

Abstract
Human rhinovirus type C (HRV-C) is a newly discovered enterovirus species frequently associated with exacerbation of asthma and other acute respiratory conditions. Until recently, HRV-C could not be propagated in vitro, hampering in-depth characterization of the virus replication cycle and preventing efficient testing of antiviral agents. Herein we describe several subgenomic RNA replicon systems and a cell culture infectious model for HRV-C that can be used for antiviral screening. The replicon constructs consist of genome sequences from HRVc15, HRVc11, HRVc24, and HRVc25 strains, with the P1 capsid region replaced by a Renilla luciferase coding sequence. Following transfection of the replicon RNA into HeLa cells, the constructs produced time-dependent increases in luciferase signal that can be inhibited in a dose-dependent manner by known inhibitors of HRV replication, including the 3C protease inhibitor rupintrivir, the nucleoside analog inhibitor MK-0608, and the phosphatidylinositol 4-kinase IIIβ (PI4K-IIIβ) kinase inhibitor PIK93. Furthermore, with the exception of pleconaril and pirodavir, the other tested classes of HRV inhibitors blocked the replication of full-length HRVc15 and HRVc11 in human airway epithelial cells (HAEs) that were differentiated in the air-liquid interface, exhibiting antiviral activities similar to those observed with HRV-16. In summary, this study is the first comprehensive profiling of multiple classes of antivirals against HRV-C, and the set of newly developed quantitative HRV-C antiviral assays represent indispensable tools for the identification and evaluation of novel panserotype HRV inhibitors.
INTRODUCTION
Human rhinoviruses (HRV) are nonenveloped positive-strand RNA viruses belonging to the genus Enterovirus in the family Picornaviridae (for a review, see reference 1). More than 160 different HRV genotypes have been isolated to date, which are divided into three major groups (HRV-A, HRV-B, and HRV-C) based on phylogenetic sequence analysis (2–5). HRV-A and -B utilize ICAM-1 or LDL-R cell surface proteins as receptors for viral entry, while the receptor for HRV-C remains to be identified (6–9). An IRES (internal ribosome entry site) element in the 5′ noncoding region (NCR) of the HRV genome is required to initiate the translation of a single polyprotein that is subsequently processed by viral 2A, 3C, and 3CD proteases (1, 10). HRV gene products efficiently shut down host cell transcription and translation (11–14) and remodel the endoplasmic reticulum (ER) and Golgi network into replication organelles that serve as the site for viral RNA synthesis by 3D polymerase (15, 16).
HRV infections of the upper and lower respiratory tract cause a broad spectrum of respiratory diseases in humans. While HRV upper respiratory tract infections (URTIs) are largely self-limiting in immunocompetent individuals and are most often associated with the common cold (17, 18), they can also cause acute otitis media and rhinosinusitis (17, 19–21). HRV lower respiratory tract infections (LRTIs) cause more severe diseases in susceptible populations and are frequently associated with asthma exacerbations in adult as well as pediatric patients (22). Moreover, HRV infections are linked to chronic obstructive pulmonary disease (COPD) and cystic fibrosis exacerbation, bronchiolitis, and community-acquired pneumonia in infants and children and with severe, sometimes fatal, pneumonia in elderly and immunocompromised adults (23–31). HRV-A and HRV-C are the predominant viruses identified in infants hospitalized with non-RSV (respiratory syncytial virus)-associated bronchiolitis (22, 23). Importantly, data from prospective studies of infants with at least one parent diagnosed with asthma have shown that wheezing due to HRV infection during infancy increases the risk of asthma development later in life (32). Thus, HRV infections not only are associated with asthma exacerbations but also may play a role in asthma development.
Despite high clinical relevance, there are currently no approved antiviral agents for the treatment of HRV infection. Different classes of HRV inhibitors have been evaluated in clinical trials, including small-molecule capsid-binding inhibitors (pleconaril and pirodavir), a 3C protease inhibitor (rupintrivir), and soluble ICAM-1. These compounds are no longer being developed as antiviral drugs due to limited efficacy in the context of natural infection, safety concerns, or potential drug interactions (33–37). In addition, the development of anti-HRV agents has been hampered by the lack of screening assays for group C viruses. Unlike HRV-A and -B serotypes, which can be easily propagated in tissue culture, HRV-C has only been shown to replicate in ex-planted sinus mucosal tissue or air-liquid interface (ALI)-differentiated sinus or bronchial epithelial cells (7, 38, 39).
In this study, we describe the development, optimization, and validation of transient subgenomic replicon systems derived from several strains of HRV-C that allow efficient compound screening. In addition, we have developed a quantitative PCR-based screening assay utilizing the human airway epithelial cell (HAE) culture model to propagate full-length infectious HRV-C and employed it together with the subgenomic replicon screening assays to characterize the antiviral activities of different classes of known HRV inhibitors against HRV-C. Our data demonstrate that these assay systems can effectively support the discovery of new types of antiviral compounds for potential treatment of respiratory diseases associated with HRV infection.
MATERIALS AND METHODS
Compounds.
The following compounds were used in testing: rupintrivir (CAS no. 223537-30-2), pleconaril (CAS no. 153168-05-9), PIK93 (CAS no. 593960-11-3), MK-0608, pirodavir (CAS no. 124436-59-5), and Win 56291 (CAS no. 107355-76-0) (Fig. 1). These compounds were synthesized internally by Gilead Medicinal Chemistry Department and subjected to standard material quality control procedures.
FIG 1.

Chemical structures of HRV inhibitors tested against HRV-C. Rupintrivir is a 3C protease inhibitor; MK-0608 is a nucleoside analog inhibitor of 3D polymerase; PIK93 is a phosphatidylinositol 4-kinase IIIβ (PI4K-IIIβ) inhibitor; and pleconaril, pirodavir, and Win 56291 are capsid inhibitors.
Cells.
The following cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA): H1-HeLa (CRL-1958), DLD-1 (CCL-221), LS-174T (CL-188), BEAS2B (CRL-9609), NCI-H522 (CRL-5810), NCI-H23 (CRL-5800), A549 (CCL-185), MRC5 (CCL-171), and WI-38 (CCL-75). H1-HeLa, A549, NCI-H23, NCI-H522, and DLD-1 cell lines were maintained in RPMI 1640 medium (Life Technologies, Carlsbad, CA). WI-38, MRC5, and LS174T were maintained in Eagle's minimum essential medium (EMEM; Life Technologies, Carlsbad, CA). Both RPMI 1640 and EMEM were supplemented with 10% heat-inactivated fetal bovine serum and 100 U/ml penicillin-streptomycin. BEAS2B cells were grown in bronchial epithelial cell growth medium (BEGM) (Lonza, Walkersville, MD) in flasks coated with 0.01 mg/ml fibronectin, 0.03 mg/ml collagen I, and 0.01 mg/ml bovine serum albumin (BSA). ALI-differentiated HAE cultures (EpiAirway system; donors 9831 and TBE-21) and media were obtained from MatTek (Ashland, MA) and maintained at 37°C with 5% CO2. Upon delivery, HAE transwells were transferred into 6-well plates containing 2 ml of fresh medium and incubated at 37°C. The viability of the HAE cultures was evaluated by visual observation of ciliary movement by microscopy. Small airway epithelial cell (SAEC) and normal human bronchial epithelial (NHBE) primary cultures were obtained from Lonza (Walkersville, MD) and maintained in small airway epithelial cell growth medium (SAGM) and BEGM, respectively.
