots). The biological significance of these insertions or amino acid changes is unknown, though they could offer possibilities for the design of trypanosomatid-specific CK1 inhibitors.
TABLE 1.
Percent identity between the six Leishmania CK1 family members and six human CK1s
| Kinase (accession no.) | % identity to LmaCK1.2a |
|
|---|---|---|
| Catalytic domain | Whole protein | |
| LmjF35.1000 (Q9NHE2) | 72 | 67 |
| LmjF30.3470 (Q4Q6U1) | 50 | 42 |
| LmjF25.1580 (Q4Q9T3) | 55 | 46 |
| LmjF04.1210 (O96999) | 37 | 32 |
| LmjF27.1780 (E9ADF5) | 36 | 33 |
| HsCK1α (P48729) | 66 | 58 |
| HsCK1ε (P49674) | 71 | 59 |
| HsCK1δ (P48730) | 71 | 60 |
| HsCK1γ1 (Q9HCP0) | 61 | 50 |
| HsCK1γ2 (P78368) | 59 | 49 |
| HsCK1γ3 (Q9Y6M4) | 61 | 52 |
The percentages were obtained from multiple-sequence alignment of the catalytic domain or the whole protein and were computed using the PSI-Coffee mode (version 8.91).
FIG 1.

Amino acid sequence alignment of Leishmania casein kinase 1 proteins. (A) The amino acid sequences of LmjF.35.1010 (L. major CK1.2), LbrM.34.1000 (L. braziliensis CK1.2), LinJ.35.1030 (L. infantum CK1.2), LdBPK_351030.1 (L. donovani CK1.2), and LmxM.34.1010 (L. mexicana CK1.2) were compared using the ClustalW2 program (53). *, amino acid residues that are invariant in all four CK1s. (B) Differential conservation of L. major protein kinases in other Leishmania species or in human. The average similarity of L. major protein kinase sequences in L. braziliensis, L. infantum, and L. mexicana is plotted against the similarity of the closest human homologs. LmaCK1.2 is indicated by a black dot in the top right corner, corresponding to the protein showing the highest percent identity (ID%) with Leishmania and human homologs. Only proteins for which a BLAST search identified a human counterpart with an E value lower than 10−10 are displayed. Vertical black line, the median of the average similarity of L. major proteins to other Leishmania spp. (95%); horizontal black line, the median of the average similarity of L. major proteins to human homologs (37%). (C) The amino acid sequences of LmjF.35.1010, Tb927.5.800 (Trypanosoma brucei CK1 [TbCK1]; accession number Q57W25), Tc00.1047053508541.240 (Trypanosoma cruzi CK1 [TcCK1]; accession number Q4DN94), TgCK1a (Toxoplasma gondii CK1a; accession number Q6QNM1), PfCK1 (Plasmodium falciparum CK1; UniProtKB accession number C6S3F7), hhp1 (Schizosaccharomyces pombe CK1 [SpCK1]; accession number P40235), CSNK1A1 (Homo sapiens CK1 alpha [HsCK1a]; accession number P48729), CSNK1D (Homo sapiens CK1 delta [HsCK1d]; accession number P48730), and CSNK1E (Homo sapiens CK1 epsilon [HsCK1e]; accession number P49674) were compared, and the alignments were computed using the PSI-Coffee mode of T-Coffee (29) that uses homology extension against NR (version 8.91). The resulting alignments were visualized using Jalview (30, 31).
rLmaCK1.2 is intrinsically active and phosphorylates the CK1-specific substrate CK-S.
To biochemically characterize Leishmania CK1.2, a tagged recombinant protein was generated, using the sequence from L. major, the only sequence available at the time that this study was initiated. However, because the whole-protein sequence of CK1.2 from L. major is more than 99% conserved in CK1.2 from L. donovani and 100% conserved if we consider only the kinase domain, we regarded LmaCK1.2 to be equivalent to L. donovani CK1.2 (LdCK1.2) for all subsequent experiments. A codon-optimized gene encoding LmaCK1.2 was synthesized in order to improve bacterial expression and cloned into the pBAD-thio-Topo vector. Additionally, a recombinant LmaCK1.2 kinase-dead mutant was generated. Because in earlier reports CK1ε-K38A was described to be kinase dead, we mutated the corresponding lysine 40 in LmaCK1.2 into an alanine (LmaCK1.2-K40A) by site-directed mutagenesis (35). The resulting proteins, tagged at the C terminus with both V5 and His6, were purified from Escherichia coli on cobalt resin (Fig. 2Aa and Ab). We first characterized the purified recombinant LmaCK1.2 (rLmaCK1.2) by an in vitro kinase assay using a known CK1 peptide substrate, CK-S (34). Whereas rLmaCK1.2 could phosphorylate CK-S very efficiently, no phosphorylation was detected with rLmaCK1.2-K40A (Fig. 2B). This indicates that CK-S phosphorylation is specifically due to rLmaCK1.2 and not to a bacterial copurified kinase. We next assessed the specificity of rLmaCK1.2 using casein, histone H1, and MBP as the substrates. As shown in Fig. 2C, all three substrates were phosphorylated by rLmaCK1.2 but not by rLmaCK1.2-K40A; casein, as expected, was the best substrate (Fig. 2C). Altogether, these data showed that rLmaCK1.2 is an intrinsically active casein kinase 1.
FIG 2.

LmaCK1.2, but not LmaCK1.2-K40A, strongly phosphorylates CK1 substrates. (A) Purification of LmaCK1.2 (a) and LmaCK1.2-K40A (b) from E. coli. Aliquots from the total extract, flowthrough, and two elutions from both samples were separated by SDS-PAGE, and the gel was stained by colloidal Coomassie (Bio-Rad). (B) Kinase assays were performed using 27 μM CK-S peptide and 0.5 μg of recombinant LmaCK1.2 or 1 μg of LmaCK1.2-K40A. The reaction samples were spotted onto P81 phosphocellulose paper (Whatman), and phosphate incorporation was measured using a scintillation counter. (C) Kinase assays were performed with 36 μg dephosphorylated casein 16 μg histone H1 or 6 μg MBP with 0.5 μg of recombinant LmaCK1.2 or 1 μg of LmaCK1.2-K40A. (a) Autoradiography; (b) Coomassie-stained gel. (D) One microgram of LmaCK1.2 was subjected to kinase assays at 30°C under increasing pH conditions, using 27 μM CK-S as the substrate. The reaction mixtures were spotted onto P81 paper, and the incorporated radioactivity was measured by a scintillation counter. (E) Recombinant LmaCK1.2 was subjected to kinase assays, as described for panel D, at pH 7.5 and either 30 or 37°C.
Leishmania faces dramatic environmental changes during the infectious cycle: promastigotes are adapted to 26°C and a neutral pH inside the sand fly vector, while amastigotes reside inside host cell phagolysosomes at 37°C and pH 5.5 (36). As LmaCK1.2 has been previously identified as an exokinase secreted into the host environment, we investigated the sensitivity of rLmaCK1.2 activity to pH and temperature. We tested the phosphorylation of CK-S by rLmaCK1.2 at pH 5.5, 6.5, 7.5, and 8.0. rLmaCK1.2 is inactive at pH 5.5; its activity is first detected at pH 6.5 and steadily increases to attain the maximum of 5.1 pmol of phosphate incorporated/min at pH 8.0 (Fig. 2D). The pH thus has a strong impact on rLmaCK1.2 kinase activity, suggesting that although LmaCK1.2 could be released by the parasite directly into the phagolysosome, it is unlikely that it would phosphorylate substrates inside this compartment. Likewise, rLmaCK1.2 kinase activity is decreased by about 60% at the temperature encountered inside the mammalian host (Fig. 2E). The increase in temperature has an influence on rLmaCK1.2 activity but does not completely inactivate the kinase, suggesting either that in the amastigote the activity of LmaCK1.2 is reduced or that LmaCK1.2 might require the presence of chaperones to maintain its normal activity.
Recombinant Leishmania CK1.2 is sensitive to known CK1 inhibitors.
