

Abstract
The increasing prevalence of drug-resistant pathogens highlights the need to identify novel antibiotics. Here we investigated the efficacies of four new antimicrobial peptides (AMPs) for potential drug development. The antibacterial activities, synergistic effects, and antibiofilm properties of the four chimeric AMPs were tested against Acinetobacter baumannii, an emerging Gram-negative, nosocomial, drug-resistant pathogen. Nineteen A. baumannii strains resistant to ampicillin, cefotaxime, ciprofloxacin, tobramycin, and erythromycin were isolated at a hospital from patients with cholelithiasis. All four peptides exhibited significant antibacterial effects (MIC = 3.12 to 12.5 μM) against all 19 strains, whereas five commercial antibiotics showed little or no activity against the same pathogens. An exception was polymyxin, which was effective against all of the strains tested. Each of the peptides showed synergy against one or more strains when administered in combination with cefotaxime, ciprofloxacin, or erythromycin. The peptides also exhibited an ability to prevent biofilm formation, which was not seen with cefotaxime, ciprofloxacin, or erythromycin, though polymyxin also inhibited biofilm formation. Indeed, when administered in combination with ciprofloxacin, the AMP HPMA exerted a potent synergistic effect against A. baumannii biofilm formation. Collectively, our findings indicate that the AMPs tested have no cytotoxicity but possess potent antibacterial and antibiofilm activities and may act synergistically with commercial antibiotics.
INTRODUCTION
The Gram-negative pathogenic bacterium Acinetobacter baumannii is known for its ability to cause severe nosocomial infections, its resistance to most antibiotics, and its ability to form biofilm (1, 2). Because this microorganism is generally associated with infections in compromised hospitalized patients, community-acquired infections with multidrug-resistant (MDR) A. baumannii are an increasing concern (3). The commonly prescribed antimicrobial products with reliable antipseudomonal activity are now limited to a few agents in three major pharmacological classes: lactams, fluoroquinolones, and aminoglycosides (4). First-line treatment of A. baumannii infections currently entails administration of a carbapenem antibiotic such as imipenem, but carbapenem resistance is increasingly common. Other treatment options include polymyxins, tigecycline, and aminoglycosides (5). However, isolation of MDR Acinetobacter has soared from 6.7% in 1993 to 29.9% by 2004, highlighting the need for newer and better drugs (6).
Natural antimicrobials, known as host defense peptides or antimicrobial peptides (AMPs), defend host organisms against microbes. Methods for designing synthetic AMPs for therapeutic application have been developed using various optimization strategies, and several products are currently being tested clinically (7, 8). One important aim is to design AMPs that target biofilm as well as free bacteria, since peptides showing an ability to inhibit biofilm formation could provide the basis for a new generation of anti-infective agents (8). It appears that chimeric peptides are essential for achieving optimal bactericidal activity, as they exhibit broader bactericidal spectra than the parental AMPs. In addition, chimeric peptides display lower MICs or minimum bactericidal concentrations (MBCs), which could potentially reduce their cost as next-generation antibiotics. A variety of AMPs, including chimeric AMPs, have been tested in vitro against MDR Acinetobacter isolates (9, 10). For example, cecropin A (CA)-melittin (ME) peptide and its parents, CA and ME, exhibit good antimicrobial activity against colistin- and pan-resistant strains (9–12). Our focus, therefore, has been on chimeric AMPs constructed from CA, ME, magainin 2 (MA), and HP(2-20), which is the N-terminal region of the Helicobacter pylori ribosomal protein L1 (RPL1) (13). CA and MA each exhibit strong bactericidal activities against both Gram-positive and Gram-negative bacteria but show less toxicity toward eukaryotic cells (e.g., erythrocytes) (14–18). CA is a cationic 37-amino-acid peptide isolated from Hyalaphora cecropia pupae (13), while MA is a cationic 23-amino-acid peptide found in the skin of the African clawed frog (Xenopus laevis) (19), and HP(2-20) exhibits antimicrobial activity reflecting its relation to cecropin-like N-terminal peptides (20). Likewise, AMPs derived from the honeybee peptide ME show strong antimicrobial activity against a wide range of pathogens. However, despite their potential as alternative antibiotics, the clinical use of these AMPs has been impeded by their relatively high toxicity toward eukaryotic cells (21).
In the present study, the antibacterial and antibiofilm activities of four chimeric AMPs were tested against 19 clinically isolated MDR A. baumannii strains. In addition, we investigated the synergistic effects of the four peptides when administered in combination with commercial antibiotics. Our findings indicate that the four AMPs tested were effective against all A. baumannii 19 strains and that HP(2-9)-MA(1-12) (HPMA) acted synergistically in combination with cefotaxime, ciprofloxacin, or erythromycin to inhibit biofilm development by A. baumannii.
