

Abstract
Antibiotic-resistant enterococci are major causes of hospital-acquired infections. All enterococci are intrinsically resistant to most cephalosporins, antibiotics in the beta-lactam family that impair peptidoglycan synthesis by inactivating the transpeptidases responsible for cross-linking. In addition, clinical isolates of enterococci often possess acquired resistance to vancomycin, a glycopeptide antibiotic that impairs peptidoglycan biosynthesis by a mechanism distinct from that of the beta-lactams, namely, by binding to the d-Ala-d-Ala termini found in peptidoglycan precursors to prevent their utilization by biosynthetic transglycosylases. Antimicrobial synergism between vancomycin and beta-lactams against vancomycin-resistant enterococci was originally described decades ago, but the genetic basis for synergy has remained unknown. Because a complete understanding of the mechanism underlying synergy between vancomycin and beta-lactams might suggest new targets or strategies for therapeutic intervention against antibiotic-resistant enterococci, we explored the genetic basis for synergy between vancomycin and cephalosporins in Enterococcus faecalis. To do so, we developed a counterselection strategy based on a dominant-negative mutant of thymidylate synthase and implemented this approach to create a panel of mutants in vancomycin-resistant E. faecalis. Our results confirm that vancomycin promotes synergy by inducing expression of the van resistance genes, as a mutant in which the van genes are expressed in the absence of vancomycin exhibits susceptibility to cephalosporins. Further, we show that peptidoglycan precursors substituted with d-Ala-d-Lac are not required for vancomycin-enhanced cephalosporin sensitivity. Instead, production of the d,d-carboxypeptidase VanYB is both necessary and sufficient to dramatically sensitize E. faecalis to cephalosporins.
INTRODUCTION
Enterococci are ubiquitous inhabitants of the gastrointestinal tract in most animals, including humans (1). However, antibiotic-resistant enterococci are also major causes of hospital-acquired infections and therefore represent a serious public health problem (2, 3). One well-known risk factor for the acquisition of enterococcal hospital-acquired infections is prior therapy with broad-spectrum cephalosporins (4), antibiotics that belong to the β-lactam family and interfere with cell wall biosynthesis by inhibiting the transpeptidase function of penicillin-binding proteins (PBPs) that cross-link peptidoglycan. Enterococci exhibit intrinsic resistance to cephalosporins, enabling them to proliferate and achieve abnormally high densities in the gastrointestinal (GI) tract of patients during cephalosporin therapy (5), thereby promoting dissemination to other sites where they cause infection. This intrinsic cephalosporin resistance is a trait shared by essentially all isolates of Enterococcus faecalis. Although the underlying basis for cephalosporin resistance in enterococci is not fully understood, one key factor that is required is a specialized “low-affinity” PBP, known as Pbp5 (6–8). Pbp5 exhibits reduced affinity for beta-lactam antibiotics relative to the other enterococcal PBPs, which enables Pbp5 to carry out peptidoglycan cross-linking to mediate growth in the presence of cephalosporins. In addition to the role of cephalosporin resistance in promoting intestinal overgrowth and hospital-acquired infections, cephalosporin resistance is also relevant in clinical settings when antibiotic-resistant hospital lineages of E. faecalis are responsible for infection in immunocompromised patients, as few therapeutic options exist to treat such infections.
Vancomycin-resistant enterococci (VRE) are particularly troublesome in this regard. Although not naturally resistant to therapeutic levels of vancomycin, E. faecalis can become resistant through the acquisition of any of several mobile genetic elements carrying a group of contiguous genes, referred to here as the “van gene cluster” (reviewed in references 9, 10, 11, and 12). One of two related clusters, the vanA cluster or the vanB cluster, is responsible for resistance in most clinically relevant contexts; each of the clusters enables the synthesis of altered peptidoglycan precursors containing d-Ala-d-lactate termini in place of the d-Ala-d-Ala moiety found in normal precursors. The d-Ala-d-Lac substitution prevents binding of vancomycin (which normally recognizes the d-Ala-d-Ala termini) but can still be used as a substrate for peptidoglycan synthesis by the biosynthetic transpeptidases (thereby conferring vancomycin resistance). Each of the vanA and vanB clusters encodes several common biological functions in 2 transcriptional units. First, each encodes a vancomycin-responsive two-component signaling system (composed of the VanS sensor kinase and the VanR response regulator in the vanA cluster; by convention, equivalent genes in the vanB cluster are denoted with a subscript “B”) that is activated when vancomycin is present in the environment and drives transcription of the signaling genes themselves, as well as genes in the second transcriptional unit. This second unit encodes the biochemical functions required for alteration of the peptidoglycan to provide vancomycin resistance: the VanH dehydrogenase that reduces pyruvate to d-Lac; the Van ligase (VanA or VanB, respectively) that catalyzes formation of an ester bond between d-Ala and d-Lac to form a depsipeptide; the VanX d,d-dipeptidase that cleaves naturally produced d-Ala-d-Ala dipeptides to prevent their incorporation into peptidoglycan precursors; and the VanY d,d-carboxypeptidase that cleaves d-Ala-d-Ala moieties that escape the action of VanX and are incorporated into peptidoglycan precursors. Each cluster also encodes an additional, cluster-specific protein often referred to as an “accessory” factor (VanZ or VanW, respectively) whose functions are not clearly defined. A recent report indicated that some vanB clusters encode an additional accessory protein, VanV, that does not detectably contribute to vancomycin resistance and whose function is unknown (13).
Widespread occurrence of cooperative antimicrobial activity between vancomycin and beta-lactam antibiotics against both vanA and vanB vancomycin-resistant enterococci was described soon after the emergence of vancomycin resistance in enterococci (14–17), although, for reasons that are unclear, some clinical isolates do not exhibit this trait (18–20). This phenomenon was referred to as “synergy” in the original publications, a convention that is followed here. The mechanism of synergy has been investigated; for example, researchers who performed studies of Enterococcus faecium (16, 17) reported that beta-lactam antibiotics do not interfere with the vancomycin-induced expression of vancomycin-resistance determinants or with their enzymatic activities. These findings led to a model in which Pbp5, the “low-affinity” PBP that catalyzes cross-linking of peptidoglycan in the presence of beta-lactams (and is therefore required for cephalosporin resistance), is unable to use peptidoglycan precursors containing d-Ala-d-Lac termini as substrates for cross-linking. In this model, when vancomycin induces expression of van genes and the cells produce precursors containing d-Ala-d-Lac, other PBPs must carry out all peptidoglycan cross-linking functions. Because the other PBPs exhibit relatively high affinity for beta-lactams compared to Pbp5 (i.e., are readily inhibited by beta-lactams), the bacteria are highly susceptible to beta-lactam agents in these circumstances (i.e., synergy is observed). However, these studies were performed prior to the complete elucidation of the genetic basis for vancomycin resistance in enterococci (i.e., delineation of the organization and content of the van clusters and of the biochemical functions of the van gene products), and this model for synergy has therefore not been tested genetically.
Because a complete understanding of the mechanism underlying synergy between vancomycin and beta-lactams may suggest new targets or strategies for therapeutic intervention against antibiotic-resistant enterococci, we explored the genetic basis for synergy in E. faecalis, exploiting a new counterselectable marker system for efficient genetic manipulation. Our findings confirm that vancomycin itself is required only to activate expression of the van gene cluster. Furthermore, we show that production of peptidoglycan precursors substituted with d-Ala-d-Lac is not required for vancomycin-enhanced cephalosporin sensitivity. Instead, production of the d,d-carboxypeptidase VanYB is both necessary and sufficient to dramatically sensitize E. faecalis to cephalosporins.