Mammalian CK1 isoforms are sensitive to different pharmacological inhibitors, such as D4476 (37), IC261 (38), and CKI-7 (39), whose structures are presented in Fig. S2 in the supplemental material. CKI-7 was the first described CK1 inhibitor and has a 50% inhibitory concentration (IC50) greater than 10 μM against most human CK1 isoforms (39). D4476 is the most specific and potent CK1 inhibitor known to date and has an IC50 in the submicromolar range (37, 40), whereas IC261 specifically inhibits CK1δ and CK1ε with an IC50 of 1 μM (38). We tested the effect of 10 μM of these inhibitors on rLmaCK1.2, using the phosphorylation of peptide substrate CK-S as the readout. At this concentration, CK-S phosphorylation by rLmaCK1.2 was reduced by 97% and 50% using D4476 and CKI-7, respectively, while IC261 had no effect on kinase activity (Fig. 3A). When we tested the effect of these inhibitors on LmaCK1.2 using casein as a substrate, we found that D4476 remained very efficient, while the other inhibitors did not have any effect (Fig. 3B). We noticed that CKI-7 partially inhibited CK1.2 activity toward peptide substrates but not toward protein substrates. Together, these data demonstrate that, like human CK1s, LmaCK1.2 is inhibited by the specific CK1 inhibitor D4476 and is therefore a druggable Leishmania kinase.
FIG 3.

LmaCK1.2 is sensitive to known CK1 inhibitors. (A) Kinase assay using 27 μM CK-S and 1 μg recombinant LmaCK1.2 in the presence of either 1% DMSO alone or D4476, IC261, and CKI-7 at 10 μM in 1% DMSO. (B) Kinase assay using 36 μg dephosphorylated casein and 1 μg of recombinant LmCK1.2 in the presence of either DMSO or inhibitors at 10 μM (as described for panel A). (a) Autoradiography; (b) Coomassie staining. (C) Determination of the IC50 of D4476 (a), CKI-7 (b), and IC261 (c) for mammalian CK1 from porcine brain (SsCK1) and recombinant LmaCK1.2. We performed kinase assays using SsCK1 or LmaCK1.2 in the presence of increasing concentrations of the respective CK1 inhibitors.
We next asked whether, despite the high degree of homology of their catalytic domains, rLmaCK1.2 and mammalian CK1s could be differentially inhibited. We thus compared the IC50s of selected inhibitors against rLmaCK1.2 and porcine brain CK1 (Sus scrofa CK1 [SsCK1] [34]). We performed kinase assays in the presence of increasing concentrations of inhibitors. For both kinases, we used the amount of protein that corresponded to an activity of 10,000 cpm and we measured the decrease in activity as stated by Reinhardt et al. (34). D4476 inhibited rLmaCK1.2 more potently than it inhibited SsCK1, with IC50s of 0.09 ± 0 μM and 0.3 ± 0.1 μM, respectively (Fig. 3Ca). CKI-7 (Fig. 3Cb) and IC261 (Fig. 3Cc) were more potent toward SsCK1 (0.47 ± 0 μM and 4.8 ± 1.5 μM, respectively) than rLmaCK1.2 (>10 μM for both). The IC50 of IC261 for rLmaCK1.2 was closer to that obtained for Schizosaccharomyces pombe ckiα (Spckiα; 16 μM) than that obtained for Spckiδ and Spckiε (1 μM), suggesting that although its protein sequence is more closely related to that of CK1δ or CK1ε, the ATP binding pocket of LmaCK1.2 might be structurally more closely related to that of CK1α (38).
Transgenic CK1.2-V5-His6 is sensitive to D4476.
To gain access to the endogenous CK1.2 and determine whether it is expressed in promastigotes as well as in amastigotes, we raised a polyclonal antibody designated SY3535 that recognizes the kinase domain of LmaCK1.2 but, on the basis of its sequence, could also recognize LmjF35.1000 and, partially, LmjF25.1580. Proteins were extracted from L. donovani promastigotes and axenic amastigotes. One microgram of purified rLmaCK1.2 or 50 μg of total protein extract was analyzed by Western blotting with SY3535 or anti-Homo sapiens CK1α (anti-HsCK1α), used as a positive control. As shown in Fig. 4A, SY3535 recognized rLmaCK1.2a (a band above the band with a molecular mass of 55 kDa, lane 1), thus showing the specificity of the SY3535 antibody. With total protein extracts from L. donovani promastigotes (Fig. 4A, lane 2) and axenic amastigotes (Fig. 4A, lane 3), a band corresponding to the endogenous LdCK1.2 (39 kDa) was detected. Similar results were obtained using anti-HsCK1α (Fig. 4A, lanes 1′ to 3′). These findings demonstrate that LdCK1.2 is also expressed in axenic amastigotes and not only in promastigotes (13, 18).
FIG 4.

Transgenic CK1.2 is sensitive to D4476. (A) One microgram of recombinant LmaCK1.2 (lanes 1 and 1′), 50 μg of promastigote total protein extract (lanes 2 and 2′), and 50 μg of axenic amastigote total protein extract (lanes 3 and 3′) were analyzed by Western blotting using SY3535 (lanes 1 to 3) or an anti-HsCK1 antibody (lanes 1′ to 3′). (B) Twenty micrograms of extracts obtained from transgenic promastigotes or axenic amastigotes expressing either pLEXSY (mock transfection) or pLEXSY-CK1.2 was analyzed by Western blotting using SY3535 (top) and stained by colloidal Coomassie to evaluate the loading (bottom). (C) As described for panel B, total extracts or immunopurified extracts were analyzed by Western blotting using an anti-V5 antibody (top and middle) and stained by colloidal Coomassie to evaluate the loading (bottom). Numbers to the left of the gels in panels B and C are molecular masses (in kilodaltons). (D) Kinase assay using 27 μM CK-S and 10 μl of V5-coated beads purified from transgenic promastigotes or axenic amastigotes expressing either pLEXSY (mock transfection) or pLEXSY-CK1.2 with or without 10 μM D4476 in 1% DMSO. The activities in the reaction mixtures were measured using a scintillation counter.
As our attempts to purify endogenous LdCK1.2 using SY3535 antibody were unsuccessful, we cloned CK1.2-V5-His6 into the leishmanial expression vector pLEXSY to overexpress LmaCK1.2-V5-His6 in parasites. Proteins were extracted from transgenic L. donovani promastigotes and axenic amastigotes containing either the pLEXSY or the pLEXSY-CK1.2-V5-His6 vector. Twenty micrograms of total protein extracts was analyzed by Western blotting with SY3535 antibody. We detected one band corresponding to endogenous CK1.2 present in all the samples and a higher band between 40 and 55 kDa, corresponding to CK1.2-V5-His6, that was absent in the mock-transfected controls (Fig. 4B, top). As judged by growth and cell cycle analysis, the overexpression of CK1.2-V5-His6 did not affect parasite proliferation in vitro (data not shown).
To test for activity, CK1.2-V5-His6 was immunoprecipitated from promastigote and amastigote total extracts containing either pLEXSY or pLEXSY-CK1.2-V5-His6, using the anti-V5 antibody. Equal amounts of total extract and purified CK1.2-V5-His6-bound beads were analyzed by Western blotting with the anti-V5 antibody. As presented in Fig. 4C, CK1.2-V5-His6 was detected in both promastigote and axenic amastigote extracts. In the promastigote extract, we could observe an additional faint band at about 40 kDa that was recognized by the anti-V5 antibody but that was absent in transgenic axenic amastigotes or in mock-transfected parasites (Fig. 4C). This protein was also enriched by immunoprecipitation and could correspond to an N-terminal degradation product of CK1.2-V5-His6.
To determine the relative activity of CK1.2V5-His6 in promastigotes and in axenic amastigotes, we incubated equal amounts of purified beads, corresponding to equal amounts of protein (Fig. 4C) from transgenic or mock-transfected promastigotes and axenic amastigotes at 30 or 37°C for 30 min in the presence of [γ-32P]ATP, 27 μM CK-S. From the mock-transfected controls, we detected a weak kinase activity—higher in promastigotes than in amastigotes—that could use CK-S as a substrate and be inhibited by D4476, suggesting that the agarose beads could directly or indirectly bind endogenous CK1 (Fig. 4D). We detected a higher phosphotransferase activity in promastigotes (2.5 pmol of phosphate incorporated/min) than in axenic amastigotes (1.7 pmol of phosphate incorporated/min). This activity decreased dramatically when the samples were treated with D4476. Similar to the recombinant CK1.2, the transgenic CK1.2 was inhibited by D4476. We also confirmed the influence of temperature on transgenic CK1.2 and showed a decrease of activity of transgenic CK1.2 upon a temperature shift from 30°C to 37°C, which could explain the observed drop in activity between promastigotes and amastigotes (see Fig. S1 in the supplemental material).
