

Abstract
The rise of resistant pathogens and chronic infections tolerant to antibiotics presents an unmet need for novel antimicrobial compounds. Identifying broad-spectrum leads is challenging due to the effective penetration barrier of Gram-negative bacteria, formed by an outer membrane restricting amphipathic compounds, and multidrug resistance (MDR) pumps. In chronic infections, pathogens are shielded from the immune system by biofilms or host cells, and dormant persisters tolerant to antibiotics are responsible for recalcitrance to chemotherapy with conventional antibiotics. We reasoned that the dual need for broad-spectrum and sterilizing compounds could be met by developing prodrugs that are activated by bacterium-specific enzymes and that these generally reactive compounds could kill persisters and accumulate over time due to irreversible binding to targets. We report the development of a screen for prodrugs, based on identifying compounds that nonspecifically inhibit reduction of the viability dye alamarBlue, and then eliminate generally toxic compounds by testing for cytotoxicity. A large pilot of 55,000 compounds against Escherichia coli produced 20 hits, 3 of which were further examined. One compound, ADC111, is an analog of a known nitrofuran prodrug nitrofurantoin, and its activity depends on the presence of activating enzymes nitroreductases. ADC112 is an analog of another known antimicrobial tilbroquinol with unknown mechanism of action, and ADC113 does not belong to an approved class. All three compounds had a good spectrum and showed good to excellent activity against persister cells in biofilm and stationary cultures. These results suggest that screening for overlooked prodrugs may present a viable platform for antimicrobial discovery.
INTRODUCTION
The need for novel antibiotics to combat drug-resistant pathogens is well understood (1). Less recognized, but no less important, is the unmet need for compounds capable of effectively killing dormant forms of pathogens (2). Biofilm infections are on the rise, largely a result of medical intervention, and form chronic, poorly treatable infections. Biofilms form readily on indwelling devices, such as catheters and prostheses. Biofilms are also responsible for infective endocarditis, recurring urinary tract infections (UTIs), infective osteomyelitis, and the incurable infection of the lungs of patients with cystic fibrosis (3). Antibiotics depend on the immune response to clear an infection, and a chronic disease often forms in immunocompromised patients. Importantly, most chronic infections recalcitrant to treatment are caused by drug-susceptible pathogens. Recalcitrance to treatment results from tolerance rather than resistance. Pathogens produce a small subpopulation of dormant persister cells that are tolerant to antibiotics (2), and the biofilm matrix protects them from the immune system. Once the concentration of the antibiotic drops, persisters resuscitate and repopulate the biofilm, causing a relapsing infection. Several mechanisms lead to dormancy in Escherichia coli and rely mostly on the action of toxin/antitoxin modules. The toxins responsible for persister formation include mRNA endonucleases (4, 5), the HipA kinase (6), and TisB, which decreases the energy level of the cell by opening an ion channel (7). Bactericidal antibiotics kill by corrupting their targets (4, 8); for example, fluoroquinolones inhibit the religation step in DNA gyrase and topoisomerase, turning the enzymes into endonucleases (9). Targets are inactive in dormant persisters, explaining their tolerance to antibiotics. The high degree of redundancy in the mechanisms of persister formation precludes development of conventional target-based inhibitors.
We considered prodrugs as a type of compounds that could kill persister cells and eradicate a chronic infection. Nitroaromatic prodrugs such as metronidazole or nitrofurantoin are benign compounds that enter into the cell and are converted into a reactive drug by nitroreductases specific to microorganisms (Fig. 1). Since the highly activated species produced hit multiple targets, this could in principle kill both growing and dormant cells. Given that redox-activated prodrugs bind covalently to their targets, this creates an irreversible sink, ensuring accumulation over time. The sink is likely to counter efflux by multidrug resistance (MDR) pumps. The dual barrier of the outer membrane and MDR pumps prevents most compounds from entering the cells of Gram-negative bacteria (10, 11) and is largely responsible for the paucity of broad-spectrum antibiotics. The last class of broad-spectrum compounds, the fluoroquinolones, was discovered over 50 years ago (12).
FIG 1.

Prodrug antibiotics. An ideal prodrug is an inactive compound that penetrates the membrane, enters the cell, and is converted by a bacterium-specific enzyme (ENZ) into a reactive molecule. The reactive form binds covalently to multiple targets, killing both regular and dormant cells. Importantly, covalent binding creates an irreversible sink, which leads to accumulation of the drug over time.
It is interesting to note that all prodrug antibiotics were discovered in the 1950s (12). It seems that subsequently developed validation tests based on determining the specificity of action of hits precluded prodrug discovery. In an effort to eliminate generally toxic compounds, the specificity test was introduced, where the ability of a test compound to inhibit label incorporation into major biopolymers is measured (13). Compounds that inhibit all biosynthesis are nonspecific and are eliminated. By this test, metronidazole is a nuisance compound. Interestingly, metronidazole is a broad-spectrum antibiotic, but its use is limited, since nitroreductases are expressed primarily under anaerobic/microaerophilic conditions. Given the potential of prodrugs for both broad-spectrum and sterilizing activity, we considered developing a screen for these compounds. The rationale for the screen is an inverted specificity test: compounds that lack specificity are desired hits. In order to make the screen practical, a vital dye is used instead of label incorporation. A subsequent cytotoxicity test against mammalian cells then differentiates between prodrug candidates and generally toxic compounds. In this study, we report development of a prodrug screen and validation of hit compounds, including their ability to kill persister cells.
MATERIALS AND METHODS
Growth of bacterial strains.
Bacterial strains used in this study are shown in Table 1. Strains of Escherichia coli, Staphylococcus aureus, Francisella tularensis, Bacillus anthracis, Yersinia pestis, Salmonella enterica serotype Typhimurium, Acinetobacter baumannii, and Pseudomonas aeruginosa were grown in cation-adjusted Mueller-Hinton II broth (CA-MHB; BD catalog no. 212322). Strains of Enterococcus faecalis, Enterococcus faecium, and Clostridium difficile were grown in brain heart infusion (BHI; BD catalog no. 211059) broth supplemented with yeast extract (5 g/liter), cysteine (1 g/liter), and hemin (15 mg/liter). All strains were grown at 37°C with aeration at 220 rpm except for E. faecium, which was grown statically.
TABLE 1.
Bacterial strains used in this study
| Strain or species | Relevant genotype | Parent or source | Reference or strain designationd |
|---|---|---|---|
| BW25113 | K-12 rrnB3 ΔlacZ4787 hsdR514 Δ(araBAD)567 Δ(rhaBAD)568 rph-1 | 15 | |
| nfsB-FRT | ΔnfsB::FRT | JW0567 | 15 |
| MV1970 | ΔnfsB::FRT ΔnfsA::kan | JW0835 into nfsB-FRT | 15 |
| tolC | ΔtolC::kan | JW5503 | 15 |
| acrB | ΔacrB::kan | JW0451 | 15 |
| nfsA+ | BW25113(pZS*24nfsA) | 17 | |
| nfsA− | ΔnfsA::kan | JW0835 | 15 |
| nfsB+ | BW25113(pZS*24nfsB) | 17 | |
| nfsB− | ΔnfsB::kan | JW0567 | 15 |
| Francisella tularensis | SchuS4a | ||
| Bacillus anthracis | Amesa | ||
| Yersinia pestis | KIMa | ||
| Escherichia coli | ATCC 25922 | ||
| Staphylococcus aureus | NCTC 8325 | ||
| Enterococcus faecalis | ATCC 47077 | ||
| Salmonella Typhimurium (LT2) | ATCC 700720 | ||
| Staphylococcus aureus (MRSA)e | NRS54b, Novobiotic Pharmaceuticals | ||
| Clostridium difficile (CD196) | Clinical isolate, CD196c | ||
| Enterococcus faecium (VRE BM4147)f | BM4147, Novobiotic Pharmaceuticals | ||
| Acinetobacter baumannii | ATCC 17978 | ||
| Pseudomonas aeruginosa (PAO1) | ATCC BAA-47 |
Strain designation from the NERCE/BEID facility.