MATERIALS AND METHODS
Bacterial strains, growth media, and chemicals.
Bacterial strains and plasmids used in the study are listed in Table 1. E. faecalis strains were grown in half-strength brain heart infusion (hBHI) for routine maintenance or in Mueller-Hinton broth (MHB) for antimicrobial susceptibility assays at 37°C. MM9YE medium (21) was used in specific cases as indicated. Escherichia coli strains were grown in lysogeny broth (LB) during selection for resistance to chloramphenicol (Cm) or hBHI during selection for resistance to erythromycin (Em). Concentrations of antibiotics were as follows: chloramphenicol, 10 μg/ml (E. faecalis and E. coli); erythromycin, 10 μg/ml (E. faecalis) or 100 μg/ml (E. coli). All antibiotics and chemicals were obtained from Sigma-Aldrich unless stated otherwise.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant genotype or descriptiona | Source or reference |
|---|---|---|
| Strains | ||
| E. coli LE392 | Cloning host for pCJK245-based plasmids | Laboratory stock |
| E. coli DH5α | Routine cloning host | Laboratory stock |
| E. faecalis OG1 | Wild-type reference strain (MLST 1); Vans | 33 |
| E. faecalis T1 (SS498) | Wild-type (MLST 21), CDC reference strain; Vans | 42 |
| E. faecalis HIP11704 | vanA-containing clinical isolate (MLST 4); Vanr | 32, 43 |
| E. faecalis V583 | vanB-containing clinical isolate (MLST 6); Vanr | 30 |
| E. faecalis CK217 | V583 ΔvanRB2 | This work |
| E. faecalis CK219 | V583 ΔvanW2 | This work |
| E. faecalis CK220 | V583 ΔvanB2 | This work |
| E. faecalis CK222 | V583 ΔvanHB2 | This work |
| E. faecalis CK223 | V583 ΔvanYB2 | This work |
| E. faecalis CK224 | V583 ΔvanXB2 | This work |
| E. faecalis CK225 | V583 vanSB T237A | This work |
| Plasmids | ||
| pCJK218 | E. faecalis allelic exchange vector (Cmr); pheS* counterselection | 25 |
| pCJK245 | E. faecalis allelic exchange vector (Cmr); thyA* counterselection | This work |
| pCJK247 | ΔvanRB2 (ΔL7-G213) in pCJK245 | This work |
| pCJK249 | ΔvanW2 (ΔT7-I270) in pCJK245 | This work |
| pCJK250 | ΔvanB2 (ΔE15-T337) in pCJK245 | This work |
| pCJK256 | ΔvanHB2 (ΔQ14-R318) in pCJK245 | This work |
| pCJK257 | ΔvanYB2 (ΔY6-G257) in pCJK245 | This work |
| pCJK258 | ΔvanXB2 (ΔF5-Y192) in pCJK245 | This work |
| pCJK259 | vanSB T237A in pCJK245 | This work |
| pBK2 | cCF10-inducible expression vector (Cmr) | 24 |
| pJLL98 | vanYB in pBK2 | This work |
| pJLL99 | vanYB3 (ΔM1-V46) in pBK2 | This work |
| pJLL107 | vanYB D195A in pBK2 | This work |
| pJLL108 | vanYB E238A in pBK2 | This work |
| pBK200 | E. faecalis expression vector, constitutive P23 promoter (Emr) | Snyder et al. (44) |
| pCJK237 | thyA R177E C197W in pBK200 | This work |
MLST, multilocus sequence type; Van, vancomycin.
Construction of plasmids.
A derivative of the expression plasmid pBK200 (pCJK237) carrying the dominant-negative allele of thyA (also known as EF1576) from E. faecalis OG1RF was constructed by first using a BsaI-based cloning scheme to seamlessly fuse two PCR amplicons encoding thyA with the R177E C197W substitutions. The resulting product was introduced into pBK200 using primer-encoded BamHI and SphI restriction sites.
To construct the markerless allelic exchange plasmid pCJK245 for genetic manipulation of E. faecalis, we first used PCR to fuse the constitutively expressed P23 promoter (22) to a synthetic thyA R177E C197W allele (synthesized at IDT Technologies) containing synonymous substitutions at the third position of most codons (to prevent recombination with thyA encoded in the genome of E. faecalis). This P23-thyA* fragment was introduced into pCJK218 between the SphI and MfeI sites by Gibson Assembly (23), thereby replacing the P-pheS* counterselection cassette of pCJK218 in its entirety. The resulting plasmid is otherwise identical to pCJK218.
Derivatives of pCJK245 carrying mutant alleles for all genes of interest were created by introducing PCR products into pCJK245 using Gibson Assembly (23). The ΔvanRB2 allele encodes a short VanRB-derived peptide that retains 6 amino acids at the N-terminal end and 7 amino acids at the C-terminal end (∼94% of the open reading frame [ORF] is deleted), which were retained in the mutant in an attempt to avoid perturbing the expression of adjacent genes. Similarly, the ΔvanW2 allele encodes a short VanW-derived peptide that retains 6 amino acids at the N-terminal end and 5 amino acids at the C-terminal end (∼96% of the ORF is deleted); the ΔvanB2 allele encodes a short VanB-derived peptide that retains 14 amino acids at the N-terminal end and 5 amino acids at the C-terminal end (∼94% of the ORF is deleted); the ΔvanHB2 allele encodes a short VanHB-derived peptide that retains 13 amino acids at the N-terminal end and 5 amino acids at the C-terminal end (∼94% of the ORF is deleted); the ΔvanYB2 allele encodes a short VanYB-derived peptide that retains 5 amino acids at the N-terminal end and 11 amino acids at the C-terminal end (∼94% of the ORF is deleted); and the ΔvanXB2 allele encodes a short VanXB-derived peptide that retains 4 amino acids at the N-terminal end and 10 amino acids at the C-terminal end (∼93% of the ORF is deleted). V583 genomic DNA was used as the template for all PCRs to construct deletion alleles.
Plasmid pBK2 contains the pheromone-responsive prgQ promoter derived from the pCF10 enterococcal plasmid upstream of a promoterless lacZ (24). Expression from pBK2 can be induced by addition of cCF10 peptide pheromone. Derivatives of pBK2 containing vanYB were constructed using Gibson assembly to replace the BamHI-EcoRI fragment encoding lacZ with PCR-amplified vanYB alleles, generated from V583 genomic DNA.
Construction of E. faecalis mutants.
Markerless allelic exchange with pCJK245-based plasmids was used to construct mutant strains of E. faecalis. The deletion mutants were in frame, and all mutants were unmarked. The procedure for mutant construction was identical to that used previously with pCJK218 (25), except that counterselection was performed by plating dilutions of cell suspensions on MH agar (no thymine supplement) at 30°C. The desired mutants were identified after counterselection by PCR analysis. For each mutant constructed, two isolates that were constructed completely independently from each other were analyzed and found to exhibit identical phenotypes.
Antibiotic susceptibility.
