

Abstract
Antimony-based drugs are still the mainstay of chemotherapy against Leishmania infections in many countries where the parasites are endemic. The efficacy of antimonials has been compromised by increasing numbers of resistant infections, the basis of which is not fully understood and likely involves multiple factors. By using a functional cloning strategy, we recently identified a novel antimony resistance marker, ARM58, from the parasite Leishmania braziliensis that protects the parasites against antimony-based antileishmanial compounds. Here we show that the Leishmania infantum homologue also confers resistance against antimony but not against other antileishmanial drugs and that its function depends critically on one of four conserved domains of unknown function. This critical domain requires at least two hydrophobic amino acids and is predicted to form a transmembrane structure. Overexpression of ARM58 in antimony-exposed parasites reduces the intracellular Sb accumulation by over 70%, indicating a role for ARM58 in Sb extrusion pathways, but without involvement of energy-dependent transporter proteins.
INTRODUCTION
Infections with protozoan parasites are among the most important neglected tropical diseases. In particular, this is true for infections with protists from the order Kinetoplastida, which belongs to the Euglenozoa, an early branch of the Eukaryota that is characterized by molecular features such as trans-splicing, RNA editing, and a largely unregulated, polycistronic RNA synthesis (1). The human-pathogenic kinetoplastid protozoa all belong to the family Trypanosomatidae, which includes the genera Trypanosoma and Leishmania. The former cause human and cattle sleeping sickness in Africa as well as Chagas disease in South America. Species of the latter are responsible for the various forms of leishmaniasis, with over 2 million new cases per year in 88 countries (2).
The most severe form of leishmaniasis, visceral leishmaniasis (VL; also called kala-azar), is lethal if left untreated or if the treatment fails. Since no safe and effective vaccine is available, and since the rate of transmission remains high, monitoring and chemotherapy are the sole control measures available.
The most common treatment of leishmaniasis and also the most affordable option are drugs based on pentavalent antimony (SbV). Although they have been in use for over 60 years, their exact mode of action is not clear. What has become clear is that SbV is a prodrug and requires reduction to SbIII (3–5). This reduction may take place inside the host cell, usually antigen-presenting cells such as macrophages and monocytes, where the parasites reside inside the phagosomes. The intracellular parasites, known as amastigotes, can also reduce SbV to SbIII. By contrast, the flagellated, arthropod-borne form, the promastigote, is impervious to SbV, since it cannot reduce the prodrug to SbIII. SbIII is toxic to promastigotes and amastigotes but also to mammalian cells and tissues, thus precluding its direct use as a drug.
Over the last 3 decades, a large percentage (>60%) of Leishmania infections in northern India and adjacent regions have become resistant to SbV-based drugs (6–9). Alternative drugs approved for treatment either are too expensive for the health services or come with severe side effects and risk factors (6, 10–13). Antimony treatment itself is associated with cardiotoxicity, in particular when repeated treatment courses and high doses are required due to primary-treatment failure.
Therefore, efforts have been made to unravel the molecular basis of antimony drug resistance by comparing drug-sensitive and -resistant field isolates and laboratory strains. Features such as increased levels of trypanothione (14) and P-glycoproteins, which can act as extrusion pumps (15, 16), were linked to SbIII resistance. Conversely, reduced levels of aquaglyceroporin, which is involved in Sb uptake, correlate with resistance (17). Another resistance mechanism operates on the host cell level. Leishmania donovani isolates with no intrinsic Sb resistance were shown to induce host cell extrusion pathways (18, 19).
The feature of drug resistance also lends itself very well to unbiased screening strategies such as functional cloning, whereby a genomic DNA library in a shuttle cosmid vector is transfected into drug-sensitive parasites that are then challenged with the drug. Only those parasites carrying cosmids with dominant drug resistance markers will survive the challenge and grow under selection. Recovering and analyzing the cosmids within the surviving parasites will then make it possible to identify resistance marker genes (20). This approach has already been used to identify novel markers of resistance against antimonials and other antileishmanial compounds (21, 22).
By its nature, the functional cloning approach is bound to identify genetic markers with suspected functionality (21, 23, 24) but also genes coding for hypothetical proteins without suspected function and defined functional domains (22, 25). In the latter case, the function of the gene product and its link to drug resistance must be established to understand the role of the marker.
Using a functional complementation screen approach, we recently identified a dominant SbIII resistance marker from the South American species Leishmania braziliensis (42). The gene encodes a 58-kDa protein, ARM58 (antimony resistance marker, 58 kDa), that is unique to the genus Leishmania, confers SbIII resistance to promastigotes and SbV resistance to intracellular amastigotes, and consists of four nonidentical but related domains of unknown function, DUF1935. No orthologous gene was found outside the leishmaniae, but a related gene, encoding ARM58rel, has orthologues in Trypanosoma spp. In order to understand its functionality, we constructed a series of truncated and otherwise mutated variants of Leishmania infantum ARM58 and tested their function. We demonstrate that a small putative transmembrane domain is crucial for function and that ARM58 overexpression is sufficient to lower intracellular antimony levels in SbIII-exposed cells.
MATERIALS AND METHODS
Parasite strains and isolates.
Leishmania infantum strain MHOM/FR/91/LEM 2259, belonging to zymodeme MON-1 Klon 3511, was described before (22, 26). L. donovani strain BPK091 was a gift from S. Decuypere and J.-C. Dujardin (27, 28).
Parasite cultivation.
Promastigotes were cultivated at 25°C in supplemented medium 199 (29). G418 (Geneticin sulfate; Carl Roth) was added to 50 μg ml−1 for recombinant cell populations.
Electroporation.