MATERIALS AND METHODS
Bacterial strains.
Nineteen clinical isolates of A. baumannii strains with resistance to ampicillin, cefotaxime, ciprofloxacin, tobramycin, and erythromycin were collected from Eulji University Hospital, Seoul, South Korea. All strains were confirmed to be A. baumannii using amplified ribosomal rRNA gene restriction analysis (ARDRA). In addition, the sequences of the 16S rRNA gene from several isolates were determined by ProBionic (Korea Research Institute of Bioscience & Biotechnology [KRIBB], Taejon, South Korea). All isolates were stored at −70°C until required. Standard strains of A. baumannii (KCTC 2508) were obtained from the Korean Collection for Type Cultures (KCTC), KRIBB, Taejon, South Korea.
Chimeric AMPs and antibiotic agents.
HP(2-9)-ME(1-12) (HPME) (AKKVFKRLGIGAVLKVLTTG), HP(2-9)-MA(1-12) (HPMA) (AKKVFKRLGIGKFLHSAKKF-NH2), CA(1-8)-ME(1-12) (CAME) (KWKLFKKIGIGAVLKVLTTG-NH2), and CA(1-8)-MA(1-12) (CAMA) (KWKLFKKIGIGKFLHSAKKF-NH2) were prepared using an automated solid-phase method by applying 9-fluorenylmethoxy carbonyl (Fmoc) active-ester chemistry. The peptides were subsequently purified using reversed-phase high-pressure liquid chromatography (RP-HPLC) (22), after which the levels of their purity were verified by analytical RP-HPLC, and the molecules were further characterized by mass spectrometry and amino acid analysis. Antibiotics were prepared in the form of standard laboratory powder and used according to the guidelines of the Clinical and Laboratory Standards Institute (CLSI) (23).
MIC values.
The antimicrobial activity of each peptide was tested in broth microdilution assays following the procedure recommended by CLSI (23). Briefly, bacteria were grown to the stationary phase overnight in Mueller-Hilton broth (MHB) (50 ml in a 250-ml Byrex flask) at 200 rpm and 37°C, after which the cultures were diluted with fresh MHB to a final concentration of 2 × 105 CFU/ml. A stock solution (400 μM) of each peptide was prepared in 0.01% acetic acid in a polypropylene microtube. The peptide solution was then subjected to a series of 2-fold dilutions in 0.01% acetic acid until reaching a final concentration of 1.56 μM. Thereafter, a 100-μl aliquot of microbial suspension was dispensed into each well of a 96-well polypropylene microtiter plate (3790; Costar, Corning, NY), and 10 μl of one of the peptide solutions was added. Several wells were left untreated as a control for monitoring bacterial growth. After 18 h of incubation at 37°C, the antibacterial activities of the peptides were assessed based on the optical density in each well. The MICs were expressed as the lowest concentration that caused complete inhibition of growth. There were three replicates of each test sample.
Evaluation of the synergistic activities of four chimeric AMPs with antibiotics.
The MIC of each antibiotic, alone or in combination, was determined in accordance with CLSI standards using cation-adjusted MHB in a modified broth microdilution checkerboard procedure (23). For combination treatments, a two-dimensional checkerboard with 2-fold dilutions of each AMP and antibiotic (cefotaxime, ciprofloxacin, or erythromycin) was set up for the combined treatment. Growth control wells containing only medium were included in each plate, and each test was performed in three independent replicates. The fractional inhibitory concentration (FIC) was calculated for the first clear well in each row of the microtiter plate containing all antimicrobial agents as follows: FIC of drug A (FIC A) = MIC of drug A in combination/MIC of drug A alone, and FIC of drug B (FIC B) = MIC of drug B in combination/MIC of drug B alone (19). The FIC index (FICi), calculated as the sum of each FIC, was interpreted as follows: FICi of <0.5, synergy; FICi of ≥0.5 and <1, partial synergy; FICi of 1, additive; FICi of ≥2 and <4, indifferent; and FICi of >4, antagonism (24, 25).
Biofilm formation and quantification.