MICs for antibiotics were determined in liquid cultures using a microtiter plate serial dilution method in a Bioscreen C plate reader (Oy Growth Curves Ab, Ltd.). Two-fold dilutions of antibiotics in the indicated growth medium were prepared in the wells of a 100-well honeycomb microtiter plate. Where indicated, the medium was supplemented with subinhibitory levels of vancomycin or with cCF10 peptide (for induction of expression from pBK2-based plasmids). Bacteria from stationary-phase cultures were inoculated into each well to a cell density of ∼105 CFU/ml. Plates were incubated at 37°C for 24 h with brief shaking, and measurement of the optical density at 600 nm (OD600) was performed at 15-min intervals. The lowest concentration of antibiotic that prevented growth was recorded as the MIC. Time-kill assays were performed by growing cells at 37°C to the exponential phase in MHB supplemented or not with vancomycin (0.25 μg/ml). The cultures were split, and ceftriaxone or ceftazidime was added to reach a final concentration of 2,048 μg/ml. Control cultures received no cephalosporins. Samples were removed immediately or after 24 h of incubation, and viable cells were enumerated on MH agar plates. Checkerboard susceptibility assays to assess combinations of antimicrobials were performed in MHB at 37°C as previously described (26, 27), except in the wells of 100-well honeycomb plates. The fractional inhibitory concentration (FIC) indices were calculated according to the following formula: FIC index = FICA + FICB, where FICA = (MIC of drug A in combination)/(MIC of drug A alone) and FICB = (MIC of drug B in combination)/(MIC of drug B alone). Conservative interpretation of the FIC index has traditionally defined synergism as an FIC index of ≤0.5.
Analysis of growth kinetics.
Generation times for exponentially growing cells were determined from growth curves performed in static MHB cultures at 37°C using a Bioscreen C plate reader.
qRTPCR analysis of gene expression.
Overnight cultures were diluted to OD600 = 0.01 in MHB supplemented or not with a subinhibitory concentration of vancomycin (0.25 μg/ml) and incubated at 37°C until the exponential phase was reached. Quantitative reverse transcription-PCR (qRTPCR) was performed on RNA extracted from exponentially growing cultures and subjected to cDNA synthesis using random hexamer primers. Calculations of fold change in gene expression used the Pfaffl method and gyrB as a reference gene.
RESULTS
Counterselection based on dominant-negative thymidylate synthase (TS).
To perform allelic exchange in E. faecalis, we and others have used a counterselection scheme based on a mutant allele of a phenylalanyl tRNA synthetase subunit (encoded by pheS) exhibiting relaxed substrate specificity (21, 28). The mutant synthetase aminoacylates tRNAPhe with the nonnative substrate, p-Cl-Phe, leading to incorporation of p-Cl-Phe into protein and inhibition of growth. To implement pheS-mediated counterselection in E. faecalis, bacteria must be grown on a specific semidefined medium (MM9YE) lacking significant amounts of phenylalanine but supplemented with high concentrations of p-Cl-Phe (10 mM). Although we have generally experienced success with this counterselection strategy, in some cases we were unable to isolate recombinants carrying the desired mutation on counterselection medium containing p-Cl-Phe (C. J. Kristich; data not shown), even though we knew from other studies that such mutants should be viable. Prompted by concern that some mutants of interest might be unable to grow on p-Cl-Phe counterselection medium, we sought an alternative strategy for counterselection that might be more flexible, with the potential to be widely applicable. We hypothesized that dominant-negative mutants of conditionally essential gene products would be well suited for use in counterselection schemes.
West and coworkers previously described a dominant-negative mutant of thymidylate synthase (TS) from Escherichia coli (29). TS is enzymatically active as a homodimer, whose subunits undergo exchange in solution. One product of TS activity, dTMP, is required for synthesis of DNA; thus, TS is essential for viability unless thymine is available for uptake in the environment. West (29) identified a catalytically inactive mutant TS bearing 2 substitutions (R126E C146W) that poisons the function of wild-type TS subunits upon coexpression in E. coli, rendering the cells unable to grow without thymine supplements. We therefore reasoned that dominant-negative TS could form the basis of an effective counterselection strategy, because only recombinants lacking dominant-negative TS should be able to grow on medium devoid of thymine.
Alignment of the amino acid sequence of E. faecalis TS (EF1576; encoded by thyA) with that of E. coli revealed that the E. faecalis enzyme possesses residues equivalent to R126 and C146 of the E. coli enzyme (R177 and C197, respectively). To test if an analogous double-mutant TS would function as a dominant negative in E. faecalis, we constructed the E. faecalis thyA R177E C197W double mutant. Expression of thyA R177E C197W in 2 distinct lineages of otherwise wild-type E. faecalis substantially inhibited growth on 2 types of nutrient media in the absence of exogenous thymine (Fig. 1), indicating that the E. faecalis R177E C197W double-mutant TS is fully capable of exerting a dominant-negative effect on wild-type TS. Although inoculation at high cell density yields some residual growth on media lacking thymine, the efficiency of plating is reduced by 4 orders of magnitude in cells expressing the double-mutant TS (Fig. 1). Thus, to exploit this property as a counterselectable marker for genetic manipulation, one need only plate a series of serial dilutions to isolate colonies lacking dominant-negative TS. To implement this approach, we constructed a modified version of our previous allelic exchange plasmid (pCJK218) in which the pheS* counterselectable marker found in pCJK218 has been replaced with the dominant-negative thyA R177E C197W double mutant (yielding pCJK245). This new allelic exchange plasmid was used to explore the genetic basis for the potent synergy between vancomycin and cephalosporins in vancomycin-resistant E. faecalis. An overview of the genetic exchange strategy used is depicted in Fig. 2.
FIG 1.
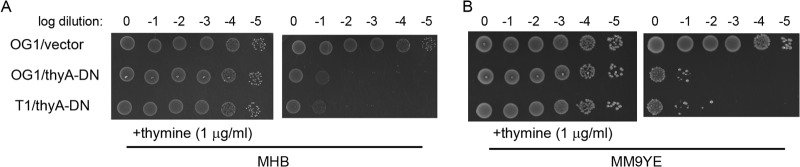
The E. faecalis thymidylate synthase R177E C197W double mutant functions as a dominant negative. Wild-type E. faecalis strains carrying either an empty expression vector or the expression vector with thyA R177E C197W were subjected to 10-fold serial dilutions and inoculated on MH agar (A) or the semidefined MM9YE agar (B) that had been supplemented with Em and thymine where indicated, followed by incubation at 37°C for ∼16 h. Wild-type E. faecalis does not require thymine for growth (top) due to the activity of TS. Expression of thyA R177E C197W impairs the function of wild-type TS, thereby preventing growth unless exogenous thymine is provided. The E. faecalis host strains were OG1 and T1. The plasmids were as follows: vector, pBK200; thyA-DN, pCJK237 carrying thyA R177E C197W.
FIG 2.