Electrotransfection of Leishmania promastigotes was carried out as described previously (30). Promastigotes were harvested during late log phase of growth, washed twice in ice-cold phosphate-buffered saline (PBS) and once in prechilled electroporation buffer, and suspended at a density of 1 × 108 ml−1 in electroporation buffer (31, 32). Circular DNA (20 μg) in an electroporation cuvette was mixed on ice with 0.4 ml of the cell suspension. The mixture was immediately subjected to electroporation using a Bio-Rad Gene Pulser apparatus. Electrotransfection of DNA was carried out by three pulses at 3.750 V/cm and 25 μF in a 4-mm electroporation cuvette. Mock transfection of Leishmania was performed in an identical fashion but without plasmid DNA, to obtain negative-control strains for antibiotic selection. Following electroporation, cells were kept on ice for 10 min before they were transferred to 10 ml drug-free medium. G418 (50 μg ml−1) was added after 24 h for selection of recombinant cells.
Dose-inhibition experiments.
Dose-inhibition curves for antimonyl tartrate (catalog no. 383376; Sigma-Aldrich), miltefosine (catalog no. M5571; Sigma-Aldrich), or pentamidine isethionate salt (catalog no. P0547; Sigma-Aldrich) were established by seeding the promastigotes at 5 × 105 ml−1 in medium 199 containing various concentrations of the drugs. Cell density was measured after 72 h using a Schaerfe system CASY cell counter. The growth was normalized against growth of the untreated cells. Verapamil or sodium vanadate (both from Sigma-Aldrich) were prepared as stock solutions in water at 10 mM and 20 mM, respectively, and added to the cultures as indicated (see the legend to Fig. 5).
FIG 5.
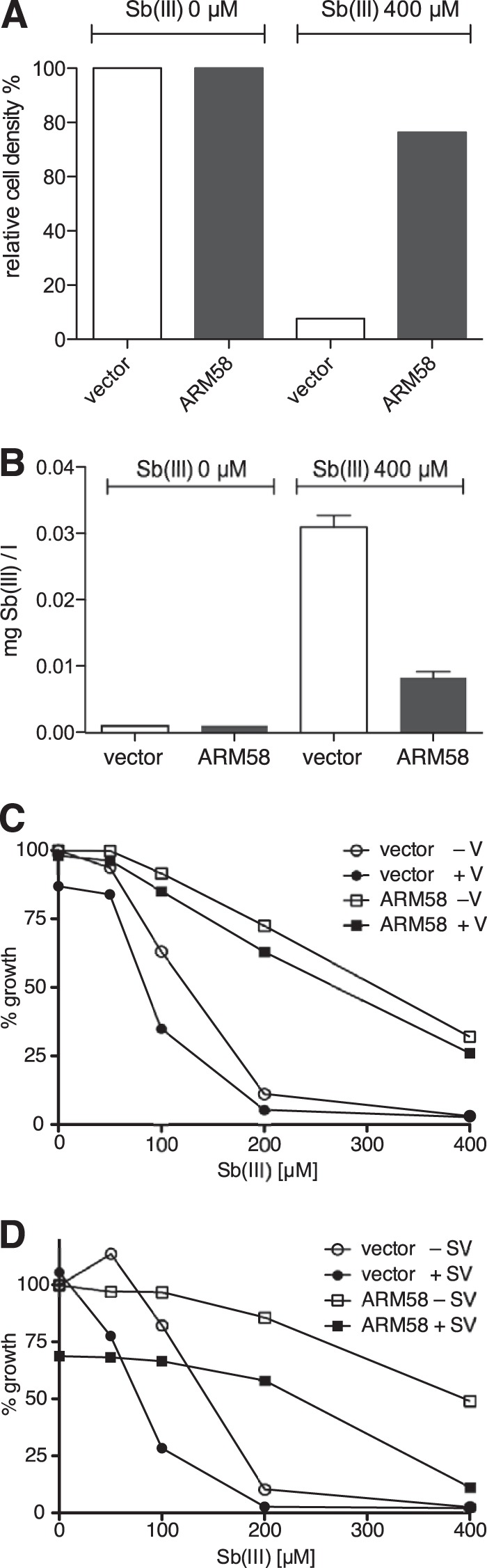
Intracellular Sb concentration following SbIII exposure. L. infantum(pCLN) and L. infantum(pCLN-ARM58) were incubated for 48 h with 0 μM or at 400 μM SbIII. Cells were then counted (A). From each culture, 5 × 107 promastigotes were precipitated, washed, and lysed in nitric acid. Sb concentration in each lysate was measured by inductively coupled plasma mass spectrometry (ICPMS) (n = 3) (B). (C and D) Induction of SbIII resistance in strains receiving 10 μM verapamil (C) or 50 μM sodium vanadate (D). Vector controls and ARM58 overexpressing L. infantum were grown for 72 h. Cell densities were measured and plotted against the indicated SbIII concentrations. Values are the medians from 4 independent experiments. V, verapamil; SV, sodium vanadate.
In vitro infections.
In vitro infections were performed as described previously (25). Bone marrow-derived macrophages (BMMs) were isolated from the femurs of C57BL/6 mice and incubated in Iscove‘s modified Dulbecco's medium (IMDM) supplemented with 10% heat-inactivated fetal calf serum (FCS), 5% horse serum, and 30% L929 cell supernatant containing macrophage colony-stimulating factor (MCSF), modified as described in reference 33. After differentiation, BMMs were harvested, washed, and seeded into 8-well chamber slides (Nunc) at a density of 4 × 105 cells/well. Macrophages were incubated for 48 h at 37°C and 9% CO2 to permit adhesion. BMMs were then infected using stationary-phase promastigotes (34) at 10 parasites per macrophage. After 4 h of incubation at 37°C in modified medium 199 (29), free parasites were washed off with PBS. Incubation was continued for another 72 h in IMDM without or with 160 μg/ml of sodium stibogluconate (Pentostam; gift from Fachbereich Tropenmedizin, Bundeswehrkrankenhaus Hamburg) at 37°C and 9% CO2. After the medium was removed, the cells were washed twice in PBS and subsequently fixed in ice-cold methanol. Intracellular parasites were quantified by nuclear staining with DAPI (4′,6′-diamidino-2-phenylindole, 1.25 μg ml−1; Sigma) and fluorescence microscopy.