Strain designation from the Network on Antimicrobial Resistance in Staphylococcus aureus (NARSA).
This clinical isolate was a gift from Linc Sonenshein at Tufts University.
Abbreviations: ATCC, American Type Culture Collection; NCTC, National Collection of Type Cultures.
MRSA, methicillin-resistant Staphylococcus aureus.
VRE, vancomycin-resistant Enterococcus faecium.
Construction of bacterial strains.
Mutations were introduced into the parental strain, E. coli K-12 BW25113, by P1 transduction (14). The kanamycin resistance cassette from the mutant alleles originated from the Keio collection (15) and was cured when needed by expressing the FLP recombinase from the helper plasmid pCP20 (16). The acrB and tolC MDR pump deletion mutant are derived from the Keio collection. Strain BW25113 pZS*24nfsA was constructed by amplifying the nfsA open reading frame (ORF) using primers nfsAfwKpn1 (fw stands for forward) (5′-GTAGTAGTAGGTACCCCGTCCACCGCAATATTCACGTT-3′) (restriction site in bold) and nfsArevCla1 (rev stands for reverse) (5′-GTAGTAGTAATCGATGGTTGGGCGACGCGCTAA-3′) and cloning into the KpnI/ClaI-digested sites of pZS*24 (17). Strain BW25113 pZS*24nfsB was constructed by amplifying the nfsB ORF using primers nfsBfwCla1 (5′-GTAGTAGTAATCGATGCTGGCACGCAAAATTACTTTCAC-3′) and nfsBrevMlu1 (5′-GTAGTAGTAACGCGTCCGGCAAGAGAGAATTACACTTCGG-3′) and cloning into the ClaI/MluI-digested sites of pZS*24 (17). Amplification of all DNA used for mutant construction and screening was performed with Phusion high-fidelity polymerase (New England BioLabs). Cloning and PCR techniques were performed in accordance with standard protocols (18–20). All restriction enzymes were purchased from New England BioLabs.
alamarBlue reduction screen.
In order to test the fidelity of the screen, Z prime (Z′) scores were determined by the method of Zhang et al. (21) by using nitrofurantoin (NFT) at 4× MIC for the positive control in six columns of a 96-well plate and 1% dimethyl sulfoxide (DMSO) as the negative control in the other six columns of the 96-well plate. The “prodrug hit” Z′ was determined from fluorescence readings after 4 h of incubation, and the direct activity Z′ was determined by analyzing optical density at 600 nm (OD600) data after 24 h of incubation. Each screening plate also contained a negative- and positive-control column at each end of the 96-well plate.
Antimicrobial agents of interest were added to separate wells of a 96-well plate with black sides and a clear bottom (catalog no. 3094; Costar). A 10% solution of alamarBlue (catalog no. PI88952; Thermo Scientific) in MHB along with antibiotics and test compounds were added to the screening plate. Broth cultures of E. coli were grown in MHB to exponential phase (∼107 CFU/ml), diluted 1:10 in MHB, and added to the screening plate containing MHB with 10% alamarBlue and antimicrobial, resulting in a final antibiotic concentration of 50 μg/ml. Control wells contained cells with alamarBlue, but no antimicrobial compound or alamarBlue and antimicrobial compound without cells. A fluorometer (Spectra MAX GeminiXS) was used to take readings at an excitation wavelength of 544 nm and emission wavelength of 590 nm. Kinetic readings were taken at 37°C every 10 min for 240 min.
MIC determination.
For killing experiments and MIC determination, bacterial cells were grown in CA-MHB. MICs were determined according to CLSI recommendations (22, 23). MIC determination for all biosafety level 3 (BSL3) agents were performed at the New England Regional Center of Excellence/Biodefense and Emerging Infectious Diseases (NERCE/BEID) facility at Harvard Medical School in Boston, MA.
Time- and concentration-dependent killing assays.
Prior to the addition of antibiotics and test compounds, overnight cultures were diluted 100-fold into 3 ml of fresh medium in 17- by 100-mm polypropylene tubes and incubated for 1.5 h with aeration at 220 rpm to a cell concentration of ∼2 × 108 CFU/ml. For determination of CFU counts, cells were washed in 1% NaCl solution, serially diluted (10-fold serial dilutions), and plated on LB (Luria-Bertani medium) agar plates supplemented with 20 mM MgSO4.
Biofilm killing.
E. coli biofilms were grown by the hanging-peg model as previously described (24). Briefly, a device containing 96 polystyrene pegs was used (catalog no. 445497; Nunc), with a single peg hanging into each well of a microtiter plate (catalog no. 269787; Nunc). For biofilm formation, the pegs were placed in a sterile 96-well plate filled with MHB and cells (105/ml) and incubated for 24 h at 37°C. Once the biofilms formed on the pegs, they were washed in MHB and placed into a sterile microtiter plate with fresh MHB for drug susceptibility testing. Following 24-h incubation in the presence of an antimicrobial agent, the pegs were washed twice in MHB. The pegs were then moved into a fresh sterile microtiter plate with MHB and incubated for 15 min in a water bath sonicator (Branson ultrasonic cleaner). For each antimicrobial concentration tested, cells were collected from four parallel pegs, serially diluted 10-fold, and plated for colony counting.
Cytotoxicity.
All mammalian cell lines were grown in vented 75-cm2 flasks treated for tissue culture (tissue culture-treated flasks) (catalog no. 353136; BD Falcon) with 5% CO2 at 37°C in Eagle's minimal essential medium (EMEM) with l-glutamine (ATCC 30-2003), supplemented with either 10% or 20% fetal bovine serum (FBS) (ATCC 30-2020). Attached cells were removed from flasks using trypsin-EDTA (0.25% trypsin, 0.53 mM EDTA) and counted using a hemocytometer. The cells (2 × 104 cells) were added to each well of a black-sided, clear flat-bottom, tissue culture-treated 96-well microtiter plate (Costar 3904) containing appropriately diluted test compounds. Amphotericin B was used as a control. The cells were incubated in the presence of compounds for 24 h. The challenged cells were then washed three times with fresh medium and left to recover in fresh EMEM with FBS for 24 h. The medium was then aspirated, and fresh medium containing 10% alamarBlue was added to each well. A fluorometer (Spectra MAX GeminiXS) was used to take readings at an excitation wavelength of 544 nm and emission wavelength of 590 nm. The cytotoxic concentration is determined as greater than or equal to 50% transmittance of the control.
RESULTS
A screen for prodrugs.
Once activated, prodrugs are expected to hit multiple targets, and we used this property to develop a whole-cell screen. We reasoned that prodrugs with a nonspecific mode of action will inhibit general metabolism and that they therefore can be identified with a viability dye such as alamarBlue (resazurin). Actively metabolizing cells reduce blue resazurin to red-colored resofurin, and there is a strong change in fluorescence as well. A cytotoxicity test would then discriminate prodrug candidates from generally toxic compounds.