Construction of expression plasmids for HRV-C replicons and infectious virus.
Subgenomic replicon DNA constructs for HRV-C genotypes HRVc24, HRVc25, HRVc15, and HRVc11 were built synthetically by DNA2.0 (Menlo Park, CA) based upon publically available sequence data. The constructs contain a T7 promoter with the 5′ end of the viral genomes cloned at the +3 transcriptional start site. Downstream of the 3′ NCR for each construct, a 50-bp poly(A) tract was added, followed by a MluI site for linearization. The cis replication element (CRE) found in the VP2 coding sequence was cis complemented by moving the CRE along with 200 bp of flanking sequence between the stop codon at the end of polymerase 3D (3Dpol) and the 3′ NCR. The location of the CRE in the picornavirus genomes is variable and is positionally independent (40). The CRE hairpin is essential for replication and serves as a binding site for the 3CD protein, which catalyzes the uridylylation of the VPg protein, which serves as the primer for genomic RNA synthesis (41–45). The P1 capsid coding (VPO-VPI) sequence was replaced with the human codon optimized coding sequence for Renilla luciferase fused to the final 21 bp of VP1, representing the N-terminal half of the 2A protease cleavage site. The native 5′ NCR sequences containing the cloverleaf RNA structure and IRES elements were retained. Following electroporation of in vitro-transcribed RNA, Renilla luciferase was translated as a fusion protein with the HRV polyprotein and released by the cis cleavage of the 2A protease. A 3Dpol replication-deficient variant of the HRVc15 replicon containing an asparagine-to-alanine substitution at amino acid position 295 (N295A) was built to control for the baseline luciferase expression and RNA levels of electroporated replicon RNA. The N295A variant was introduced by site-directed mutagenesis of positions 4985 and 4986, converting the dinucleotide AA to GC in the HRVc15 replicon using the QuikChange Lightning kit (Stratagene, La Jolla, CA) (5′-TCTGGTATCTGTGGCACAAGTATTTTCGCTACCATGATCAATAATATAATCATTAG-3′ and CTAATGATTATATTATTGATCATGGTAGCGAAAATACTTGTGCCACAGATACCAGA). Full-length reverse genetic constructs for HRVc15, HRVc24, and HRVc11 were also built synthetically by DNA2.0 (Menlo Park, CA), using the same design parameters for the T7 promoter, poly(A) tract, and MluI linearization site.
The HRV16 reverse genetic construct pR16.11 (46) was obtained from ATCC and further optimized by removing the extraneous 19 bp between the poly(A) tract and the SacI linearization site. pR16.11 was digested with AfeI-SacI, and a synthetic fragment containing a complete HRV16 3′ NCR with an MluI site immediately 3′ of a 50-bp poly(A) tract was inserted. To build the HRV16 replicon, the P1 region of pR16.11 was removed by digestion with SalI-ClaI and replaced with a synthetic fragment containing Renilla luciferase using the same design parameters as the HRV-C replicons.
To produce in vitro RNA for electroporation, replicon and full-length constructs were linearized with MluI, ethanol precipitated, and in vitro transcribed using the T7 RiboMax Express large-scale RNA production system (Promega, Madison, WI), with the resulting RNA purified using the MegaClear kit (Life Technologies, Carlsbad, CA).
Replicon assay.
For HRV replicon assays, log-phase H1-HeLa cells were trypsinized, washed twice with 10 ml phosphate-buffered saline (PBS), resuspended at 1 × 107 cells/ml in PBS, and stored on ice. A total of 5 × 106 cells were electroporated (900 V, 25 μF, ∞ resistance) with 1 to 10 μg of in vitro-transcribed RNA and allowed to rest on ice for 10 min. Electroporated cells were then diluted using RPMI 1640 media and seeded in 96-well plates at 4 × 104 cells/well in a 150-μl volume. Serial dilutions of tested compounds were added using an HP D300 digital titration instrument (Hewlett Packard, Corvallis, OR), and the cells were incubated at 33°C for 48 to 72 h. Compounds were tested in triplicate, and 50% effective concentrations (EC50s) represent the mean of multiple (n ≥ 3) experiments. At the time of harvest, medium was removed from the well, and the cells were lysed with 30 μl Renilla lysis buffer; 100 μl/well of Renilla luciferase assay buffer (Promega, Madison, WI) was added, and the plate was immediately read with a luminometer.
The HRVc15 replicon was adapted to the 384-well format for high-throughput screening. Compound titrations at 100 nl/well were spotted in low-volume 3826 plates (Corning, Tewksbury, MA) using the Echo acoustic liquid handler (Labcyte, Sunnyvale CA), and the plates were heat sealed and stored at −20°C until use. On the day of assay, prespotted plates were equilibrated to room temperature, and HeLa cells were trypsinized and electroporated as described above. Electroporated cells were seeded at 2,000 cells/well in 20 μl and incubated at 33°C for 48 h. To measure luciferase activity, Renilla-Glo (16 μl/well; Promega, Madison, WI) was added, the mixture was incubated for 20 min, and luminescence was read by EnVision (PerkinElmer, Waltham MA). Z′ and the signal-to-background ratio were calculated for 12 experiments.
Generation of viral stocks.
RNAs transcribed in vitro from the full-length HRVc15, HRVc11, HRVc24, or HRV16.11 cDNA constructs were electroporated into H1-HeLa cells using the electroporation conditions described above. Electroporated cells were diluted with RPMI 1640 medium to 5 × 106 cells/10 ml, seeded into a T75 flask, and incubated at 33°C for 72 h. Viral supernatants were then collected, centrifuged for 5 min at 1,500 × g to remove cell debris, and stored at −80°C. Cell lines evaluated for permissivity to HRVc15 infection were seeded in 12-well plates and grown to 50 to 75% confluence prior to infection. Cells were incubated for 3 h at 33°C with either 100 μl or 10 μl of HRVc15 (3.8 × 109 RNA copies/ml) or HRV16.11 (1.2 × 1010 RNA copies/ml) viral supernatant. After the 3 h of incubation, the cells were washed with 1 ml PBS and fresh medium with dimethyl sulfoxide (DMSO) or 1 μM rupintrivir was added. Cells were incubated for 5 days at 33°C; total RNA was purified from the cells and the level of HRV replication measured by reverse transcription-quantitative PCR (RT-qPCR).
RNA isolation and RT-qPCR analysis.
Viral RNA was purified using the RNeasy96 kit (Qiagen, Valencia, CA); for supernatant samples carrier RNA (Qiagen, Valencia, CA) was added to the RLT lysis buffer at 10 μg/ml. Viral RNA copy number in purified samples was measured using the One Step SYBR RT-qPCR kit (TaKaRa, Madison, WI) following the manufacturer's protocol using a ViiA 7 real-time PCR instrument (Applied Biosystems, Foster City CA). Viral copy number data were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) RNA levels using a HuGAPDH PCR assay with the TaqMan fast virus one-step master mix (Applied Biosystems, Foster City, CA). The following primers were used for amplification: for HRVc15, 5′-TGGTGAGTTGCTTGCTTTTG-3′ and 5′-ACCAGAGGGCATACCTCCTT-3′; for HRVc11 5′-CCTGGGATACTGTCACTTTCCT-3′ and 5′-ACAGGGACGGATCGAACTTT-3′; and for HRV16 5′-CCCTCTGGTTGTTCTGGAAC-3′ and 5′-GGCTATGGCTTCCATGTCTAG-3′. HRVc15, HRVc11, and HRV16 in vitro-transcribed RNA was quantified by spectrophotometry and used to generate standard curves for quantitation of RT-qPCR data.
