

Abstract
Cryptosporidium parasites are a major cause of diarrhea and malnutrition in the developing world, a frequent cause of waterborne disease in the developed world, and a potential bioterrorism agent. Currently, available treatment is limited, and Cryptosporidium drug discovery remains largely unsuccessful. As a result, the pharmacokinetic properties required for in vivo efficacy have not been established. We have been engaged in a Cryptosporidium drug discovery program targeting IMP dehydrogenase (CpIMPDH). Here, we report the activity of eight potent and selective inhibitors of CpIMPDH in the interleukin-12 (IL-12) knockout mouse model, which mimics acute human cryptosporidiosis. Two compounds displayed significant antiparasitic activity, validating CpIMPDH as a drug target. The best compound, P131 (250 mg/kg of body weight/day), performed equivalently to paromomycin (2,000 mg/kg/day) when administered in a single dose and better than paromomycin when administered in three daily doses. One compound, A110, appeared to promote Cryptosporidium infection. The pharmacokinetic, uptake, and permeability properties of the eight compounds were measured. P131 had the lowest systemic distribution but accumulated to high concentrations within intestinal cells. A110 had the highest systemic distribution. These observations suggest that systemic distribution is not required, and may be a liability, for in vivo antiparasitic activity. Intriguingly, A110 caused specific alterations in fecal microbiota that were not observed with P131 or vehicle alone. Such changes may explain how A110 promotes parasitemia. Collectively, these observations suggest a blueprint for the development of anticryptosporidial therapy.
INTRODUCTION
Cryptosporidium parasites, especially C. parvum and C. hominis, are frequent causes of diarrheal disease (1–4). Cryptosporidium oocysts are highly resistant to most methods of water treatment, so outbreaks occur with regularity even in the developed world. In fact, Cryptosporidium was identified as the cause of 87% of cases of waterborne illness in the United States in 2007 (5). Disease is self-limiting in healthy adults but can be chronic and fatal in immunocompromised individuals. Small children, especially infants, are also highly susceptible. The recent GEMS epidemiological study found Cryptosporidium second only to rotavirus as a cause of childhood diarrhea (6). Cryptosporidium was highly associated with moderate to severe diarrhea and death in infants over the study period. Cryptosporidium infection can also cause an unrecoverable growth deficit in young children, making these parasites a major cause of the “vicious cycle” of diarrhea and malnutrition in the developing world (7). C. parvum oocysts can be obtained with relative ease, and the water supply is readily accessed, so there is also a credible concern that these organisms could be used maliciously (8). The 1993 natural Milwaukee outbreak illustrates the potential damage of such an act of bioterrorism: contaminated drinking water resulted in approximately 403,000 cases of disease, the hospitalization of 4,400 patients, and an estimated 69 deaths (9).
Although hundreds of antiparasitic and antimicrobial drugs have been evaluated for anticryptosporidial activity, the current treatment options are limited to one approved drug, nitazoxanide, which hastens the resolution of symptoms in immunocompetent patients (10). Nitazoxanide is less efficacious in malnourished children and shows no benefit in immunocompromised patients (11). Importantly, the target of nitazoxanide is undefined in Cryptosporidium, so no clinically validated targets exist for the treatment of cryptosporidiosis (12). Paromomycin is effective in rodents and has also been used to treat cryptosporidiosis, though a significant benefit has not been demonstrated in humans (13). Approximately 20 additional compounds have displayed at least some anticryptosporidial activity in animal models (14–26). Unfortunately, as with nitazoxanide, the targets of most of these compounds have not been identified, which makes further development difficult. Several promising targets have emerged with the sequencing of the C. parvum and C. hominis genomes (27–37), but only two target-based drug discovery programs have reported activity in an animal model (26, 37). Adding to the challenge, given the limited efficacy of these compounds, the pharmacokinetic and physicochemical properties required for in vivo efficacy have not been established. Clearly, new strategies are needed to combat cryptosporidiosis in immunocompetent and especially immunocompromised patients.
Cryptosporidium spp. are obligate intracellular parasites (38, 39). Infections can occur when as few as 1 to 10 oocysts are ingested. Oocysts release sporozoites in the intestine, where infections are predominately localized to the jejunum and ileum but can extend to other parts of the gastrointestinal tract in immunocompromised patients. Biliary and other organ involvement also occurs in approximately 20% of immunocompromised patients (39–41). The parasite resides within a parasitophorous vacuole that protrudes out of the host cytoplasm into the intestinal lumen. The routes of nutrient and drug uptake, whether direct from the intestinal lumen or via the host cell, are largely unknown. Unfortunately, Cryptosporidium parasites cannot be cultured continuously in vitro and genetic tools do not yet exist to construct transgenic reporter parasites that would greatly facilitate screening efforts. Tissue culture models of Cryptosporidium infection provide an imperfect window to measure drug effects and certainly do not recapitulate the complex environment of the gastrointestinal tract, which includes a myriad of commensal organisms that may influence infection (42). Several animal models exist that mimic either acute or chronic human disease, though these generally require immunosuppression to permit Cryptosporidium infection. These conditions constrain drug discovery efforts.
We have been engaged in a program to develop inhibitors of C. parvum IMP dehydrogenase (CpIMPDH) as potential treatment for cryptosporidiosis. This enzyme is a promising target because Cryptosporidium relies on CpIMPDH for the biosynthesis of guanine nucleotides (note that C. hominis contains the identical enzyme and the same guanine biosynthetic pathway [27–29]). Moreover, the CpIMPDH gene was obtained from an epsilonproteobacterium by lateral gene transfer and thus is very different from host IMPDHs (30, 43). We have developed several structurally distinct classes of CpIMPDH inhibitors, achieving nanomolar potency and selectivity in excess of 1,000-fold versus the human enzymes (44–50). Here, we report the antiparasitic activity of our eight most promising compounds in the interleukin-12 (IL-12) knockout mouse model. Infection is confined to the intestine in this model and resolves in 2 weeks, which is similar to acute human cryptosporidiosis (51). One compound, P131, displays greater antiparasitic activity than paromomycin, validating CpIMPDH as a target. Our results also provide insights into the properties required for anticryptosporidial activity.
MATERIALS AND METHODS
Materials.
Compounds were synthesized as previously described (45, 48). Properties were calculated using ChemBioDraw Ultra version 12.0.3.1216 (Cambridgesoft).
Cell culture model of C. parvum infection.
In vitro evaluation was performed as described previously (52). C. parvum oocysts were kindly supplied by Michael Arrowood (Centers for Disease Control and Prevention). C. parvum oocysts (Iowa bovine isolate) were collected, purified through discontinuous sucrose and cesium chloride gradients, and stored as previously described (53). Before use, purified C. parvum oocysts were washed free of 2.5% aqueous potassium dichromate (K2Cr2O7, a storage buffer) with phosphate-buffered saline (PBS, pH 7.4). Oocysts were then resuspended in Dulbecco's modified Eagle's medium (DMEM) base with 0.75% sodium taurocholate and incubated for 10 min at 37°C. The excystation mixture was diluted with Ultraculture medium (BioWhittaker Inc., Walkersville, MD), and approximately 1 × 105 oocysts and sporozoites were allowed to infect confluent human ileocecal adenocarcinoma epithelial cells (HCT-8) or Madin-Darby canine kidney cells (MDCK). The monolayer was washed with PBS after 3 h and incubated with fresh Ultraculture medium with or without test compounds, inhibitor and media were refreshed after 24 h, and the parasites were cultured for a total of 48 h. Cultures were fixed and counted using an anti-C. parvum fluorescein-labeled monoclonal antibody (C3C3-fluorescein isothiocyanate [FITC]) or a high-content imaging assay (54). The 50% effective concentration (EC50) values were calculated using the Hill-Slope model using Prism v5 (GraphPad Software Inc., La Jolla, CA): % growth = (maximum − minimum)/{1 + (EC50/[I])n}, where n is the Hill coefficient.
In vivo toxicity evaluation.
Compound toxicity was evaluated at 250 mg/kg of body weight in uninfected C57BL/6 mice treated for 7 days (5 mice/group). Toxicity was assessed by weight loss and signs of distress (e.g., ruffled fur, hunched shoulders, and decreased appetite). No overt signs of toxicity were observed for any of the compounds. No significant changes in weight were observed between treated and vehicle control mice.
Mouse model of C. parvum infection.
The anticryptosporidial activity of the CpIMPDH inhibitors was assessed in the IL-12 knockout mouse model that resembles the acute human disease (51, 55). The protocol was approved by the Institutional Animal Care and Use Committees of Emory University, the Atlanta VA Medical Center, and Brandeis University. Mice (6 to 10 per group) were inoculated with 1,000 purified C. parvum oocysts (Iowa isolate, from cattle). Treatment by gavage began 4 h postinfection with either vehicle (5% dimethyl sulfoxide [DMSO] in corn oil), 250 mg/kg compound, or 2,000 mg/kg paromomycin. Compounds were given for 7 days, and mice were sacrificed on day 8 (peak infection). Parasite load was quantified by fluorescence-activated cell sorting (FACS) assays for the presence of the oocysts in the feces at days 0, 4, and 7. Fecal pellets from individual mice were routinely collected daily and homogenized in adjusted volumes of 2.5% potassium dichromate. Samples were processed individually. Aliquots (200 μl) of vortexed samples were processed over microscale sucrose gradients as previously described (56). The oocyst-containing fraction was collected, washed, and treated with monoclonal antibody (OW5O-FITC) for 20 min. Samples were adjusted to 600 μl, and a portion (100 μl) was assayed with a 102-s sampling interval using logical gating of forward/side scatter and OW5O-FITC fluorescence signal on a Becton, Dickinson FACScan flow cytometer (57). Flow cytometry data were evaluated by analysis of variance (KaleidaGraph [Synergy Software, Reading PA]; Microsoft Excel [Microsoft Corporation, Redmond, WA]).