Endogenous CK1 activity is sensitive to D4476.
We took advantage of the CK1-specific peptide substrate and the CK1-specific inhibitor D4476 to assess CK1 activity in total protein extracts; this unique combination allowed us to gain access to endogenous CK1 activity. We first tested whether CK1 inhibitors could prevent the phosphorylation of CK-S by endogenous CK1 activities. Twenty micrograms of total protein extracts was incubated in the presence of 1% DMSO or IC261, D4476, or CKI-7, each at 10 μM (Fig. 5A). Only the most potent inhibitor of rLmaCK1.2, D4476, prevented CK-S phosphorylation (Fig. 5A). The CK1 activity detected in Leishmania protein extracts showed a sensitivity to D4476, IC261, and CKI-7 comparable to that of the recombinant LmaCK1.2. Because CK1.2 seems to be the main casein kinase 1 expressed in Leishmania (13, 15–17), we could estimate the contribution of CK1.2 to the total kinase activity. We thus performed a kinase assay with total protein extracts as a source of kinases but no substrate other than endogenous substrates. The reactions were performed with or without 10 μM D4476, IC261, or CKI-7. We observed a decrease in the phosphorylation of numerous substrates upon addition of D4476 (Fig. 5B) but not upon addition of IC261 or CKI-7. The level of inhibition of D4476 on endogenous protein phosphorylation was subsequently estimated by quantitative imaging to be about 46% of the total endogenous kinase activity in promastigotes and 58% in amastigotes (Fig. 5B). Neither IC261 nor CKI-7 showed a significant effect. Altogether, these results suggest that CK1 might be one of the major protein kinase families expressed and active in Leishmania at both stages.
FIG 5.
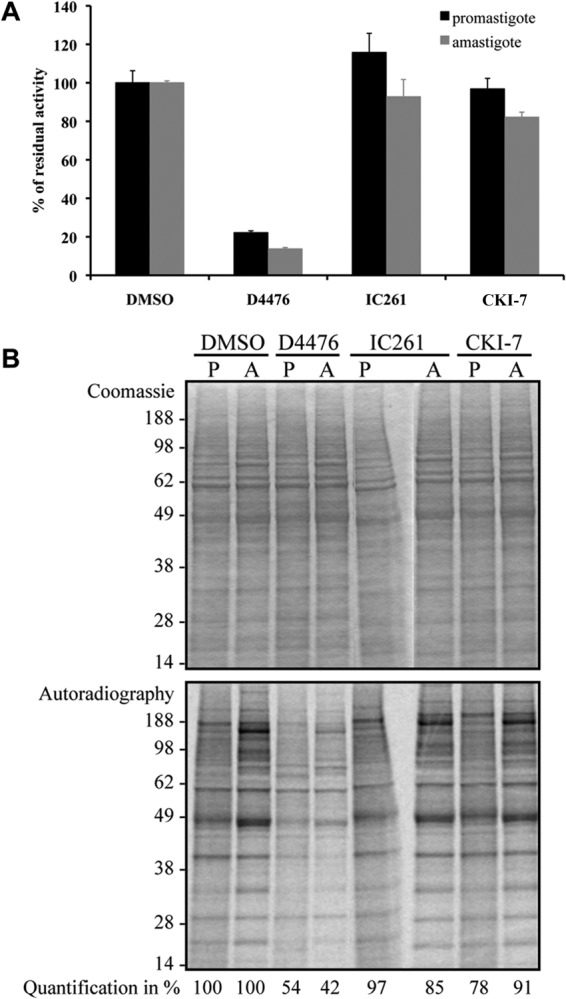
CK1 represents a bulk of the total kinase activity. (A) Kinase assays using 27 μM CK-S and 20 μg of total protein extract from promastigotes or axenic amastigotes were performed in the presence of either 1% DMSO or IC261, D4476, and CKI-7 at 10 μM in 1% DMSO. The activities in the reaction mixtures, spotted on P81 paper, were measured using a scintillation counter. For IC261 and CKI-7, there were no statistically significant differences between the inhibition obtained in promastigotes or in amastigotes (P = 0.510 and P = 0.111, respectively). There were no statistically significant differences between DMSO and IC261 (P = 0.573) and DMSO and CKI-7 (P = 0.286) in promastigotes or between DMSO and IC261 (P = 0.237) in amastigotes, but there was a significant difference between DMSO and CKI-7 (P < 0.001) in amastigotes. (B) Endogenous phosphorylation of 20 μg total protein extract from promastigotes (lanes P) and axenic amastigotes (lanes A) in the presence of either 1% DMSO or D4476, IC261, and CKI-7 at 10 μM in 1% DMSO. The reactions were analyzed by SDS-PAGE, and the gels were stained by colloidal Coomassie for loading assessment (top) and autoradiography (bottom). Quantification was estimated by the quantitative imaging software ImageJ.
Inhibition of CK1 is cytostatic in promastigotes and cytotoxic in axenic amastigotes.
We have shown thus far that D4476 inhibits recombinant, transgenic, and endogenous CK1.2. To validate this kinase as a therapeutic target, we investigated the effect of its inhibition on parasite viability. Previously, Allocco et al. showed that the LmaCK1.2 inhibitors pyrrole and imidazopyridine strongly reduced promastigote growth (13), but they did not investigate their effect on the viability of amastigotes. We thus assessed the effect of our three CK1 inhibitors on L. donovani promastigotes as well as on axenic amastigotes by measuring the percentage of metabolically active cells using the alamarBlue method (26). D4476, the most efficient CK1 inhibitor, was also the most efficient compound against parasites, with an EC50 of 30 μM on promastigotes and an EC50 of 40 μM on axenic amastigotes (Table 2; see Fig. S3A in the supplemental material). The EC50s of D4476 for promastigotes of L. amazonensis, L. braziliensis, and L. major, determined in triplicate in three independent experiments, were 25 ± 2, 20 ± 3, and 47 ± 6 µM, respectively, suggesting that D4476 has an impact on the causative agents of cutaneous, mucocutaneous, and visceral leishmaniasis. IC261 had a weak effect on promastigotes, with an EC50 of 70 μM, and no effect on axenic amastigotes (EC50 > 100 μM; Table 2; see Fig. S3B in the supplemental material). CKI-7 did not reduce the percentage of metabolically active cells in promastigotes or axenic amastigotes, which is consistent with the absence of inhibition toward the recombinant kinase (EC50 > 100 μM; Table 2; see Fig. S3C in the supplemental material). These results show that targeting CK1 with D4476 has an impact on promastigote and, more importantly, axenic amastigote biosynthetic activity. Each compound showed stage-specific potency, demonstrating the necessity to test compounds on amastigotes and not only on promastigotes. To determine whether the effect of D4476 is cytostatic or cytotoxic, we followed the effect of the treatment on the growth and the cell survival of promastigotes and axenic amastigotes. We first treated promastigote cultures with a 1× EC50 dose (30 μM) of D4476 or the equivalent volume of DMSO and followed cell growth for 8 days, also measuring cell death by propidium iodide staining and fluorescence-activated cells sorter analysis. As seen in Fig. 6A, treated promastigotes stopped their growth after 48 h for 24 h (Fig. 6A) and then resumed growth to reach stationary phase with a density higher than that of promastigotes treated with DMSO. The percentage of dead cells remained below 12% (data not shown). This finding indicates that CK1.2 is dispensable in promastigotes, suggesting that its function might not be essential. Second, we followed the effect of a 1× EC50 dose (40 μM) of D4476 or the equivalent volume of DMSO at different time points during axenic amastigote differentiation. We treated the cells at 0 h, when they were first exposed to environmental changes; at 24 h, when morphological changes occurred; and at 48 h, when axenic amastigotes began to maturate and multiply. The untreated cells proliferated for 72 h (Fig. 6Ba, closed circles) and then entered into stationary phase, which correlated with an increase in the percentage of dead cells (Fig. 6Ba, open circles). For all the treated samples, we observed an increase in the percentage of dead cells about 24 h after the treatment was administered. When samples were treated at 0 h, we did not observe an increase in cell density (Fig. 6Bb, closed circles) but observed an elevated percentage of dead cells, which reached up to 80% (Fig. 6Bb, open circles). A similar phenomenon was observed when samples were treated after 24 h or 48 h into the differentiation process (Fig. 6Bc and Bd, respectively, closed circles), but with a percentage of dead cells of only 40% (Fig. 6Bc and Bd, respectively, open circles). This difference suggests that CK1.2 activity could be crucial in the early differentiation of axenic amastigotes but remains essential throughout this process. Altogether, we demonstrated that the effect of the EC50 dose is only cytostatic in promastigotes, whereas it is cytotoxic in amastigotes. This confirms that CK1.2 is a promising drug target for antileishmanial treatment.
