

Abstract
Although approved by the U.S. Food and Drug Administration, enfuvirtide is rarely used in combination antiretroviral therapies (cART) to treat HIV-1 infection, primarily because of its intense dosing schedule that requires twice-daily subcutaneous injection. Here, we describe the development of enfuvirtide-loaded, degradable poly(lactic-co-glycolic) acid microparticles that provide linear in vitro release of the drug over an 18-day period. This sustained-release formulation could make enfuvirtide more attractive for use in cART.
TEXT
Enfuvirtide (Fuzeon) is a 36-amino-acid synthetic peptide that inhibits HIV-1 replication through binding to the heptad repeat 1 domain of the viral gp41 protein, preventing the conformational changes that are required for virus-cell fusion (1, 2). Although enfuvirtide has been approved by the U.S. Food and Drug Administration, its intense dosing schedule—which requires twice-daily subcutaneous injection—has limited its use to only HIV-1-infected individuals for whom all other treatments have failed (i.e., salvage therapy). In this regard, the development of a sustained-release formulation, which would allow for less-frequent dosing, could make enfuvirtide more attractive for use in first- and second-line combination antiretroviral therapies, particularly given that it exhibits a mechanism of action that is distinct from all other approved anti-HIV drugs.
Our group has recently developed several models of controlled release from biodegradable polymer matrices, including microparticles of poly(lactic-co-glycolic) acid (PLGA), a biodegradable polyester approved by the U.S. Food and Drug Administration for human use (3–5). Briefly, these models compute the diffusion of drug out of a degradable matrix via ever-developing networks of interconnected pores formed by heterogeneous degradation and erosion using the following equation:
where D is the diffusivity of drug (CA) in the polymer matrix, erf is the error function, t is time, tε is mean time until pore formation, and σ is the variance of that time. Constants, such as D and tε, are linked empirically to the drug and matrix properties based on data from more than 50 formulations. Using these mathematical models, we determined that 10 kDa 50:50 PLGA fabricated into 10- to 60-μm diameter, nonporous microparticles were predicted to effectively sustain linear enfuvirtide (obtained through the NIH AIDS Reagent Repository) release for >2 weeks. We also determined, based on molecular weight, charge, and hydrophobicity, that a 3-kDa dextran labeled 1:2 with Texas Red molecules (dex-TR; Invitrogen) would serve as an appropriate surrogate for enfuvirtide, thereby enabling direct characterization of microparticle size, morphology, drug loading, and drug release.
All of the degradable microparticles described in this study were produced using a standard double-emulsion fabrication procedure, as described previously (6). Briefly, a 40:1 emulsion of 50 mg/ml PLGA in dichloromethane (oil phase) and 2 mg/ml aqueous enfuvirtide or dex-TR were mixed for 10 s using a probe sonicator (Sonics & Materials, Inc.). This single emulsion was then homogenized (200 rpm, 60 s, Silverson L4RT-A) to form a water-in-oil-in-water emulsion. After 3 h, precipitated PLGA particles were collected by centrifugation (1,500 × g, 10 min, 4°C) and washed 4 times in deionized water before being lyophilized (Virtis Benchtop K freeze dryer, operating at 60 mTorr) for storage until use. Dissolution of microparticles by a 1:5 solution of dimethyl sulfoxide to aqueous 0.05 N sodium hydroxide and 0.5% sodium dodecyl sulfate confirmed encapsulation of dexTR at 25.2 ± 5.1 μg per mg of formulation. For both dex-TR and enfuvirtide, size measurements by the volume impedance method revealed a volume-average particle diameter of 32.2 ± 10.2 μm. These dimensions are consistent with other FDA-approved PLGA microparticle medications, including risperidone long-acting injection (Risperdal Consta) and exenatide entended release (Bydureon), which establish stable subcutaneous depots in vivo for systemic sustained release.
Surface morphology and internal distribution of dex-TR in our rationally designed PLGA microparticles were assessed by scanning electron microscopy (SEM) and confocal microscopy, respectively. All particles appeared to have a spherical shape with uniformly smooth surfaces (Fig. 1A). A seemingly identical morphology was observed for our rationally designed enfuvirtide-loaded microparticles (data not shown). Confocal microscopy revealed that dex-TR was occluded in small pockets randomly scattered throughout the polymer matrix, as would be expected (Fig. 1B). This morphology (along with the size and hydrophilicity of the encapsulated agent) is consistent with polymer degradation and erosion-driven controlled-release processes. In vitro dissolution assays were used to verify the release kinetics of dex-TR release from the microparticles. Assays were conducted at 37°C with end-over rotation and pseudoperfect sink conditions, as described previously (6). The concentration of dex-TR was measured by fluorescence spectrometry and used to calculate cumulative dex-TR release for the study's duration as a percentage of the formulation's loading. As indicated in Fig. 2A, we found that dex-TR was released from the microparticles in accordance with our model predictions. In light of this finding, we next assessed the dissolution kinetics of enfuvirtide-loaded microparticles. Enfuvirtide concentration was measured by a high-pressure liquid chromatographic (HPLC) assay, as described previously (7). Releasate for analysis from incubating particles was collected at set time points and replaced with fresh phosphate-buffered saline to maintain sink conditions. Our data show that enfuvirtide was released in vitro from the PLGA microparticles, as predicted by our mathematical models (Fig. 2B). More specifically, a steady-state concentration of peptide ranging from 60 to 140 ng/ml was maintained for up to 18 days. Because the hydrolysis of PLGA microparticles can produce an acidic environment which can denature or degrade peptides, we next assessed the stability and antiviral activity of the enfuvirtide that was released from the microparticles. HPLC analyses revealed that the enfuvirtide used in these studies was ∼94.5% pure prior to the encapsulation procedure. Following encapsulation and release from the polymer matrix, peptide purity was maintained at 83% ± 19% (data not shown), which was not statistically different from the starting material. Most importantly, the enfuvirtide that was released from the PLGA microparticles retained its ability to effectively inhibit HIV-1LAI infection of TZM-bl cells. This assay was implemented as done previously, and the resulting 50% inhibitory concentration (IC50) values (11.5 ± 3.7 ng/ml) compared favorably with those in published data (8). By comparing serially diluted releasate to an enfuvirtide control in the TZM antiviral assays (i.e., enfuvirtide prior to encapsulation), we were able to back-calculate the concentration of active peptide that was released from the microparticles, which very closely followed the enfuvirtide release measured using HPLC (Fig. 2B).