Infection of HAE cultures.
Prior to infection, HAE Transwell apical layers were washed twice with 500 μl PBS to remove accumulated mucus and placed in new 6-well plates containing 2 ml of fresh medium and compound. HAE cultures were infected apically for 3 h at 33°C with viral supernatants in a total volume of 100 μl using PBS (3.8 × 107copies); the Transwells were then washed with 500 μl PBS and incubated at 33°C. The apical surface was washed at selected time points with 200 μl PBS, and the wash was retained for further analysis. RNA was then purified from cells or from 100 μl of the apical wash, and HRV copy number was assayed by RT-qPCR. The remaining apical wash was stored at −80°C for use as an inoculum for reinfection. Unless otherwise noted, antiviral evaluations were conducted in triplicate, and EC50s were calculated from multiple (n ≥ 3) experiments.
HRV-C sequences.
HRV-C sequences used to synthesize replicon and full-length reverse genetic constructs were obtained from the GenBank database with the following accession numbers: HRVc24, EF582385; HRVc25, EF582386; HRVc15, GU219984; HRVny074, DQ875932; and HRVc11, EU840952.
RESULTS
Characterization of multiple HRV-C replicons.
Monocistronic subgenomic HRV replicons for species A and B have been previously developed (47, 48) and were used as a design template for the HRV-C replicons. HRV-C genotypes were selected based upon genetic diversity of the publically available full-length sequences. Putative CRE sequences in VP2 were identified by sequence analysis and cloned into the 3′ NCR (42). The capsid coding region for each construct was replaced with a human-codon-optimized sequence for Renilla luciferase while retaining the 2A cleavage site (Fig. 2). The replicon genes are expressed as a single open reading frame (ORF) with Renilla luciferase cotranslated as part of the polyprotein and released by 2A-mediated proteolysis. The resulting luciferase protein contains 7 amino acids at the C terminus corresponding to the N-terminal half of the 2A cleavage site and retains enzymatic activity. In vitro-transcribed RNA from each of the replicon constructs (HRVc15, HRVc11, HRVc24, HRVc25, and HRV16) was electroporated into H1-HeLa cells (5 × 106 cells) and incubated at 33°C. Renilla luciferase expression was quantified as a marker of the replication activity of the HRV replicons. For each replicon, the incubation time (24 to 72 h) and amount of input RNA (0.1 to 10 μg) were varied to optimize replication-dependent luciferase signal (data not shown). The 3C protease inhibitor rupintrivir (49–51) and the nucleoside analog MK-0608 (52), a putative 3D polymerase inhibitor, were used to control for luciferase background signal emanating from the first round of the translation of the electroporated RNA. The replicons exhibited optimal activity and signal-to-background ratios at 1 to 10 μg input RNA at 48 h postelectroporation (Fig. 3).
FIG 2.

Diagrams for HRV replicon constructs. Replicons with identical gene arrangements were built for HRV-C genotypes HRVc15, HRVc11, HRVc24, and HRVc25 and for the species A genotype HRV16. Full-length constructs were made using the coding sequences for HRVc15 and HRV16. All constructs contain a T7 promoter at the 5′ end for in vitro transcription and an ∼50-bp poly(A) tract upstream of MluI site following the 3′ NCR. For the HRV-C replicons, the cis replication element (CRE) normally found in VP2 was moved to the 3′ NCR. HRVc15-RepMut, an HRVc15 replicon containing the 3Dpol N295A substitution, was constructed to assess the replicon baseline luciferase expression and RNA levels.
FIG 3.
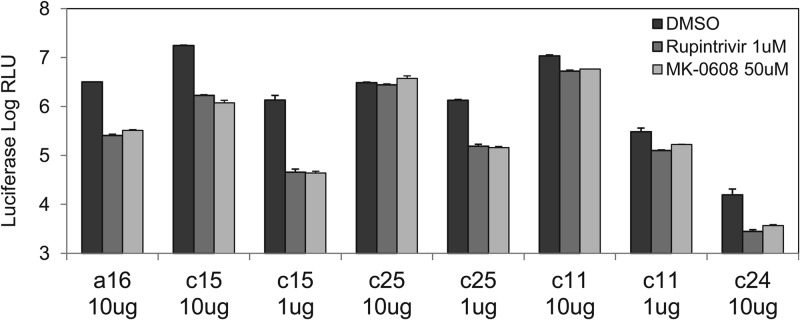
Replication of HRV-C replicons. H1-HeLa cells were electroporated with 10 μg or 1 μg of in vitro-transcribed RNA from subgenomic replicon constructs for HRV-C genotypes c15, c24, c25, and c11 and for the species A virus HRV16. Cells were treated with DMSO or HRV inhibitors at the concentrations shown, incubated at 33°C for 48 h, and assayed for Renilla luciferase activity. The 1-μg samples for the HRVc24 replicon were consistently at background levels and are therefore not included in the figure. Error bars represent the standard deviations of the means from triplicate tests.
The HRVc15 and HRV16 replicons showed robust luciferase expression at all RNA concentrations tested and could be effectively inhibited by rupintrivir and MK-0608 with 5- to 20-fold suppression of the luciferase signal in the presence of the inhibitors (Fig. 3). The HRVc25 replicon RNA electroporated at 10 μg/5 × 106 cells showed luciferase expression equivalent to that of the HRVc15 replicon; however, addition of the inhibitors had no effect on luciferase activity. Reducing the RNA input to 1 μg/5 × 106 cells did not affect the luciferase signal for the HRVc25 replicon but, importantly, sensitized the replicon to both rupintrivir and MK-0608. The HRVc11 replicon also demonstrated robust baseline luciferase expression; however, it exhibited a limited inhibition response to rupintrivir and MK-0608 (signal-to-background ratio, <3) at both input RNA concentrations. We attempted to further optimize the HRVc11 replicon by varying input RNA (10, 3, 1, 0.3, and 0.1 μg) and incubation time (24 to 120 h) with no success (data not shown). The HRVc24 replicon demonstrated a relatively low luciferase signal even at the largest amount of electroporated RNA but retained sensitivity to the inhibitors. We hypothesize that this lower baseline activity may be due to attenuated translation initiation from the HRVc24 IRES.
To confirm that the time-dependent increase in the luciferase activity of the replicon constructs correlated with the subgenomic RNA replication, the RNA was directly quantified by RT-qPCR against a standard curve generated using known quantities of in vitro transcribed HRVc15 RNA. To measure baseline RNA and luciferase levels originating directly from the RNA electroporation input, a replication-deficient HRVc15 replicon containing a N295A substitution in the 3Dpol that impairs its enzymatic function was constructed (53). Following the electroporation of HRVc15 replicon RNA, Renilla luciferase expression and RNA copy number increased over time and were inhibited 900-fold with rupintrivir (Fig. 4). Furthermore, luciferase expression and RNA copy number in cells electroporated with the N295A mutant HRVc15 replicon remained constant over time and showed RNA and luciferase levels similar to those of the wild-type replicon treated with rupintrivir, confirming that the increases in both luciferase signal and replicon RNA levels are driven by a bona fide replication of subgenomic RNA. Identical experiments conducted with the HRVc25 replicon demonstrated the same correlation between the kinetics of the luciferase activity and the RNA level (data not shown). It should be noted that electroporation of replicon RNA resulted in morphological changes (cell rounding) in approximately 50 to 70% of the cells by 48 to 72 h postelectroporation. The effect appeared to be caused by the initial RNA electroporation combined with culture at 33°C and was not the result of high levels of replication, since the observed phenotypes were comparable for the HRVc15 and N295A replicons (data not shown).