PK.
Pharmacokinetics (PK) was assessed at either Stony Brook Translation Experimental Laboratory Therapeutics (Stony Brook, NY) or GVK Biosciences (Hyderabad, India). C57BL/6 mice (12 per analysis) were treated with a single dose of 250 mg/kg compound in 5% DMSO-corn oil by oral gavage. Plasma samples were collected from 3 mice predose and 0.25, 0.5, 1, 2, 4, 6, and 24 h after administration. Two samples were collected from each mouse. Samples were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) on a Thermo TSQ Quantum Access (Thermo-Fisher) triple quadrupole mass spectrometer.
Tissue concentrations of P131.
C57BL/6 and IL-12 knockout mice were treated with three daily doses of 83 mg/kg P131 in 5% DMSO-corn oil by oral gavage. Tissues were collected 24 h after the initial dose and stored at −80°C. Before analysis, the tissues were thawed, weighed, and transferred into a 12- by 75-mm heavy-wall glass test tube. A methanol solution (1.0 ml) containing internal standard (0.5 μM formononetin) was added to the test tube. The tissue was processed (about 20 s) with a Tissue Tearor homogenizer (Biospec Products, Bartlesville, OK) into a homogeneous mixture and transferred carefully into a 1.5-ml microcentrifuge tube. The tube was washed twice (200 μl each) with methanol. All solutions were then combined in the centrifuge tube and centrifuged for 15 min at 15,500 rpm. Then, 1.0 ml of supernatant was taken out and dried under nitrogen. The residual was reconstituted in 200 μl of methanol. A standard curve was prepared in blood with the same procedure, and this curve was used for quantification of P131 in blood and tissues.
Cell culture.
Caco-2 TC7 cells were originally a gift of Monique Rousset of INSERM U178 (Villejuif, France). Briefly, a cell monolayer was prepared by seeding 250,000 cells per insert (Nunc; surface area of 4.2 cm2, 3-μm pore size). Cells were maintained at 37°C under 90% humidity and 5% CO2. Monolayers were used between 19 and 22 days after seeding. The integrity of each monolayer was checked by measuring the transepithelial electrical resistance (TEER) (Millicell-ERS voltohmmeter) before the experiment. The normal TEER values obtained were between 500 and 750 Ω·cm2. Cell monolayers with TEER values of <500 Ω·cm2 were not used. Hanks balanced salt solution (HBSS) (9.8 g/ml) supplemented with NaHCO3 (0.37 g/liter), HEPES (5.96 g/liter), and glucose (3.5 g/liter) was used for all experiments after the pH had been adjusted to a desired value. Cells were used at passages 41 to 49.
Transport experiments using the Caco-2 cell culture model.
The experiment protocol and calculation were described in our previous reports (58). Briefly, an HBSS containing the compound(s) of interest was loaded onto the apical or basolateral (donor) side. Five donor samples (500 μl) and five receiver samples (500 μl) were taken at 0, 1, 2, 3, and 4 h followed by the addition of 500 μl of fresh donor solution to the donor side or 500 μl of fresh buffer to the receiver side. The samples were then analyzed by ultrahigh-performance liquid chromatography (UPLC)–MS/MS. The apparent permeability coefficient (P) was determined by the equation P = (dQ/dt)/(A × Co), where dQ/dt is the drug permeation rate (μmol/s), A is the surface area of the epithelium (cm2), and Co is the initial concentration in the donor compartment at time zero (mM).
Uptake experiments.
The uptake of tested compounds was determined according to our previous protocol (58). Briefly, after the transport study, the cell membranes were rinsed three times with ice-cold HBSS, pH 7.4. The cells attached to the polycarbonate membranes were cut off from the inserts with a sharp blade. The latter was immersed into 1 ml of HBSS and sonicated for half an hour at low temperature (iced water bath) to break up the cells. The mixture was centrifuged at 15,000 rpm for 15 min, and 500 μl of the supernatant was taken, which was subjected to LC-MS analysis as described in Table S2 in the supplemental material.
DNA isolation and microbiota sequencing.
Fecal samples from 30 individual mice (uninfected IL-12 knockout mice) were freshly collected and stored at −20°C prior to treatment (day 0). Mice were randomly assigned to three groups (10/group) and treated with either vehicle alone, A110, or P131 (250 mg/kg/day in 5% DMSO-corn oil). The fresh fecal pellets from each individual mouse were collected into sterile Eppendorf tubes at day 0 (prior to treatment) and day 7 (after 7 days of treatment) and stored at −20°C. The total genomic DNA was isolated from individual fecal pellets using the Maxwell automated DNA isolation method as implemented in a Promega genomic DNA isolation kit. The 16S rRNA genes were amplified using individual genomic DNA template with the universal primer pair 27f (AGAGTTTGATCCTGGCTCAG) and 534r (ATTACCGCGGCTGCTGG), which produce an amplicon containing variable regions V1 to V3. The primers were anchored with adapters and barcodes to identify each sample in a multiplexed 454 sequencing reaction. PCR amplification was performed with a FastStart Hifidelity PCR system (Roche) using 0.5 μM primer concentrations. The PCR cycling conditions were 95°C for 5 min, followed by 30 cycles of 94°C for 30 s, 56°C for 30 s, and 72°C for 1 min and 30 s with a final extension period of 8 min at 72°C. Each PCR was performed in triplicate and pooled for gel purification. The PCR amplicon products were pooled and purified using Qiagen gel purification columns. The amplicon pool was quantified using a QuatiT Picogreen kit (Invitrogen). The pooled purified amplicons were sequenced using a 454Roche Jr instrument according to the manufacturer's protocols.
Microbiota sequence analysis.
The bacterial 16S rRNA gene sequence analysis was performed using the QIIME pipeline (QIIME 1.6.0, www.qiime.org) developed and maintained by Rob Knight's group (59). Briefly, the quality sequences (200- to 650-bp lengths) were demultiplexed based on their barcodes. The 16S rRNA operational taxonomic units (OTUs) were picked based on 97% sequence identity using UCLUST (60) against the GreenGenes 16S rRNA database (http://greengenes.lbl.gov/cgi-bin/nph-citation.cgi) (gg_otus-12_10) (61). The GreenGene taxonomies were used to generate the taxon summaries at different levels of phylogeny (phylum, order, class, family, genus, and species). Each OTU was represented by a single sequence that was aligned by Python Nearest Alignment Space Termination (PyNAST [62]) for phylogenetic tree-based analyses. To standardize the sequences across the samples with uneven sampling, the sequences were rarified at 1,000 randomly selected sequences per sample. The phylogenetic tree was built with FastTree (63). The beta diversity (diversity between the samples) was measured using both weighted and unweighted UniFrac measurements (64, 65). The detailed analytical protocols and scripts can be found at www.qiime.org (66).
RESULTS
Compound selection.
The unique intracellular, extracytosolic localization of Cryptosporidium within intestinal epithelial cells presents a special challenge to drug design. We reasoned that a successful drug must traverse the gastrointestinal tract and cross both host and parasite membranes to reach the parasite target, much like an orally active drug must cross the intestinal epithelium to reach systemic circulation. Therefore, medicinal chemistry optimization chiefly followed the guidelines for oral bioavailability, e.g., Lipinski's and Veber's rules, with respect to molecular weight, hydrogen bond donors and acceptors, hydrophobicity (CLogP), polarity (topological polar surface area [TPSA]), and number of rotatable bonds (67, 68). One compound, P131, was designed to be retained in the gastrointestinal tract and therefore exceeded the recommended TPSA (TPSA of ≤140 Å2). Compounds were evaluated for enzyme inhibition and antiparasitic activity against a reporter Toxoplasma gondii strain (T. gondii/CpIMPDH) engineered to rely on CpIMPDH for the production of guanine nucleotides (54). Compounds that performed well in these two assays, with 50% inhibitory concentrations (IC50) of ≤20 nM and EC50s of ≤2 μM, were candidates for testing in the IL-12 knockout mouse model of acute cryptosporidiosis.
Several additional assays were performed to further prioritize compounds for testing in the mouse model. Compounds were evaluated for stability in mouse liver microsomes, which serve as a convenient model for liver metabolism. However, since the tissue distribution required for in vivo antiparasitic activity has not been defined, this information was not used to eliminate candidates. Instead, compounds were initially selected to have a range of metabolic stabilities. Antiparasitic activity was also assessed in a tissue culture model of C. parvum infection (Table 1). However, since parasite proliferation is limited and does not recapitulate the full life cycle in vitro, candidates were eliminated only if the EC50 exceeded 20 μM. It is worth noting that the efficacies of nitazoxanide and paromomycin vary depending upon which stage of the life cycle is assayed, further justifying using a “loose filter” in the in vitro C. parvum assay (69, 70).
TABLE 1.