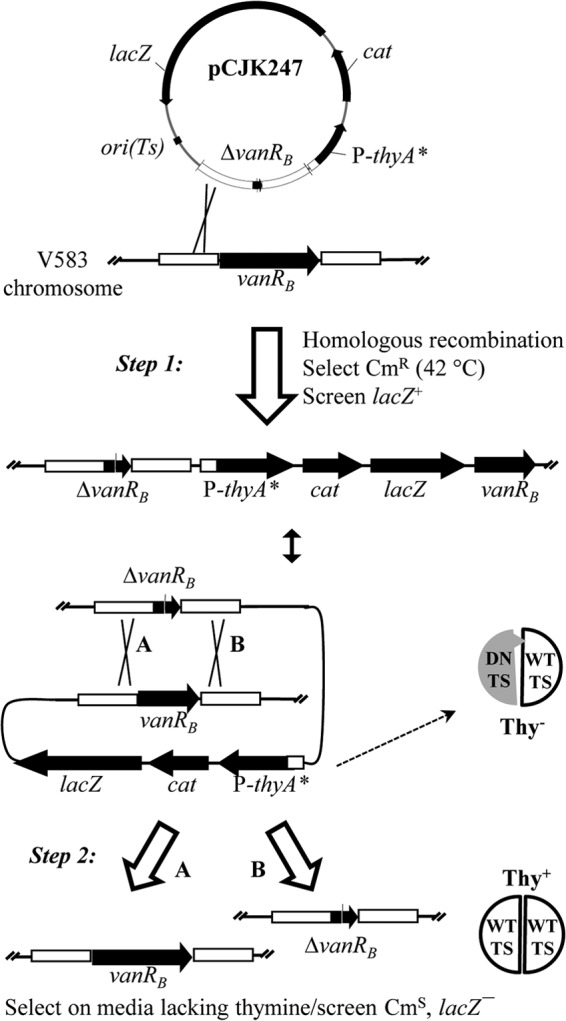
Schematic of dominant-negative TS-mediated genetic exchange. A derivative of pCJK245 carrying an allele of interest (e.g., pCJK247) is introduced into the target strain at a permissive temperature (30°C). Selection for resistance to Cm at a nonpermissive temperature (42°C) yields recombinants in which the plasmid has integrated into the chromosome via a region of cloned chromosomal DNA (step 1). Production of dominant-negative TS (DN TS) renders the cells Thy negative (Thy−) by interfering with wild-type TS (WT TS) activity. Plasmid excision (step 2) leaves either the wild-type allele (A) or the mutant allele (B) on the chromosome and renders the cells Thy+ (capable of robust growth on medium lacking thymine). An equivalent sequence of events can be drawn for plasmid integration at either the upstream or downstream fragment.
Synergy between vancomycin and cephalosporins requires expression of genes in the van gene cluster.
Previous studies describing synergy between vancomycin and beta-lactams in clinical isolates of enterococci reported a correlation between synergy and vancomycin resistance of the strain and suggested that synergy occurs only if vancomycin resistance genes are expressed (14, 16). We examined 3 strains of E. faecalis for synergy between subinhibitory levels of vancomycin and ceftriaxone, a broad-spectrum cephalosporin: V583, a vancomycin-resistant clinical isolate whose genome contains a vanB cluster (30, 31); HIP11704, a vancomycin-resistant clinical isolate whose genome contains a vanA cluster (32); and OG1, a vancomycin-susceptible strain whose genome lacks a van gene cluster (33). Indeed, subinhibitory levels of vancomycin led to a drastic reduction in ceftriaxone resistance for both V583 and HIP11704 (Table 2), demonstrating synergism in accordance with previous studies on vancomycin-resistant enterococci. For V583, the extent of reduction in ceftriaxone resistance correlated in a dose-dependent manner with the amount of vancomycin present. This synergism was also apparent when evaluated with checkerboard susceptibility assays, in which both V583 and HIP11704 exhibited fractional inhibitory concentration (FIC) indices of ≤0.25, well below the conservative cutoff value of 0.5 that typically defines synergism. Synergism was also observed for V583 against ceftazidime, another broad-spectrum cephalosporin, but not for an antibiotic (chloramphenicol) with a distinct cellular target, indicating that the synergism is not the result of a nonspecific general stress response. Time-kill studies were performed to determine if either ceftriaxone or ceftazidime at 2,048 μg/ml was bactericidal to V583 cells treated with vancomycin at a concentration (0.25 μg/ml) sufficient to substantially reduce the MIC (Table 2), but no killing was observed whether or not vancomycin was present (not shown). Thus, vancomycin does not promote bactericidal activity of the cephalosporins but instead acts to prevent growth at substantially lower cephalosporin concentrations than in its absence. In contrast to the strains containing van clusters, subinhibitory levels of vancomycin did not exhibit synergy with ceftriaxone toward E. faecalis OG1; instead, it led to a substantial increase in cephalosporin resistance by an as-yet-unknown mechanism. Thus, these results are consistent with a model in which expression of one or more genes in the van cluster is required for synergy with cephalosporins.
TABLE 2.
Median MICs for various antibiotics in the presence of subinhibitory vancomycin levels
| Strain or antibiotic | MIC (µg/ml)a |
||||
|---|---|---|---|---|---|
| No Van | + Van (0.25 μg/ml) | + Van (0.5 μg/ml) | + Van (1 μg/ml) | + Van (2 μg/ml) | |
| V583 | |||||
| Ceftriaxone | 512 | 8 | 2 | 1 | 0.5 |
| Ceftazidime | 1,024 | 32 | 16 | 4 | 2 |
| Chloramphenicol | 8 | 8 | 8 | 8 | 8 |
| HIP11704 | |||||
| Ceftriaxone | 512 | ND | ND | ND | 2 |
| OG1 | |||||
| Ceftriaxone | 32 | 128 | 256 | ND | ND |
MICs were determined in MHB after 24 h at 37°C from a minimum of 3 independent experiments. Vancomycin (Van) MICs were as follows: for V583, 16; for HIP11704, 512; and for OG1, 2. ND, not determined.
To test this hypothesis, we constructed a mutant of E. faecalis V583 lacking the VanRB response regulator. VanRB is the DNA-binding transcription factor that drives expression of the vancomycin resistance genes in the vanB cluster (34); thus, mutants lacking VanRB are unable to express any of the cluster genes. We confirmed these previous observations by qRTPCR analysis of a distal gene in the van cluster (vanXB), which was chosen as an indicator of expression for the vanYBWHBBXB operon, revealing that essentially no induction of van expression occurred in our ΔvanRB mutant upon treatment with vancomycin (Table 3). As expected from previous studies (35), loss of VanRB rendered V583 susceptible to vancomycin (Table 4). Furthermore, and in contrast to wild-type V583, inclusion of a subinhibitory level of vancomycin did not yield synergy with ceftriaxone for the ΔvanRB mutant (Table 4), indicating that VanRB-mediated van gene expression is necessary for synergism.
TABLE 3.
qRTPCR analysis of vanXB gene expression in van mutants
| E. faecalis strainb | Fold change (±SE)a |
|
|---|---|---|
| Uninduced | Induced (+ Van [0.25 μg/ml]) | |
| ΔvanRB | 0.78 (±0.15) | 0.02 (±0.0005) |
| vanSB T237A | 105.24 (±3.65) | 14.85 (±3.54) |
| ΔvanYB | 1.32 (±0.03) | 0.56 (±0.13) |
| ΔvanB | 0.76 (±0.1) | 1.44 (±0.39) |
Data represent fold change in vanXB transcript levels expressed relative to wild-type V583 ± standard error. Data represent the averages of the results of 2 independent experiments, each performed in triplicate.
Strains were as follows: ΔvanRB, E. faecalis CK217; vanSB T237A, E. faecalis CK225; ΔvanYB, E. faecalis CK223; ΔvanB, E. faecalis CK220.
TABLE 4.
Median MICs for van mutants
| E. faecalis strainb | MIC (µg/ml)a |
||
|---|---|---|---|
| Vancomycin | Ceftriaxone | Ceftriaxone (+ Van [0.25 μg/ml]) | |
| V583 | 16 | 512 | 8 |
| ΔvanRB | 1 | 512 | 512 |
| vanSB T237A | 128 | 4 | ND |
| ΔvanYB | 2 | 512 | 512 |
| ΔvanW | 16 | 512 | 4 |
| ΔvanHB | 2 | 512 | 4 |
| ΔvanB | 2 | 512 | 2 |
| ΔvanXB | 4 | 512 | 4 |
MICs were determined in MHB after 24 h at 37°C from a minimum of 3 independent experiments. ND, not determined.