Fluorescence microscopy.
L. infantum(pCL2N-mCH::ARM58) promastigotes were fixed and costained with DAPI as described previously (25), with the exception that fluorescence was detected in the Cy5 channel.
Intracellular Sb concentration.
Recombinant cells were incubated for 48 h at 0 μM or at 400 μM SbIII. From each culture, 5 × 107 promastigotes were precipitated, washed, and lysed in 100 μl nitric acid. After 24 h, 2.9 ml double-distilled water (ddH2O) was added. The Sb concentration was measured using inductively coupled plasma mass spectrometry (ICP-MS) at Eurofins WEJ Contaminants GmbH, Hamburg, according to standard protocols (35).
Plasmids.
Both LinARM58 (LinJ34.0220) and LinARM58rel (LinJ34.0210) were amplified using primers that created NdeI and BamHI sites at the 5′ and 3′ ends, respectively. The PCR products were then ligated into NdeI- and BamHI-cut pUC19 (36). From these templates, deletions and point mutations of ARM58 and ARM58rel coding sequences were created by primer-directed mutagenesis according to the protocol described in reference 37. A listing of the primers used for each mutation is available upon request.
For the domain swapping between ARM58 and ARM58rel, we first deleted the third DUF1935 coding region from either gene, using primer-directed deletion PCR followed by recircularization and leaving SnaBI and NarI sites in place. Then, we amplified the coding sequences for the third DUF1935 from either ARM58 or ARM58rel with in-frame SnaBI and NarI sites at the 5′ and 3′ ends, respectively. We could then insert either the homologous or the paralogous DUF1935 coding sequences in ARM58 ΔD3 and ARM58rel ΔD3, resulting in ARM58rel, ARM58rel_D3, ARM58, and ARM58_D3rel. These constructs were then inserted into the pCLN expression plasmid.
The pCLN vector was described previously (37). ARM58, ARM58rel, and all mutant variants thereof were excised from the pUC19 vector with NdeI and BamHI sites and ligated into NdeI- and BglII-cut pCLN.
For expression of a mCherry-ARM58 fusion protein, new restriction sites were inserted into the multiple cloning site of pCLN to create pCL2N. Then the mCherry coding sequence was amplified from the pOB90mCHERRY vector (gift from V. Heussler) and ligated between the KpnI and NdeI restriction sites of pCL2N. ARM58 coding sequences were amplified with primers introducing NdeI and BamHI sites at the 5′ and 3′ ends, respectively. The PCR products were then digested with NdeI and BamHI and ligated into the NdeI- and BglII-digested pCL2N-mCH to create the episomal expression vector pCL2N-mCH::ARM58.
RESULTS
Structural features of LinARM58 and LinARM58rel.
The putative L. infantum homologue of L. braziliensis ARM58 (Lbr20.0210) (42) is located on chromosome 34 (LinJ34.0220), but in a syntenic array of genes, including the ARM58rel gene (LinJ34.0210). The putative products of both genes share the tetrapartite arrangement of 4 DUF1935 domains. As in L. braziliensis, ARM58 is distinguished from ARM58rel by an ∼30-bp insertion between DUF1935-3 and DUF1935-4 and by the presence of a putative transmembrane domain (TMD) sequence (Fig. 1A).
FIG 1.

Validation of LinARM58. (A) Putative domain structure of LinARM58 and LinARM58rel. The boxes represent 4 putative domains of unknown function (DUF1935) for each protein. TMD, transmembrane domain; insertion, 31-amino-acid sequence absent from ARM58rel. The numbers below the structures show the positions within the amino acid sequence. (B) Dose-inhibition experiment for growth of L. infantum carrying the indicated transgenes at the indicated antimonyl tartrate (SbIII) concentrations. Growth over 72 h was normalized to that of the 0 μM samples (100%). The dotted lines indicate the respective IC50s. **, P < 0.01; ***, P < 0.001 (n = 8). (C) As for panel B, but with L. donovani 1SR as the acceptor strain. (D) In vitro infection of bone marrow-derived macrophages and treatment with sodium stibogluconate. L. infantum transfected with vector or with the ARM58 transgene was used to infect BMMs at a 10:1 ratio. After 4 h, extracellular parasites were removed. Sodium stibogluconate was added to the cultures at 160 μg/ml, and incubation was continued for 72 h. After DAPI staining, infection rates relative to the untreated control infections (data not shown) were determined by fluorescence microscopy (10 experiments with 100 macrophages each). The horizontal bars depict the median values. **, P < 0.01. (E) As for panel D, except that L. donovani 1SR served as the acceptor strain. *, P < 0.05 (n = 6). (F) As for panel B, except that parasites were treated with the indicated concentrations of miltefosine. (G) As for panel B, except that parasites were treated with the indicated concentrations of pentamidine.
To ascertain that the LinARM58 is a true homologue of LbrARM58, we overexpressed LinARM58 and LinARM58rel both in L. infantum and in L. donovani to assess the effects on SbIII resistance. We performed dose-inhibition experiments against controls bearing the empty expression vector pCLN. Overexpression of LinARM58 in L. infantum increases the 50% inhibitory concentration (IC50) for SbIII from 115 μM to 330 μM, with LinARM58rel showing no such effect (Fig. 1B). The effect was slightly more pronounced when L. donovani was used as the acceptor—the IC50 increased from 180 μM to 680 μM (Fig. 1C). This confirms LinARM58 as a structural and functional homologue of LbrARM58.