Compound physicochemical properties and performance in preliminary assays
| Compound | CLogPa | TPSA (Å2)a | Enzyme inhibition, IC50 (nM) |
T. gondii/CpIMPDH assaye |
C. parvum assay, EC50 (μM) | Mouse liver microsomal stability, t1/2 (min) | |
|---|---|---|---|---|---|---|---|
| EC50 (nM) | Selectivity | ||||||
| A110 | 4.4 | 72 | 18b,c | 500f | 33f | <0.8f; 11g | 320 |
| A119 | 4.0 | 62 | 1.2b | 14 | 360 | 12–25f | 4.6 |
| P25 | 1.9 | 100 | 59b,d | 380d | >66d | 12g,h | ≥600k |
| P32 | 2.3 | 117 | 6.7b,d | 1,900d | >12d | 6g,i | 190 |
| P82 | 4.3 | 74 | 1.1b,d | 10d | 250d | 15g; 8j; 7j; 9j | 88d |
| P83 | 4.6 | 63 | 2.9b,d | 16d | 86d | 9j; 10j; 8j | 33d |
| P96 | 3.6 | 125 | 1.3b,d | 6d | 670d | 8g,i | 11d |
| P131 | 3.7 | 141 | 18b,d | 510d | 20d | 7g,i | 120d |
Calculated with ChemDraw software (Cambridgesoft).
IC50 of >5 μM versus human IMPDH2 in all cases.
Values from reference 45.
Values from reference 48.
Antiparasitic activity was assayed in a Toxoplasma gondii strain (T. gondii/CpIMPDH) engineered to rely on CpIMPDH for the production of guanine nucleotides (54). Wild-type T. gondii contains a eukaryotic enzyme similar to human IMPDH that should be resistant to CpIMPDH inhibitors. Selectivity is the ratio of the values of EC50 for wild-type and T. gondii/CpIMPDH strains. High selectivity confirms that the compounds target CpIMPDH and serves as a proxy for host cell effects.
Values from reference 54.
HCT-8 host cells.
High-content imaging assay; 27% inhibition at 12.5 μM, 83% inhibition at 25 μM.
High-content imaging assay; same antiparasitic activity as that of 1 mM paromomycin.
MDCK host cells.
No metabolism observed over 45 min. Assuming conservatively that 5% is the minimum metabolism reliably observed, then the lower limit of t1/2 is 600 min.
Eight compounds, A110, A119, P25, P32, P82, P83, P96, and P131, were chosen to test in the mouse model (Fig. 1). These compounds represent a wide range of hydrophobicity (CLogP), polarity (TPSA), and metabolic stability values. We expected that these variations would produce very different pharmacokinetic behaviors and systemic exposures and would therefore provide insight into the requirements for antiparasitic activity (Fig. 1 and Table 1).
FIG 1.

Structures of the compounds used in this study.
Antiparasitic activity in an immunosuppressed mouse model of acute cryptosporidiosis.
In vivo antiparasitic activity was evaluated in the IL-12 knockout mouse model of acute disease (51, 55). IL-12 knockout mice are highly susceptible to C. parvum. Infection results in moderate to heavy shedding of oocysts beginning 3 days after infection with a peak at days 4 to 7. No hepatobiliary involvement has been observed in this model. Mice resolve the infection and recover within 2 to 3 weeks, which closely resembles infections in immunocompetent humans. Up to three compounds can be evaluated in a single experiment together with vehicle and paromomycin control groups. Since these experiments were the initial evaluation of in vivo antiparasitic activity, the highest feasible dose was administered (250 mg/kg). Toxicity was assessed prior to antiparasitic activity by orally administering compounds at 250 mg/kg daily to uninfected C57BL/6 mice for 5 days. Animal weight and behavior (e.g., grooming) were noted daily. No overt signs of acute toxicity or significant weight loss were observed.
The CpIMPDH inhibitors were evaluated in seven separate experiments (Fig. 2A to G; see also Table S1 in the supplemental material). IL-12 knockout mice were infected with 1,000 purified C. parvum oocysts (Iowa strain, isolated from cattle). Treatment was initiated after 4 h and continued for 7 days. Mice were treated daily via oral gavage, with either 250 mg/kg of CpIMPDH inhibitor, vehicle, or 2,000 mg/kg paromomycin (note that nitazoxanide has little to no efficacy in mouse models [71]). Infections were monitored by counting fecal oocysts from individual mouse samples at peak infection using flow cytometry. The distribution of fecal oocysts was not Gaussian, so the nonparametric Mann-Whitney test was used to assess significance (Fig. 2; see also Table S1). Paromomycin reliably reduced oocyst numbers by approximately 90%.
FIG 2.

Antiparasitic activity of CpIMPDH inhibitors administered in a single daily dose. IL-12 knockout mice were infected with 1,000 C. parvum oocysts and treated with single daily doses of vehicle (Veh), paromomycin (Prm) (2,000 mg/kg), or compound by oral gavage. All compounds were dosed at 250 mg/kg, except A110 in panel B, which was dosed at 100 mg/kg. Fecal oocysts were counted on day 7 postinfection. Each panel displays an independent experiment. Each point represents the fecal oocyst count from an individual mouse in “bee swarm” format. The box plot denotes the median and quartiles. Significance was determined via the Mann-Whitney test using Prism software. Only significant P values are shown (or when nonsignificance is interesting, as in the comparison of the single-dose P131 and paromomycin). All P values and oocyst counts can be found in Table S1 in the supplemental material.
Most of the compounds failed to significantly reduce the numbers of fecal oocysts (Fig. 2A to G; see also Table S1 in the supplemental material). A110, A119, P25, P32, and P83 did not display any signs of in vivo antiparasitic activity. Surprisingly, A110 significantly increased the numbers of oocysts (Fig. 2A), suggesting that this compound promoted parasite growth. Fecal oocyst counts also appeared higher when mice were treated with A119 and P32, although in these cases the differences were not statistically significant (Fig. 2C and F). Compound P82 decreased oocyst numbers, but the difference with vehicle-treated animals was not quite significant at the 95% confidence level (P = 0.054, Fig. 2D). Compound P96 did display significant, albeit weak, antiparasitic activity in two of three trials (P = 0.047 and 0.043 versus vehicle; Fig. 2E and F), though this compound was less effective than paromomycin in both instances (P = 0.012 and 0.0005).
In contrast, P131 displayed substantial antiparasitic activity. Treatment with P131 significantly decreased oocyst numbers relative to the vehicle control group (P = 0.0039) to an extent that was indistinguishable from the paromomycin treatment group (P = 0.22; Fig. 2G). The antiparasitic activity of P131 was somewhat surprising because this compound is a 10-fold-less-potent inhibitor of CpIMPDH than P96 (Table 1). This observation suggested that the parasite must be exposed to greater concentrations of P131 to compensate for its decreased in vitro biochemical potency. Taken together, the antiparasitic activities of P96 and P131 provide proof-of-concept in an animal model and validate CpIMPDH as a target for the treatment of cryptosporidiosis.
Plasma pharmacokinetic properties.
In order to investigate the relationship between anticryptosporidial activity and systemic exposure, we measured the plasma pharmacokinetics for five compounds, A110, P82, P83, P96, and P131, after 250-mg/kg single oral doses (Table 2). P82 is the primary product of P83 metabolism, so the concentration of P82 in plasma was also measured during P83 treatment. As hoped, the five compounds displayed a broad range of plasma pharmacokinetic behavior. Maximal plasma concentrations (Cmax) varied from 9 to 250 μM while plasma half-life (t1/2) varied from 4 h to more than 24 h. The values of plasma Cmax exceeded the values of IC50 for enzyme inhibition by at least 400-fold for all five compounds. The values of plasma Cmax also exceeded the values of EC50 for the Toxoplasma reporter assay by at least a factor of 9. Thus, if the plasma concentrations were comparable to concentrations within the parasite, all five compounds should have displayed antiparasitic activity. Compound A110 had excellent systemic pharmacokinetics, with the greatest values of Cmax and t1/2, for an overall area under the curve (AUC) in excess of 2,000 h · μg/ml. The other two inactive compounds, P82 and P83, had the next highest exposures, with AUCs of 41 and 79 h · μg/ml, respectively. The two compounds that displayed antiparasitic activity, P96 and P131, had the lowest exposure, with AUCs of 15 and 13 h · μg/ml, respectively. The most active compound, P131, displayed the lowest plasma Cmax and shortest t1/2. Thus, good plasma distribution did not correlate with antiparasitic activity. From a toxicological perspective, higher systemic distribution may actually be a liability.
TABLE 2.
Pharmacokinetic parameters for CpIMPDH inhibitorsa
| Compound | Mouse activity | Plasma value |
||||
|---|---|---|---|---|---|---|
| Cmax (μg/ml) | Tmax (h) | t1/2 (h) | AUCinf (h · μg/ml) | CL (ml/h · kg) | ||
| A110 | − (!) | 100 (270 μM) | 1 | >24 | >2,000 | 11 |
| P82 | + | 9 (21 μM) | 0.3 | 12 | 41 | 6,100 |
| P83 | − | 22 (51 μM) | 1 | 4 | 79 | 3,100 |
| P82 ex P83 (*) | − | 3.6 (8.7 μM) | 2 | 4 | ND | ND |
| P96 | + | 5 (13 μM) | 0.3 | 8 | 15 | 17,000 |
| P131 | + | 4 (9.2 μM) | 1 | 4 | 13 | 20,000 |
C57BL/6 mice received a single dose of 250 mg/kg in 5% DMSO-corn oil by oral gavage. Mouse activity: +, significant antiparasitic activity in at least one trial; −, no antiparasitic activity; − (!), promotes parasitemia. *, measured levels of P82 generated by metabolism of P83; ND, not determined.