FIG 4.

Characterization of the HRVc15 replicon replication kinetics. H1-HeLa cells were electroporated with 10 μg in vitro-transcribed RNA from HRVc15 wild-type or 3Dpol N295A replicons. The HRVc15 wild-type replicon was treated with DMSO or 1 μM rupintrivir. The cells were incubated at 33°C and assayed for replicon RNA levels by RT-qPCR (A) or Renilla luciferase activity (B). Error bars represent the standard deviations of the means from triplicate tests.
Using the established assays, dose-dependent antiviral activity was determined for rupintrivir and MK-0608 using the luciferase and RT-qPCR assays. Notably, the EC50s calculated with either Renilla luciferase or RT-qPCR readouts gave comparable results for the two tested inhibitors (Fig. 5). The EC50s for MK-0608 were 9.1 and 2.3 μM, while the EC50s for rupintrivir were 58 and 29 nM using luciferase and RT-qPCR as the viral replication readout, respectively.
FIG 5.

Inhibition of HRVc15-replicon in the presence of rupintrivir and MK-0608. H1-HeLa cells were electroporated with 10 μg in vitro-transcribed RNA including the HRVc15 replicon and treated with serially diluted concentrations of rupintrivir or MK-0608. The cells were incubated at 33°C for 48 h and assayed for Renilla luciferase activity and replicon RNA levels by RT-qPCR. Error bars represent the standard deviations of the means from triplicate tests.
Inhibition of multiple HRV-C replicons by antivirals.
To determine the breadth of the inhibition across multiple replicons derived from diverse HRV-C strains, the dose-dependent antiviral activities of MK-0608, rupintrivir, PIK93, and pleconaril were measured against HRVc15, HRVc11, HRVc24, HRVc25, and HRV16 replicons in a 96-well format assay. While the first three compounds have been shown to inhibit virus replication in host cells, pleconaril interferes with virus uncoating by binding to the virus capsid during entry and inhibiting capsid disassembly (33, 54–57). The three replication inhibitors tested were active against both HRV-A and HRV-C replicons as well as against HRV-A (Table 1). Each of these three inhibitors showed consistent antiviral activity across all assays. In contrast, pleconaril did not inhibit any of the HRV replicons but, as expected, was active against infectious HRV-A, which requires functional capsid protein for replication. The HRVc11 replicon was included in the screening despite its poor signal-to-background ratio (2- to 3-fold) since it maintained a Z′ value of >0.5.
TABLE 1.
Activities of known HRV inhibitors against subgenomic HRV repliconsa
| Antiviral | EC50 (nM) |
CC50 (nM) | |||||
|---|---|---|---|---|---|---|---|
| HRVc15 | HRVc11 | HRVc25 | HRVc24 | HRVA16 replicon | HRVA16 virus | ||
| Rupintrivir | 56 ± 30 | 44 ± 9.9 | 37 ± 37 | 9.2 ± 5 | 2.8 ± 1 | 57 ± 18 | >100,000 |
| MK-0608 | 5,803 ± 1,876 | 11,931 ± 4,038 | 10,105 ± 3,436 | 5,123 ± 5,063 | 3,376 ± 2,019 | 12,020 ± 3,953 | >100,000 |
| PIK93 | 285 ± 258 | 90 ± 10 | 57 ± 33 | 75 ± 9 | 172 ± 66 | 574 ± 115 | >10,000 |
| Pleconaril | >10,000 | >10,000 | >10,000 | >10,000 | >10,000 | 722 ± 291 | >10,000 |
EC50s are means and standard deviations from multiple experiments (n ≥ 3). Compound activity against HRV16 virus (EC50s) and cytotoxicity (CC50s) were determined in HeLa cells with a 5-day incubation.
Characterization of HRV-C replication in HAE culture.
The replicon assays represent useful antiviral screening tools for identifying inhibitors of HRV-C replication but are not suitable for evaluating compounds that inhibit infection by replication-independent mechanisms. Evaluating compounds effecting virion entry, packaging, or egress would require an assay system capable of supporting multicycle HRV-C infection. To this end, we independently developed and characterized a full-length infectious HRV-C assay using air-liquid interface-differentiated bronchial-tracheal epithelial (HAE) cultures. Recently, two other groups published reports describing similar assay systems for HRV-C replication (38, 39). Viral stocks of HRVc15, HRVc11, HRVc24, and HRV16 generated by the transfection of H1-HeLa cells with in vitro-transcribed RNA were tested in a selected set of primary cells and cell lines to determine their permissivity to HRV-C infection (see Fig. S1 in the supplemental material). Viral replication kinetics was measured and found to be consistent with multicycle virus replication. HAEs infected with HRVc15, HRVc11, or HRV16 stocks shed newly synthesized virus progeny into the apical compartment of the differentiated culture as measured by RT-qPCR, with a peak viral titer being observed between 48 and 72 h after infection (Fig. 6). In contrast, undifferentiated normal human bronchial epithelial cells (NHBE) from the same donor were unable to support HRVc15 infection but remained permissive to HRV16 (data not shown). The increase in viral copy number for HRVc15 in differentiated HAE cultures reached 30,000-fold relative to the first time point measured. Addition of 1 μM rupintrivir inhibited the HRVc15 replication approximately 800-fold, while the corresponding HRVc15 replicon inhibited it only 5- to 20-fold, suggesting that the full-length HRVc15 HAE infection is multicycle compared to that with the replicon in which replication is restricted to single cells. Similarly, HRVc11 HAE infections demonstrated a 106,000-fold increase in apical copy number over time with rupintrivir treatment at 48 h, resulting in a 1,946-fold inhibition. As a comparison, the HRVc24 replicon showed weak baseline expression and replication of the full-length HRVc24 in HAE culture could not be detected (data not shown). Secondary infection of HAE cultures using apical wash material containing 5.4 × 106 copies of HRVc15 produced in the primary HAE infection showed viral replication kinetics similar to that seen during the primary infection of HAE cultures with supernatants from electroporated HeLa cells (see Fig. S2 in the supplemental material). In contrast, the infection of NHBE and SAEC cells with HRVc15 produced in HAE cultures did not result in active virus replication (data not shown). Infection of HAE cultures derived from a second healthy donor (donor TBE-21) with HRVc15 resulted in productive infections with similar virus yields and replication kinetics compared to the HAE cultures established from donor 9831. Importantly, the replication of HRVc15 in the HAE cultures can be blocked by rupintrivir, MK-0608, and PIK93 treatment (see below). These data confirm that differentiated HAE cell cultures are permissive to HRV-C infection in a donor-independent manner and emphasize the in vitro differentiation state as an important factor for permissive HRV-C infection.
FIG 6.

Growth curves of HRVc15 (A), HRV16 (B), and HRVc11 (C) in the HAE cultures. HAE cells in a 24-well format were infected with HRVc15 or HRV16 produced in H1-HeLa cells at 3.8 × 107 copies of RNA/well in the presence of 0.5% DMSO or 1 μM rupintrivir, incubated for 3 h, and washed to remove the virus inoculum. Cultures were subsequently incubated at 33°C. Virus production into the HAE apical compartment was determined by RT-qPCR. Error bars represent the standard deviations of the means from triplicate tests.