Known nitrofuran prodrugs nitrofurazone (NFZ) and nitrofurantoin (NFT) rapidly inhibited alamarBlue reduction by E. coli (Fig. 2). The protonophore carbonyl cyanide m-chlorophenyl hydrazine (CCCP) had a similar effect. In contrast, ciprofloxacin, a specific inhibitor of DNA gyrase/topoisomerase, and kanamycin, a protein synthesis inhibitor, had no effect on the initial reduction of resazurin during the first 3 h. Similar results were obtained with Bacillus anthracis (not shown). This differential action of nonspecific versus specific compounds enabled development of a prodrug screen. We determined that with a starting inoculum of ∼2 × 106 CFU/ml, the optimal time point to read fluorescence and differentiate between the rapid shutdown of metabolism by compounds with nonspecific targets and target-specific antibiotics was 4 h. The Z′ for the screen was >0.9 (data not shown), showing that the approach had high fidelity (see Materials and Methods).
FIG 2.
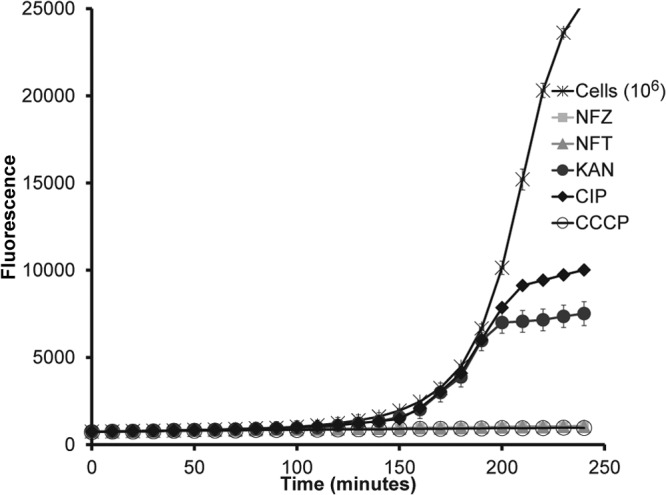
alamarBlue reduction as a basis for a prodrug screen. The prodrugs nitrofurazone (NFZ) and nitrofurantoin (NFT), protonophore CCCP, ciprofloxacin (CIP), and kanamycin (KAN) were added at 50 μg/ml to wells of a microtiter plate containing alamarBlue and E. coli cells. Fluorescence was detected using a excitation wavelength at 544 nm and emission wavelength at 590 nm. Values are means ± standard deviations (SD) (error bars). Data are representative of three independent trials.
A screen was then performed against E. coli, measuring alamarBlue reduction at the 4-h time point after delivering compounds at 35 μg/ml. We started with a pilot screen of 11,000 structurally diverse compounds from the ChemBridge library (E-set). The hit rates for nonspecific compounds inhibiting alamarBlue reduction were 0.05% for E. coli (Table 2) and 14% for B. anthracis. This stark contrast in susceptibility reflects the known differences in the permeability barriers between the Gram-negative E. coli and the Gram-positive B. anthracis. A hit rate of 14% is not useful, and a direct screen against B. anthracis would have to be performed at a considerably lower concentration of compounds. Screening a total of 55,000 compounds against E. coli produced a cumulative hit rate of approximately 0.1%. On the basis of potential medicinal chemistry properties, including low molecular weight (<500) and low lipophilicity (cLogP < 5), and after removal of reactive electrophilic and known promiscuous species, 20 synthetically tractable compounds were selected for further analysis. Of these 20 compounds, three compounds are described in this paper (Fig. 3).
TABLE 2.
Results of the prodrug screena
| Chemical library (no. of compounds) | No. of prodrug hits for E. coli | Hit rate (%) |
|---|---|---|
| ChemBridge library (11,280) | 4 | 0.05 |
| ChemDiv, MPEX library (23,040) | 41 | 0.18 |
| Enamine library (20,160) | 5 | 0.025 |
Compounds were tested against E. coli at 35 μg/ml in wells of a microtiter plate with Mueller-Hinton broth containing alamarBlue. Fluorescence changes were detected after incubation for 4 h. The overall E. coli hit rate was 0.09%. The total number of compounds screened was 54,480. The total number of hits was 50.
FIG 3.
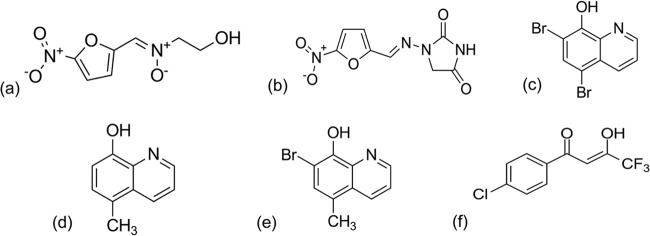
Prodrugs and hit compounds. ADC111 (a) and nitrofurantoin (b) are nitrofurans. ADC112 (c) is an analog of tiliquinol (d) and tilbroquinol (e). ADC113 (f) is a β-diketone.
ADC111 is a nitrofuran resembling nitrofurantoin, a known prodrug that is used to treat urinary tract infections (UTIs) caused by E. coli and other pathogens (25). ADC112 is an analog of tiliquinol and tilbroquinol, the two 8-hydroxyquinoline compounds of Intetrix. ADC112 differs from tilbroquinol only in the R7 group, having a bromine instead of a methyl group. The mechanism of action for 8-hydroxyquinolines remains unknown, although it has been proposed that these compounds chelate metals necessary for multiple enzymatic catalysis reactions, including DNA synthesis (26, 27). ADC113 is a β-diketone and does not belong to an approved class of antimicrobials.
Hit validation.
The aim of the prodrug screen was to find bactericidal compounds that are not generally toxic without a specific target in the bacteria. We therefore examined the cytotoxicity of the hits against mammalian cells. Compounds with a therapeutic index (TI) of ≥10 were considered desirable hits. ADC111 was less toxic against mammalian cells and considerably more active against E. coli compared to its approved analog nitrofurantoin, which translated into an excellent TI (Table 3). For example, the TI of ADC111 with FaDu cells was 320 compared to the nitrofurantoin TI of only 10. ADC112 was also considerably less toxic than its analog tilbroquinol, with a TI of 21/42 versus 4. ADC113 had a TI ranging from 5 to 16 depending on the cell type, a reasonable number for a hit compound. The cytotoxicity data suggested that further evaluation of these compounds was warranted.
TABLE 3.
Cytotoxicity of the hit compoundsa
| Compound | MIC (μg/ml) with 10% FBS | Cytotoxicity (µg/ml) in the following cell lines: |
TI | ||
|---|---|---|---|---|---|
| FaDu | Caco2 | HepG2 | |||
| ADC111 | 0.78 | 250 | 31.25 | 31.25 | 320/40 |
| Nitrofurantoin | 12.5 | 125 | 125 | 125 | 10 |
| ADC112 | 1.5 | 31.25 | 62.5 | 62.5 | 21/42 |
| Tilbroquinol | 25 | 100 | 100 | 100 | 4 |
| ADC113 | 6.25–12.5 | 100 | 100 | 62.5 | 8–16/5–10 |
MICs listed are for wild-type E. coli. Compounds were considered cytotoxic at concentrations where there was less than 50% survival compared to that of the untreated control. Multiple TI values correspond to the two different cytotoxicity values.