We also tested the effects of ARM58 overexpression on the survival of intracellular amastigotes undergoing sodium stibogluconate treatment. Bone marrow-derived macrophages were infected with stationary-phase promastigotes of L. infantum (Fig. 1D) or L. donovani (Fig. 1E) carrying the empty vector or the ARM58 transgene and then subjected to sodium stibogluconate treatment for 72 h. In both species, the ARM58 transgenes increase the median parasite numbers after the drug treatment >2-fold, proving that the SbIII resistance of promastigotes translates into SbV resistance of amastigotes.
We also tested the effects of ARM58 overexpression in L. infantum against two other anti-leishmanial agents, miltefosine and pentamidine. ARM58 induces resistance against neither miltefosine (Fig. 1F) nor pentamidine (Fig. 1G). The effects of LinARM58, therefore, specifically interfere with the efficacy of antimony compounds.
Domain deletion analysis.
The tetrapartite structure of ARM58 raised the question of whether the functionality of the DUF-1935 domains is specific or redundant. We therefore constructed a series of ARM58 transgenes lacking one or more putative domains (Fig. 2A) and tested their ability to confer elevated SbIII resistance to L. infantum promastigotes. Dose-inhibition experiments were performed in quadruplicate to determine the IC50 for strains overexpressing the ARM58 variants. The first set of data (Fig. 2B) shows that DUF1935-3 is essential for ARM58 function, while DUF1935-1 and DUF1935-2 are required for full activity. Only DUF1935-4 and the insertion sequence between DUF1935-3 and DUF1935-4 that is unique to ARM58 are dispensable for mediating SbIII resistance.
FIG 2.

(A) Schematic depiction of ARM58 deletion constructs used for panels B and C. The shaded boxes represent the four DUF1935 putative domains. The black box signifies the putative transmembrane domain (TMD). The numbers indicate the positions relative to the wild-type amino acid sequence; insertion, 31-amino-acid sequence absent from ARM58rel. (B) SbIII IC50s (μM) for L. infantum carrying the indicated ARM58 variants. *, P < 0.05 (n = 4) relative to full-length ARM58. (C) As for panel B, but with further shortened ARM58 variants (n = 4).
Next we determined the minimal domain combination required for ARM58 function. Based upon the ARM58_ΔD4 mutant, which has full activity, we deleted either DUF1935-1 (ARM58_ΔD14), DUF1935-2 (ARM58_ΔD24), or both (ARM58_ΔD124) and tested the ability of the truncated transgenes to confer SbIII resistance to L. infantum promastigotes (Fig. 2C). Any combination of DUF1935-3 with either DUF1935-1 or DUF1935-2 retains a minimal effect, but DUF1935-3 alone, although essential, is not sufficient to mitigate SbIII toxicity.
Mutagenic analysis of DUF1935-3.
In the previous experiment, an essential role for the DUF1935-3 in SbIII resistance was established. The domain contains a putative 21-amino-acid transmembrane domain (TMD) with the sequence N-GFVAAVEVLPLATVPFLVGAP-C. Computer simulation using the TMpred algorithm (38) identified the residues Val336 and Val368 (underlined) as critical for TMD formation. We therefore produced three transgenes based on ARM58: (i) ARM58_ΔTMD, which lacks the 21-amino-acid TMD entirely; (ii) ARM58_V336/338-N, in which the aliphatic valine side chains are replaced with polar asparagines; and (iii) ARM58_V336/338-L, in which the aliphatic valines are replaced with equally aliphatic leucines (Fig. 3A).
FIG 3.

(A) Schematic depiction of mutated ARM58 constructs used for panel B. The shaded boxes represent the 4 DUF1935 putative domains. The black box signifies the putative transmembrane domain (TMD). The numbers indicate the positions relative to the wild-type amino acid sequence. (B) SbIII IC50s (μM) for L. infantum carrying the indicated ARM58 mutants. *, P < 0.05 (n = 4) relative to full-length ARM58. (C) Schematic depiction of ARM58rel, ARM58, and two DUF1935-III domain swapping mutants, ARM58_3Drel and ARM58rel_D3. (D) As for panel B, but testing domain swapping mutants of ARM58 and AMR58rel (n = 4).
L. infantum parasites expressing ARM58 and the three mutants were then challenged with SbIII, and the IC50s for each recombinant parasite line were established in quadruplicate. The results implicate the putative TMD in SbIII resistance. Both the deletion of the TMD and the replacement of the two critical valines with asparagines caused an ∼50% loss of function. By contrast, the replacement of the two valines with leucine residues, which are compatible with a TMD, did not cause any loss of function (Fig. 3C). While this is not proof of a TMD, the results show the importance of aliphatic and hydrophobic side chains in DUF1935-3.
Domain swapping experiments.
We next made use of the structural similarity between ARM58 and ARM58rel to establish the impact of DUF1935-3. We created two domain swapping mutants that either place the DUF1935-3 of ARM58 in the context of the ARM58rel sequence (ARM58rel_D3) or replace the DUF1935-3 in ARM58 with its ARM58rel counterpart DUF1935-3rel (ARM58_D3rel) (Fig. 3C). By expressing both mosaic genes and the two wild-type genes in L. infantum we determined their effects on SbIII resistance. As shown in Fig. 3D, replacing DUF1935-3 with DUF1935-3rel completely abolishes ARM58 functionality. By contrast, the DUF1935-3 in the ARM58rel context exhibits almost full resistance functionality. This underscores the importance of DUF1935-3 and may explain how ARM58 developed from the ancestral ARM58rel gene by gaining a hydrophobic sequence stretch inside DUF1935-3. Moreover, it shows that DUF1935-1rel and DUF1935-2rel are not specific and can complement DUF1935-3. Conversely, DUF1935-1 and DUF1935-2 cannot produce the resistance phenotype without the original DUF1935-3.
Subcellular localization of the mCH::LinARM58 fusion protein.