Inhibition of infectious HRV-C by antivirals.
We evaluated the dose-dependent antiviral activities of rupintrivir, MK-0608, PIK93, and pleconaril in HAE cultures infected with several HRV-C strains, including HRVc15, HRVc11, and HRV16. In parallel, the EC50s of tested compounds were also determined in HeLa cells against their respective replicons, as well as HRV16. The antiviral activities of rupintrivir, MK-0608, and PIK93 in the HAE culture assay were comparable to those observed in the HRV replicon assays in HeLa cells (Table 2). Interestingly, the HRVc15 replicon was 20-fold less sensitive to rupintrivir treatment than the HRV16 replicon. As expected, pleconaril was inactive in all HRV replicon assays. Surprisingly, HRVc15 and HRVc11 infections in HAE cultures were also insensitive to inhibition by pleconaril, while this compound exhibited the expected activity against HRV16 in both HAE and HeLa cell cultures. To further characterize this observation, the antiviral activity of two other capsid-binding inhibitors, Win 56291 and pirodavir (34, 58–61), were also tested in the HAE infection assay. While both compounds retained antiviral activity against HRV16, pirodavir was inactive and Win 56291 was 88-fold and 110-fold less active against HRVc15 and HRVc11, respectively, than HRV16 (Table 2). In addition, pleconaril and Win 56291 had no effect on the infectivity of HRVc15 grown in the presence of compounds (data not shown). This implies that the capsid inhibitors were unable to bind or inhibit newly formed HRV-C capsid protomers.
TABLE 2.
Antiviral activity of HRV inhibitors in HAE culturesa
| Antiviral | EC50 (nM) |
|||
|---|---|---|---|---|
| HRVc15 (HAE culture) | HRVc11 (HAE culture) | HRV16 |
||
| HAE culture | HeLa culture | |||
| Rupintrivir | 58 ± 39 | 56 ± 27 | 22 ± 16 | 57 ± 18 |
| MK-0608 | 2,416 ± 556 | 4,176 ± 519 | 1,847 ± 479 | 12,020 ± 3,953 |
| PIK93 | 225 ± 103 | 342 ± 81 | 127 ± 50 | 574 ± 115 |
| Pleconaril | >10,000 | >10,000 | 899 ± 465 | 722 ± 291 |
| Win 56291 | 4,762 ± 3,296 | 5,107 ± 1,538 | 54 ± 18 | 160 ± 52 |
| Pirodavir | >20,000 | >10,000 | 402 ± 103 | ND |
Values are means and standard deviations from multiple (n ≥ 3) experiments, with the exception of those for Win 56291 and pirodavir, which were tested in 2 experiments.
Development of a high-throughput HRV-C inhibitor screening assay.
The identification and optimization of novel inhibitors that can be safely used to treat HRV-C infection in the clinic require access to a robust and reproducible assay for HRV-C replication suitable for use in a high-throughput format. Since the HAE infection technique is laborious and can be used only in a very restricted low-throughput format, we adapted the HRVc15 replicon assay to a 384-well-plate format by reoptimizing the cell number, plate type, RNA input, incubation time, and luciferase reagent. A 48-hour assay in the optimized 384-well format displayed a robust and reproducible luciferase signal with a Z′ value of 0.61 ± 0.16, and signal-to-background ratio of 13.2 ± 6.35, for mock-treated control samples (0% inhibition) versus samples treated with 1 μM rupintrivir (100% inhibition) (n = 12). These characteristics support the use of the HRVc15 replicon as a high-throughput tool to facilitate the identification of novel panserotype HRV inhibitors that also target HRV-C.
DISCUSSION
Design and implementation of assay systems to measure HRV-C replication will facilitate the discovery and development of panserotype antiviral drugs to treat HRV infection. Unlike HRV-A and -B, HRV-C does not replicate in the conventional transformed or primary cell lines (HeLa, WI-38, MRC5, NHBE, or SAEC) used to study HRV type A and B biology. In this study, assays using transient subgenomic replicon and infectious virus replication were developed for HRV-C and used to evaluate the antiviral activity of compounds known to inhibit rhinovirus replication. The replicon systems described here are amenable to high-throughput screening and represent novel tools to facilitate the characterization of HRV-C replication and to quantify antiviral activity of potential inhibitors.
HRV-C replicons were designed based upon data from previous studies using HRV-A and B subgenomic replicons that were constructed to study the function of the CRE in uridylylation of VPg for priming of RNA synthesis (40, 43, 47, 48). Expression of luciferase from the HRV subgenomic replicon constructs correlated with viral RNA levels measured by qRT-PCR and was used to quantify viral RNA replication. HRV replication causes cytopathic effects in most cell types, and several viral proteins are known to be cytotoxic, making the prospect of developing stable HRV replicon cell lines more challenging (11–14, 16). Stable replicon systems have been developed for antiviral testing for other positive-strand RNA viruses, such as HCV and dengue virus (62, 63). These systems are easier to manipulate and have less variability than transient-replication assays. Creating a stable replicon for HRV would likely require the removal of the 2A protease which is known to be cytotoxic. However, the removal of the 2A protease function would prevent virus-mediated inhibition of host cell cap-dependent translation and would significantly reduce IRES-mediated translation of the viral genome, impairing replicon replication efficiency. Furthermore, a stable HRV replicon would likely be bicistronic, expressing separate marker and genomic ORFs, and would require selection of a cell line that is resistant to the toxic effects of 3C and 3CD proteases required for replication. The advantage of the transient-replicon assays is that the endogenous HRV IRES elements are sufficient to drive expression of reporter and viral genes and do not require multiple rounds of host cell and replicon selection.
One of the inherent limitations of the replicon assay is that the complete viral life cycle is not articulated and thus cannot be used to evaluate compounds affecting virion entry, packaging, or virus release. Following the initial discovery of HRV-C, suitable cell lines supporting high levels of virus replication were not readily available. The first report of in vitro HRV-C replication was in primary sinus-mucosal tissue explants obtained from nasal biopsies of individuals with chronic sinusitis (7). While this system supports HRV-C replication, the explant model has significant limitations due to low assay throughput and tissue availability. Recently, human airway epithelial (HAE) cultures were shown to support HRV-C replication in vitro and were used by two independent groups to grow clinical HRV-C isolates and HRV-C produced in vitro from cells transfected with genomic HRV-C RNA (38, 39). HAE cultures are composed of polarized, fully differentiated airway epithelial cells with active cilia on the apical surface. The cultures also contain goblet cells that secrete mucus on the apical side of the monolayer, forming a natural tissue barrier resembling respiratory epithelium (64). We have independently developed and validated the HRV-C infection system in HAE cultures and utilized it for antiviral testing of known HRV inhibitors. Consistent with data from the replicon assays in HeLa cells, infection of HAE cultures with full-length HRVc15 or HRVc11 was inhibited by rupintrivir and MK-0608. These inhibitors target the 3C protease and 3D polymerase, respectively, and exhibit panserotype antiviral activity with rupintrivir EC50s consistent with antiviral activity previously observed for HRV-A and -B (49). Interestingly, the capsid-binding compounds pleconaril and pirodavir were not active against HRVc15 in the HAE model but were fully active against HRV16, with EC50s comparable to those obtained in HeLa cells. Furthermore, an analog of pleconaril, Win 56291, was more than 100-fold less active against HRVc15 and HRVc11 than HRV16 infection. In addition, HRVc15 grown in the presence of either pleconaril or Win 56291 retained infectivity in subsequent reinfection experiments. Together, these results suggest that HRV-C may be less susceptible to the current class of capsid-binding compounds and are consistent with the hypothesis that HRV-C utilizes a receptor pathway for viral entry that is distinct from that used by other HRV groups (7, 39).