P131 is more effective than paromomycin in a multiple-dosing regimen.
If intestinal exposure is the key to antiparasitic activity, then the efficacy of P131 might increase if administered in multiple doses. Therefore, we evaluated the antiparasitic activity of P131 (250 mg/kg) and paromomycin (2,000 mg/kg) when administered in 3 daily doses, the most frequent dosing schedule practical. The infection protocol was modified to accommodate the increased dosing by shortening the treatment period and increasing the parasite inocula from 1,000 to 10,000 oocysts. Fecal oocysts were counted on day 4 postinfection. Multiple dosing did not improve the efficacy of paromomycin in two separate trials (approximately 90% reduction of fecal oocysts; Fig. 3). In contrast, the efficacy of P131 improved with multiple dosing, reducing fecal oocysts more than paromomycin did in both trials (P = 0.045 and 0.024; Fig. 3). No oocysts were found in the samples of 3/9 mice in the first trial and 6/10 mice in the second trial, while oocysts were found in all of the paromomycin-treated mice (see Table S1 in the supplemental material).
FIG 3.

Antiparasitic activity of P131 administered in multiple daily doses. IL-12 knockout mice were infected with 10,000 C. parvum oocysts and treated with three daily doses of either vehicle, paromomycin (670 mg/kg), or P131 (83 mg/kg). Fecal oocysts were counted on day 5 postinfection. Each panel represents an independent experiment. Each point represents the fecal oocyst count from an individual mouse in “bee swarm” format. The box plot denotes the median and quartiles. Significance was determined via the Mann-Whitney test using Prism software. Oocyst counts can be found in Table S1 in the supplemental material.
Compound P131 accumulates in Caco-2 cells.
Since plasma exposure did not appear to be required for in vivo antiparasitic activity, we hypothesized that the intestinal accumulation might determine efficacy. Therefore, we measured the permeability and uptake of the CpIMPDH inhibitors in Caco-2 cells, a widely used model of human intestinal epithelial cells (72). Permeability varied 20-fold among the compounds, although in all cases it was greater than 10−5 cm · s−1 (Table 3). Therefore, all of the compounds are considered highly permeable. In all cases, permeabilities were similar in both the apical-to-basal (A→B) and basal-to-apical (B→A) directions, suggesting that none of the compounds were substrates for an efflux pump (efflux ratio of ≤2 for all compounds). Thus, differences in permeability do not explain the differences in antiparasitic activity.
TABLE 3.
CpIMPDH inhibitor permeability and uptake in Caco-2 TC7 cellsa
| Compound | Cellular uptake (nmol/4.2 cm2) |
Intracellular concnb (μM) |
Permeability (cm/s) (105) |
Efflux ratio | |||
|---|---|---|---|---|---|---|---|
| A→B | B→A | A→B | B→A | A→B | B→A | ||
| A110 | 0.42 ± 0.03 | 0.39 ± 0.03 | 100 | 96 | 2.3 ± 0.4 | 2.6 ± 0.2 | 1.1 |
| A119 | 2.9 ± 0.3 | 3.0 ± 0.4 | 710 | 740 | 3.5 ± 0.4 | 3.1 ± 0.3 | 0.89 |
| P25 | 0.0021 ± 0.0002 | 0.0025 ± 0.0002 | 0.53 | 0.64 | 6 ± 1 | 9.6 ± 0.9 | 1.7 |
| P32 | 0.018 ± 0.002 | 0.031 ± 0.001 | 4.4 | 7.7 | 29 ± 5 | 60 ± 10 | 2.0 |
| P82 | 0.4 ± 0.1 | 0.15 ± 0.04 | 98 | 38 | 18 ± 3 | 11 ± 3 | 0.61 |
| P83 | 0.5 ± 0.2 | 0.43 ± 0.09 | 120 | 110 | 2.6 ± 0.6 | 1.4 ± 0.3 | 0.52 |
| P96 | 0.11 ± 0.04 | 0.1 ± 0.02 | 28 | 29 | 33 ± 5 | 28 ± 2 | 0.85 |
| P131 | 5 ± 2 | 7 ± 1 | 1,300 | 1,900 | 1.7 ± 0.1 | 1.2 ± 0.2 | 0.71 |
All compounds at 10 μM, pH 7.4, on both sides. Solid recovery of >80% in all cases.
Approximate intracellular concentration, assuming 4 μl of total cellular water for 1 mg of cellular protein.
In contrast, Caco-2 cell uptake varied over a range of 2,000-fold among the eight CpIMPDH inhibitors (Table 3). The lowest uptake was observed for the two inactive compounds, P25 and P32. The intracellular concentrations of these compounds did not reach the extracellular concentration (10 μM). All of the other compounds accumulated intracellular concentrations in excess of the extracellular concentration. P131 had the highest uptake, reaching millimolar concentrations. Only A119 reached an intracellular concentration comparable to that of P131. The concentrations of all the other compounds were lower by at least a factor of 10. The increased uptake of P131 relative to the other P compounds may explain its antiparasitic activity.
The high accumulation of P131 suggested that the efflux of P131 from Caco-2 cells might be unusually slow. We preloaded Caco-2 cells with 5, 20, or 100 μM P131 and then measured efflux (Fig. 4). The initial intracellular concentrations of P131 were dose dependent and scaled with the extracellular concentration for the 5 and 20 μM preincubation conditions. Some cytotoxicity was noted when cells were preincubated with 100 μM P131, which may account for the somewhat lower than expected intracellular concentration at this dose. P131 efflux was very slow under all three conditions, with t1/2 values of 1 to 2 h. In contrast, the values of t1/2 for the efflux of amino acids and other nutrients are on the order of 15 to 30 min (73).
FIG 4.
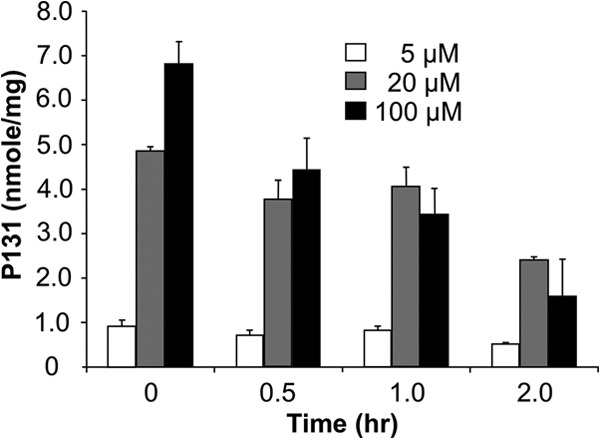
Accumulation of P131 in Caco-2 cells. Amount of P131 remaining in Caco-2 TC7 cells after the cells were loaded with different concentrations of P131 for 2 h at 37°C in HBSS. The efflux t1/2 is ∼1 h for loading at 100 μM and ∼2 h for 20 and 5 μM.
Compound P131 accumulates in intestinal tissues.
The above observations prompted a more thorough investigation of the tissue distribution of P131 in both C57BL/6 and IL-12 knockout mice. Tissues were harvested 24 h after the first dose of an oral 250-mg/kg dose, split, and given as 3 daily doses (83-mg/kg dose). No significant difference was noted between the two mouse strains, justifying the use of C57BL/6 mice for routine pharmacokinetic evaluations. As expected from the plasma pharmacokinetic experiments, little P131 was found in plasma after 24 h (Fig. 5). Higher concentrations were found in the liver, suggesting that liver metabolism, or perhaps enterohepatic recycling, might limit the systemic distribution of P131. The highest concentrations of P131 were present in the intestine and feces (3 to 6 μM). These concentrations were more than 100-fold higher than the IC50 for CpIMPDH inhibition. These findings suggest that in vivo antiparasitic activity requires the accumulation of CpIMPDH inhibitors in the gastrointestinal tract.
FIG 5.
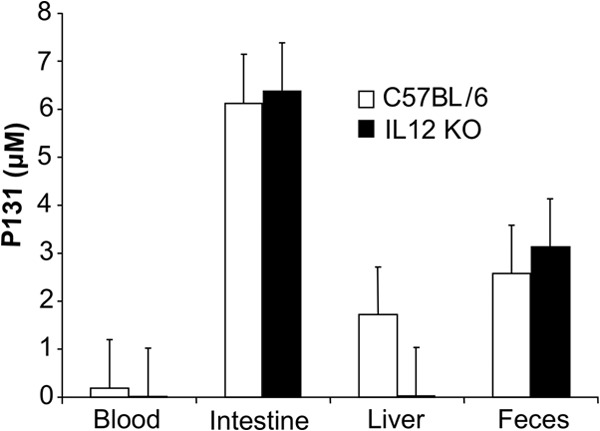
Accumulation of P131 in mouse tissues. Concentration of P131 in various tissues 24 h after the first of three daily 83-mg/kg doses (250 mg/kg total). Concentration in tissue is reported assuming that 1 g of tissue is equivalent to 1 ml of liquid. An alternative way to express concentration is nmol/g. KO, knockout.
A110 perturbs the gut microbiota.
We hypothesized that the ability of compounds such as A110 to promote C. parvum infection might result from perturbation of gut microbiota. CpIMPDH inhibitors do inhibit some bacterial IMPDHs, and at least one CpIMPDH inhibitor has antibacterial activity (74). Many commensal bacteria contain IMPDHs that should be sensitive to CpIMPDH inhibitors. Whether this sensitivity translates into antibacterial activity will be determined by uptake as well as the ability of the bacteria to salvage purines and bypass IMPDH, so it is impossible to predict which bacteria may be affected.