Next, we examined the spectrum of activity of the hits. All three compounds showed a reasonably broad spectrum with good activity against Gram-positive and Gram-negative species (Table 4). ADC111 was considerably more active than its analog nitrofurantoin. For example, the E. coli MIC of ADC111 is 0.78 μg/ml, which compares favorably to the nitrofurantoin MIC of 12.5 μg/ml. The compounds showed excellent activity against important drug-resistant pathogens, including methicillin-resistant S. aureus and vancomycin-resistant E. faecalis. The compounds also showed good activity against pathogens important for biodefense, such as B. anthracis, Y. pestis, and F. tularensis.
TABLE 4.
Spectrum of activity for ADC compoundsa
| Speciesb | MIC (μg/ml)c |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| ADC111 | ADC112 | ADC113 | CIP | STR | DOX | RIF | VAN | TIG | |
| Francisella tularensis (SchuS4)* | ND | 0.097 | 0.195–0.34 | ND | 8 | ND | ND | ND | ND |
| Bacillus anthracis (Ames)* | 0.35 | 1.5 | 0.78 | 0.015 | ND | ND | ND | ND | ND |
| Yersinia pestis (KIM)* | ≤0.78 | 1.5 | 0.78–1.56 | ND | ND | 9 | ND | ND | ND |
| Escherichia coli (ATCC 25922) | 0.78 | 1.5 | 6.25–12.5 | 0.015 | ND | ND | ND | ND | ND |
| Staphylococcus aureus (NCTC 8325) | 1.56 | ≤1.5 | 3.12 | 0.15 | ND | ND | ≤0.06 | 1 | ND |
| Enterococcus faecalis (OG1RF, ATCC 47077) | 3.12 | ≤1.5 | 6.25 | 0.031 | ND | ND | ND | ND | ND |
| Salmonella Typhimurium (LT2) | 5 | ND | ND | ND | 2 | ND | ND | ND | ND |
| Staphylococcus aureus (MRSA, NRS54) | 6.25 | ≤1.5 | 6.25 | ND | ND | ND | ≤0.06 | 4 | ND |
| Clostridium difficile (CD196) | 6.25 | ≤1.5 | 6.25 | ND | ND | ND | ND | 1.5 | ND |
| Enterococcus faecium (VRE BM4147) | 12.5 | 6.25 | 6.25 | ND | ND | ND | ND | ND | 0.125 |
| Acinetobacter baumannii (AB17978) | 12.5 | 6.25 | 12.5 | 0.06 | ND | ND | ND | ND | ND |
| Pseudomonas aeruginosa (PAO1) | 25 | >50 | >50 | 0.031 | ND | ND | ND | ND | ND |
MICs were determined according to CLSI guidelines.
BSL3 agents are denoted by an asterisk and were tested at the NERCE/BEID facility in Boston, MA.
Abbreviations: CIP, ciprofloxacin; STR, streptomycin; DOX, doxycycline; RIF, rifampin; VAN, vancomycin; TIG, tigecycline; ND, not determined.
One of the expected features of a prodrug mode of action is good permeability. Since activated prodrugs bind covalently to their targets, this will create an accumulation sink, countering multidrug efflux pumps. The activity of hit compounds was therefore tested in strains lacking the transenvelope pump AcrB (28) or lacking TolC, a common porin with a gated channel that services several MDR pumps (11, 29).
In a control experiment, the activity of erythromycin against E. coli increases 128-fold in a tolC mutant (Table 5), reflecting the prominent role of efflux in protecting the cells from antibiotics. In contrast, there is only a 2- to 4-fold increase in the potency of the known prodrugs nitrofurazone and nitrofurantoin in a tolC strain, and a similar 4-fold increase in the potency of the nitrofuran ADC111. Other candidate prodrugs tested, ADC112 and ADC113, also showed moderate increases in potency against the MDR pump mutant strains.
TABLE 5.
Activity of test compounds against MDR pump mutant strainsa
| Strain | MIC (μg/ml)b |
|||||
|---|---|---|---|---|---|---|
| ADC111 | ADC112 | ADC113 | NFT | NFZ | ERY | |
| BW25113 | 1.25 | 2.5 | 12.5 | 12.5 | 6.25 | 200 |
| ΔtolC mutant | 0.4 | 0.67 | 1.25 | 3.125 | 3.12 | 1.56 |
| ΔacrB mutant | 1.25 | 2.5 | 3.12 | 6.25 | 3.12 | 1.56 |
MIC was determined by broth microdilution.
NFT, nitrofurantoin; NFZ, nitrofurazone; ERY, erythromycin.
Given that ADC111 is a nitrofuran, we examined the activity of this compound against strains lacking the activating enzymes for this class of prodrugs, the nitroreductases NfsA and NfnB. In the alamarBlue test, ADC111 and nitrofurantoin rapidly inhibited reduction of the dye (Fig. 4A). This inhibition was largely relieved in a ΔnfsA ΔnfnB double mutant (Fig. 4B). Similarly, the MIC of ADC11 increased 8-fold in the ΔnfsA ΔnfnB strain compared to the MIC in the wild type (Table 6), consistent with previously published resistance to nitrofuran compounds (30). The nitrofurantoin MIC increased 4-fold in the ΔnfsA ΔnfnB strain. There are likely additional, not-yet characterized nitroreductases in E. coli (31) accounting for residual activity of the prodrugs in the ΔnfsA ΔnfnB mutant. Overexpression of the activating enzymes produced the opposite effect, an increase in potency (Table 6). This behavior is the opposite of what is expected from conventional inhibitors of targets, where downregulation causes increased susceptibility and overexpression leads to reduced activity, but is consistent with prodrugs. This contrasting behavior of prodrugs may serve as a good validation tool for this type of compound. For ADC111, MIC dropped to 15 ng/ml in a strain overexpressing NfsA, showing the potential of a prodrug as a potent therapeutic agent. We were unable to obtain mutants resistant to ADC112 and ADC113 so far. This may indicate the presence of more than one activating enzyme/target.
FIG 4.
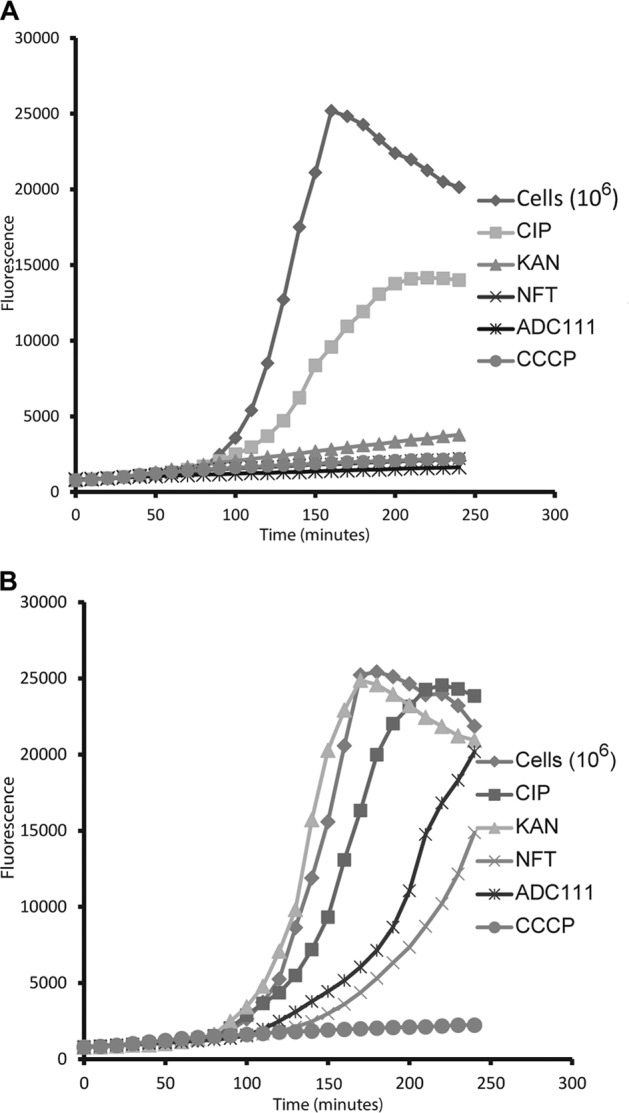
Inhibition of alamarBlue reduction by nitrofurans. (A) Wild-type E. coli; (B) ΔnfsA ΔnfsB double mutant (KAN resistant). The strong inhibition of alamarBlue reduction by kanamycin, as shown in panel A, is due to its high concentration (50 µg/ml). The experiment was performed in a microtiter plate, and fluorescence was measured every 10 min with an excitation wavelength of 544 nm and an emission wavelength of 590 nm.