Strains were as follows: ΔvanRB, E. faecalis CK217; vanSB T237A, E. faecalis CK225; ΔvanYB, E. faecalis CK223; ΔvanW, E. faecalis CK219; ΔvanHB, E. faecalis CK222; ΔvanB, E. faecalis CK220; ΔvanXB, E. faecalis CK224.
al-Obeid and coworkers (17) previously studied a strain of Enterococcus faecium with an unknown mutation that constitutively expresses vancomycin resistance proteins (i.e., in the absence of vancomycin). This mutant was hypersusceptible to beta-lactams, suggesting that expression of the van gene cluster is sufficient for synergism and that vancomycin itself is required for synergy only to induce expression of the van gene cluster. However, the mutation conferring constitutive vancomycin resistance in this strain was not identified, and thus some question remained about the effect of the mutation on susceptibility to beta-lactams. To directly test if van gene expression is sufficient for synergism, we took advantage of previous findings (36) showing that mutations at Thr237 of the VanSB sensor kinase led to constitutive activation of the VanRB response regulator, even in the absence of vancomycin. We constructed an E. faecalis mutant that encodes a VanSB T237A mutant at its native location in the vanB cluster. qRTPCR analysis revealed that the mutant exhibited high levels of van gene expression in the absence of vancomycin, as expected (Table 3). Furthermore, induction by vancomycin led to levels of van expression that were ∼15-fold higher than those observed in wild-type V583. Susceptibility analyses (Table 4) revealed that the mutant exhibited elevated levels of resistance to vancomycin, presumably as a result of the unusually high-level expression of the van genes. However, resistance to ceftriaxone was substantially diminished in the vanSB T237A mutant (in the absence of vancomycin), which is consistent with the hypothesis that van gene expression—but not vancomycin per se—is necessary and sufficient for synergy with cephalosporins.
Synergy requires VanYB but not production of peptidoglycan precursors containing d-Ala-d-Lac.
To define which of the genes in the vanB cluster promote synergism between vancomycin and cephalosporins, we constructed a series of additional in-frame deletion mutants in E. faecalis, each lacking one of the van genes. Deletion of the vanW accessory factor had no effect on resistance to either vancomycin or ceftriaxone, and the function of VanW remains unknown. As expected, mutations in the genes known to be important for vancomycin resistance (vanYB, vanHB, vanB, vanXB) all led to substantial reductions in the MIC for vancomycin but did not affect resistance toward ceftriaxone (in the absence of vancomycin; Table 4). We confirmed by qRTPCR that in-frame deletions of several of the van genes in the vanYBWHBBXB transcriptional unit did not significantly perturb expression of the van locus (vanXB expression was within a factor of 2 of that of wild-type V583 for both the vanYB and vanB mutants; Table 3). Mutations in most of the genes in the vanYBWHBBXB transcriptional unit, including those genes required for production of peptidoglycan precursors containing d-Ala-d-Lac (vanHB and vanB), did not impair vancomycin-ceftriaxone synergy, as the mutants exhibited robust synergism between subinhibitory levels of vancomycin and ceftriaxone (Table 4). Thus, production of precursors containing d-Ala-d-Lac is not required for synergy, and neither are the functions of VanXB and VanW. In contrast, subinhibitory levels of vancomycin no longer promoted susceptibility to ceftriaxone in a mutant lacking VanYB, defining vanYB as the key determinant in the vanB locus mediating synergy between vancomycin and cephalosporins.
VanYB is sufficient to render E. faecalis susceptible to cephalosporins.
The results presented above indicate that VanYB is necessary for synergy between vancomycin and cephalosporins. To test whether VanYB is sufficient for synergy, we cloned vanYB into an expression plasmid containing an inducible promoter and introduced the recombinant plasmid into E. faecalis. Induction of VanYB production restored vancomycin resistance to the ΔvanYB mutant (the MIC for vancomycin was 8 μg/ml upon induction, compared with 1 μg/ml under uninduced conditions), indicating that the expression plasmid is functional and that the ΔvanYB mutation is not polar on downstream genes. In both E. faecalis V583 and OG1 hosts, production of VanYB led to a substantial decrease in resistance to ceftriaxone (in the absence of vancomycin) that did not occur in the absence of inducer or in strains carrying an empty vector (Table 5). Thus, vancomycin-mediated induction of VanYB is both necessary and sufficient for the synergy observed between vancomycin and cephalosporins in vanB-containing E. faecalis, and indeed, VanYB production alone is capable of significantly impairing the intrinsic ability of even vancomycin-susceptible E. faecalis to resist cephalosporins.
TABLE 5.
Median MICs for plasmid-bearing strains versus ceftriaxone
| Strain/plasmidb | MIC (µg/ml)a |
|
|---|---|---|
| − cCF10 | + cCF10c | |
| OG1/vector | 32 | 16 |
| OG1/vanYB | 32 | 4 |
| V583/vector | 512 | 256 |
| V583/vanYB | 512 | 4 |
| V583/vanYB Δ1-46 | 512 | 512 |
| V583/vanYB D195A | 512 | 512 |
| V583/vanYB E238A | 512 | 512 |
MICs were determined in MHB supplemented with Cm (no vancomycin) after 24 h at 37°C from a minimum of 3 independent experiments.
The plasmids used were as follows: vector, pBK2; vanYB, pJLL98; vanYB Δ-46, pJLL99; vanYB D195A, pJLL107; vanYB E238A, pJLL108.
cCF10 was used to induce plasmid-borne gene expression at 10 ng/ml for OG1 host strains or at 0.5 ng/ml for V583 host strains.
Arthur and coworkers previously described a mutant form of VanY (VanY Δ1–45) lacking a predicted N-terminal membrane anchor (37). VanY Δ1–45 retains normal levels of d,d-carboxypeptidase activity but is unable to localize properly to the membrane. To test if membrane localization is required for the ability of VanYB to sensitize E. faecalis to cephalosporins, we constructed an analogous mutant lacking the N-terminal membrane anchor of VanYB (VanYB Δ1–46). Expression of VanYB Δ1–46 in the ΔvanYB mutant failed to restore resistance to vancomycin (MIC of 1 μg/ml after induction), indicating that proper membrane localization of VanYB is required for vanB-mediated vancomycin resistance. Unlike wild-type VanYB, production of VanYB Δ1–46 did not enhance ceftriaxone susceptibility (Table 5), indicating that proper membrane localization is also required for VanYB to promote synergy with cephalosporins.