We next tried to determine the subcellular localization of ARM58 in L. infantum as an indicator of possible functions. Unfortunately, we were unable to raise antibodies against recombinantly expressed ARM58 protein. While the recombinant antigens were recognized with specificity, the antibodies did not work in either Western blots or indirect immunofluorescence microscopy (C. Schäfer, unpublished data). We therefore resorted to the expression of chromophore fusion proteins, placing emphasis on the functionality of such fusion proteins. Fusion of green fluorescent protein (GFP) to the C terminus of ARM58 resulted in a loss of SbIII resistance-mediating function (data not shown). We next tried an ARM58 with an N-terminal mCherry (RFP) fusion domain. The construct is shown in Fig. 4A. Expression of the mCH::ARM58 fusion protein indeed raises the IC50 for SbIII in the same way the ARM58 overexpression does (Fig. 4B), indicating that the fusion protein is the functional equivalent of the overexpressed ARM58 and lending credibility to any localization observed.
FIG 4.

Subcellular localization of mCH::ARM58 fusion proteins. (A) Schematic depiction of the episomal expression plasmid pCL2N-mCH::ARM58. (B) Functional test of mCherry::ARM58 fusion protein. L. infantum overexpressing either ARM58 (solid squares) or mCH::ARM58 (diamonds) was subjected to a 72-h dose effect growth experiment with various SbIII concentrations and compared to a control strain transfected with pCLN (vector). Dotted lines show IC50s; error bars show standard deviations (n = 4). (C to F) Fluorescence microscopy imaging of L. infantum(pCL2N-mCH::ARM58) showing differential inference contrast (DIC) (C), DAPI (D), and mCherry (E) channels plus overlay of DAPI and mCherry (F). n, nucleus; k, kinetoplast. (G to K) Confocal laser microscopy imaging of L. infantum(pCL2N-mCH::ARM58) showing DIC (G), DAPI (H), and mCherry (I) channels plus overlay of DAPI and mCherry (K). n, nucleus; k, kinetoplast. Size bar, 5 μm.
The subcellular localization of this functional fusion protein was then determined using fluorescence microscopy (Fig. 4C to F) or confocal laser fluorescence microscopy (Fig. 4G to K). Both technologies deliver very similar results. The mCH::ARM58 fusion proteins localize mostly to the anterior half of the parasite, with intense fluorescence around the flagellar pocket, the interface for exo- and endocytotic pathways. In addition, fluorescence can be seen around but excluding the nucleus and in a few focal spots dispersed over the cell body. Expression of the mCherry domain alone yielded the usual even cytoplasmic distribution (data not shown).
The main localization near the flagellar pocket hints at an involvement in cross-membrane traffic and together with the putative transmembrane domain suggests an involvement with Sb transport.
Intracellular Sb concentration.
Next, we determined the effect of ARM58 overexpression on the intracellular Sb concentration before and after exposure to high (400 μM) concentrations of SbIII in vitro. A L. infantum vector control strain and the L. infantum(pCLN-ARM58) strain were cultivated in vitro, for 48 h, either at 0 μM or at 400 μM antimonyl tartrate. We determined both the growth-inhibitory effects of SbIII exposure and the amount of extractable antimony from 5 × 107 cells.
Measurements show a 95% reduction of cell density for the vector control strain treated with SbIII and only a 25% reduction for the ARM58-overexpressing parasites (Fig. 5A). After extraction, we measured 30 μg/liter from the vector control cells but only 8 μg/liter from ARM58-overexpressing parasites (Fig. 5B). Factoring in the average cell volume of L. infantum promastigotes in culture (2.4 × 10−14 liter), the cell number (5 × 107), and the total extraction volume (3 ml), we calculate intracellular concentrations of 625 μM (vector control) and 165 μM (ARM58 overexpression). We conclude that the intracellular SbIII concentration is at least as high as the extracellular concentration, while overexpression of ARM58 lowers the intracellular SbIII concentration by 74%, thus facilitating proliferation at an otherwise inhibiting SbIII concentration.
ARM58 shows no signature sequences for P-glycoproteins or for ATPases. Nevertheless, it is conceivable that ARM58 may depend in its function on P-glycoproteins or other energy-dependent transporters. Therefore, we tested the effectiveness of ARM58 overexpression both in the presence of the P-glycoprotein inhibitor verapamil and in the presence of sodium vanadate, an ATPase inhibitor. At 10 μM verapamil, both the control strain (vector) and ARM58-overexpressing parasites showed slightly lowered growth at the various SbIII concentrations. However, the advantage of ARM58 overexpression during SbIII treatment remained unchanged (Fig. 5C). We find no indication of a functional dependence of ARM58 on P-glycoproteins.
In the presence of 50 μM sodium vanadate, L. infantum growth is more sensitive to SbIII. This is true both for the vector control and for the ARM58-overexpressing parasites (Fig. 5D). However, ARM58 overexpression still protects L. infantum growth against SbIII inhibition in the presence of sodium vanadate. Therefore, ATPase inhibition does not abrogate the effect of ARM58, meaning that we have no indication that ATP hydrolysis is critical for ARM58-mediated antimony resistance.
DISCUSSION
In spite of decades of active research, the molecular basis of antimony drug action and antimony drug resistance in Leishmania spp. is not fully understood. The latter is likely due to a multitude of mechanisms by which Leishmania spp. can counteract exposure to and impact of antimonials. Among the chief strategies employed are the induction of antimony extrusion either by host cell proteins (19) or by the parasite (35). It was shown by Brochu and colleagues that intracellular SbIII accumulation can be lower in resistant strains and that the difference to sensitive strains is dependent on ATP hydrolysis—an indicator of an energy-dependent extrusion system.
ARM58 does not display any sequence motifs of known ATP binding sites. The DUF1935 (domains of unknown function 1935) of which ARM58 and ARM58rel are entirely composed are not known to be part of energy-dependent transporter proteins. The other proteins containing DUF1935 signatures are several calpain-like cysteine peptidases and a receptor-type adenylate cyclase, which all contain only single DUF1935 motifs.