TABLE 6.
Activity of ADC111 against strains lacking and overexpressing activating enzymesa
| Strain | MIC (μg/ml) |
|
|---|---|---|
| ADC111 | NFT | |
| BW25113 | 2 | 12.5 |
| BW25113ΔnfsAΔnfsB | 16 | 50 |
| BW25113ΔnfsA | 4 | 12.5 |
| BW25113ΔnfsB | 2 | 12.5 |
| nfsA++ | 0.0156 | 0.025 |
| nfsB++ | 1 | 12.5 |
MIC was determined by broth microdilution.
Bactericidal activity of hit compounds.
An ability to hit unrelated targets suggests that prodrugs may be able to effectively kill both growing and dormant cells. Both ADC111 and nitrofurantoin were highly bactericidal against an exponentially growing culture of E. coli. The killing was biphasic, with persisters surviving considerably better than regular cells (Fig. 5).
FIG 5.

Time- and concentration-dependent killing of E. coli BW25113 (wild type) in exponential phase. Cells were challenged with various concentrations (the MIC, 4× MIC, 8× MIC, etc.) of ADC111 (MIC of 2 μg/ml), NFT (MIC of 12.5 μg/ml), and ciprofloxacin (Cipro) (MIC of 0.01 μg/ml). Cell count was determined by plating on nutrient agar. The limit of detection is the x axis. Values are means ± SD. Data are representative of three independent trials. WT, wild type.
Next, we tested the ability of ADC111 to kill cells growing in a biofilm. E. coli produces biofilms on indwelling catheters (3) and forms intracellular biofilms in bladder epithelial cells (32). ADC111 was considerably more effective than nitrofurantoin used to treat UTI in killing a biofilm within a 24-h period (Fig. 6).
FIG 6.
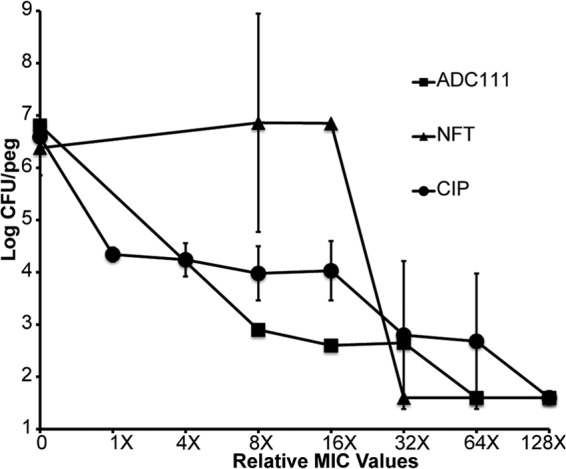
Biofilm killing by ADC111, NFT, and CIP. E. coli biofilms (24 h old) were grown on pegs and then moved into fresh medium containing antimicrobials for 24 h. The cells were dislodged and plated for colony count. Values are means ± SD. Data are representative of three independent trials.
Unlike slow-growing biofilms, a stationary culture produces more persisters and is harder to eradicate (33, 34). This is clear from observing the effect of ciprofloxacin on a stationary culture. Even at high concentrations, the killing is less than 3 log units. Ciprofloxacin reaches a very high concentration in the bladder, up to 400 μg/ml (35). This, however, does not help eradicate the pathogen due to the presence of persister cells and because of the paradoxical relationship between killing efficiency and concentration, which inverts at higher levels of this drug (36). Nitrofurantoin was considerably more effective than ciprofloxacin in killing a stationary culture, and ADC111 had an even greater effect than nitrofurantoin, decreasing the cell count by 7 orders of magnitude (Fig. 7).
FIG 7.
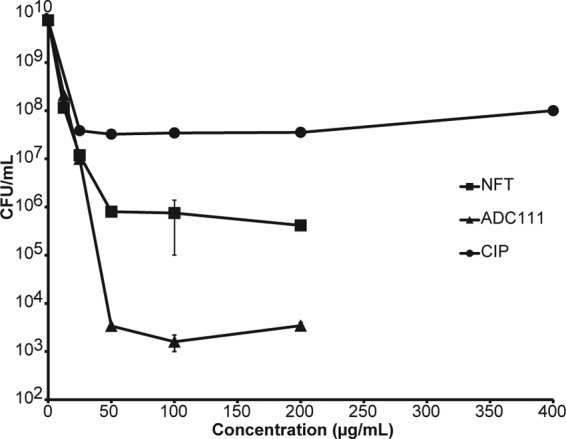
Concentration-dependent killing of wild-type E. coli in stationary phase. The cells were challenged for 24 h. The limit of detection is the x axis. Values are means ± SD. Data are representative of three independent trials.
Next, we tested the ability of ADC112 to kill biofilm and stationary cultures of E. coli. Tilbroquinol, an analog of ADC112, killed well compared to ciprofloxacin but showed a distinct paradoxical effect at higher concentrations (Fig. 8). ADC112 was similar in effectiveness to tilbroquinol but did not exhibit the paradoxical effect, showing a gradual increase in killing at higher concentrations.
FIG 8.
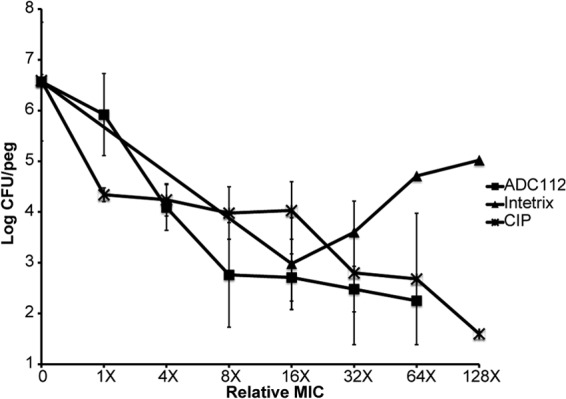
Biofilm killing by ADC112 and tilbroquinol. Biofilms (24 h old) were treated with compounds for 24 h. The number of surviving cells per peg was determined by colony count. The limit of detection is log 1.6 (CFU/ml). Values are means ± SD. Data are representative of three independent trials.
ADC112 was effective in killing stationary cells of E. coli, achieving complete sterilization (Fig. 9).
FIG 9.