To test if the d,d-carboxypeptidase activity of VanYB is required for its role in sensitizing E. faecalis to cephalosporins, we sought to construct VanYB point mutants that lacked catalytic activity. To our knowledge, site-directed mutagenesis has not been used to probe for specific amino acids required for catalysis by VanYB; however, VanYB contains consensus sequence motifs that are known to be critical for catalytic activity of the dipeptidase VanX, and these motifs in VanYB have been predicted to be essential for the divalent cation-dependent d,d-carboxypeptidase activity of VanYB (38, 39). We therefore constructed 2 full-length VanYB mutants with single alanine substitutions either at D195 (predicted to be essential for coordination of the divalent metal cofactor) or at E238 (predicted to act as the catalytic base during hydrolysis) and expressed them in E. faecalis. Unfortunately, an antibody specific for VanYB is not available to confirm that the mutants were stably expressed, but given the relatively benign nature of the mutations introduced and the fact that 2 independent mutants with lesions at different sites behave in the same manner, we anticipate that the VanYB mutants are indeed stably expressed. As expected, these VanYB mutants were unable to restore vancomycin resistance to the ΔvanYB mutant (the MIC for vancomycin was 1 μg/ml upon induction), consistent with the prediction that they are catalytically inactive. In contrast to wild-type VanYB, production of the VanYB point mutants did not enhance ceftriaxone susceptibility (Table 5). Thus, the acidic residues D195 and E238 in VanYB are required for the ability of VanYB to promote synergy with cephalosporins, likely reflecting a critical role for VanYB enzymatic activity in this process.
Compared to other PBPs, growth mediated by Pbp5 is more sensitive to the activity of VanYB.
The results presented above are consistent with a model in which Pbp5 cannot perform cross-linking (and therefore cannot mediate growth) upon depletion of peptidoglycan precursors containing d-Ala-d-Ala termini by the carboxypeptidase activity of VanYB. To examine whether depletion of peptidoglycan precursors containing d-Ala-d-Ala termini by VanYB also adversely affected growth mediated by other PBPs, we measured growth rates for exponentially growing cells of the ΔvanB mutant in the presence and absence of subinhibitory levels of vancomycin (0.25 μg/ml). This mutant was chosen for analysis to ensure that all peptidoglycan synthesis was completely dependent on normal precursors (i.e., the mutant cannot make precursors containing d-Ala-d-Lac). Importantly, we note that 0.25 μg/ml of vancomycin is fully capable of inducing VanYB synthesis in the mutant to levels that are sufficient to prevent Pbp5-mediated growth, as the ΔvanB mutant exhibits marked synergy with ceftriaxone under the same conditions (Table 4). We found that the generation times for the mutant were nearly identical regardless of the presence of vancomycin: 86 ± 1 min (average ± standard deviation) in the absence of vancomycin compared to 88 ± 2 min in its presence. Furthermore, microscopic examination of wild-type or ΔvanB mutant cells, cultured in either the presence or the absence of vancomycin (0.25 μg/ml), revealed no apparent differences in cellular morphology or organization (not shown). We infer that depletion of precursors containing d-Ala-d-Ala by VanYB preferentially impairs Pbp5-mediated growth but is not sufficient to impair growth mediated by other transpeptidases.
DISCUSSION
A dominant-negative mutant as a counterselectable marker.
We describe here the first example, to our knowledge, of using a dominant-negative mutant of a conditionally essential enzyme as part of a counterselection strategy for genetic manipulation. We used thymidylate synthase as the dominant-negative enzyme in our counterselection scheme, although in principle such a strategy can be adapted to essentially any enzyme for which dominant-negative mutants can be obtained. The key requirement for counterselection is that the growth conditions must be identified under which the chosen enzyme is conditionally essential. For E. faecalis (Fig. 1), for E. coli (29), and likely for many other microbes as well, TS satisfies that requirement easily: supplementation of growth medium with thymine enables cells to synthesize nucleotides for DNA replication, thereby rendering TS dispensable. The availability of a characterized TS mutant allele that is now shown to behave as a dominant negative in phylogenetically diverse organisms suggests that TS is an attractive choice for the development of new counterselectable markers in other bacteria as well. One potentially significant benefit of counterselection mediated by dominant-negative TS is the flexibility in the composition of growth media it provides. Namely, counterselection can be performed on a wide variety of nutrient growth media (dictated by the nutritional requirements of a particular bacterial host or by other specific experimental constraints), as long as the chosen medium lacks thymine. Furthermore, counterselection does not require the addition of potentially toxic or inhibitory chemicals to the medium that might impair the growth of mutants of interest.
VanYB confers susceptibility to cephalosporins.
Although synergy between glycopeptides and beta-lactam antibiotics against vancomycin-resistant enterococci has been known for decades (14–17), the genetic basis for synergy has remained unknown. Early studies to explore the mechanism of synergy (16, 17) produced a model in which the low-affinity Pbp5, normally responsible for peptidoglycan cross-linking in the presence of cephalosporins, is unable to use peptidoglycan precursors containing d-Ala-d-Lac termini as substrates following induction of van resistance genes by vancomycin. Consequently, growth is inhibited as the other PBPs become inactivated at relatively low concentrations of cephalosporins and unable to mediate cross-linking. Our results are consistent with the hypothesis that Pbp5 cannot use precursors containing d-Ala-d-Lac as substrates for cross-linking. However, production of precursors containing d-Ala-d-Lac is not required for synergy with cephalosporins, as revealed by analysis of the ΔvanB and ΔvanHB mutants (Table 4) and the studies employing inducible expression of VanYB in the absence of vancomycin (Table 5). Instead, our results are consistent with a model in which removal of the normal d-Ala-d-Ala termini from peptidoglycan precursors by VanYB-mediated hydrolysis is a critical factor that renders E. faecalis especially susceptible to cephalosporins, presumably because Pbp5 is inactive under these conditions.
Strikingly, our results suggest that precursor hydrolysis by VanYB preferentially impairs peptidoglycan synthesis by Pbp5 without significantly affecting synthesis by other PBPs. Treatment of the ΔvanB mutant with subinhibitory levels of vancomycin resulted in marked susceptibility to ceftriaxone (MIC of 2 μg/ml; Table 4) that nearly equals the level observed for an E. faecalis mutant lacking Pbp5 completely (MIC of 1 μg/ml [8]), suggesting that Pbp5 is almost completely inactive under these conditions. In contrast, in the absence of cephalosporins, the mutant is capable of growth at nearly identical rates whether or not vancomycin is present to induce VanYB, and no differences in cellular morphology or organization were observed upon induction of VanYB. These results argue that the other PBPs are fully active and capable of carrying out transpeptidation even when VanYB is actively depleting the normal precursor pool. One possibility to account for these observations could be that Pbp5 exhibits reduced affinity, relative to the affinity of other PBPs, for binding of normal free d-Ala-d-Ala termini. In this scenario, the other PBPs would carry out cross-linking under normal growth circumstances, and Pbp5 would become active in transpeptidation only if uncrosslinked d-Ala-d-Ala termini accumulate to a sufficiently high level (after inhibition of other PBPs by beta-lactam antibiotics). In this context, hydrolysis of d-Ala-d-Ala termini by VanYB would be predicted to reduce the concentration of free d-Ala-d-Ala termini sufficiently that Pbp5 cannot bind them efficiently enough to perform cross-linking and mediate growth. This defect would be apparent only if the other PBPs were inhibited by beta-lactams; hence, the defect in Pbp5 activity manifests as substantially enhanced susceptibility to cephalosporins upon VanYB production. In the absence of cephalosporins, the other PBPs would be fully capable of efficiently using the remaining (reduced) pool of d-Ala-d-Ala termini for cross-linking by virtue of their higher binding affinity, thereby enabling nearly wild-type growth kinetics despite expression of VanYB.