This observation underscores the challenge of developing broadly active HRV capsid inhibitors, since the P1 capsid region exhibits a much higher degree of sequence diversity among HRV groups than the more conserved viral proteins that support intracellular RNA replication (56). Further studies using a larger set of HRV-C serotypes should be conducted to better understand the susceptibility of the whole HRV-C group to the capsid inhibitor class (pleconaril, pirodavir, WIN compounds, and vapendavir) (54–57). In contrast to the P1 capsid region, the active sites of the 3C protease and 3D polymerase are highly conserved throughout the Picornaviridae (49, 53). This is reflected in the panserotypic activity of rupintrivir and MK-0608 in both the replicon and HAE assays and suggests that the 3C protease and likely the 3D polymerase are viable targets for the development of panserotype HRV inhibitors.
The use of the HAE model for HRV-C antiviral screening has several important limitations. Culture of differentiated HAEs is laborious, and the infection and sample processing are low throughput, requiring multiple washes to remove the mucous layer to establish infection and RNA purification to quantify virus yield. Our attempts to transfer the assay from a 24-well to a 96-well format have been unsuccessful due to a substantial well-to-well variability in infection. This is likely due to the heterogeneity of the degree of differentiation and variable number of permissive cell types in each sample well. Due to these constraints, this assay is not suitable for using in a higher-throughput format to identify novel HRV-C inhibitors. In contrast, the HRV-C replicon systems are fully amenable to higher throughput, including both a 96-well and a 384-well format.
In summary, we have developed and fully validated a series of novel assays using transient subgenomic-replicon and infectious-virus replication that can be effectively used to screen and characterize the antiviral activities of HRV inhibitors. Results of this study represent the first comprehensive and quantitative profiling of multiple known classes of antivirals against HRV-C, and these assays represent valuable tools for the identification and characterization of novel broad-spectrum inhibitors of HRV replication.
Supplementary Material
Footnotes
Published ahead of print 23 December 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.01746-13.
REFERENCES
- 1.Jacobs SE, Lamson DM, St George K, Walsh TJ. 2013. Human rhinoviruses. Clin. Microbiol. Rev. 26:135–162. 10.1128/CMR.00077-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Palmenberg AC, Spiro D, Kuzmickas R, Wang S, Djikeng A, Rathe JA, Fraser-Liggett CM, Liggett SB. 2009. Sequencing and analyses of all known human rhinovirus genomes reveal structure and evolution. Science 324:55–59. 10.1126/science.1165557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McErlean P, Shackelton LA, Andrews E, Webster DR, Lambert SB, Nissen MD, Sloots TP, Mackay IM. 2008. Distinguishing molecular features and clinical characteristics of a putative new rhinovirus species, human rhinovirus C (HRV C). PLoS One 3:e1847. 10.1371/journal.pone.0001847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McIntyre CL, McWilliam Leitch EC, Savolainen-Kopra C, Hovi T, Simmonds P. 2010. Analysis of genetic diversity and sites of recombination in human rhinovirus species C. J. Virol. 84:10297–10310. 10.1128/JVI.00962-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wisdom A, Leitch EC, Gaunt E, Harvala H, Simmonds P. 2009. Screening respiratory samples for detection of human rhinoviruses (HRVs) and enteroviruses: comprehensive VP4-VP2 typing reveals high incidence and genetic diversity of HRV species C. J. Clin. Microbiol. 47:3958–3967. 10.1128/JCM.00993-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greve JM, Davis G, Meyer AM, Forte CP, Yost SC, Marlor CW, Kamarck ME, McClelland A. 1989. The major human rhinovirus receptor is ICAM-1. Cell 56:839–847. 10.1016/0092-8674(89)90688-0 [DOI] [PubMed] [Google Scholar]
- 7.Bochkov YA, Palmenberg AC, Lee WM, Rathe JA, Amineva SP, Sun X, Pasic TR, Jarjour NN, Liggett SB, Gern JE. 2011. Molecular modeling, organ culture and reverse genetics for a newly identified human rhinovirus C. Nat. Med. 17:627–632. 10.1038/nm.2358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hofer F, Gruenberger M, Kowalski H, Machat H, Huettinger M, Kuechler E, Blaas D. 1994. Members of the low density lipoprotein receptor family mediate cell entry of a minor-group common cold virus. Proc. Natl. Acad. Sci. U. S. A. 91:1839–1842. 10.1073/pnas.91.5.1839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith TJ, Kremer MJ, Luo M, Vriend G, Arnold E, Kamer G, Rossmann MG, McKinlay MA, Diana GD, Otto MJ. 1986. The site of attachment in human rhinovirus 14 for antiviral agents that inhibit uncoating. Science 233:1286–1293. 10.1126/science.3018924 [DOI] [PubMed] [Google Scholar]
- 10.Fuchs R, Blaas D. 2010. Uncoating of human rhinoviruses. Rev. Med. Virol. 20:281–297. 10.1002/rmv.654 [DOI] [PubMed] [Google Scholar]
- 11.de Breyne S, Bonderoff JM, Chumakov KM, Lloyd RE, Hellen CU. 2008. Cleavage of eukaryotic initiation factor eIF5B by enterovirus 3C proteases. Virology 378:118–122. 10.1016/j.virol.2008.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dougherty JD, White JP, Lloyd RE. 2011. Poliovirus-mediated disruption of cytoplasmic processing bodies. J. Virol. 85:64–75. 10.1128/JVI.01657-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuyumcu-Martinez NM, Van Eden ME, Younan P, Lloyd RE. 2004. Cleavage of poly(A)-binding protein by poliovirus 3C protease inhibits host cell translation: a novel mechanism for host translation shutoff. Mol. Cell. Biol. 24:1779–1790. 10.1128/MCB.24.4.1779-1790.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghildyal R, Jordan B, Li D, Dagher H, Bardin PG, Gern JE, Jans DA. 2009. Rhinovirus 3C protease can localize in the nucleus and alter active and passive nucleocytoplasmic transport. J. Virol. 83:7349–7352. 10.1128/JVI.01748-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arita M, Kojima H, Nagano T, Okabe T, Wakita T, Shimizu H. 2011. Phosphatidylinositol 4-kinase III beta is a target of enviroxime-like compounds for antipoliovirus activity. J. Virol. 85:2364–2372. 10.1128/JVI.02249-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greninger AL, Knudsen GM, Betegon M, Burlingame AL, Derisi JL. 2012. The 3A protein from multiple picornaviruses utilizes the Golgi adaptor protein ACBD3 to recruit PI4KIIIbeta. J. Virol. 86:3605–3616. 10.1128/JVI.06778-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Makela MJ, Puhakka T, Ruuskanen O, Leinonen M, Saikku P, Kimpimaki M, Blomqvist S, Hyypia T, Arstila P. 1998. Viruses and bacteria in the etiology of the common cold. J. Clin. Microbiol. 36:539–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arruda E, Pitkaranta A, Witek TJ, Jr, Doyle CA, Hayden FG. 1997. Frequency and natural history of rhinovirus infections in adults during autumn. J. Clin. Microbiol. 35:2864–2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gwaltney JM, Jr, Phillips CD, Miller RD, Riker DK. 1994. Computed tomographic study of the common cold. N. Engl. J. Med. 330:25–30. 10.1056/NEJM199401063300105 [DOI] [PubMed] [Google Scholar]