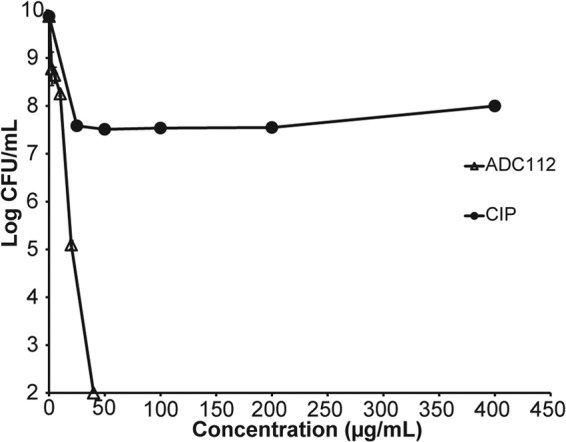
Killing of stationary-phase E. coli with ADC112. Cells were treated for 24 h and then plated for colony count. The limit of detection is the x axis. Values are means ± SD. Data are representative of three independent trials.
ADC113 does not belong to a known class of antimicrobials, and we started by performing a limited structure-activity relationship (SAR) analysis with the aim of obtaining a more potent compound. Three analogs were synthesized (1690, 1689, and 1650 [Fig. 10]), and additional analogous compounds were obtained from the Enamine library. None of the compounds were more potent against E. coli than ADC113 was (Table 7). The two compounds with the best MICs had electronegative halogens on the aryl ring and a diketone functionality (ADC113 and B020549). Replacement of the halogen with an electron-donating group in the aryl ring or removal of fluorogroups from the trifluoromethyl ketone had a negative impact on the MIC. ADC113 had excellent activity against exponentially growing E. coli cells (Fig. 11).
FIG 10.
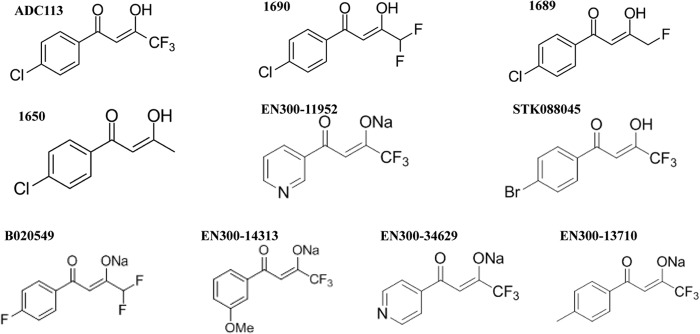
Structural analogs of ADC113. 1690, 1689, and 1650 were synthesized, and EN300-11952, STK08845, B020549, EN300-14313, EN300-34629, and EN300-13710 were obtained from the Enamine library. Me, methyl group.
TABLE 7.
Activity of ADC113 and structural analogs against E. coli
| Compound | MIC (μg/ml) against E. coli |
|---|---|
| ADC113 | 6.25/12 |
| 1690 | 25 |
| EN300-13710 | 50 |
| STK088045 | 50 |
| B020549 | 50 |
| EN300-14313 | 100 |
| 1689 | >100 |
| 1650 | >100 |
| EN300-34629 | >100 |
| EN300-11952 | >100 |
FIG 11.
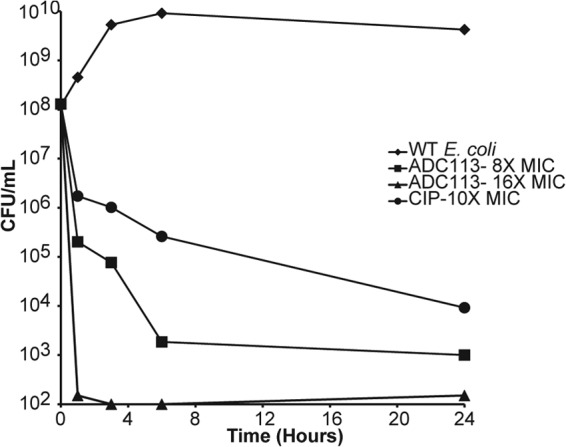
Time- and concentration-dependent killing of exponentially growing wild-type E. coli. Cells were incubated with ADC113, and the cell count was determined by plating. The limit of detection is the x axis. Values are means ± SD. Data are representative of three independent trials.
ADC113 had good killing against biofilms (Fig. 12) and was comparable to ciprofloxacin in killing stationary cells (Fig. 13).
FIG 12.
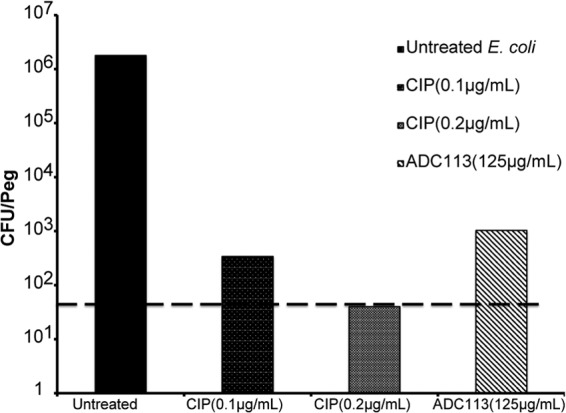
E. coli biofilm killing with ADC113. Biofilms (24 h old) were treated with 10× MIC of ADC113 and 10× and 20× MIC of CIP for 24 h. The number of surviving cells per peg was determined by colony count. The limit of detection is 101.6 CFU/peg and is indicated by the broken line.
FIG 13.
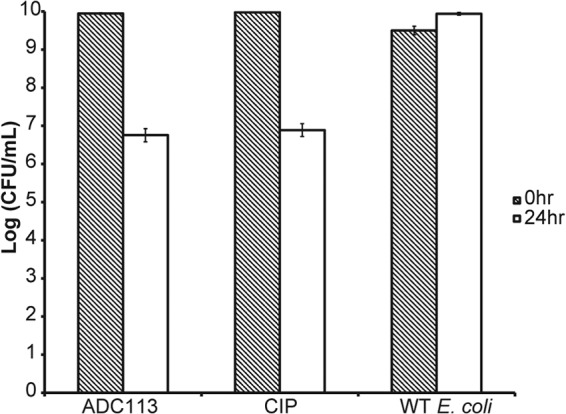
Killing of E. coli in stationary phase. Cells were challenged for 24 h with 32× MIC of ADC113 and CIP.
DISCUSSION
The antibiotic crisis we are facing is due to the lack of good starting compounds (37), a rapid rise of resistance that makes existing antibiotics ineffective, and otherwise successful medical interventions that prolong the lives of patients with deficient immune responses, leading to the rise of recalcitrant chronic infections. The major reason for the lack of new compounds is well understood—most antibiotics are produced by soil actinomycetes, and overmining of this limited resource lead to the end of the golden era of discovery (38). However, most classes of synthetic compounds were also discovered during the golden era of the 1950s and 1960s, and this is harder to understand, given enormous advances in chemistry and biology (12). A particularly puzzling case is that of antibiotics with a prodrug mode of action. As mentioned in the introduction, all of these compounds were originally discovered in the 1950s. Additional attractive compounds of the nitrofuran class have been recently reported (39). Prodrugs have the features of a theoretically “ideal” antibiotic, a compound that is broad spectrum, nontoxic, and able to kill both growing cells and dormant persister cells (40) (Fig. 1).