Other known mechanisms of Sb resistance appear to involve general stress resistance effectors, such as heat shock proteins and thiol pathways. ARM58 bears no structural resemblance to any known stress response effector proteins. While the amplification and overexpression of heat shock proteins have been reported to correlate with Sb resistance (39, 40), we did not select any stress gene loci in our original functional cloning screen (42). It may well be that Hsp-mediated resistance plays a role in intracellular amastigotes challenged with SbV, thus escaping our selection strategy. Conversely, the relative stress resistance of ARM58 overexpressing parasites should also be investigated, since it was shown recently (41) that drug resistance in L. donovani is linked to general virulence and fitness.
In this context, it may be interesting to test the overexpression of ARM58 in the background of a trypanothione synthesis-defective strain of L. infantum or L. donovani. This should tell whether ARM58 is functionally linked to the antioxidant pathways.
Nevertheless, our results mostly indicate that ARM58 participates in an extrusion pathway. Its dependence on a putative transmembrane domain, its main localization close to the flagellar pocket, and its effect on intracellular antimony levels all point at such a role. We assume for now that ARM58 may interact with and modulate the activity of actual extrusion proteins. However, the inhibition of P-glycoproteins by verapamil or the inhibition of ATPase activity cannot abrogate the effect of ARM58 overexpression. Therefore, we have no indication if and what partners are stimulated by ARM58.
Alternatively, ARM58 could be involved in the intracellular or intravesicular sequestration of antimony. Sequestration would explain the protective effects even during inhibition of ATPase-dependent pathways. However, such sequestration must be subsequently linked to extrusion pathways, since the nitric acid extraction used to determine the intracellular antimony concentration will also pick up a sequestered drug.
We can also conclude that the localization of ARM58 is not dependent on N-terminal signal sequences. This is evidenced by the specific localization of the fusion protein with the N-terminal mCherry domain. The mCH::ARM58 fusion protein also induces SbIII resistance with the same efficiency as ARM58 itself, meaning that it is fully functional in spite of an N-terminal mCherry domain. This argues against a Golgi-dependent transport of ARM58.
We repeatedly tried to raise ARM58-specific antibodies. While the IgY faithfully recognized the bacterially expressed ARM58, no such detection was observed in Western blotting or indirect immune fluorescence microscopy, in spite of a substantial overexpression in the recombinant parasites. By contrast, expression as an mCH::ARM58 fusion protein yielded good detectability. We have to assume that ARM58 may be modified in vivo, thus escaping detection by antibodies raised against bacterially expressed protein. We plan to perform a more thorough analysis, applying deglycosylation prior to antibody detection. The inability to raise effective anti-ARM58 antibodies is also a hurdle for the identification of interacting protein factors. The availability of the mCH::ARM58 fusion protein, however, should enable us to look into the subcellular localization using high-resolution immune electron microscopy.
The structure of ARM58 and ARM58rel with four semiconserved DUF1935 modules is so far unique and found only within the kinetoplastid protozoa. The domain swapping experiment whose results are shown in Fig. 3C and D indicates a close relationship between both genes, a notion that is further supported by a phylogenetic analysis. ARM58 is present in all Leishmania species, of both Old World and New World origin. Syntenic copies of ARM58rel are found both in Leishmania spp. and in African trypanosomes. This indicates a specific function of ARM58 in Leishmania parasites.
We have repeatedly attempted to generate ARM58-null mutants in L. infantum by homologous recombination. We could indeed obtain single-allele gene replacement mutants with three different selection marker genes that were flanked by 1 kb each of ARM58 5′ and 3′ flanking sequences. The single allele gene replacement mutants already showed morphological irregularities, including incomplete cell division and an enlarged flagellar pocket. All attempts to replace the second alleles did not result in selectable cells (D. Zander, unpublished data), leading to the conclusion that ARM58 plays a crucial role in Leishmania spp. apart from antimony detoxification.
Our results further underscore the notion that gene amplification is one of the mechanisms by which Leishmania parasites counter the effect of drugs. The original functional cloning screen used cosmid-based genomic DNA libraries from drug-resistant and drug-sensitive field isolates. The ARM58 gene locus was selected in both cases, showing that at least in laboratory settings, episomal gene amplification is more powerful than possible sequence variations between isolates.
The next step will have to be the analysis of ARM58 expression rates in field isolates from resistance hot spots, such as northeastern India, to test whether modulated ARM58 expression correlates with either resistance or sensitivity.
ACKNOWLEDGMENTS
We acknowledge the technical assistance of Andrea Macdonald and the help of Tobias Spielmann with confocal laser microscopy. We thank Eurofins, Hamburg, for performing the quantitative antimony analysis, Neena Goyal (Lucknow, India) for helpful discussions, and Laura Jade Lee for a careful reading of the manuscript.
During her stay at the BNI, P.T. was supported by the European Lifelong Learning Programme Leonardo da Vinci.