We analyzed fecal bacteria in IL-12 knockout mice to investigate the effects of CpIMPDH inhibitors on gut microbiota. Groups of 10 mice were treated orally with vehicle, A110, or P131 for 7 days. Individual fecal samples were collected prior to treatment (day 0) and on day 7. Total genomic DNA was isolated, and 16S rRNA gene libraries were constructed from each individual sample and sequenced. Phyla were identified using the GreenGenes 16S rRNA database with UCLUST (60). A summary of phylum distribution among the three treatment groups is given in Table 4 and Fig. 6A.
TABLE 4.
Effects of vehicle, A110, and P131 on fecal microbiotaa
| Phylum | Vehicle |
P131 |
A110 |
|||
|---|---|---|---|---|---|---|
| Day 0 | Day 7 | Day 0 (n = 4) | Day 7 | Day 0 | Day 7 (n = 9) | |
| Bacteroidetes | 73 ± 3 | 84 ± 3 | 82 ± 4 | 78 ± 3 | 82 ± 1 | 67 ± 3 |
| Cyanobacteria | 0.01 ± 0.01 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.01 ± 0.01 | 0.01 ± 0.01 | 0.00 ± 0.00 |
| Firmicutes | 25 ± 3 | 8 ± 2 | 17 ± 4 | 12 ± 2 | 17 ± 1 | 3.8 ± 0.9 |
| Proteobacteria | 0.01 ± 0.01 | 0.00 ± 0.00 | 0.00 ± 0.00 | 0.01 ± 0.00 | 0.00 ± 0.00 | 0.00 ± 0.00 |
| Tenericutes | 0.5 ± 0.1 | 0.09 ± 0.03 | 0.19 ± 0.07 | 0.05 ± 0.02 | 0.7 ± 0.2 | 0.04 ± 0.03 |
| Verrucomicrobia | 0.08 ± 0.03 | 7 ± 1 | 0.00 ± 0.00 | 10 ± 2 | 0.01 ± 0.01 | 28 ± 4 |
| Bacteria, other | 0.74 ± 0.09 | 0.58 ± 0.06 | 0.53 ± 0.08 | 0.80 ± 0.09 | 0.84 ± 0.09 | 0.82 ± 0.08 |
Mice (10 per group, except as noted) were treated with single daily doses of vehicle or 250 mg/kg A110 or P131. Fecal samples were collected, and amplicons of the bacterial 16S rRNA gene were constructed, sequenced with 454 pyrosequencing, and analyzed with the QIIME platform (59). The relative abundance of phyla is shown as the percentage and standard error of the mean of total reads.
FIG 6.

Effects of vehicle, A110, and P131 on gut microbiota. IL-12 knockout mice were treated with vehicle, A110, or P131 (250 mg/kg) for 7 days. Each treatment group contained 10 mice. Fecal samples were collected, and the 16S rRNA gene content was sequenced from each individual sample using 454 pyrosequencing and analyzed with the QIIME platform. (A) The change in phylum distribution of the three treatment groups is shown. Statistical analysis was performed using the Mann-Whitney U test (***, P < 0.001; ns, not significant). (B) Beta diversity plots comparing the bacterial taxa between the treatments at day 0 and day 7. Weighted UniFrac measurements were used to generate a matrix of pairwise distances between communities, and scatter plots were generated from a matrix of distances using principal component analysis (PCA). The P values listed in the text were generated from the original UniFrac distance matrix using QIIME scripts.
Treatment with vehicle alone induced significant changes in two phyla (Table 4 and Fig. 6A). Firmicutes bacteria decreased from 25% to 8% between day 0 and day 7, accompanied by a remarkable increase of ∼90-fold in Verrucomicrobia (from 0.08 to 7%, P < 0.0001). Analysis at the species level revealed that the increase in Verrucomicrobia resulted from expansion of Akkermansia muciniphila, while the decrease in Firmicutes derived from a reduction of members of the Lachnospiraceae family (P < 0.0001; see Fig. S1 in the supplemental material). Similar changes were observed when mice were treated with P131, suggesting that this compound has no additional impact on fecal microbiota.
In contrast, treatment with A110 induced significant changes in three phyla (Table 4 and Fig. 6A). Firmicutes, in particular members of the Lachnospiraceae family, decreased to a similar extent as was observed in the vehicle alone, suggesting that like P131, A110 has little effect on these bacteria (see Fig. S1 in the supplemental material). In contrast, A. muciniphila increased by 2,800-fold (from 0.01% to 28%). This change was 30-fold more than that observed with either vehicle or P131 (P < 0.0001). A corresponding decrease from 82% to 67% was observed in Bacteroidetes family S-24-7. This decrease was not observed in either the vehicle alone or the P131 treatment (P < 0.0001; see Fig. S1).
Beta diversity analysis confirmed that the A110 treatment had significantly different effects on the fecal microbiota than did either the vehicle or P131 (Fig. 6B). The 16S rRNA gene sequences were aligned using Fast Tree2 to generate a phylogenetic tree (59, 62, 63). The proportion of the branch length on the tree, which is unique to each bacterial community in each treatment pair, was quantified to generate UniFrac distances using QIIME tools. Principal component analysis (PCA) plots were generated using UniFrac distances using the weighted data (Fig. 6B). The weighted UniFrac PCA plots represent the relative abundance of OTUs of the samples. As shown in Fig. 6B, the fecal bacterial populations in mice treated with either vehicle or P131 display substantial overlap, while the samples from A110-treated mice formed a distinct cluster on day 7. The significance of the differences between pairs was tested using distance-based PERMANOVA. The difference between the A110 and vehicle treatments was significant with a P value of 0.001 in both the weighted and unweighted data. Likewise, the difference between the A110 and P131 treatments was significant with a P value of 0.001 in both the weighted and unweighted data. This initial study suggests that the A110-specific alterations of gut microbiota may account for the ability of this compound to promote Cryptosporidium infection.
DISCUSSION
Validation of CpIMPDH as a target for the treatment of cryptosporidiosis.
Our results demonstrate that two compounds, P96 and P131, have anticryptosporidial activity in the IL-12 knockout mouse model of acute disease. At present, no clinically validated, and few experimentally validated, targets exist for Cryptosporidium treatment, so the demonstration of in vivo efficacy for CpIMPDH inhibitors represents a significant achievement. Impressively, P131 is more effective than paromomycin when administered in split doses. Further optimization of the formulation and dosing schedule may improve the efficacy of P131 and additional CpIMPDH inhibitors.
Distribution requirements of anticryptosporidial activity.
In the absence of guidance from effective drugs, many anticryptosporidial discovery programs have assumed that good systemic exposure is required for in vivo antiparasitic activity (e.g., see reference 26). We have found that the inactive compounds A110 and P83 displayed the best plasma pharmacokinetic behavior, while the active compounds P96 and P131 have relatively poor systemic exposure. In vivo anticryptosporidial activity has also been reported for several other compounds with poor systemic bioavailability, most notably paromomycin, and also including pyrvinium pamoate, dication carbazoles, and trifluoromethylthymidine (15, 20, 25). While nitazoxanide exhibits good systemic exposure, its active metabolite tizoxanide is efficiently recycled to the intestine via glucuronidation (12). Interestingly, the glucuronidated tizoxanide has antiparasitic activity comparable to that of nitazoxanide in vitro (69). Thus, intestinal exposure appears to be a common feature in compounds with in vivo efficacy. Systemic exposure clearly does not correlate with in vivo activity and may actually be a liability from a toxicology standpoint. It is worth noting that if systemic exposure is not required, liver microsomal stability is not relevant. Indeed, the very poor stability of P96 in mouse liver microsomes would usually preclude testing this compound in animal models. We suspect that the presumption of systemic exposure may have wrongfully eliminated other promising candidates.
P131 accumulates to high concentrations in intestinal tissue, strongly suggesting that accumulation in intestinal epithelial cells is the key to antiparasitic activity. Both active compounds attain concentrations in Caco-2 cells that far exceed their IC50 values for enzyme inhibition. The accumulation of P131 is especially high, reflecting its unusually slow efflux. Paromomycin also displays high accumulation in Caco-2 cells (75). We suggest that the accumulation of compounds in Caco-2 cells is a useful filter for inhibitor optimization. We further propose that the design of anticryptosporidial drugs should focus on retention in intestinal tissue rather than the usual metrics for systemic bioavailability.
Systemic exposure may be required to treat the biliary infections that arise in approximately 20% of immunocompromised patients. We believe that this is currently an open question. Nitazoxanide is only marginally superior to paromomycin in treating biliary infections in the immunosuppressed gerbil model despite its systemic distribution (76). Moreover, the activity of nitazoxanide against biliary infections had been attributed to efficient enterohepatic recycling rather than systemic exposure (1, 76). Indeed, bile contains 10-fold-higher concentrations of the active nitazoxanide metabolites than does plasma (12). It may be possible to exploit enterohepatic recycling in the further development of CpIMPDH inhibitors.
Dosing requirements of anticryptosporidial activity.
Compound P131 was most effective when administered in three daily doses. The gastrointestinal transit time in mice is approximately 4.5 h (77, 78). Given that diarrhea decreases gastrointestinal transit times, efficacy may be improved with more frequent dosing, or in combination with antimotility agents such as loperamide (79). It is worth noting that, under the three-times-daily dosing protocol, the concentration of P131 in intestinal tissues was more than 100-fold higher than the IC50 for enzyme inhibition, which suggests that lower doses of P131 may also be effective.