An alternative possibility might be that VanYB production reduces the concentration of free d-Ala-d-Ala termini sufficiently that none of the PBPs can mediate efficient cross-linking but that an alternative transpeptidase assumes responsibility for cross-linking under these conditions. For example, Mainardi and coworkers previously identified a beta-lactam-insensitive l,d-transpeptidase that carries out peptidoglycan cross-linking under certain conditions in Enterococcus faecium (40) and described a homolog encoded in E. faecalis (40, 41) that could conceivably be responsible for cross-linking upon production of VanYB. However, in this model such an alternative transpeptidase would have to be sufficiently robust to promote growth at a rate matching that provided by the normal repertoire of d,d-transpeptidases (i.e., the PBPs), which seems unlikely. More sophisticated and focused studies to characterize the composition of peptidoglycan produced under these conditions will be required to begin understanding the detailed molecular basis for these phenomena.
ACKNOWLEDGMENTS
We thank Michael Gilmore for providing bacterial strains, Gary Dunny for providing plasmid pBK2, and members of the Kristich laboratory for critical reading of the manuscript.
This work was supported by grants R01 AI081692 and DP2 OD006447 from the NIH.
The content is solely our responsibility and does not necessarily represent the official views of the NIH.
Footnotes
Published ahead of print 23 December 2013
REFERENCES
- 1.Tannock GW, Cook G. 2002. Enterococci as members of the intestinal microflora of humans, p 101–132 In Gilmore MS, Clewell DB, Courvalin P, Dunny GM, Murray BE, Rice LB. (ed), The enterococci: pathogenesis, molecular biology, and antibiotic resistance. ASM Press, Washington, DC [Google Scholar]
- 2.Hidron AI, Edwards JR, Patel J, Horan TC, Sievert DM, Pollock DA, Fridkin SK. 2008. NHSN annual update: antimicrobial-resistant pathogens associated with healthcare-associated infections: annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006–2007. Infect. Control Hosp. Epidemiol. 29:996–1011. 10.1086/591861 [DOI] [PubMed] [Google Scholar]
- 3.Richards MJ, Edwards JR, Culver DH, Gaynes RP. 2000. Nosocomial infections in combined medical-surgical intensive care units in the United States. Infect. Control Hosp. Epidemiol. 21:510–515. 10.1086/501795 [DOI] [PubMed] [Google Scholar]
- 4.Shepard BD, Gilmore MS. 2002. Antibiotic-resistant enterococci: the mechanisms and dynamics of drug introduction and resistance. Microbes Infect. 4:215–224. 10.1016/S1286-4579(01)01530-1 [DOI] [PubMed] [Google Scholar]
- 5.Donskey CJ, Chowdhry TK, Hecker MT, Hoyen CK, Hanrahan JA, Hujer AM, Hutton-Thomas RA, Whalen CC, Bonomo RA, Rice LB. 2000. Effect of antibiotic therapy on the density of vancomycin-resistant enterococci in the stool of colonized patients. N. Engl. J. Med. 343:1925–1932. 10.1056/NEJM200012283432604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Signoretto C, Boaretti M, Canepari P. 1994. Cloning, sequencing and expression in Escherichia coli of the low-affinity penicillin binding protein of Enterococcus faecalis. FEMS Microbiol. Lett. 123:99–106. 10.1111/j.1574-6968.1994.tb07207.x [DOI] [PubMed] [Google Scholar]
- 7.Arbeloa A, Segal H, Hugonnet JE, Josseaume N, Dubost L, Brouard JP, Gutmann L, Mengin-Lecreulx D, Arthur M. 2004. Role of class A penicillin-binding proteins in PBP5-mediated beta-lactam resistance in Enterococcus faecalis. J. Bacteriol. 186:1221–1228. 10.1128/JB.186.5.1221-1228.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kristich CJ, Little JL. 2012. Mutations in the beta subunit of RNA polymerase alter intrinsic cephalosporin resistance in enterococci. Antimicrob. Agents Chemother. 56:2022–2027. 10.1128/AAC.06077-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Courvalin P. 2006. Vancomycin resistance in gram-positive cocci. Clin. Infect. Dis. 42(Suppl 1):S25–S34. 10.1086/491711 [DOI] [PubMed] [Google Scholar]
- 10.Arthur M, Courvalin P. 1993. Genetics and mechanisms of glycopeptide resistance in enterococci. Antimicrob. Agents Chemother. 37:1563–1571. 10.1128/AAC.37.8.1563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arthur M, Reynolds PE, Depardieu F, Evers S, Dutka-Malen S, Quintiliani R, Jr, Courvalin P. 1996. Mechanisms of glycopeptide resistance in enterococci. J. Infect. 32:11–16. 10.1016/S0163-4453(96)80003-X [DOI] [PubMed] [Google Scholar]
- 12.Reynolds PE. 1998. Control of peptidoglycan synthesis in vancomycin-resistant enterococci: D,D-peptidases and D,D-carboxypeptidases. Cell. Mol. Life Sci. 54:325–331. 10.1007/s000180050159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ribeiro T, Santos S, Marques MI, Gilmore M, de Fatima Silva Lopes M. 2011. Identification of a new gene, vanV, in vanB operons of Enterococcus faecalis. Int. J. Antimicrob. Agents 37:554–557. 10.1016/j.ijantimicag.2011.01.024 [DOI] [PubMed] [Google Scholar]
- 14.Leclercq R, Bingen E, Su QH, Lambert-Zechovski N, Courvalin P, Duval J. 1991. Effects of combinations of beta-lactams, daptomycin, gentamicin, and glycopeptides against glycopeptide-resistant enterococci. Antimicrob. Agents Chemother. 35:92–98. 10.1128/AAC.35.1.92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shlaes DM, Etter L, Gutmann L. 1991. Synergistic killing of vancomycin-resistant enterococci of classes A, B, and C by combinations of vancomycin, penicillin, and gentamicin. Antimicrob. Agents Chemother. 35:776–779. 10.1128/AAC.35.4.776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gutmann L, al-Obeid S, Billot-Klein D, Guerrier ML, Collatz E. 1994. Synergy and resistance to synergy between beta-lactam antibiotics and glycopeptides against glycopeptide-resistant strains of Enterococcus faecium. Antimicrob. Agents Chemother. 38:824–829. 10.1128/AAC.38.4.824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.al-Obeid S, Billot-Klein D, van Heijenoort J, Collatz E, Gutmann L. 1992. Replacement of the essential penicillin-binding protein 5 by high-molecular mass PBPs may explain vancomycin-beta-lactam synergy in low-level vancomycin-resistant Enterococcus faecium D366. FEMS Microbiol. Lett. 70:79–84 [DOI] [PubMed] [Google Scholar]
- 18.Cercenado E, Eliopoulos GM, Wennersten CB, Moellering RC., Jr 1992. Absence of synergistic activity between ampicillin and vancomycin against highly vancomycin-resistant enterococci. Antimicrob. Agents Chemother. 36:2201–2203. 10.1128/AAC.36.10.2201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Green M, Binczewski B, Pasculle AW, Edmund M, Barbadora K, Kusne S, Shlaes DM. 1993. Constitutively vancomycin-resistant Enterococcus faecium resistant to synergistic beta-lactam combinations. Antimicrob. Agents Chemother. 37:1238–1242. 10.1128/AAC.37.6.1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ravizzola G, Cabibbo E, Peroni L, Longo M, Pollara PC, Corulli M, Pizzi R, Dima F, Fallacara C, Turano A. 1997. In-vitro study of the synergy between beta-lactam antibiotics and glycopeptides against enterococci. J. Antimicrob. Chemother. 39:461–470. 10.1093/jac/39.4.461 [DOI] [PubMed] [Google Scholar]