Footnotes
Published ahead of print 23 December 2013
REFERENCES
- 1.Clayton CE. 2002. Life without transcriptional control? From fly to man and back again. EMBO J. 21:1881–1888. 10.1093/emboj/21.8.1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alvar J, Velez ID, Bern C, Herrero M, Desjeux P, Cano J, Jannin J, den Boer M. 2012. Leishmaniasis worldwide and global estimates of its incidence. PLoS One 7:e35671. 10.1371/journal.pone.0035671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shaked-Mishan P, Ulrich N, Ephros M, Zilberstein D. 2001. Novel intracellular SbV reducing activity correlates with antimony susceptibility in Leishmania donovani. J. Biol. Chem. 276:3971–3976. 10.1074/jbc.M005423200 [DOI] [PubMed] [Google Scholar]
- 4.Ephros M, Bitnun A, Shaked P, Waldman E, Zilberstein D. 1999. Stage-specific activity of pentavalent antimony against Leishmania donovani axenic amastigotes. Antimicrob. Agents Chemother. 43:278–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ashutosh Sundar S, Goyal N. 2007. Molecular mechanisms of antimony resistance in Leishmania. J. Med. Microbiol. 56:143–153. 10.1099/jmm.0.46841-0 [DOI] [PubMed] [Google Scholar]
- 6.Croft SL, Sundar S, Fairlamb AH. 2006. Drug resistance in leishmaniasis. Clin. Microbiol. Rev. 19:111–126. 10.1128/CMR.19.1.111-126.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sundar S. 2003. Indian kala-azar—better tools needed for diagnosis and treatment. J. Postgrad Med. 49:29–30. 10.4103/0022-3859.931 [DOI] [PubMed] [Google Scholar]
- 8.Sundar S. 2001. Drug resistance in Indian visceral leishmaniasis. Trop. Med. Int. Health 6:849–854. 10.1046/j.1365-3156.2001.00778.x [DOI] [PubMed] [Google Scholar]
- 9.Sundar S, Pai K, Kumar R, Pathak-Tripathi K, Gam AA, Ray M, Kenney RT. 2001. Resistance to treatment in kala-azar: speciation of isolates from northeast India. Am. J. Trop. Med. Hyg. 65:193–196 http://www.ajtmh.org/content/65/3/193.long [DOI] [PubMed] [Google Scholar]
- 10.Chappuis F, Sundar S, Hailu A, Ghalib H, Rijal S, Peeling RW, Alvar J, Boelaert M. 2007. Visceral leishmaniasis: what are the needs for diagnosis, treatment and control? Nat. Rev. Microbiol. 5:873–882. 10.1038/nrmicro1748 [DOI] [PubMed] [Google Scholar]
- 11.Agrawal S, Rai M, Sundar S. 2005. Management of visceral leishmaniasis: Indian perspective. J. Postgrad Med. 51(Suppl)1:S53-S57. http://www.jpgmonline.com/text.asp?2005/51/5/53/19816 [PubMed]
- 12.Sundar S, Rai M. 2005. Treatment of visceral leishmaniasis. Expert Opin. Pharmacother. 6:2821–2829. 10.1517/14656566.6.16.2821 [DOI] [PubMed] [Google Scholar]
- 13.Olliaro PL, Guerin PJ, Gerstl S, Haaskjold AA, Rottingen JA, Sundar S. 2005. Treatment options for visceral leishmaniasis: a systematic review of clinical studies done in India, 1980–2004. Lancet Infect. Dis. 5:763–774. 10.1016/S1473-3099(05)70296-6 [DOI] [PubMed] [Google Scholar]
- 14.Haimeur A, Guimond C, Pilote S, Mukhopadhyay R, Rosen BP, Poulin R, Ouellette M. 1999. Elevated levels of polyamines and trypanothione resulting from overexpression of the ornithine decarboxylase gene in arsenite-resistant Leishmania. Mol. Microbiol. 34:726–735. 10.1046/j.1365-2958.1999.01634.x [DOI] [PubMed] [Google Scholar]
- 15.Ouellette M, Haimeur A, Grondin K, Legare D, Papadopoulou B. 1998. Amplification of ABC transporter gene pgpA and of other heavy metal resistance genes in Leishmania tarentolae and their study by gene transfection and gene disruption. Methods Enzymol. 292:182–193 [DOI] [PubMed] [Google Scholar]
- 16.Grondin K, Haimeur A, Mukhopadhyay R, Rosen BP, Ouellette M. 1997. Co-amplification of the gamma-glutamylcysteine synthetase gene gsh1 and of the ABC transporter gene pgpA in arsenite-resistant Leishmania tarentolae. EMBO J. 16:3057–3065. 10.1093/emboj/16.11.3057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gourbal B, Sonuc N, Bhattacharjee H, Legare D, Sundar S, Ouellette M, Rosen BP, Mukhopadhyay R. 2004. Drug uptake and modulation of drug resistance in Leishmania by an aquaglyceroporin. J. Biol. Chem. 279:31010–31017. 10.1074/jbc.M403959200 [DOI] [PubMed] [Google Scholar]
- 18.Mukherjee B, Mukhopadhyay R, Bannerjee B, Chowdhury S, Mukherjee S, Naskar K, Allam US, Chakravortty D, Sundar S, Dujardin JC, Roy S. 2013. Antimony-resistant but not antimony-sensitive Leishmania donovani up-regulates host IL-10 to overexpress multidrug-resistant protein 1. Proc. Natl. Acad. Sci. U. S. A. 110:E575–582. 10.1073/pnas.1213839110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mookerjee Basu J, Mookerjee A, Banerjee R, Saha M, Singh S, Naskar K, Tripathy G, Sinha PK, Pandey K, Sundar S, Bimal S, Das PK, Choudhuri SK, Roy S. 2008. Inhibition of ABC transporters abolishes antimony resistance in Leishmania Infection. Antimicrob. Agents Chemother. 52:1080–1093. 10.1128/AAC.01196-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clos J, Choudhury K. 2006. Functional cloning as a means to identify Leishmania genes involved in drug resistance. Mini Rev. Med. Chem. 6:123–129. 10.2174/138955706775476028 [DOI] [PubMed] [Google Scholar]
- 21.Coelho AC, Beverley SM, Cotrim PC. 2003. Functional genetic identification of PRP1, an ABC transporter superfamily member conferring pentamidine resistance in Leishmania major. Mol. Biochem. Parasitol. 130:83–90. 10.1016/S0166-6851(03)00162-2 [DOI] [PubMed] [Google Scholar]
- 22.Choudhury K, Zander D, Kube M, Reinhardt R, Clos J. 2008. Identification of a Leishmania infantum gene mediating resistance to miltefosine and SbIII. Int. J. Parasitol. 38:1411–1423. 10.1016/j.ijpara.2008.03.005 [DOI] [PubMed] [Google Scholar]