Cryptosporidium and gut microbiota.
The protective effects of gut microbiota are widely recognized in other infections (80, 81). Although relatively little explored, microbiota have also been proposed to influence Cryptosporidium infection (42). The importance of gut microbiota is illustrated by the susceptibility of gnotobiotic animals (82) and reports of successful probiotic treatment (83, 84). Moreover, germfree SCID mice are more susceptible to C. parvum infection than are SCID mice with normal flora (85), and gut microbiota are required for a poly(I·C) treatment to afford protection against Cryptosporidium infection in neonatal mice (86). We hypothesized that the puzzling ability of A110 and several other CpIMPDH inhibitors to promote Cryptosporidium infection might result from a unique perturbation of gut microbiota. Intriguingly, in this initial experiment, A110 caused a significantly different perturbation of the gut microbiota than did P131 and the vehicle. Many commensal bacteria contain IMPDHs that are likely to be sensitive to CpIMPDH inhibitors, and at least one CpIMPDH inhibitor has been shown to have antibacterial activity (74). Therefore, it is possible that this perturbation derives from the “on-target” inhibition of bacterial IMPDHs. Cryptosporidium contains several other prokaryotic enzymes that have attracted interest as potential drug targets (29, 87). We suggest that it will be important to monitor gut microbiota as these drug discovery programs advance.
These experiments also suggested that the vehicle alone caused substantive changes in gut microbiota. These changes could also influence Cryptosporidium infection. To the best of our knowledge, no systematic investigation of the effects of formulation on Cryptosporidium infection has been performed. We suggest that the effects of both drug candidates and formulations on gut microbiota should be evaluated as part of the drug discovery pipeline. Clearly, the interactions between Cryptosporidium, gut microbiota, drug candidates, and formulations warrant further investigation.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by funding from the National Institute of Allergy and Infectious Diseases (UO1 AI075466 and RO1 AI093459) to L.H. G.D.C. thanks the New England Regional Center of Excellence for Biodefense and Emerging Infectious Diseases (NERCE/BEID, NIH/NIAID U54 AI057159) and the Harvard NeuroDiscovery Center for financial support. B.S. is a Georgia Research Alliance Distinguished Investigator. M.H. is supported by NIH R01GM070737.
Work performed by N.N.M. and J.R.M. was conducted at the Atlanta VA Medical Center. We thank Mandapati Kavitha and Minjia Zhang for technical support and Arifa Ahsan and Suraj Rao for assistance in producing P96.
Footnotes
Published ahead of print 23 December 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02075-13.
REFERENCES
- 1.Chalmers RM, Davies AP. 2010. Minireview: clinical cryptosporidiosis. Exp. Parasitol. 124:138–146. 10.1016/j.exppara.2009.02.003 [DOI] [PubMed] [Google Scholar]
- 2.Desai NT, Sarkar R, Kang G. 2012. Cryptosporidiosis: an under-recognized public health problem. Trop. Parasitol. 2:91–98. 10.4103/2229-5070.105173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shirley DA, Moonah SN, Kotloff KL. 2012. Burden of disease from cryptosporidiosis. Curr. Opin. Infect. Dis. 25:555–563. 10.1097/QCO.0b013e328357e569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Striepen B. 2013. Parasitic infections: time to tackle cryptosporidiosis. Nature 503:189–191. 10.1038/503189a [DOI] [PubMed] [Google Scholar]
- 5.Hlavsa MC, Roberts VA, Anderson AR, Hill VR, Kahler AM, Orr M, Garrison LE, Hicks LA, Newton A, Hilborn ED, Wade TJ, Beach MJ, Yoder JS. 2011. Surveillance for waterborne disease outbreaks and other health events associated with recreational water—United States, 2007–2008. MMWR Surveill. Summ. 60:1–32 [PubMed] [Google Scholar]
- 6.Kotloff KL, Nataro JP, Blackwelder WC, Nasrin D, Farag TH, Panchalingam S, Wu Y, Sow SO, Sur D, Breiman RF, Faruque AS, Zaidi AK, Saha D, Alonso PL, Tamboura B, Sanogo D, Onwuchekwa U, Manna B, Ramamurthy T, Kanungo S, Ochieng JB, Omore R, Oundo JO, Hossain A, Das SK, Ahmed S, Qureshi S, Quadri F, Adegbola RA, Antonio M, Hossain MJ, Akinsola A, Mandomando I, Nhampossa T, Acacio S, Biswas K, O'Reilly CE, Mintz ED, Berkeley LY, Muhsen K, Sommerfelt H, Robins-Browne RM, Levine MM. 2013. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet 382:209–222. 10.1016/S0140-6736(13)60844-2 [DOI] [PubMed] [Google Scholar]
- 7.Bartelt LA, Lima AA, Kosek M, Penataro Yori P, Lee G, Guerrant RL. 2013. “Barriers” to child development and human potential: the case for including the “neglected enteric protozoa” (NEP) and other enteropathy-associated pathogens in the NTDs. PLoS Negl. Trop. Dis. 7:e2125. 10.1371/journal.pntd.0002125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hagen RM, Loderstaedt U, Frickmann H. 3 December 2013. An evaluation of the potential use of Cryptosporidium species as agents for deliberate release. J. R. Army Med. Corps 10.1136/jramc-2013-000186 [DOI] [PubMed] [Google Scholar]
- 9.MacKenzie WR, Hoxie NJ, Proctor ME, Gradus MS, Blair KA, Peterson DE, Kazmierczak JJ, Addiss DG, Fox KR, Rose JB, Davis JP. 1994. A massive outbreak in Milwaukee of cryptosporidium infection transmitted through the public water supply. N. Engl. J. Med. 331:161–167. 10.1056/NEJM199407213310304 [DOI] [PubMed] [Google Scholar]
- 10.Rossignol JF, Kabil SM, el-Gohary Y, Younis AM. 2006. Effect of nitazoxanide in diarrhea and enteritis caused by Cryptosporidium species. Clin. Gastroenterol. Hepatol. 4:320–324. 10.1016/j.cgh.2005.12.020 [DOI] [PubMed] [Google Scholar]
- 11.Amadi B, Mwiya M, Musuku J, Watuka A, Sianongo S, Ayoub A, Kelly P. 2002. Effect of nitazoxanide on morbidity and mortality in Zambian children with cryptosporidiosis: a randomised controlled trial. Lancet 360:1375–1380. 10.1016/S0140-6736(02)11401-2 [DOI] [PubMed] [Google Scholar]
- 12.Hemphill A, Mueller J, Esposito M. 2006. Nitazoxanide, a broad-spectrum thiazolide anti-infective agent for the treatment of gastrointestinal infections. Expert Opin. Pharmacother. 7:953–964. 10.1517/14656566.7.7.953 [DOI] [PubMed] [Google Scholar]
- 13.Cabada MM, White AC., Jr 2010. Treatment of cryptosporidiosis: do we know what we think we know? Curr. Opin. Infect. Dis. 23:494–499. 10.1097/QCO.0b013e32833de052 [DOI] [PubMed] [Google Scholar]
- 14.Rehg JE. 1995. The activity of halofuginone in immunosuppressed rats infected with Cryptosporidium parvum. J. Antimicrob. Chemother. 35:391–397. 10.1093/jac/35.3.391 [DOI] [PubMed] [Google Scholar]
- 15.Downey AS, Chong CR, Graczyk TK, Sullivan DJ. 2008. Efficacy of pyrvinium pamoate against Cryptosporidium parvum infection in vitro and in a neonatal mouse model. Antimicrob. Agents Chemother. 52:3106–3112. 10.1128/AAC.00207-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kimata I, Uni S, Iseki M. 1991. Chemotherapeutic effect of azithromycin and lasalocid on Cryptosporidium infection in mice. J. Protozool. 38:232S–233S [PubMed] [Google Scholar]
- 17.Chen F, Huang K. 2012. Effects of the Chinese medicine matrine on experimental C. parvum infection in BALB/c mice and MDBK cells. Parasitol. Res. 111:1827–1832. 10.1007/s00436-012-3030-7 [DOI] [PubMed] [Google Scholar]
- 18.Rehg JE, Hancock ML. 1990. Effectiveness of arprinocid in the reduction of cryptosporidial activity in immunosuppressed rats. Am. J. Vet. Res. 51:1668–1670 [PubMed] [Google Scholar]
- 19.Moreno B, Bailey BN, Luo S, Martin MB, Kuhlenschmidt M, Moreno SN, Docampo R, Oldfield E. 2001. (31)P NMR of apicomplexans and the effects of risedronate on Cryptosporidium parvum growth. Biochem. Biophys. Res. Commun. 284:632–637. 10.1006/bbrc.2001.5009 [DOI] [PubMed] [Google Scholar]
- 20.Blagburn BL, Drain KL, Land TM, Kinard RG, Moore PH, Lindsay DS, Patrick DA, Boykin DW, Tidwell RR. 1998. Comparative efficacy evaluation of dicationic carbazole compounds, nitazoxanide, and paromomycin against Cryptosporidium parvum infections in a neonatal mouse model. Antimicrob. Agents Chemother. 42:2877–2882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fayer R, Fetterer R. 1995. Activity of benzimidazoles against cryptosporidiosis in neonatal BALB/c mice. J. Parasitol. 81:794–795. 10.2307/3283980 [DOI] [PubMed] [Google Scholar]