- 20.Winther B. 2011. Rhinovirus infections in the upper airway. Proc. Am. Thorac. Soc. 8:79–89. 10.1513/pats.201006-039RN [DOI] [PubMed] [Google Scholar]
- 21.Winther B, Alper CM, Mandel EM, Doyle WJ, Hendley JO. 2007. Temporal relationships between colds, upper respiratory viruses detected by polymerase chain reaction, and otitis media in young children followed through a typical cold season. Pediatrics 119:1069–1075. 10.1542/peds.2006-3294 [DOI] [PubMed] [Google Scholar]
- 22.Bizzintino J, Lee WM, Laing IA, Vang F, Pappas T, Zhang G, Martin AC, Khoo SK, Cox DW, Geelhoed GC, McMinn PC, Goldblatt J, Gern JE, Le Souef PN. 2011. Association between human rhinovirus C and severity of acute asthma in children. Eur. Respir. J. 37:1037–1042. 10.1183/09031936.00092410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee WM, Lemanske RF, Jr, Evans MD, Vang F, Pappas T, Gangnon R, Jackson DJ, Gern JE. 2012. Human rhinovirus species and season of infection determine illness severity. Am. J. Respir. Crit. Care Med. 186:886–891. 10.1164/rccm.201202-0330OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Message SD, Laza-Stanca V, Mallia P, Parker HL, Zhu J, Kebadze T, Contoli M, Sanderson G, Kon OM, Papi A, Jeffery PK, Stanciu LA, Johnston SL. 2008. Rhinovirus-induced lower respiratory illness is increased in asthma and related to virus load and Th1/2 cytokine and IL-10 production. Proc. Natl. Acad. Sci. U. S. A. 105:13562–13567. 10.1073/pnas.0804181105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tapparel C, Cordey S, Junier T, Farinelli L, Van Belle S, Soccal PM, Aubert JD, Zdobnov E, Kaiser L. 2011. Rhinovirus genome variation during chronic upper and lower respiratory tract infections. PLoS One 6:e21163. 10.1371/journal.pone.0021163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tapparel C, L'Huillier AG, Rougemont AL, Beghetti M, Barazzone-Argiroffo C, Kaiser L. 2009. Pneumonia and pericarditis in a child with HRV-C infection: a case report. J. Clin. Virol. 45:157–160. 10.1016/j.jcv.2009.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arden KE, Faux CE, O'Neill NT, McErlean P, Nitsche A, Lambert SB, Nissen MD, Sloots TP, Mackay IM. 2010. Molecular characterization and distinguishing features of a novel human rhinovirus (HRV) C, HRVC-QCE, detected in children with fever, cough and wheeze during 2003. J. Clin. Virol. 47:219–223. 10.1016/j.jcv.2010.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Busse WW, Lemanske RF, Gern JE. 2010. Role of viral respiratory infections in asthma and asthma exacerbations. Lancet 376:826–834. 10.1016/S0140-6736(10)61380-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fujitsuka A, Tsukagoshi H, Arakawa M, Goto-Sugai K, Ryo A, Okayama Y, Mizuta K, Nishina A, Yoshizumi M, Kaburagi Y, Noda M, Tashiro M, Okabe N, Mori M, Yokota S, Kimura H. 2011. A molecular epidemiological study of respiratory viruses detected in Japanese children with acute wheezing illness. BMC Infect. Dis. 11:168. 10.1186/1471-2334-11-168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hayden FG. 2004. Rhinovirus and the lower respiratory tract. Rev. Med. Virol. 14:17–31. 10.1002/rmv.406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lau SK, Yip CC, Tsoi HW, Lee RA, So LY, Lau YL, Chan KH, Woo PC, Yuen KY. 2007. Clinical features and complete genome characterization of a distinct human rhinovirus (HRV) genetic cluster, probably representing a previously undetected HRV species, HRV-C, associated with acute respiratory illness in children. J. Clin. Microbiol. 45:3655–3664. 10.1128/JCM.01254-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jartti T, Gern JE. 2011. Rhinovirus-associated wheeze during infancy and asthma development. Curr. Respir. Med. Rev. 7:160–166. 10.2174/157339811795589423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Palma AM, Vliegen I, De Clercq E, Neyts J. 2008. Selective inhibitors of picornavirus replication. Med. Res. Rev. 28:823–884. 10.1002/med.20125 [DOI] [PubMed] [Google Scholar]
- 34.Hayden FG, Andries K, Janssen PA. 1992. Safety and efficacy of intranasal pirodavir (R77975) in experimental rhinovirus infection. Antimicrob. Agents Chemother. 36:727–732. 10.1128/AAC.36.4.727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hayden FG, Herrington DT, Coats TL, Kim K, Cooper EC, Villano SA, Liu S, Hudson S, Pevear DC, Collett M, McKinlay M. 2003. Efficacy and safety of oral pleconaril for treatment of colds due to picornaviruses in adults: results of 2 double-blind, randomized, placebo-controlled trials. Clin. Infect. Dis. 36:1523–1532. 10.1086/375069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hayden FG, Turner RB, Gwaltney JM, Chi-Burris K, Gersten M, Hsyu P, Patick AK, Smith GJ, III, Zalman LS. 2003. Phase II, randomized, double-blind, placebo-controlled studies of ruprintrivir [sic] nasal spray 2-percent suspension for prevention and treatment of experimentally induced rhinovirus colds in healthy volunteers. Antimicrob. Agents Chemother. 47:3907–3916. 10.1128/AAC.47.12.3907-3916.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Turner RB, Wecker MT, Pohl G, Witek TJ, McNally E, St George R, Winther B, Hayden FG. 1999. Efficacy of tremacamra, a soluble intercellular adhesion molecule 1, for experimental rhinovirus infection: a randomized clinical trial. JAMA 281:1797–1804. 10.1001/jama.281.19.1797 [DOI] [PubMed] [Google Scholar]
- 38.Ashraf S, Brockman-Schneider R, Bochkov YA, Pasic TR, Gern JE. 2013. Biological characteristics and propagation of human rhinovirus-C in differentiated sinus epithelial cells. Virology 436:143–149. 10.1016/j.virol.2012.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hao W, Bernard K, Patel N, Ulbrandt N, Feng H, Svabek C, Wilson S, Stracener C, Wang K, Suzich J, Blair W, Zhu Q. 2012. Infection and propagation of human rhinovirus C in human airway epithelial cells. J. Virol. 86:13524–13532. 10.1128/JVI.02094-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McKnight KL. 2003. The human rhinovirus internal cis-acting replication element (cre) exhibits disparate properties among serotypes. Arch. Virol. 148:2397–2418. 10.1007/s00705-003-0177-7 [DOI] [PubMed] [Google Scholar]