Poor penetration across the complex envelope of Gram-negative bacteria is the main reason synthetic approaches have not been successful in obtaining broad-spectrum compounds (41), with the sole exception of fluoroquinolones. The irreversible binding of activated prodrugs to their targets creates a sink, ensuring good accumulation over time. In this study, we tested these assumptions and also developed a screen for prodrug compounds. Prodrugs such as metronidazole do not have a specific target (42, 43), and historically, once validation steps such as specificity testing were introduced, these types of antimicrobials were excluded from the discovery process. We decided to revisit prodrugs and developed a screen based on the lack of specificity of mechanism of action, essentially looking at compounds that are discarded in conventional high-throughput screening (HTS) campaigns. Using the vital dye alamarBlue, we identified hits acting against E. coli that inhibited reduction of this reporter of general metabolism and then tested the compounds for cytotoxicity. Testing for cytotoxicity in mammalian cells eliminates general target-specific energy poisons, but not energy poisons that are desirable compounds. The screen successfully differentiates target-specific compounds from known prodrugs such as nitrofurantoin. A pilot HTS with 55,000 compounds produced 50 hits, and 3 of these with low cytotoxicity were examined. Importantly, one of the compounds, ADC111, is a nitrofurazone analog of nitrofurantoin which is used to treat urinary tract infections (UTIs) (25, 44). The activity of ADC111 depended on the presence of nitroreductases in E. coli. This discovery of a prodrug validates the screen. ADC112 is an analog of another antimicrobial that has been used in the clinic, tilbroquinol. The mode of action of tilbroquinol is unknown. We also identified ADC113, a compound that has been previously reported to be a putative dehydratase inhibitor in mycobacteria (45); however, we find it to be acting as a nonspecific compound, possibly as a prodrug. Taken together, the results of the pilot screen suggest that a larger HTS is likely to produce a number of novel prodrug leads.
We also examined the main predictions of the prodrug model—the ability of compounds to have a broad spectrum of action and kill dormant cells. There was little effect of MDR pumps on the MICs of prodrugs, in contrast to erythromycin, a model substrate of MDR pumps. This suggests that irreversible binding to targets may indeed impart prodrugs with good penetration properties, enabling a broad spectrum of action. Importantly, prodrugs were able to effectively kill biofilms of E. coli and stationary cells that produce large amounts of dormant persisters. Biofilms of E. coli are often associated with recurring UTIs. E. coli forms intracellular biofilms in the bladder epithelial cells (32), and these are likely to contain persister cells. In some patients with recurring UTIs, E. coli forms “quiescent intracellular reservoirs” (QIRs) residing in Lamp1+ endosomes of bladder epithelial cells (46). QIRs are protected from the immune system and are not killed by antibiotics (47). When bladder epithelial turnover occurs and antibiotic treatment has ceased, viable E. coli cells reemerge and cause a recurrence of the UTI (48). QIRs may be equivalent to persisters. The pathways of persister formation are highly redundant, precluding development of specific anti-persister compounds. Activation of the ClpP protease in S. aureus by acyldepsipeptide leads to the degradation of proteins and death of persisters, resulting in pathogen eradication in vitro and in vivo (49). Target-specific antibiotics such as ciprofloxacin kill the bulk of the population, leaving approximately 1% of surviving persisters. Increasing the concentration of ciprofloxacin does not lead to more killing. Nitrofurantoin killed stationary cells much better than ciprofloxacin did, and ADC111 almost completely eradicated a stationary population. An irreversibly binding compound will actually be more effective against nongrowing dormant cells; in rapidly propagating bacteria, a prodrug will be diluted, diminishing the sink effect.
Taken together, results of this study suggest that prodrugs are a promising type of antimicrobials capable of sterilizing and broad-spectrum activity. The screen we developed provides a platform for the discovery of prodrugs that have been overlooked in conventional screening campaigns.
ACKNOWLEDGMENTS
This study was supported by NIH grant T-RO1 AI085585 to K.L. and the American Lebanese Syrian Associated Charities (ALSAC), St. Jude Children's Research Hospital, to R.E.L.
We thank Marin Vulić for assistance with E. coli strain construction. We thank the NERCE/BEID facility at Harvard Medical School in Boston, MA, for MIC and minimal bactericidal concentration (MBC) testing against BSL3 agents. We thank Novobiotic Pharmaceuticals for sharing bacterial strains. We thank Linc Sonenshein at Tufts University for sharing bacterial strains.
Footnotes
Published ahead of print 16 December 2013
REFERENCES
- 1.Bush K, Courvalin P, Dantas G, Davies J, Eisenstein B, Huovinen P, Jacoby GA, Kishony R, Kreiswirth BN, Kutter E, Lerner SA, Levy S, Lewis K, Lomovskaya O, Miller JH, Mobashery S, Piddock LJ, Projan S, Thomas CM, Tomasz A, Tulkens PM, Walsh TR, Watson JD, Witkowski J, Witte W, Wright G, Yeh P, Zgurskaya HI. 2011. Tackling antibiotic resistance. Nat. Rev. Microbiol. 9:894–896. 10.1038/nrmicro2693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lewis K. 2010. Persister cells. Annu. Rev. Microbiol. 64:357–372. 10.1146/annurev.micro.112408.134306 [DOI] [PubMed] [Google Scholar]
- 3.Costerton JW, Stewart PS, Greenberg EP. 1999. Bacterial biofilms: a common cause of persistent infections. Science 284:1318–1322. 10.1126/science.284.5418.1318 [DOI] [PubMed] [Google Scholar]
- 4.Keren I, Shah D, Spoering A, Kaldalu N, Lewis K. 2004. Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. J. Bacteriol. 186:8172–8180. 10.1128/JB.186.24.8172-8180.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maisonneuve E, Shakespeare LJ, Jorgensen MG, Gerdes K. 2011. Bacterial persistence by RNA endonucleases. Proc. Natl. Acad. Sci. U. S. A. 108:13206–13211. 10.1073/pnas.1100186108 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 6.Schumacher MA, Piro KM, Xu W, Hansen S, Lewis K, Brennan RG. 2009. Molecular mechanisms of HipA-mediated multidrug tolerance and its neutralization by HipB. Science 323:396–401. 10.1126/science.1163806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dorr T, Vulić M, Lewis K. 2010. Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLoS Biol. 8:e1000317. 10.1371/journal.pbio.1000317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keren I, Wu Y, Inocencio J, Mulcahy LR, Lewis K. 2013. Killing by bactericidal antibiotics does not depend on reactive oxygen species. Science 339:1213–1216. 10.1126/science.1232688 [DOI] [PubMed] [Google Scholar]
- 9.Hooper DC. 2001. Mechanisms of action of antimicrobials: focus on fluoroquinolones. Clin. Infect. Dis. 15:S9–S15 [DOI] [PubMed] [Google Scholar]
- 10.Lomovskaya O, Lewis K. 1992. Emr, an Escherichia coli locus for multidrug resistance. Proc. Natl. Acad. Sci. U. S. A. 89:8938–8942. 10.1073/pnas.89.19.8938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li XZ, Nikaido H. 2004. Efflux-mediated drug resistance in bacteria. Drugs 64:159–204. 10.2165/00003495-200464020-00004 [DOI] [PubMed] [Google Scholar]
- 12.Lewis K. 2013. Platforms for antibiotic discovery. Nat. Rev. Drug Discov. 12:371–387. 10.1038/nrd3975 [DOI] [PubMed] [Google Scholar]