TABLE 2.
EC50s of known CK1 inhibitors on L. donovani promastigotes and axenic amastigotes as well as macrophages (when required)
| Inhibitor | EC50 (μM)a |
||
|---|---|---|---|
| Promastigotes | Amastigotes | Macrophages | |
| D4476 | 30 ± 3 | 42 ± 3 | >100b |
| IC261 | 78 ± 7 | >100b | NA |
| CKI-7 | >100b | >100b | NA |
EC50s were determined in triplicate in three independent experiments, unless indicated otherwise. NA, not applicable.
EC50s were determined in triplicate in two independent experiments.
FIG 6.
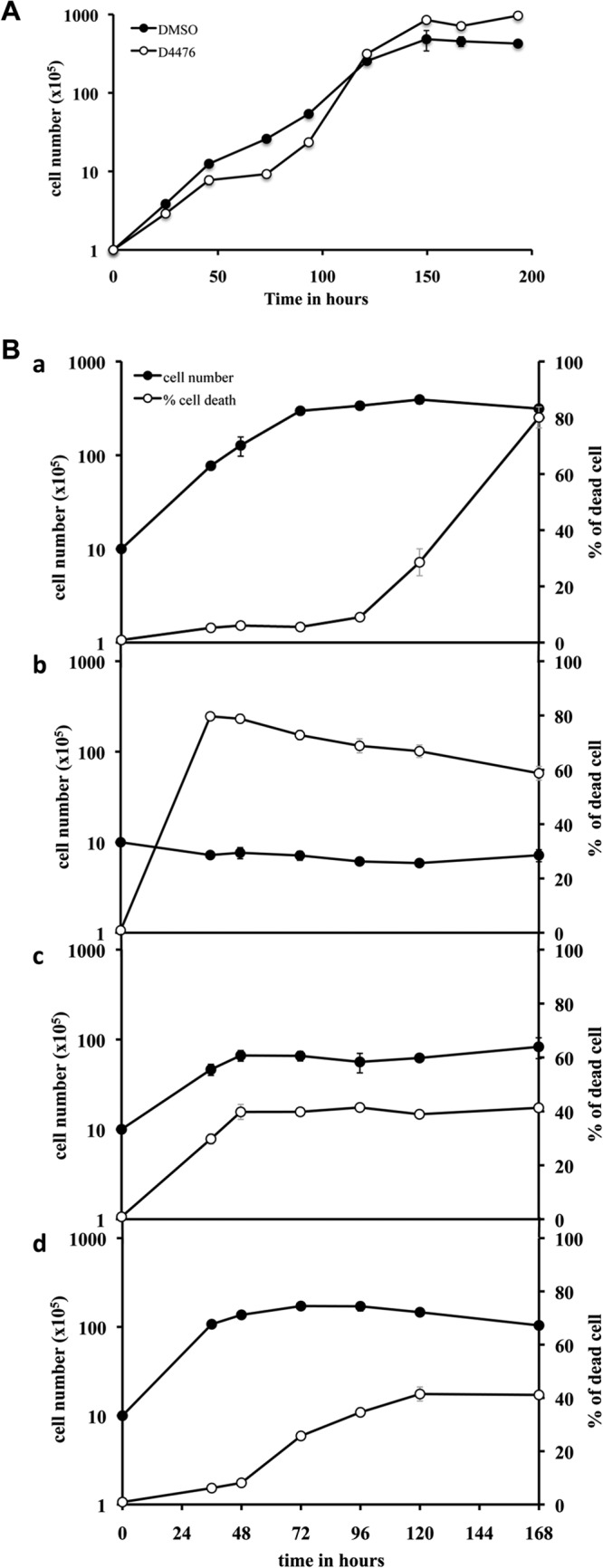
D4476 is cytostatic for L. donovani promastigotes and cytotoxic for L. donovani axenic amastigotes at the EC50 dose. (A) D4476 is cytostatic for L. donovani promastigotes. A total of 105 L. donovani promastigotes/ml culture were treated either with DMSO or with 30 μM D4476 to follow cell growth and the percentage of cells that died for 8 days. (B) A total of 106 L. donovani promastigotes/ml were transferred at pH 5.5 and 37°C, and then the culture was treated either with DMSO (a) or with 40 μM D4476 at different time points during differentiation: 0 h (b), 24 h (c), and 48 h (d). The percentage of cells that died was analyzed using a FACScalibur flow cytometer.
The effect of D4476 on parasites is mediated by inhibition of the CK1.2 isoform.
Leishmania CK1.2 is the only isoform that has been identified experimentally, by proteomics, suggesting that it may be the most abundant casein kinase 1 (13, 15–18, 41, 42). Consequently, the effect of D4476 on cells could be mediated mainly through inhibition of CK1.2. To investigate this hypothesis, we used two complementary approaches: a drug competition assay and a biological assay. First, we investigated whether D4476 was sufficient to prevent the binding of CK1.2 to an ATP binding column, as described in the Materials and Methods. In the untreated sample, endogenous CK1.2 was mainly found in the elution fraction, and only a small amount was found in the flowthrough (Fig. 7A, bottom). In contrast, in the competition sample, endogenous CK1.2 was exclusively found in the flowthrough and not in the elution fraction (Fig. 7A, bottom). These data indicate that upon competition with D4476, endogenous CK1.2 no longer binds to the ATP-beads, suggesting that CK1.2 has a high affinity for this inhibitor that cannot be easily displaced by ATP. As judged by the elution profile revealed by SYPRO Ruby staining, we did not observe a difference between the proteins eluted from the treated or the untreated samples, despite the excess of D4476, suggesting that it has no effect on the most abundant ATP binding proteins (Fig. 7A, top).
FIG 7.
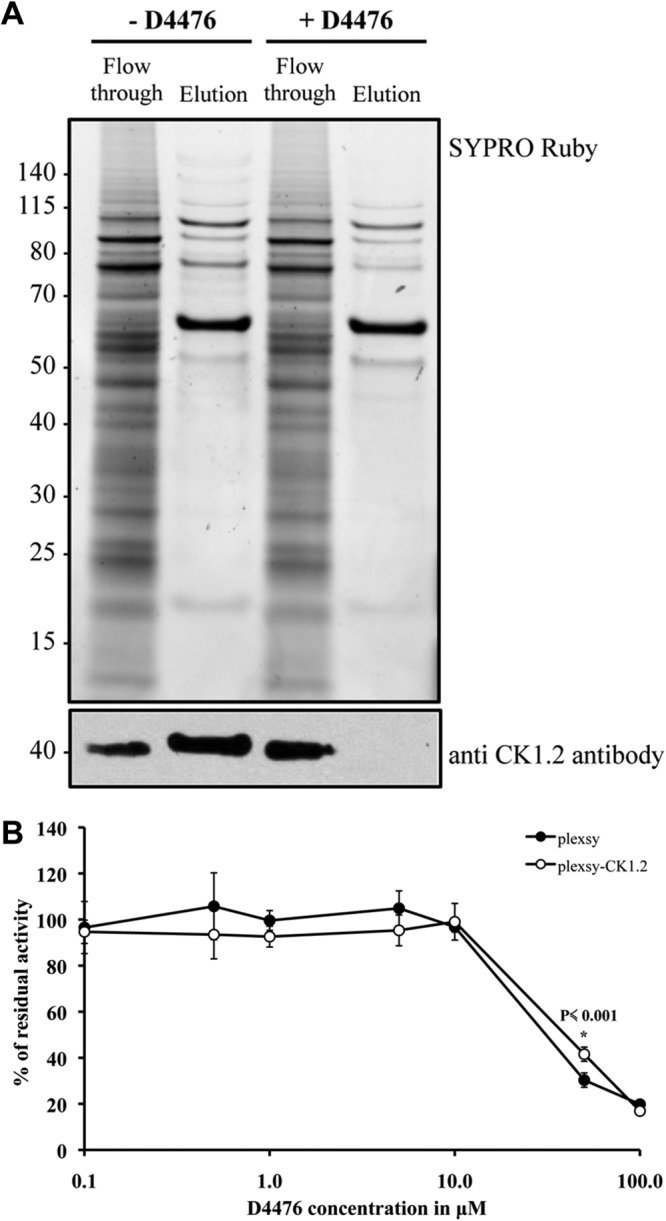
D4476 could be mainly targeting CK1.2. (A) D4476 prevents CK1.2 from binding to the ATP column. One milligram of axenic amastigote total protein extract was either untreated (assay) or incubated with an excess of 250 μM D4476 (competition) prior to loading on an ATP column. The flowthrough and the eluate were separated by SDS-PAGE, and the gel was either stained by SYPRO Ruby (top) or analyzed by Western blotting with SY3535 antibody (bottom). Numbers to the left are molecular masses (in kilodaltons). (B) CK1.2 overexpression slightly increases the EC50 of D4476. Transgenic axenic amastigotes expressing CK1.2 or the mock transfectant were mixed with an equal volume of medium containing increasing concentrations of D4476 in 1% DMSO and analyzed by the alamarBlue method. *, P value for that particular time point.