- 22.Stachulski AV, Berry NG, Lilian Low AC, Moores SL, Row E, Warhurst DC, Adagu IS, Rossignol JF. 2006. Identification of isoflavone derivatives as effective anticryptosporidial agents in vitro and in vivo. J. Med. Chem. 49:1450–1454. 10.1021/jm050973f [DOI] [PubMed] [Google Scholar]
- 23.Kayser O, Waters WR, Woods KM, Upton SJ, Keithly JS, Laatsch H, Kiderlen AF. 2002. Evaluation of in vitro and in vivo activity of benzindazole-4,9-quinones against Cryptosporidium parvum. J. Antimicrob. Chemother. 50:975–980. 10.1093/jac/dkf199 [DOI] [PubMed] [Google Scholar]
- 24.Rueda C, Fenoy S, Simon F, Del Aguila C. 2008. Bobel-24 activity against Cryptosporidium parvum in cell culture and in a SCID mouse model. Antimicrob. Agents Chemother. 52:1150–1152. 10.1128/AAC.01019-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun XE, Sharling L, Muthalagi M, Mudeppa DG, Pankiewicz KW, Felczak K, Rathod PK, Mead J, Striepen B, Hedstrom L. 2010. Prodrug activation by Cryptosporidium thymidine kinase. J. Biol. Chem. 285:15916–15922. 10.1074/jbc.M110.101543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Castellanos-Gonzalez A, White AC, Jr, Ojo KK, Vidadala RS, Zhang Z, Reid MC, Fox AM, Keyloun KR, Rivas K, Irani A, Dann SM, Fan E, Dustin J, Van Voorhis WC. 2013. A novel calcium-dependent protein kinase inhibitor as a lead compound for treating cryptosporidiosis. J. Infect. Dis. 208:1342–1348. 10.1093/infdis/jit327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abrahamsen MS, Templeton TJ, Enomoto S, Abrahante JE, Zhu G, Lancto CA, Deng M, Liu C, Widmer G, Tzipori S, Buck GA, Xu P, Bankier AT, Dear PH, Konfortov BA, Spriggs HF, Iyer L, Anantharaman V, Aravind L, Kapur V. 2004. Complete genome sequence of the apicomplexan, Cryptosporidium parvum. Science 304:441–445. 10.1126/science.1094786 [DOI] [PubMed] [Google Scholar]
- 28.Xu P, Widmer G, Wang Y, Ozaki LS, Alves JM, Serrano MG, Puiu D, Manque P, Akiyoshi D, Mackey AJ, Pearson WR, Dear PH, Bankier AT, Peterson DL, Abrahamsen MS, Kapur V, Tzipori S, Buck GA. 2004. The genome of Cryptosporidium hominis. Nature 431:1107–1112. 10.1038/nature02977 [DOI] [PubMed] [Google Scholar]
- 29.Striepen B, Pruijssers AJ, Huang J, Li C, Gubbels MJ, Umejiego NN, Hedstrom L, Kissinger JC. 2004. Gene transfer in the evolution of parasite nucleotide biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 101:3154–3159. 10.1073/pnas.0304686101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Umejiego NN, Li C, Riera T, Hedstrom L, Striepen B. 2004. Cryptosporidium parvum IMP dehydrogenase: identification of functional, structural, and dynamic properties that can be exploited for drug design. J. Biol. Chem. 279:40320–40327. 10.1074/jbc.M407121200 [DOI] [PubMed] [Google Scholar]
- 31.Nelson RG, Rosowsky A. 2001. Dicyclic and tricyclic diaminopyrimidine derivatives as potent inhibitors of Cryptosporidium parvum dihydrofolate reductase: structure-activity and structure-selectivity correlations. Antimicrob. Agents Chemother. 45:3293–3303. 10.1128/AAC.45.12.3293-3303.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murphy RC, Ojo KK, Larson ET, Castellanos-Gonzalez A, Perera BG, Keyloun KR, Kim JE, Bhandari JG, Muller NR, Verlinde CL, White AC, Jr, Merritt EA, Van Voorhis WC, Maly DJ. 2010. Discovery of potent and selective inhibitors of calcium-dependent protein kinase 1 (CDPK1) from C. parvum and T. gondii. ACS Med. Chem. Lett. 1:331–335. 10.1021/ml100096t [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Artz JD, Dunford JE, Arrowood MJ, Dong A, Chruszcz M, Kavanagh KL, Minor W, Russell RG, Ebetino FH, Oppermann U, Hui R. 2008. Targeting a uniquely nonspecific prenyl synthase with bisphosphonates to combat cryptosporidiosis. Chem. Biol. 15:1296–1306. 10.1016/j.chembiol.2008.10.017 [DOI] [PubMed] [Google Scholar]
- 34.Zhu G, Shi X, Cai X. 2010. The reductase domain in a type I fatty acid synthase from the apicomplexan Cryptosporidium parvum: restricted substrate preference towards very long chain fatty acyl thioesters. BMC Biochem. 11:46. 10.1186/1471-2091-11-46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fritzler JM, Zhu G. 2012. Novel anti-Cryptosporidium activity of known drugs identified by high-throughput screening against parasite fatty acyl-CoA binding protein (ACBP). J. Antimicrob. Chemother. 67:609–617. 10.1093/jac/dkr516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lei C, Rider SD, Jr, Wang C, Zhang H, Tan X, Zhu G. 2010. The apicomplexan Cryptosporidium parvum possesses a single mitochondrial-type ferredoxin and ferredoxin:NADP+ reductase system. Protein Sci. 19:2073–2084. 10.1002/pro.487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yarlett N, Waters WR, Harp JA, Wannemuehler MJ, Morada M, Bellcastro J, Upton SJ, Marton LJ, Frydman BJ. 2007. Activities of DL-alpha-difluoromethylarginine and polyamine analogues against Cryptosporidium parvum infection in a T-cell receptor alpha-deficient mouse model. Antimicrob. Agents Chemother. 51:1234–1239. 10.1128/AAC.01040-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fayer R. 2004. Cryptosporidium: a water-borne zoonotic parasite. Vet. Parasitol. 126:37–56. 10.1016/j.vetpar.2004.09.004 [DOI] [PubMed] [Google Scholar]
- 39.Huang DB, White AC. 2006. An updated review on Cryptosporidium and Giardia. Gastroenterol. Clin. North Am. 35:291–314. 10.1016/j.gtc.2006.03.006 [DOI] [PubMed] [Google Scholar]
- 40.Snelling WJ, Xiao L, Ortega-Pierres G, Lowery CJ, Moore JE, Rao JR, Smyth S, Millar BC, Rooney PJ, Matsuda M, Kenny F, Xu J, Dooley JS. 2007. Cryptosporidiosis in developing countries. J. Infect. Dev. Ctries. 1:242–256. 10.3855/jidc.360 [DOI] [PubMed] [Google Scholar]
- 41.Hunter PR, Nichols G. 2002. Epidemiology and clinical features of Cryptosporidium infection in immunocompromised patients. Clin. Microbiol. Rev. 15:145–154. 10.1128/CMR.15.1.145-154.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rehg J. 1994. New potential therapies for cryptosporidiosis: an analysis of variables affecting drug efficacy. Folia Parasitol. (Praha) 41:23–26 [PubMed] [Google Scholar]
- 43.Striepen B, White MW, Li C, Guerini MN, Malik SB, Logsdon JM, Jr, Liu C, Abrahamsen MS. 2002. Genetic complementation in apicomplexan parasites. Proc. Natl. Acad. Sci. U. S. A. 99:6304–6309. 10.1073/pnas.092525699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Umejiego NN, Gollapalli D, Sharling L, Volftsun A, Lu J, Benjamin NN, Stroupe AH, Riera TV, Striepen B, Hedstrom L. 2008. Targeting a prokaryotic protein in a eukaryotic pathogen: identification of lead compounds against cryptosporidiosis. Chem. Biol. 15:70–77. 10.1016/j.chembiol.2007.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maurya SK, Gollapalli DR, Kirubakaran S, Zhang M, Johnson CR, Benjamin NN, Hedstrom L, Cuny GD. 2009. Triazole inhibitors of Cryptosporidium parvum inosine 5′-monophosphate dehydrogenase. J. Med. Chem. 52:4623–4630. 10.1021/jm900410u [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.MacPherson IS, Kirubakaran S, Gorla SK, Riera TV, D'Aquino JA, Zhang M, Cuny GD, Hedstrom L. 2010. The structural basis of Cryptosporidium-specific IMP dehydrogenase inhibitor selectivity. J. Am. Chem. Soc. 132:1230–1231. 10.1021/ja909947a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kirubakaran S, Gorla SK, Sharling L, Zhang M, Liu X, Ray SS, Macpherson IS, Striepen B, Hedstrom L, Cuny GD. 2012. Structure-activity relationship study of selective benzimidazole-based inhibitors of Cryptosporidium parvum IMPDH. Bioorg. Med. Chem. Lett. 22:1985–1988. 10.1016/j.bmcl.2012.01.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gorla SK, Kavitha M, Zhang M, Liu X, Sharling L, Gollapalli DR, Striepen B, Hedstrom L, Cuny GD. 2012. Selective and potent urea inhibitors of Cryptosporidium parvum inosine 5′-monophosphate dehydrogenase. J. Med. Chem. 55:7759–7771. 10.1021/jm3007917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johnson CR, Gorla SK, Kavitha M, Zhang M, Liu X, Striepen B, Mead JR, Cuny GD, Hedstrom L. 2013. Phthalazinone inhibitors of inosine-5′-monophosphate dehydrogenase from Cryptosporidium parvum. Bioorg. Med. Chem. Lett. 23:1004–1007. 10.1016/j.bmcl.2012.12.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gorla SK, Kavitha M, Zhang M, Chin JE, Liu X, Striepen B, Makowska-Grzyska M, Kim Y, Joachimiak A, Hedstrom L, Cuny GD. 2013. Optimization of benzoxazole-based inhibitors of Cryptosporidium parvum inosine 5′-monophosphate dehydrogenase. J. Med. Chem. 56:4028–4043. 10.1021/jm400241j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ehigiator HN, Romagnoli P, Borgelt K, Fernandez M, McNair N, Secor WE, Mead JR. 2005. Mucosal cytokine and antigen-specific responses to Cryptosporidium parvum in IL-12p40 KO mice. Parasite Immunol. 27:17–28. 10.1111/j.1365-3024.2005.00736.x [DOI] [PubMed] [Google Scholar]