- 13.Hilliard JJ, Goldschmidt RM, Licata L, Baum EZ, Bush K. 1999. Multiple mechanisms of action for inhibitors of histidine protein kinases from bacterial two-component systems. Antimicrob. Agents Chemother. 43:1693–1699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller JH. 1992. A short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 15.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2:2006.0008. 10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645. 10.1073/pnas.120163297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lutz R, Bujard H. 1997. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 25:1203–1210. 10.1093/nar/25.6.1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. (ed). 1987. Current protocols in microbiology. John Wiley & Sons, Inc., New York, NY [Google Scholar]
- 19.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. (ed). 1992. Short protocols in microbiology, 2nd ed. John Wiley & Sons, Inc., New York, NY [Google Scholar]
- 20.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual. Cold Spring Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 21.Zhang JH, Chung TD, Oldenburg KR. 1999. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 4:67–73. 10.1177/108705719900400206 [DOI] [PubMed] [Google Scholar]
- 22.Clinical and Laboratory Standards Institute 2012. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard-ninth edition. CLSI document M07-A9. Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 23.Clinical and Laboratory Standards Institute 2012. Methods for antimicrobial susceptibility testing of anaerobic bacteria; approved standard-eighth edition. CLSI document M11-A8. Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 24.Ceri H, Olson ME, Stremick C, Read RR, Morck D, Buret A. 1999. The Calgary Biofilm Device: new technology for rapid determination of antibiotic susceptibilities of bacterial biofilms. J. Clin. Microbiol. 37:1771–1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guay DR. 2001. An update on the role of nitrofurans in the management of urinary tract infections. Drugs 61:353–364. 10.2165/00003495-200161030-00004 [DOI] [PubMed] [Google Scholar]
- 26.Bryskier A. (ed). 2005. Antimicrobial agents: antibacterials and antifungals. ASM Press, Washington, DC [Google Scholar]
- 27.Gershon H, Parmegiani R. 1963. Antimicrobial activity of 8-quinolinol, its salts with salicylic acid and 3-hydroxy-2-naphthoic acid, and the respective copper (II) chelates in liquid culture. Appl. Microbiol. 11:62–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okusu H, Ma D, Nikaido H. 1996. AcrAB efflux pump plays a major role in the antibiotic resistance phenotype of Escherichia coli multiple-antibiotic-resistance (Mar) mutants. J. Bacteriol. 178:306–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tegos G, Stermitz FR, Lomovskaya O, Lewis K. 2002. Multidrug pump inhibitors uncover remarkable activity of plant antimicrobials. Antimicrob. Agents Chemother. 46:3133–3141. 10.1128/AAC.46.10.3133-3141.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCalla DR, Kaiser C, Green MH. 1978. Genetics of nitrofurazone resistance in Escherichia coli. J. Bacteriol. 133:10–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Whiteway J, Koziarz P, Veall J, Sandhu N, Kumar P, Hoecher B, Lambert IB. 1998. Oxygen-insensitive nitroreductases: analysis of the roles of nfsA and nfsB in development of resistance to 5-nitrofuran derivatives in Escherichia coli. J. Bacteriol. 180:5529–5539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anderson GG, Palermo JJ, Schilling JD, Roth R, Heuser J, Hultgren SJ. 2003. Intracellular bacterial biofilm-like pods in urinary tract infections. Science 301:105–107. 10.1126/science.1084550 [DOI] [PubMed] [Google Scholar]
- 33.Keren I, Kaldalu N, Spoering A, Wang Y, Lewis K. 2004. Persister cells and tolerance to antimicrobials. FEMS Microbiol. Lett. 230:13–18. 10.1016/S0378-1097(03)00856-5 [DOI] [PubMed] [Google Scholar]
- 34.Spoering AL, Lewis K. 2001. Biofilms and planktonic cells of Pseudomonas aeruginosa have similar resistance to killing by antimicrobials. J. Bacteriol. 183:6746–6751. 10.1128/JB.183.23.6746-6751.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gilbert DN. 2006. Urinary tract infections in patients with chronic renal insufficiency. Clin. J. Am. Soc. Nephrol. 1:327–331. 10.2215/CJN.01931105 [DOI] [PubMed] [Google Scholar]
- 36.Lewin CS, Morrissey I, Smith JT. 1991. The mode of action of quinolones: the paradox in activity of low and high concentrations and activity in the anaerobic environment. Eur. J. Clin. Microbiol. Infect. Dis. 10:240–248. 10.1007/BF01966996 [DOI] [PubMed] [Google Scholar]
- 37.Payne DJ, Gwynn MN, Holmes DJ, Pompliano DL. 2007. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 6:29–40. 10.1038/nrd2201 [DOI] [PubMed] [Google Scholar]
- 38.Lewis K. 2012. Antibiotics: recover the lost art of drug discovery. Nature 485:439–440. 10.1038/485439a [DOI] [PubMed] [Google Scholar]
- 39.Miyamoto Y, Kalisiak J, Korthals K, Lauwaet T, Cheung DY, Lozano R, Cobo ER, Upcroft P, Upcroft JA, Berg DE, Gillin FD, Fokin VV, Sharpless KB, Eckmann L. 2013. Expanded therapeutic potential in activity space of next-generation 5-nitroimidazole antimicrobials with broad structural diversity. Proc. Natl. Acad. Sci. U. S. A. 110:17564–17569. 10.1073/pnas.1302664110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lewis K. 2007. Persister cells, dormancy and infectious disease. Nat. Rev. Microbiol. 5:48–56. 10.1038/nrmicro1557 [DOI] [PubMed] [Google Scholar]
- 41.Alekshun MN, Levy SB. 2007. Molecular mechanisms of antibacterial multidrug resistance. Cell 128:1037–1050. 10.1016/j.cell.2007.03.004 [DOI] [PubMed] [Google Scholar]
- 42.Edwards DI. 1993. Nitroimidazole drugs–action and resistance mechanisms I. Mechanism of action. J. Antimicrob. Chemother. 31:9–20. 10.1093/jac/31.1.9 [DOI] [PubMed] [Google Scholar]
- 43.Müller M. 1986. Reductive activation of nitroimidazoles in anaerobic microorganisms. Biochem. Pharmacol. 35:37–41. 10.1016/0006-2952(86)90552-6 [DOI] [PubMed] [Google Scholar]
- 44.Shah RR, Wade G. 1989. Reappraisal of the risk/benefit of nitrofurantoin: review of toxicity and efficacy. Adverse Drug React. Acute Poisoning Rev. 8:183–201 [PubMed] [Google Scholar]
- 45.Bhowruth V, Brown AK, Besra GS. 2008. Synthesis and biological evaluation of NAS-21 and NAS-91 analogues as potential inhibitors of the mycobacterial FAS-II dehydratase enzyme Rv0636. Microbiology 154:1866–1875. 10.1099/mic.0.2008/017434-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mysorekar IU, Hultgren SJ. 2006. Mechanisms of uropathogenic Escherichia coli persistence and eradication from the urinary tract. Proc. Natl. Acad. Sci. U. S. A. 103:14170–14175. 10.1073/pnas.0602136103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schilling JD, Hultgren SJ. 2002. Recent advances into the pathogenesis of recurrent urinary tract infections: the bladder as a reservoir for uropathogenic Escherichia coli. Int. J. Antimicrob. Agents 19:457–460. 10.1016/S0924-8579(02)00098-5 [DOI] [PubMed] [Google Scholar]
- 48.Silverman JA, Schreiber HL, IV, Hooton TM, Hultgren SJ. 2013. From physiology to pharmacy: developments in the pathogenesis and treatment of recurrent urinary tract infections. Curr. Urol. Rep. 14:448–456. 10.1007/s11934-013-0354-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Conlon BP, Nakayasu ES, Fleck LE, Lafleur MD, Isabella VM, Coleman K, Leonard SN, Smith RD, Adkins JN, Lewis K. 2013. Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature 503:365–370. 10.1038/nature12790 [DOI] [PMC free article] [PubMed] [Google Scholar]