We next asked whether even a modest increase in CK1.2 protein levels could lead to an increase in the EC50. We assessed the effect of D4476 on L. donovani axenic amastigotes overexpressing or not overexpressing CK1.2-V5-His6 by measuring the percentage of metabolically active cells using the alamarBlue method (as done for the assay whose results are presented in Table 1) (26). We found EC50s of 31 ± 2 μM for the mock-transfected control and 44 ± 2 μM for transgenic parasites (Fig. 7B). Thus, the overexpression of CK1.2-V5-His6 led to a modest, but significant, increase in the EC50, which is consistent with the fact that CK1.2-V5-His6 is weakly expressed compared to the level of expression of endogenous CK1.2 (Fig. 4B). While we cannot completely exclude the possibility that D4476 targets other kinases, theses results strengthen the idea that CK1.2 might be the main target.
D4476 reduces the parasite burden in macrophages.
Compounds that are efficient against cultured parasites are not necessarily efficient against intracellular parasites, as previously reported (43, 44). Moreover, the contribution of CK1 kinases to parasite survival within macrophages is largely unknown. We therefore evaluated the effect of D4476, the most efficient CK1 inhibitor, on intracellular amastigote survival. We did not determine the effect of IC261 and CKI-7 on intracellular parasites, as they were inefficient against axenic amastigotes (Table 2). First, we determined the activity of D4476 on L. donovani-infected PEMs. Preliminary experiments were performed to assess compound toxicity on macrophages using the alamarBlue method, and we measured an EC50 close to 100 μM (Table 2). Thus, we chose to treat the infected macrophages at 50 μM D4476, corresponding to half of the macrophage EC50, and asked whether this treatment could efficiently trigger intracellular parasite death. As a positive control, cells were treated with 1 μM amphotericin B (AMB), a reference antileishmanial drug. The treatment was performed for 24 h and 48 h, and the number of infected macrophages was determined microscopically, counting 300 cells in two independent triplicate experiments. As expected, AMB was extremely efficient, as less than 1% of macrophages remained infected after 24 h of treatment. Compared to the results for the vehicle (1% DMSO)-treated control, there was a significant decrease in the number of infected macrophages after treatment with 50 μM D4476, with only 45% of the macrophages remaining infected at 24 h posttreatment and 38% remaining infected at 48 h posttreatment (Fig. 8A). This observation suggests that the inhibition of Leishmania CK1 kinases has an impact on intracellular L. donovani survival. To confirm these results, we used a high-content cell-based assay to determine the activity of D4476 on intramacrophagic L. amazonensis amastigotes (27). In this assay, a complete clearance of fluorescent parasitophorous vacuoles (PVs) reflects the presence of a very active antileishmanial molecule. Additionally, the analysis of macrophage nuclei with Hoechst 33342 allows determination of the health status of compound-treated macrophages. Infected macrophages were treated with either 1% DMSO, 1 μM D4476, 10 μM D4476, 0.5 μM AMB, or 180 μM cycloheximide (CHX) (27). We show in Fig. 8Ba that the macrophage viability index of samples treated with 1 or 10 μM D4476 was identical to that of samples treated with DMSO or AMB (noncytotoxic agents) but different from that of samples treated with CHX (a cytotoxic agent). D4476 is thus not toxic to host cells at these concentrations. Noticeably, 1% DMSO had no effect on macrophage viability indexes, PV counts, or amastigote loads, as shown by Aulner et al. (27). We next determined the antileishmanial activity of D4476 on infected macrophages by establishing the ratio between the number of PVs and the number of healthy macrophages (Fig. 8Bb). DMSO-treated samples gave an SSMD* close to 0 (no effect), whereas amphotericin B gave an SSMD* of about −15 (maximum effect). With D4476, we observed a modest but significant effect at 1 μM, with an SSMD* of about −2 and an even stronger effect at 10 μM (SSMD*, about −7; Fig. 8Bb). In Fig. 8C, we show representative images of infected macrophages incubated with DMSO, 1 μM D4476, or 10 μM D4476. In the DMSO-treated control, most of the macrophages were infected, as judged by the presence of numerous PVs stained green by LysoTracker dye. In contrast, infected macrophages treated with D4476 showed a decrease in the number of PVs, especially with D4476 at 10 μM. Altogether, these data demonstrate a significant concentration-dependent decrease in parasite load on macrophages treated with D4476. Moreover, these results are in accordance with those obtained with L. donovani-infected macrophages and suggest that D4476 is efficient against intracellular parasites down to a concentration of 10 μM. These findings reveal, through a pharmacological approach, the essential biological functions of CK1 kinases at the amastigote stage in two different species and highlight the potential of this protein kinase family as drug targets for antileishmanial therapy.
FIG 8.

Inhibition of CK1 reduces the parasite burden of the infected macrophages. (A) PEMs were incubated with opsonized L. donovani promastigotes, and after 48 h, 1% DMSO (control), 50 μM D4476 in 1% DMSO, or 1 μM AMB in 1% DMSO was added to the culture. Intracellular parasites were detected by nuclear staining with Hoechst 33342 in paraformaldehyde (PFA)-fixed cells after 24 h or 48 h of contact with the drugs. Three hundred macrophages were analyzed per coverslip, and the experiment was performed twice in triplicate. Results are expressed as percentages of the value for the DMSO control. (B) Bone marrow-derived mouse macrophages were infected with L. amazonensis lesion-derived amastigotes and treated for 3 days with DMSO (final concentration, 1%), amphotericin B (0.5 μM), CHX (180 μM), and D4476 (1 and 10 μM). Results of compound activity are expressed as SSMD*s, calculated using DMSO-treated macrophages as a reference group for macrophage viability (a), and antileishmanial indexes (the ratio between the number of PVs and the number of healthy macrophages) (b). About 10,000 macrophages were analyzed per well, and the SSMD*s for replicate wells (4, 32, 16, and 16 replicate wells for D4476-, DMSO-, amphotericin B-, and cycloheximide-treated macrophage cultures, respectively) are shown in a box-and-whisker plot. *, P ≤ 0.001 comparing the DMSO control to D4476. (C) Representative merged images for DMSO- and D4476-treated infected macrophages are shown. A zoomed view of the DMSO control culture is displayed to illustrate more clearly the PVs (green), the macrophage nuclei (blue), and the amastigotes (red). Bars, 50 μm.
DISCUSSION
In this study, we have characterized LmjF.35.1010 (LmaCK1.2) using bioinformatics, biochemical, and pharmacological approaches. Analyzing the effect of CK1-specific inhibitors on (i) recombinant LmaCK1.2, (ii) transgenic CK1.2, (iii) endogenous CK1 activity detected in parasite extracts, (iv) free parasites, as well as (v) bona fide intracellular L. donovani and L. amazonensis amastigotes, we provide here the first evidence that CK1 protein kinases are required for intracellular parasite survival and infectivity and thus represent important drug targets in Leishmania.