- 41.Cheney IW, Naim S, Shim JH, Reinhardt M, Pai B, Wu JZ, Hong Z, Zhong W. 2003. Viability of poliovirus/rhinovirus VPg chimeric viruses and identification of an amino acid residue in the VPg gene critical for viral RNA replication. J. Virol. 77:7434–7443. 10.1128/JVI.77.13.7434-7443.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cordey S, Gerlach D, Junier T, Zdobnov EM, Kaiser L, Tapparel C. 2008. The cis-acting replication elements define human enterovirus and rhinovirus species. RNA 14:1568–1578. 10.1261/rna.1031408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Steil BP, Barton DJ. 2009. Cis-active RNA elements (CREs) and picornavirus RNA replication. Virus Res. 139:240–252. 10.1016/j.virusres.2008.07.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang Y, Rijnbrand R, McKnight KL, Wimmer E, Paul A, Martin A, Lemon SM. 2002. Sequence requirements for viral RNA replication and VPg uridylylation directed by the internal cis-acting replication element (cre) of human rhinovirus type 14. J. Virol. 76:7485–7494. 10.1128/JVI.76.15.7485-7494.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang Y, Rijnbrand R, Watowich S, Lemon SM. 2004. Genetic evidence for an interaction between a picornaviral cis-acting RNA replication element and 3CD protein. J. Biol. Chem. 279:12659–12667. 10.1074/jbc.M312992200 [DOI] [PubMed] [Google Scholar]
- 46.Lee WM, Wang W. 2003. Human rhinovirus type 16: mutant V1210A requires capsid-binding drug for assembly of pentamers to form virions during morphogenesis. J. Virol. 77:6235–6244. 10.1128/JVI.77.11.6235-6244.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McKnight KL, Lemon SM. 1996. Capsid coding sequence is required for efficient replication of human rhinovirus 14 RNA. J. Virol. 70:1941–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McKnight KL, Lemon SM. 1998. The rhinovirus type 14 genome contains an internally located RNA structure that is required for viral replication. RNA 4:1569–1584. 10.1017/S1355838298981006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Binford SL, Maldonado F, Brothers MA, Weady PT, Zalman LS, Meador JW, III, Matthews DA, Patick AK. 2005. Conservation of amino acids in human rhinovirus 3C protease correlates with broad-spectrum antiviral activity of rupintrivir, a novel human rhinovirus 3C protease inhibitor. Antimicrob. Agents Chemother. 49:619–626. 10.1128/AAC.49.2.619-626.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patick AK, Binford SL, Brothers MA, Jackson RL, Ford CE, Diem MD, Maldonado F, Dragovich PS, Zhou R, Prins TJ, Fuhrman SA, Meador JW, Zalman LS, Matthews DA, Worland ST. 1999. In vitro antiviral activity of AG7088, a potent inhibitor of human rhinovirus 3C protease. Antimicrob. Agents Chemother. 43:2444–2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Patick AK, Brothers MA, Maldonado F, Binford S, Maldonado O, Fuhrman S, Petersen A, Smith GJ, III, Zalman LS, Burns-Naas LA, Tran JQ. 2005. In vitro antiviral activity and single-dose pharmacokinetics in humans of a novel, orally bioavailable inhibitor of human rhinovirus 3C protease. Antimicrob. Agents Chemother. 49:2267–2275. 10.1128/AAC.49.6.2267-2275.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Olsen DB, Eldrup AB, Bartholomew L, Bhat B, Bosserman MR, Ceccacci A, Colwell LF, Fay JF, Flores OA, Getty KL, Grobler JA, LaFemina RL, Markel EJ, Migliaccio G, Prhavc M, Stahlhut MW, Tomassini JE, MacCoss M, Hazuda DJ, Carroll SS. 2004. A 7-deaza-adenosine analog is a potent and selective inhibitor of hepatitis C virus replication with excellent pharmacokinetic properties. Antimicrob. Agents Chemother. 48:3944–3953. 10.1128/AAC.48.10.3944-3953.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gong P, Peersen OB. 2010. Structural basis for active site closure by the poliovirus RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 107:22505–22510. 10.1073/pnas.1007626107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kaiser L, Crump CE, Hayden FG. 2000. In vitro activity of pleconaril and AG7088 against selected serotypes and clinical isolates of human rhinoviruses. Antiviral Res. 47:215–220. 10.1016/S0166-3542(00)00106-6 [DOI] [PubMed] [Google Scholar]
- 55.Ledford RM, Collett MS, Pevear DC. 2005. Insights into the genetic basis for natural phenotypic resistance of human rhinoviruses to pleconaril. Antiviral Res. 68:135–138. 10.1016/j.antiviral.2005.08.003 [DOI] [PubMed] [Google Scholar]
- 56.Ledford RM, Patel NR, Demenczuk TM, Watanyar A, Herbertz T, Collett MS, Pevear DC. 2004. VP1 sequencing of all human rhinovirus serotypes: insights into genus phylogeny and susceptibility to antiviral capsid-binding compounds. J. Virol. 78:3663–3674. 10.1128/JVI.78.7.3663-3674.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pevear DC, Hayden FG, Demenczuk TM, Barone LR, McKinlay MA, Collett MS. 2005. Relationship of pleconaril susceptibility and clinical outcomes in treatment of common colds caused by rhinoviruses. Antimicrob. Agents Chemother. 49:4492–4499. 10.1128/AAC.49.11.4492-4499.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Andries K, Dewindt B, Snoeks J, Willebrords R, van Eemeren K, Stokbroekx R, Janssen PA. 1992. In vitro activity of pirodavir (R 77975), a substituted phenoxy-pyridazinamine with broad-spectrum antipicornaviral activity. Antimicrob. Agents Chemother. 36:100–107. 10.1128/AAC.36.1.100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Badger J, Minor I, Kremer MJ, Oliveira MA, Smith TJ, Griffith JP, Guerin DM, Krishnaswamy S, Luo M, Rossmann MG, et al. 1988. Structural analysis of a series of antiviral agents complexed with human rhinovirus 14. Proc. Natl. Acad. Sci. U. S. A. 85:3304–3308. 10.1073/pnas.85.10.3304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim KH, Willingmann P, Gong ZX, Kremer MJ, Chapman MS, Minor I, Oliveira MA, Rossmann MG, Andries K, Diana GD, et al. 1993. A comparison of the anti-rhinoviral drug binding pocket in HRV14 and HRV1A. J. Mol. Biol. 230:206–227. 10.1006/jmbi.1993.1137 [DOI] [PubMed] [Google Scholar]
- 61.Zhao R, Pevear DC, Kremer MJ, Giranda VL, Kofron JA, Kuhn RJ, Rossmann MG. 1996. Human rhinovirus 3 at 3.0 A resolution. Structure 4:1205–1220. 10.1016/S0969-2126(96)00128-1 [DOI] [PubMed] [Google Scholar]
- 62.Uprichard SL. 2010. Hepatitis C virus experimental model systems and antiviral drug research. Virol. Sin. 25:227–245. 10.1007/s12250-010-3134-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ng CY, Gu F, Phong WY, Chen YL, Lim SP, Davidson A, Vasudevan SG. 2007. Construction and characterization of a stable subgenomic dengue virus type 2 replicon system for antiviral compound and siRNA testing. Antiviral Res. 76:222–231. 10.1016/j.antiviral.2007.06.007 [DOI] [PubMed] [Google Scholar]
- 64.Crystal RG, Randell SH, Engelhardt JF, Voynow J, Sunday ME. 2008. Airway epithelial cells: current concepts and challenges. Proc. Am. Thorac. Soc. 5:772–777. 10.1513/pats.200805-041HR [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.