- 23.Dobson DE, Scholtes LD, Valdez KE, Sullivan DR, Mengeling BJ, Cilmi S, Turco SJ, Beverley SM. 2003. Functional identification of galactosyltransferases (SCGs) required for species-specific modifications of the lipophosphoglycan adhesin controlling Leishmania major-sand fly interactions. J. Biol. Chem. 278:15523–15531. 10.1074/jbc.M301568200 [DOI] [PubMed] [Google Scholar]
- 24.Descoteaux A, Avila HA, Zhang K, Turco SJ, Beverley SM. 2002. Leishmania LPG3 encodes a GRP94 homolog required for phosphoglycan synthesis implicated in parasite virulence but not viability. EMBO J. 21:4458–4469. 10.1093/emboj/cdf447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reiling L, Chrobak M, Schmetz C, Clos J. 2010. Overexpression of a single Leishmania major gene is sufficient to enhance parasite infectivity in vivo and in vitro. Mol. Microbiol. 76:1175–1190. 10.1111/j.1365-2958.2010.07130.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garin YJ, Sulahian A, Pratlong F, Meneceur P, Gangneux JP, Prina E, Dedet JP, Derouin F. 2001. Virulence of Leishmania infantum is expressed as a clonal and dominant phenotype in experimental infections. Infect. Immun. 69:7365–7373. 10.1128/IAI.69.12.7365-7373.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Decuypere S, Vanaerschot M, Brunker K, Imamura H, Muller S, Khanal B, Rijal S, Dujardin JC, Coombs GH. 2012. Molecular mechanisms of drug resistance in natural Leishmania populations vary with genetic background. PLoS neglected tropical diseases 6:e1514. 10.1371/journal.pntd.0001514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Decuypere S, Rijal S, Yardley V, De Doncker S, Laurent T, Khanal B, Chappuis F, Dujardin JC. 2005. Gene expression analysis of the mechanism of natural Sb(V) resistance in Leishmania donovani isolates from Nepal. Antimicrob. Agents Chemother. 49:4616–4621. 10.1128/AAC.49.11.4616-4621.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hubel A, Krobitsch S, Horauf A, Clos J. 1997. Leishmania major Hsp100 is required chiefly in the mammalian stage of the parasite. Mol. Cell. Biol. 17:5987–5995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krobitsch S, Brandau S, Hoyer C, Schmetz C, Hübel A, Clos J. 1998. Leishmania donovani heat shock protein 100: characterization and function in amastigote stage differentiation. J. Biol. Chem. 273:6488–6494. 10.1074/jbc.273.11.6488 [DOI] [PubMed] [Google Scholar]
- 31.Laban A, Wirth DF. 1989. Transfection of Leishmania enriettii and expression of chloramphenicol acetyltransferase gene. Proc. Natl. Acad. Sci. U. S. A. 86:9119–9123. 10.1073/pnas.86.23.9119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kapler GM, Coburn CM, Beverley SM. 1990. Stable transfection of the human parasite Leishmania major delineates a 30-kilobase region sufficient for extrachromosomal replication and expression. Mol. Cell. Biol. 10:1084–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Racoosin EL, Swanson JA. 1989. Macrophage colony-stimulating factor (rM-CSF) stimulates pinocytosis in bone marrow-derived macrophages. J. Exp. Med. 170:1635–1648. 10.1084/jem.170.5.1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Racoosin EL, Beverley SM. 1997. Leishmania major: promastigotes induce expression of a subset of chemokine genes in murine macrophages. Exp. Parasitol. 85:283–295. 10.1006/expr.1996.4139 [DOI] [PubMed] [Google Scholar]
- 35.Brochu C, Wang J, Roy G, Messier N, Wang XY, Saravia NG, Ouellette M. 2003. Antimony uptake systems in the protozoan parasite Leishmania and accumulation differences in antimony-resistant parasites. Antimicrob. Agents Chemother. 47:3073–3079. 10.1128/AAC.47.10.3073-3079.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yanisch-Perron C, Vieira J, Messing J. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33:103–119. 10.1016/0378-1119(85)90120-9 [DOI] [PubMed] [Google Scholar]
- 37.Hombach A, Ommen G, Chrobak M, Clos J. 2013. The Hsp90-Sti1 interaction is critical for Leishmania donovani proliferation in both life cycle stages. Cell. Microbiol. 15:585–600. 10.1111/cmi.12057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hofmann K, Stoffel W. 1993. TMbase—a database of membrane spanning proteins segments. Biol. Chem. Hoppe-Seyler 374:166 [Google Scholar]
- 39.Brochu C, Haimeur A, Ouellette M. 2004. The heat shock protein HSP70 and heat shock cognate protein HSC70 contribute to antimony tolerance in the protozoan parasite leishmania. Cell Stress Chaperones 9:294–303. 10.1379/CSC-15R1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vergnes B, Gourbal B, Girard I, Sundar S, Drummelsmith J, Ouellette M. 2007. A proteomics screen implicates HSP83 and a small kinetoplastid calpain-related protein in drug resistance in Leishmania donovani clinical field isolates by modulating drug-induced programmed cell death. Mol. Cell. Proteomics 6:88–101. 10.1074/mcp.M600319-MCP200 [DOI] [PubMed] [Google Scholar]
- 41.Vanaerschot M, De Doncker S, Rijal S, Maes L, Dujardin JC, Decuypere S. 2011. Antimonial resistance in Leishmania donovani is associated with increased in vivo parasite burden. PLoS One 6:e23120. 10.1371/journal.pone.0023120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nühs A, Schäfer C, Zander D, Trübe L, Tejera Nevado P, Schmidt S, Arevalo J, Adaui V, Maes L, Dujardin J-C, Clos J. 2014. A novel marker, ARM58, confers antimony resistance to Leishmania spp. Int. J. Parasitol: Drugs and Drug Resist. 4:37–47. 10.1016/j.ijpddr.2013.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]