Although we cannot completely exclude the possibility of an off-target effect, the antileishmanial activity of D4476 seems to be mediated mainly through CK1.2 inhibition for the following reasons. First, D4476 is a specific human CK1 inhibitor that targets different CK1 isoforms in the nanomolar range with a similar efficiency (40, 45). It has initially been identified as an inhibitor of activin receptor-like kinase 5 (ALK5), a member of the type I transforming growth factor β receptor family (46). Subsequent assessment of its specificity on a panel of over 70 protein kinases revealed that D4476 is most potent toward CK1. This compound affects only two other protein kinases, PKD1 and p38α, with inhibitory activity 10- and 30-fold lower than that for CK1, respectively (40). Neither ALK5 nor PKD1 has an ortholog in Leishmania, limiting any potential off-target effect of D4476 on p38α. Second, four out of the five other Leishmania CK1 isoforms are only weakly related to CK1.2, with major differences observed in their amino acid sequences and significant sequence insertions for two isoforms (data not shown). Consequently, the difference in the structures of their ATP binding pockets suggests that their sensitivity to D4476 could be lower than that of CK1.2. Third, CK1.2 is the only isoform for which expression has been detected in Leishmania. This protein kinase has been identified in several proteomic studies, suggesting that CK1.2 could be the most abundant CK1 and could therefore represent the main CK1 activity detected in Leishmania (15–18). Moreover, Allocco et al. have shown that CK1.2 is the only isoform responsible for CK1 activity in promastigotes (13). This finding is in agreement with our data, as each inhibitor that we have tested has the same potency toward recombinant LmaCK1.2, endogenous CK1 kinases, or free parasites. Fourth, the binding of CK1.2 to an immobilized ATP matrix could be abolished by preincubation with an excess of D4476, suggesting a strong affinity of this inhibitor for CK1.2 without visible changes in the general profile of the ATP binding proteins. Finally, increasing even slightly the expression of CK1.2 in transgenic axenic amastigotes led to a small but significant increase in the EC50 obtained for D4476.
Our pharmacological approach revealed stage-specific biological functions of CK1.2. In promastigotes, CK1.2 does not seem to be essential. Indeed, treatment at the EC50 dose merely led to a delay in growth, from which cells recovered within 48 h to reach a cell density higher than that for the control treated with DMSO. Only treatment at twice the EC50 dose has a cytotoxic effect on promastigotes, causing annexin V-independent cell death (47). In contrast, treatment of axenic amastigotes at the EC50 dose is sufficient to observe a dramatic effect on cell survival, as within 24 h of treatment the percentage of cells that died strongly increased. Moreover, if cells are treated within the first 24 h after shifting the temperature and pH, the effect on cell survival is further increased. Altogether, our data suggest that CK1.2 plays a critical role within the first 24 h of differentiation but remains essential throughout differentiation. On the basis of the results published by Barak et al., promastigotes start to transform into amastigote-shaped cells in the first hours after exposure to elevated temperatures, a process that occurs synchronously as the parasites undergo cell cycle arrest at G1 (48). The acidification of the medium allows the release of the cells from this arrest (48). Several reports have shown the involvement of CK1 in cell cycle progression, suggesting that Leishmania CK1.2 could be involved in the regulation of cell cycle arrest in G1 or its release during the first 24 h following exposure of promastigotes to differentiation signals (49). Eukaryote CK1s have also been shown to be involved in the stress response following DNA damage or viral infection (50), suggesting that the strong effect of CK1.2 inhibition in the first hours of differentiation could also reflect the requirement for CK1.2 for the parasite to resist the stress caused by pH and temperature shock. In addition, it seems that the results obtained with axenic amastigotes could be extrapolated to intracellular amastigotes, as the inhibition of CK1.2 also impaired intracellular parasite survival.
LmaCK1.2, the only Leishmania CK1 identified in intracellular parasites, shows a high degree of identity to human CK1s, which attains 71% for the kinase domain (16). Given the evolutionary distance of Leishmania and humans, the question of the biological significance of this remarkable similarity arises. For successful host cell infection, Leishmania has evolved numerous survival strategies (i) to secure nutrients by exploiting the host cell metabolic pathways for its own benefit, (ii) to defend itself against host cell cytolytic activities by interfering with, for example, vesicular trafficking or cytokine signaling, and (iii) to subvert host immunity by modulating host cell cytokine production (51). From recent studies, it has emerged that proteins released by parasites into host cells, either free or in vesicles, could regulate these processes (15, 17, 18, 42). Indeed, parasite-derived exosomes have been shown to modulate the immune system by attenuating the cytokine response and IFN-γ treatment, suggesting that parasite proteins released by exocytosis, which include kinases such as MAPKs and CK1.2, could play a major role in the pathogenesis of leishmaniasis (17, 42). Conceivably, the release of Leishmania protein kinases into the host cytoplasm could be a potential and potent mechanism by which the parasite modulates the host cell immune response and metabolism for its own needs. A similar phenomenon has been observed in malaria infection. P. falciparum-infected red blood cells show an increase in phosphorylated proteins compared to uninfected ones. The majority of these proteins are of parasite origin, but several are of human origin, suggesting that the released parasite kinases phosphorylate host substrates (52). Likewise, exocytosis of Leishmania CK1.2 may regulate essential host cell processes through phosphorylation, which would explain the high level of conservation between parasite CK1.2 and human CK1 kinases due to the evolutionary pressure to recognize host cell substrates (15, 17). This hypothesis is supported by several reports showing that Leishmania CK1.2 phosphorylates human proteins such as IFNAR1 and the human complement component C3a (18, 19). Hence, inhibition of parasite CK1.2 could have dramatic effects on Leishmania intracellular survival and could contribute to rescue an antileishmanial immune response. Moreover, because these vital external CK1.2 functions rely on the capacity of this kinase to recognize and phosphorylate host proteins, the risk for selecting drug-resistant parasites expressing a mutated CK1.2 is extremely low.
For those reasons, Leishmania CK1.2 represents a good drug target, but the high degree of conservation between the parasite and human CK1 isoforms may challenge the identification of parasite-specific CK1 inhibitors with limited side effects on host kinases. On the contrary, our data provide important evidence for specific inhibition. First, comparison of the efficacy of different CK1 inhibitors on mammalian CK1 and LmaCK1.2 revealed that the parasite kinase is more sensitive to D4476. Second, although they were inefficient against recombinant CK1.2, IC261 and CKI-7 are quite potent against mammalian CK1, suggesting that Leishmania and mammalian CK1 have significant structural differences that can be discriminated by compounds. Finally, we have previously published the finding that purvalanol B has a higher affinity to Leishmania CK1.2 than to the human homolog, demonstrating yet again differences in ATP binding pocket conformation (20). This divergence in affinity could be exploited in the future by high-throughput, target-based screening campaigns for the identification of parasite-specific inhibitory compounds.
In conclusion, we have shown for the first time the effects of CK1 kinase inhibition on axenic and intracellular amastigotes, clearly establishing the essential role of Leishmania CK1 in host infection and thus validating members of the CK1 kinase family, which have essential biological functions in the survival of the disease-relevant, intracellular amastigote form, as an important target for antileishmanial drug development.
Supplementary Material
ACKNOWLEDGMENTS
We thank all members of the FP7 LEISHDRUG consortium for fruitful discussions, in particular, Geneviève Milon. Additional thanks go to Olivier Helynck and Hélène Munier-Lehmann for providing access to the TECAN Freedom EVOware platform for automatic distribution of cells, parasites, and chemicals in a biosafety level 2 facility and to Anne Danckaert, Pierre-Henri Commere, and Spencer L. Shorte from Imagopole.
This work was supported by the 7th Framework Program of the European Commission through grants to the LEISHDRUG project (223414), ANR-11-RPIB-0016 TRANSLEISH, the French Government's Investissements d'Avenir program Laboratoire d'Excellence Integrative Biology of Emerging Infectious Diseases (grant no. ANR-10-LABX-62-IBEID), the Conseil de la Région Ile-de-France (program S.E.S.A.M.E. 2007 Imagopole to Spencer L. Shorte) in the framework of France bioimaging (grant no. ANR-10-INSB-04-01, Investments for the Future, to Imagopole), the Fondation Française pour la Recherche Médicale (FRM; Grands Équipements program, to Nathalie Aulner), and Chemical Library projects I06-222/R and I09-1739/R to Hélène Munier-Lehmann.