- 52.Mead JR, McNair N. 2006. Antiparasitic activity of flavonoids and isoflavones against Cryptosporidium parvum and Encephalitozoon intestinalis. FEMS Microbiol. Lett. 259:153–157. 10.1111/j.1574-6968.2006.00263.x [DOI] [PubMed] [Google Scholar]
- 53.Arrowood MJ, Sterling CR. 1987. Isolation of Cryptosporidium oocysts and sporozoites using discontinuous sucrose and isopycnic Percoll gradients. J. Parasitol. 73:314–319. 10.2307/3282084 [DOI] [PubMed] [Google Scholar]
- 54.Sharling L, Liu X, Gollapalli DR, Maurya SK, Hedstrom L, Striepen B. 2010. A screening pipeline for antiparasitic agents targeting cryptosporidium inosine monophosphate dehydrogenase. PLoS Negl. Trop. Dis. 4:e794. 10.1371/journal.pntd.0000794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Campbell LD, Stewart JN, Mead JR. 2002. Susceptibility to Cryptosporidium parvum infections in cytokine- and chemokine-receptor knockout mice. J. Parasitol. 88:1014–1016. 10.1645/0022-3395(2002)088[1014:STCPII]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- 56.Arrowood MJ, Donaldson K. 1996. Improved purification methods for calf-derived Cryptosporidium parvum oocysts using discontinuous sucrose and cesium chloride gradients. J. Eukaryot. Microbiol. 43:89S. 10.1111/j.1550-7408.1996.tb05015.x [DOI] [PubMed] [Google Scholar]
- 57.Arrowood MJ, Hurd MR, Mead JR. 1995. A new method for evaluating experimental cryptosporidial parasite loads using immunofluorescent flow cytometry. J. Parasitol. 81:404–409. 10.2307/3283822 [DOI] [PubMed] [Google Scholar]
- 58.Yang Z, Wang JR, Niu T, Gao S, Yin T, You M, Jiang ZH, Hu M. 2012. Inhibition of P-glycoprotein leads to improved oral bioavailability of compound K, an anticancer metabolite of red ginseng extract produced by gut microflora. Drug Metab. Dispos. 40:1538–1544. 10.1124/dmd.111.044008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7:335–336. 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. 10.1093/bioinformatics/btq461 [DOI] [PubMed] [Google Scholar]
- 61.DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. 2006. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72:5069–5072. 10.1128/AEM.03006-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. 2010. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26:266–267. 10.1093/bioinformatics/btp636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Price MN, Dehal PS, Arkin AP. 2009. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26:1641–1650. 10.1093/molbev/msp077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lozupone C, Knight R. 2005. UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71:8228–8235. 10.1128/AEM.71.12.8228-8235.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R. 2011. UniFrac: an effective distance metric for microbial community comparison. ISME J. 5:169–172. 10.1038/ismej.2010.133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lozupone C, Hamady M, Knight R. 2006. UniFrac—an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics 7:371. 10.1186/1471-2105-7-371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. 2001. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 46:3–26. 10.1016/S0169-409X(00)00129-0 [DOI] [PubMed] [Google Scholar]
- 68.Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. 2002. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 45:2615–2623. 10.1021/jm020017n [DOI] [PubMed] [Google Scholar]
- 69.Gargala G, Delaunay A, Li X, Brasseur P, Favennec L, Ballet JJ. 2000. Efficacy of nitazoxanide, tizoxanide and tizoxanide glucuronide against Cryptosporidium parvum development in sporozoite-infected HCT-8 enterocytic cells. J. Antimicrob. Chemother. 46:57–60. 10.1093/jac/46.1.57 [DOI] [PubMed] [Google Scholar]
- 70.Griffiths JK, Theodos C, Paris M, Tzipori S. 1998. The gamma interferon gene knockout mouse: a highly sensitive model for evaluation of therapeutic agents against Cryptosporidium parvum. J. Clin. Microbiol. 36:2503–2508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Theodos CM, Griffiths JK, D'Onfro J, Fairfield A, Tzipori S. 1998. Efficacy of nitazoxanide against Cryptosporidium parvum in cell culture and in animal models. Antimicrob. Agents Chemother. 42:1959–1965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hidalgo IJ. 2001. Assessing the absorption of new pharmaceuticals. Curr. Top. Med. Chem. 1:385–401. 10.2174/1568026013395010 [DOI] [PubMed] [Google Scholar]
- 73.Hu M, Borchardt RT. 1992. Transport of a large neutral amino acid in a human intestinal epithelial cell line (Caco-2): uptake and efflux of phenylalanine. Biochim. Biophys. Acta 1135:233–244. 10.1016/0167-4889(92)90226-2 [DOI] [PubMed] [Google Scholar]
- 74.Gollapalli DR, Macpherson IS, Liechti G, Gorla SK, Goldberg JB, Hedstrom L. 2010. Structural determinants of inhibitor selectivity in prokaryotic IMP dehydrogenases. Chem. Biol. 17:1084–1091. 10.1016/j.chembiol.2010.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Griffiths JK, Balakrishnan R, Widmer G, Tzipori S. 1998. Paromomycin and geneticin inhibit intracellular Cryptosporidium parvum without trafficking through the host cell cytoplasm: implications for drug delivery. Infect. Immun. 66:3874–3883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Baishanbo A, Gargala G, Duclos C, Francois A, Rossignol JF, Ballet JJ, Favennec L. 2006. Efficacy of nitazoxanide and paromomycin in biliary tract cryptosporidiosis in an immunosuppressed gerbil model. J. Antimicrob. Chemother. 57:353–355. 10.1093/jac/dki456 [DOI] [PubMed] [Google Scholar]
- 77.Schwarz R, Kaspar A, Seelig J, Kunnecke B. 2002. Gastrointestinal transit times in mice and humans measured with 27Al and 19F nuclear magnetic resonance. Magn. Reson. Med. 48:255–261. 10.1002/mrm.10207 [DOI] [PubMed] [Google Scholar]
- 78.Marona HR, Lucchesi MB. 2004. Protocol to refine intestinal motility test in mice. Lab. Anim. 38:257–260. 10.1258/002367704323133637 [DOI] [PubMed] [Google Scholar]
- 79.Casburn-Jones AC, Farthing MJ. 2004. Management of infectious diarrhoea. Gut 53:296–305. 10.1136/gut.2003.022103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kelly D, Delday MI, Mulder I. 2012. Microbes and microbial effector molecules in treatment of inflammatory disorders. Immunol. Rev. 245:27–44. 10.1111/j.1600-065X.2011.01079.x [DOI] [PubMed] [Google Scholar]
- 81.Kamada N, Seo SU, Chen GY, Nunez G. 2013. Role of the gut microbiota in immunity and inflammatory disease. Nat. Rev. Immunol. 13:321–335. 10.1038/nri3430 [DOI] [PubMed] [Google Scholar]
- 82.Tzipori S, Rand W, Griffiths J, Widmer G, Crabb J. 1994. Evaluation of an animal model system for cryptosporidiosis: therapeutic efficacy of paromomycin and hyperimmune bovine colostrum-immunoglobulin. Clin. Diagn. Lab. Immunol. 1:450–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Travers MA, Florent I, Kohl L, Grellier P. 2011. Probiotics for the control of parasites: an overview. J. Parasitol. Res. 2011:610769. 10.1155/2011/610769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pickerd N, Tuthill D. 2004. Resolution of cryptosporidiosis with probiotic treatment. Postgrad. Med. J. 80:112–113. 10.1136/pmj.2003.014175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Harp JA, Chen W, Harmsen AG. 1992. Resistance of severe combined immunodeficient mice to infection with Cryptosporidium parvum: the importance of intestinal microflora. Infect. Immun. 60:3509–3512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lantier L, Drouet F, Guesdon W, Mancassola R, Metton C, Lo-Man R, Werts C, Laurent F, Lacroix-Lamandé S. 15 October 2013. Poly(I:C)-induced-protection of neonatal mice against intestinal Cryptosporidium parvum infection requires an additional TLR5 signal provided by the gut flora. J. Infect. Dis. 10.1093/infdis/jit432 [DOI] [PubMed] [Google Scholar]
- 87.Huang J, Mullapudi N, Lancto CA, Scott M, Abrahamsen MS, Kissinger JC. 2004. Phylogenomic evidence supports past endosymbiosis, intracellular and horizontal gene transfer in Cryptosporidium parvum. Genome Biol. 5:R88. 10.1186/gb-2004-5-11-r88 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.