FIG 1.

SEM and confocal microscopy images of dex-TR-loaded PLGA microparticles. (A) SEM images of the microparticles revealing a uniform shape and smooth surface. (B) Confocal microscopy images revealing that dex-TR is randomly and heterogeneously dispersed throughout the polymer matrix.
FIG 2.
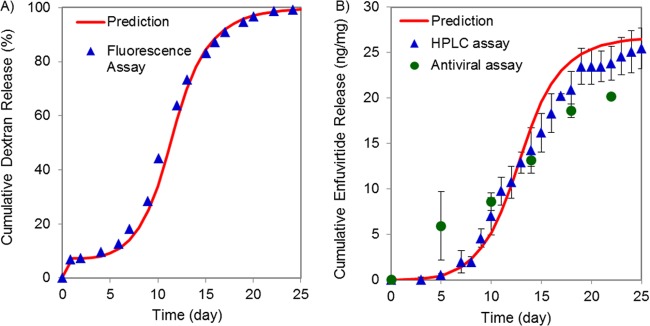
In vitro dissolution assays of dex-TR and enfuvirtide-loaded PLGA microparticles. (A) In vitro dissolution assays of DexTR from PLGA microparticles as measured by fluorescence spectroscopy (n = 3). (B) In vitro dissolution assays of enfuvirtide from PLGA microparticles as measured by HPLC (7) and antiviral (8) assays (n = 2). Error bars indicate one standard deviation from the mean.
In conclusion, our data suggest that model-aided design of sustained-release formulation of enfuvirtide can indeed generate a formulation that provides a linear release of the drug over an 18-day period. PLGA microparticles do not appear to impact either enfuvirtide stability or antiviral activity, although preclinical studies in a suitable animal model are required to further validate these findings. Importantly, the successful development of a sustained-release formulation of enfuvirtide could reposition this antiviral such that it is more widely used in combination antiretroviral therapies for the treatment of HIV-1 infection.
ACKNOWLEDGMENTS
Steven R. Little invented, and has a right to receive licensing proceeds generated from, the mathematical modeling technology that was evaluated in this research study. Steven R. Little and the University of Pittsburgh also hold equity in Qrono, Inc., which has a license to this technology from the University.
Footnotes
Published ahead of print 23 December 2013
REFERENCES
- 1.Dietrich U. 2001. HIV-1 entry inhibitors. AIDS Rev. 3:89–97 [Google Scholar]
- 2.Reeves JD, Gallo SA, Ahmad N. 2002. Sensitivity of HIV-1 to entry inhibitors correlates with envelope/coreceptor affinity, receptor density and fusion kinetics. Proc. Natl. Acad. Sci. U. S. A. 99:16249–16254. 10.1073/pnas.252469399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rothstein SN, Federspiel WJ, Little SR. 2008. A simple model framework for the prediction of controlled release from bulk eroding polymer matrices. J. Mater. Chem. 18:1873–1880. 10.1039/b718277e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rothstein SN, Federspiel WJ, Little SR. 2009. A unified mathematical model for the prediction of controlled release from surface and bulk eroding polymer matrices. Biomaterials 30:1657–1664. 10.1016/j.biomaterials.2008.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rothstein SN, Little SR. 2011. A “tool box” for rational design of degradable controlled release formulations. J. Mater. Chem. 21:29–39. 10.1039/c0jm01668c [DOI] [Google Scholar]
- 6.Rothstein SN, Kay JE, Schopfer FJ, Freeman BA, Little SR. 2012. A retrospective mathematical analysis of controlled release design and experimentation. Mol. Pharm. 9:3003–3011. 10.1021/mp300388w [DOI] [PubMed] [Google Scholar]
- 7.D'Avolio A, Sciandra M, de Requena DG, Ibañez A, Bonora S, Di Perri G. 2006. An improved HPLC fluorimetric method for the determination of enfuvirtide plasma levels in HIV-infected patients. Ther. Drug Monit. 28:110–115. 10.1097/01.ftd.0000179421.25337.62 [DOI] [PubMed] [Google Scholar]
- 8.Ingallinella P, Bianchia E, Ladwaa NA, Wang Y-J, Hrin R, Veneziano M, Bonelli F, Ketas TJ, Moore JP, Miller MD, Pessi A. 2009. Addition of a cholesterol group to an HIV-1 peptide fusion inhibitor dramatically increases its antiviral potency. Proc. Natl. Acad. Sci. U. S. A. 106(14):5801–5806. 10.1073/pnas.0901007106 [DOI] [PMC free article] [PubMed] [Google Scholar]