- 21.Kristich CJ, Chandler JR, Dunny GM. 2007. Development of a host-genotype-independent counterselectable marker and a high-frequency conjugative delivery system and their use in genetic analysis of Enterococcus faecalis. Plasmid 57:131–144. 10.1016/j.plasmid.2006.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Y, Staddon JH, Dunny GM. 2007. Specificity determinants of conjugative DNA processing in the Enterococcus faecalis plasmid pCF10 and the Lactococcus lactis plasmid pRS01. Mol. Microbiol. 63:1549–1564. 10.1111/j.1365-2958.2007.05610.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, III, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6:343–345. 10.1038/nmeth.1318 [DOI] [PubMed] [Google Scholar]
- 24.Shokeen S, Johnson CM, Greenfield TJ, Manias DA, Dunny GM, Weaver KE. 2010. Structural analysis of the Anti-Q.-Qs interaction: RNA-mediated regulation of E. faecalis plasmid pCF10 conjugation. Plasmid 64:26–35. 10.1016/j.plasmid.2010.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vesić D, Kristich CJ. 2013. A Rex family transcriptional repressor influences H2O2 accumulation by Enterococcus faecalis. J. Bacteriol. 195:1815–1824. 10.1128/JB.02135-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elion GB, Singer S, Hitchings GH. 1954. Antagonists of nucleic acid derivatives. VIII. Synergism in combinations of biochemically related antimetabolites. J. Biol. Chem. 208:477–488 [PubMed] [Google Scholar]
- 27.Pillai SK, Moellering RC, Jr, Eliopoulos GM. 2005. Antimicrobial combinations, p 365–440 In Lorian V. (ed), Antibiotics in laboratory medicine. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 28.Kast P. 1994. pKSS—a second-generation general purpose cloning vector for efficient positive selection of recombinant clones. Gene 138:109–114. 10.1016/0378-1119(94)90790-0 [DOI] [PubMed] [Google Scholar]
- 29.West DK, Porter DC, Saxl RL, Maley F. 2004. A Trojan horse approach for silencing thymidylate synthase. Biochemistry 43:9177–9184. 10.1021/bi049105v [DOI] [PubMed] [Google Scholar]
- 30.Sahm DF, Kissinger J, Gilmore MS, Murray PR, Mulder R, Solliday J, Clarke B. 1989. In vitro susceptibility studies of vancomycin-resistant Enterococcus faecalis. Antimicrob. Agents Chemother. 33:1588–1591. 10.1128/AAC.33.9.1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paulsen IT, Banerjei L, Myers GS, Nelson KE, Seshadri R, Read TD, Fouts DE, Eisen JA, Gill SR, Heidelberg JF, Tettelin H, Dodson RJ, Umayam L, Brinkac L, Beanan M, Daugherty S, DeBoy RT, Durkin S, Kolonay J, Madupu R, Nelson W, Vamathevan J, Tran B, Upton J, Hansen T, Shetty J, Khouri H, Utterback T, Radune D, Ketchum KA, Dougherty BA, Fraser CM. 2003. Role of mobile DNA in the evolution of vancomycin-resistant Enterococcus faecalis. Science 299:2071–2074. 10.1126/science.1080613 [DOI] [PubMed] [Google Scholar]
- 32.Palmer KL, Carniol K, Manson JM, Heiman D, Shea T, Young S, Zeng Q, Gevers D, Feldgarden M, Birren B, Gilmore MS. 2010. High-quality draft genome sequences of 28 Enterococcus sp. isolates. J. Bacteriol. 192:2469–2470. 10.1128/JB.00153-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gold OG, Jordan HV, van Houte J. 1975. The prevalence of enterococci in the human mouth and their pathogenicity in animal models. Arch. Oral Biol. 20:473–477. 10.1016/0003-9969(75)90236-8 [DOI] [PubMed] [Google Scholar]
- 34.Evers S, Courvalin P. 1996. Regulation of VanB-type vancomycin resistance gene expression by the VanS(B)-VanR (B) two-component regulatory system in Enterococcus faecalis V583. J. Bacteriol. 178:1302–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hancock LE, Perego M. 2004. Systematic inactivation and phenotypic characterization of two-component signal transduction systems of Enterococcus faecalis V583. J. Bacteriol. 186:7951–7958. 10.1128/JB.186.23.7951-7958.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baptista M, Rodrigues P, Depardieu F, Courvalin P, Arthur M. 1999. Single-cell analysis of glycopeptide resistance gene expression in teicoplanin-resistant mutants of a VanB-type Enterococcus faecalis. Mol. Microbiol. 32:17–28. 10.1046/j.1365-2958.1999.01308.x [DOI] [PubMed] [Google Scholar]
- 37.Arthur M, Depardieu F, Cabanie L, Reynolds P, Courvalin P. 1998. Requirement of the VanY and VanX D,D-peptidases for glycopeptide resistance in enterococci. Mol. Microbiol. 30:819–830. 10.1046/j.1365-2958.1998.01114.x [DOI] [PubMed] [Google Scholar]
- 38.McCafferty DG, Lessard IA, Walsh CT. 1997. Mutational analysis of potential zinc-binding residues in the active site of the enterococcal D-Ala-D-Ala dipeptidase VanX. Biochemistry 36:10498–10505. 10.1021/bi970543u [DOI] [PubMed] [Google Scholar]
- 39.Lessard IA, Walsh CT. 1999. Mutational analysis of active-site residues of the enterococcal D-Ala-D-Ala dipeptidase VanX and comparison with Escherichia coli D-Ala-D-Ala ligase and D-Ala-D-Ala carboxypeptidase VanY. Chem. Biol. 6:177–187. 10.1016/S1074-5521(99)89009-7 [DOI] [PubMed] [Google Scholar]
- 40.Mainardi JL, Fourgeaud M, Hugonnet JE, Dubost L, Brouard JP, Ouazzani J, Rice LB, Gutmann L, Arthur M. 2005. A novel peptidoglycan cross-linking enzyme for a beta-lactam-resistant transpeptidation pathway. J. Biol. Chem. 280:38146–38152. 10.1074/jbc.M507384200 [DOI] [PubMed] [Google Scholar]
- 41.Magnet S, Arbeloa A, Mainardi JL, Hugonnet JE, Fourgeaud M, Dubost L, Marie A, Delfosse V, Mayer C, Rice LB, Arthur M. 2007. Specificity of L,D-transpeptidases from Gram-positive bacteria producing different peptidoglycan chemotypes. J. Biol. Chem. 282:13151–13159. 10.1074/jbc.M610911200 [DOI] [PubMed] [Google Scholar]
- 42.Maekawa S, Yoshioka M, Kumamoto Y. 1992. Proposal of a new scheme for the serological typing of Enterococcus faecalis strains. Microbiol. Immunol. 36:671–681. 10.1111/j.1348-0421.1992.tb02070.x [DOI] [PubMed] [Google Scholar]
- 43.McBride SM, Fischetti VA, Leblanc DJ, Moellering RC, Jr, Gilmore MS. 2007. Genetic diversity among Enterococcus faecalis. PLoS One 2:e582. 10.1371/journal.pone.0000582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Snyder H, Kellogg SL, Skarda LM, Little JL, Kristich CJ. 2014. Nutritional control of antibiotic resistance via an interface between the phosphotransferase system and a two-component signaling system. Antimicrob. Agents Chemother. 58:957–965. 10.1128/AAC.01919-13 [DOI] [PMC free article] [PubMed] [Google Scholar]