Footnotes
Published ahead of print 23 December 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02022-13.
REFERENCES
- 1.Murray HW, Berman JD, Davies CR, Saravia NG. 2005. Advances in leishmaniasis. Lancet 366:1561–1577. 10.1016/S0140-6736(05)67629-5 [DOI] [PubMed] [Google Scholar]
- 2.Olliaro PL, Guerin PJ, Gerstl S, Haaskjold AA, Rottingen JA, Sundar S. 2005. Treatment options for visceral leishmaniasis: a systematic review of clinical studies done in India, 1980-2004. Lancet Infect. Dis. 5:763–774. 10.1016/S1473-3099(05)70296-6 [DOI] [PubMed] [Google Scholar]
- 3.Eglen R, Reisine T. 2011. Drug discovery and the human kinome: recent trends. Pharmacol. Ther. 130:144–156. 10.1016/j.pharmthera.2011.01.007 [DOI] [PubMed] [Google Scholar]
- 4.Doerig C. 2004. Protein kinases as targets for anti-parasitic chemotherapy. Biochim. Biophys. Acta 1697:155–168. 10.1016/j.bbapap.2003.11.021 [DOI] [PubMed] [Google Scholar]
- 5.Morales MA, Renaud O, Faigle W, Shorte SL, Spath GF. 2007. Over-expression of Leishmania major MAP kinases reveals stage-specific induction of phosphotransferase activity. Int. J. Parasitol. 37:1187–1199. 10.1016/j.ijpara.2007.03.006 [DOI] [PubMed] [Google Scholar]
- 6.Grant KM, Dunion MH, Yardley V, Skaltsounis AL, Marko D, Eisenbrand G, Croft SL, Meijer L, Mottram JC. 2004. Inhibitors of Leishmania mexicana CRK3 cyclin-dependent kinase: chemical library screen and antileishmanial activity. Antimicrob. Agents Chemother. 48:3033–3042. 10.1128/AAC.48.8.3033-3042.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Q, Melzer IM, Kruse M, Sander-Juelch C, Wiese M. 2005. LmxMPK4, a mitogen-activated protein (MAP) kinase homologue essential for promastigotes and amastigotes of Leishmania mexicana. Kinetoplastid Biol. Dis. 4:6. 10.1186/1475-9292-4-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Budini M, Jacob G, Jedlicki A, Perez C, Allende CC, Allende JE. 2009. Autophosphorylation of carboxy-terminal residues inhibits the activity of protein kinase CK1alpha. J. Cell. Biochem. 106:399–408. 10.1002/jcb.22019 [DOI] [PubMed] [Google Scholar]
- 9.Barik S, Taylor RE, Chakrabarti D. 1997. Identification, cloning, and mutational analysis of the casein kinase 1 cDNA of the malaria parasite, Plasmodium falciparum. Stage-specific expression of the gene. J. Biol. Chem. 272:26132–26138 [DOI] [PubMed] [Google Scholar]
- 10.Calabokis M, Kurz L, Wilkesman J, Galan-Caridad JM, Moller C, Gonzatti MI, Bubis J. 2002. Biochemical and enzymatic characterization of a partially purified casein kinase-1 like activity from Trypanosoma cruzi. Parasitol. Int. 51:25–39. 10.1016/S1383-5769(01)00104-0 [DOI] [PubMed] [Google Scholar]
- 11.Spadafora C, Repetto Y, Torres C, Pino L, Robello C, Morello A, Gamarro F, Castanys S. 2002. Two casein kinase 1 isoforms are differentially expressed in Trypanosoma cruzi. Mol. Biochem. Parasitol. 124:23–36. 10.1016/S0166-6851(02)00156-1 [DOI] [PubMed] [Google Scholar]
- 12.Donald RG, Zhong T, Meijer L, Liberator PA. 2005. Characterization of two T. gondii CK1 isoforms. Mol. Biochem. Parasitol. 141:15–27. 10.1016/j.molbiopara.2005.01.011 [DOI] [PubMed] [Google Scholar]
- 13.Allocco JJ, Donald R, Zhong T, Lee A, Tang YS, Hendrickson RC, Liberator P, Nare B. 2006. Inhibitors of casein kinase 1 block the growth of Leishmania major promastigotes in vitro. Int. J. Parasitol. 36:1249–1259. 10.1016/j.ijpara.2006.06.013 [DOI] [PubMed] [Google Scholar]
- 14.Urbaniak MD. 2009. Casein kinase 1 isoform 2 is essential for bloodstream form Trypanosoma brucei. Mol. Biochem. Parasitol. 166:183–185. 10.1016/j.molbiopara.2009.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Silverman JM, Chan SK, Robinson DP, Dwyer DM, Nandan D, Foster LJ, Reiner NE. 2008. Proteomic analysis of the secretome of Leishmania donovani. Genome Biol. 9:R35. 10.1186/gb-2008-9-2-r35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paape D, Barrios-Llerena ME, Le Bihan T, Mackay L, Aebischer T. 2010. Gel free analysis of the proteome of intracellular Leishmania mexicana. Mol. Biochem. Parasitol. 169:108–114. 10.1016/j.molbiopara.2009.10.009 [DOI] [PubMed] [Google Scholar]
- 17.Silverman JM, Clos J, de'Oliveira CC, Shirvani O, Fang Y, Wang C, Foster LJ, Reiner NE. 2010. An exosome-based secretion pathway is responsible for protein export from Leishmania and communication with macrophages. J. Cell Sci. 123:842–852. 10.1242/jcs.056465 [DOI] [PubMed] [Google Scholar]
- 18.Vieira LL, Sacerdoti-Sierra N, Jaffe CL. 2002. Effect of pH and temperature on protein kinase release by Leishmania donovani. Int. J. Parasitol. 32:1085–1093. 10.1016/S0020-7519(02)00067-X [DOI] [PubMed] [Google Scholar]
- 19.Liu J, Carvalho LP, Bhattachariya S, Carbone CJ, Kumar KG, Leu NA, Yau PM, Donald RG, Weiss MJ, Baker DP, McLaughlin KJ, Scott P, Fuchs SY. 2009. Mammalian casein kinase 1alpha and its leishmanial ortholog regulate stability of IFNAR1 and type I interferon signaling. Mol. Cell. Biol. 29:6401–6412. 10.1128/MCB.00478-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Knockaert M, Gray N, Damiens E, Chang YT, Grellier P, Grant K, Fergusson D, Mottram J, Soete M, Dubremetz JF, Le Roch K, Doerig C, Schultz P, Meijer L. 2000. Intracellular targets of cyclin-dependent kinase inhibitors: identification by affinity chromatography using immobilised inhibitors. Chem. Biol. 7:411–422. 10.1016/S1074-5521(00)00124-1 [DOI] [PubMed] [Google Scholar]
- 21.De Muylder G, Ang KKH, Chen S, Arkin MR, Engel JC, McKerrow JH. 2011. A screen against Leishmania intracellular amastigotes: comparison to a promastigote screen and identification of a host cell-specific hit. PLoS Negl. Trop. Dis. 5:e1253. 10.1371/journal.pntd.0001253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morales MA, Watanabe R, Laurent C, Lenormand P, Rousselle JC, Namane A, Spath GF. 2008. Phosphoproteomic analysis of Leishmania donovani pro- and amastigote stages. Proteomics 8:350–363. 10.1002/pmic.200700697 [DOI] [PubMed] [Google Scholar]
- 23.Morales MA, Watanabe R, Dacher M, Chafey P, Osorio y Fortea J, Scott DA, Beverley SM, Ommen G, Clos J, Hem S, Lenormand P, Rousselle JC, Namane A, Spath GF. 2010. Phosphoproteome dynamics reveal heat-shock protein complexes specific to the Leishmania donovani infectious stage. Proc. Natl. Acad. Sci. U. S. A. 107:8381–8386. 10.1073/pnas.0914768107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saar Y, Ransford A, Waldman E, Mazareb S, Amin-Spector S, Plumblee J, Turco SJ, Zilberstein D. 1998. Characterization of developmentally-regulated activities in axenic amastigotes of Leishmania donovani. Mol. Biochem. Parasitol. 95:9–20. 10.1016/S0166-6851(98)00062-0 [DOI] [PubMed] [Google Scholar]
- 25.Goyard S, Segawa H, Gordon J, Showalter M, Duncan R, Turco SJ, Beverley SM. 2003. An in vitro system for developmental and genetic studies of Leishmania donovani phosphoglycans. Mol. Biochem. Parasitol. 130:31–42. 10.1016/S0166-6851(03)00142-7 [DOI] [PubMed] [Google Scholar]
- 26.Shimony O, Jaffe CL. 2008. Rapid fluorescent assay for screening drugs on Leishmania amastigotes. J. Microbiol. Methods 75:196–200. 10.1016/j.mimet.2008.05.026 [DOI] [PubMed] [Google Scholar]
- 27.Aulner N, Danckaert A, Rouault-Hardoin E, Desrivot J, Helynck O, Commere PH, Munier-Lehmann H, Spath GF, Shorte SL, Milon G, Prina E. 2013. High content analysis of primary macrophages hosting proliferating Leishmania amastigotes: application to anti-leishmanial drug discovery. PLoS Negl. Trop. Dis. 7:e2154. 10.1371/journal.pntd.0002154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang XD. 2011. Illustration of SSMD, z score, SSMD*, z* score, and t statistic for hit selection in RNAi high-throughput screens. J. Biomol. Screen. 16:775–785. 10.1177/1087057111405851 [DOI] [PubMed] [Google Scholar]
- 29.Kemena C, Notredame C. 2009. Upcoming challenges for multiple sequence alignment methods in the high-throughput era. Bioinformatics 25:2455–2465. 10.1093/bioinformatics/btp452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clamp M, Cuff J, Searle SM, Barton GJ. 2004. The Jalview Java alignment editor. Bioinformatics 20:426–427. 10.1093/bioinformatics/btg430 [DOI] [PubMed] [Google Scholar]
- 31.Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. 2009. Jalview version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191. 10.1093/bioinformatics/btp033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402. 10.1093/nar/25.17.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Altschul SF, Wootton JC, Gertz EM, Agarwala R, Morgulis A, Schaffer AA, Yu YK. 2005. Protein database searches using compositionally adjusted substitution matrices. FEBS J. 272:5101–5109. 10.1111/j.1742-4658.2005.04945.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reinhardt J, Ferandin Y, Meijer L. 2007. Purification of CK1 by affinity chromatography on immobilised axin. Protein Expr. Purif. 54:101–109. 10.1016/j.pep.2007.02.020 [DOI] [PubMed] [Google Scholar]
- 35.Eide EJ, Vielhaber EL, Hinz WA, Virshup DM. 2002. The circadian regulatory proteins BMAL1 and cryptochromes are substrates of casein kinase Iepsilon. J. Biol. Chem. 277:17248–17254. 10.1074/jbc.M111466200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zilberstein D, Blumenfeld N, Liveanu V, Gepstein A, Jaffe CL. 1991. Growth at acidic pH induces an amastigote stage-specific protein in Leishmania promastigotes. Mol. Biochem. Parasitol. 45:175–178. 10.1016/0166-6851(91)90040-D [DOI] [PubMed] [Google Scholar]
- 37.Rena G, Bain J, Elliott M, Cohen P. 2004. D4476, a cell-permeant inhibitor of CK1, suppresses the site-specific phosphorylation and nuclear exclusion of FOXO1a. EMBO Rep. 5:60–65. 10.1038/sj.embor.7400048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mashhoon N, DeMaggio AJ, Tereshko V, Bergmeier SC, Egli M, Hoekstra MF, Kuret J. 2000. Crystal structure of a conformation-selective casein kinase-1 inhibitor. J. Biol. Chem. 275:20052–20060. 10.1074/jbc.M001713200 [DOI] [PubMed] [Google Scholar]
- 39.Chijiwa T, Hagiwara M, Hidaka H. 1989. A newly synthesized selective casein kinase I inhibitor, N-(2-aminoethyl)-5-chloroisoquinoline-8-sulfonamide, and affinity purification of casein kinase I from bovine testis. J. Biol. Chem. 264:4924–4927 [PubMed] [Google Scholar]
- 40.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, Klevernic I, Arthur JS, Alessi DR, Cohen P. 2007. The selectivity of protein kinase inhibitors: a further update. Biochem. J. 408:297–315. 10.1042/BJ20070797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sacerdoti-Sierra N, Jaffe CL. 1997. Release of ecto-protein kinases by the protozoan parasite Leishmania major. J. Biol. Chem. 272:30760–30765. 10.1074/jbc.272.49.30760 [DOI] [PubMed] [Google Scholar]
- 42.Silverman JM, Clos J, Horakova E, Wang AY, Wiesgigl M, Kelly I, Lynn MA, McMaster WR, Foster LJ, Levings MK, Reiner NE. 2010. Leishmania exosomes modulate innate and adaptive immune responses through effects on monocytes and dendritic cells. J. Immunol. 185:5011–5022. 10.4049/jimmunol.1000541 [DOI] [PubMed] [Google Scholar]
- 43.Siqueira-Neto JL, Song OR, Oh H, Sohn JH, Yang G, Nam J, Jang J, Cechetto J, Lee CB, Moon S, Genovesio A, Chatelain E, Christophe T, Freitas-Junior LH. 2010. Antileishmanial high-throughput drug screening reveals drug candidates with new scaffolds. PLoS Negl. Trop. Dis. 4:e675. 10.1371/journal.pntd.0000675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Muylder G, Ang KKH, Chen S, Arkin MR, Engel JC, McKerrow JH. 2011. A screen against Leishmania intracellular amastigotes: comparison to a promastigote screen and identification of a host cell-specific hit. PLoS Negl. Trop. Dis. 5:e1253. 10.1371/journal.pntd.0001253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huart AS, MacLaine NJ, Meek DW, Hupp TR. 2009. CK1alpha plays a central role in mediating MDM2 control of p53 and E2F-1 protein stability. J. Biol. Chem. 284:32384–32394. 10.1074/jbc.M109.052647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Callahan JF, Burgess JL, Fornwald JA, Gaster LM, Harling JD, Harrington FP, Heer J, Kwon C, Lehr R, Mathur A, Olson BA, Weinstock J, Laping NJ. 2002. Identification of novel inhibitors of the transforming growth factor beta1 (TGF-beta1) type 1 receptor (ALK5). J. Med. Chem. 45:999–1001. 10.1021/jm010493y [DOI] [PubMed] [Google Scholar]
- 47.Foucher AL, Rachidi N, Gharbi S, Blisnick T, Bastin P, Pemberton IK, Spath GF. 2013. Apoptotic marker expression in the absence of cell death in staurosporine-treated Leishmania donovani. Antimicrob. Agents Chemother. 57:1252–1261. 10.1128/AAC.01983-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barak E, Amin-Spector S, Gerliak E, Goyard S, Holland N, Zilberstein D. 2005. Differentiation of Leishmania donovani in host-free system: analysis of signal perception and response. Mol. Biochem. Parasitol. 141:99–108. 10.1016/j.molbiopara.2005.02.004 [DOI] [PubMed] [Google Scholar]
- 49.Knippschild U, Gocht A, Wolff S, Huber N, Lohler J, Stoter M. 2005. The casein kinase 1 family: participation in multiple cellular processes in eukaryotes. Cell Signal. 17:675–689. 10.1016/j.cellsig.2004.12.011 [DOI] [PubMed] [Google Scholar]
- 50.MacLaine NJ, Oster B, Bundgaard B, Fraser JA, Buckner C, Lazo PA, Meek DW, Hollsberg P, Hupp TR. 2008. A central role for CK1 in catalyzing phosphorylation of the p53 transactivation domain at serine 20 after HHV-6B viral infection. J. Biol. Chem. 283:28563–28573. 10.1074/jbc.M804433200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaye P, Scott P. 2011. Leishmaniasis: complexity at the host-pathogen interface. Nat. Rev. Microbiol. 9:604–615. 10.1038/nrmicro2608 [DOI] [PubMed] [Google Scholar]
- 52.Wu Y, Nelson MM, Quaile A, Xia D, Wastling JM, Craig A. 2009. Identification of phosphorylated proteins in erythrocytes infected by the human malaria parasite Plasmodium falciparum. Malar. J. 8:105. 10.1186/1475-2875-8-105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. 10.1093/bioinformatics/btm404 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.