For this study, 19 antibiotic-resistant bacterial strains were isolated from patients with cholelithiasis and cultured in MHB supplemented with 0.2% glucose. Biofilm formation was identified and quantified using the tissue culture plate (TCP) method as described previously with a few modifications (26). Briefly, 100 μl of bacterial cells suspended in MHB supplemented with 0.2% glucose (1 × 106 CFU/ml) was added to the wells of sterile polystyrene 96-well flat-bottom TCPs. As a control, medium without cells was added to the wells. The cells were then incubated for 24 h at 37°C to determine whether they would form a biofilm. To quantify the biofilms formed, the culture medium was discarded from the wells following the 24-h incubation, and any free-floating planktonic cells were removed by washing the wells with phosphate-buffered saline (PBS). The biofilms formed on the well bottoms were then fixed by adding 100 μl of 100% methanol and incubating for 15 min. After removal of the methanol, the biofilm in each plate was stained with 0.4% (wt/vol) crystal violet solution for 30 min. The excess stain was then thoroughly rinsed away with distilled water, and the plates were left to dry. Once dry, 100% ethanol was added to each well, and the optical density at 595 nm (OD595) of the stained biofilm was measured using a Versa-Max microplate enzyme-linked immunosorbent assay (ELISA) reader (Molecular Devices, Sunnyvale, CA, USA). These optical density values were considered to be a measure of the bacteria adhering to the surface and forming biofilms (27–29). Experiments were performed in triplicate, and the data were averaged.
Assay of susceptibilities of biofilm-forming strains to AMPs and conventional antibiotics.
To examine the inhibitory effect of chimeric peptides and conventional antibiotics on biofilm growth, the TCP method was employed with a few modifications (26). Each test agent (antimicrobial peptide [AMP] and antibiotic) was diluted in 0.01% acetic acid and 0.2% bovine serum albumin. AMPs (10 μl) at concentrations ranging from 200 μM to 1.56 μM or antibiotics (10 μl) at 400 μM to 0.78 μM were added to 90 μl of bacterial cells suspended in MHB supplemented with 0.2% glucose (1 × 106 CFU/ml). Wells in which no test agents were added served as controls. After incubation for 24 h, inhibition of biofilm growth was quantified using crystal violet staining as described above, and the percentage of biofilm inhibition was calculated as [1 − (OD595 of cells treated with test agent/OD595 of untreated control)] × 100 (27–29). The minimum biofilm-inhibitory concentration (MBIC) was defined as the lowest concentration that showed 90% inhibition of biofilm formation. Experiments were performed in triplicate, and the data were averaged.
Evaluation of the synergistic effects of HPMA with antibiotics on MDR A. baumannii biofilm.
The TCP method was used to assess the biofilm-inhibiting activity of HPMA in combination with conventional antibiotics (26). In this study, four A. baumannii stains were selected, and the MBIC of the peptide alone or in combination with an antibiotic was determined using the broth microdilution method in accordance with CLSI standards for the broth microdilution checkerboard procedure (23).
Cell cultures and cytotoxicity.
The HaCaT human keratinocyte cell line was a gift from N. E. Fusenig (Heidelberg, Germany). The cells were cultured in 75-cm2 plastic flasks containing Dulbecco's modified Eagle medium (DMEM) supplemented with antibiotics (100-U/ml penicillin, 100-μg/ml streptomycin), 10% fetal calf serum, 1 mM pyruvate, and 4 mM l-glutamine. The cultures were incubated at 37°C in a humidified chamber containing 5% CO2 and 95% air.
Percent survival was determined using 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) assays (Sigma). Briefly, cells were seeded into a 96-well plate to a density of 4 × 103 cells/well and incubated for 24 h. Selected concentrations of the test peptides were then added to the wells, and the cells were incubated for an additional 24 h at 37°C. This was followed by addition of 10 μl of MTT (5 mg/ml) to each well, after which cells were incubated for another 4 h. The supernatant was then aspirated, and 100 μl of dimethyl sulfoxide (DMSO) was added to the wells to dissolve any precipitate present. Finally, the absorbance was measured at a wavelength of 570 nm using an ELX800 reader (Bio-Tek instruments, Inc., Winooski, VT). There were three replicates of each test sample.
RESULTS AND DISCUSSION
Effects of antibiotic agents on MDR A. baumannii cells.
The 19 MDR strains used in this study were isolated from patients with cholelithiasis (Table 1) and were identified as A. baumannii based on their biological characteristics (data not shown). We then confirmed the identities of four of the isolates by sequencing their 16S rRNA genes, which were found to be 100% homologous with the GenBank reference strain of A. baumannii. In an antimicrobial susceptibility test, only a nonresistant control strain was susceptible to any of the five antibiotics tested (ampicillin, cefotaxime, ciprofloxacin, tobramycin, and erythromycin), confirming that all of the clinically isolated A. baumannii strains were resistant to antibiotics at concentrations of >400 μM (Table 1). On the other hand, polymyxins were effective against all the A. baumannii strains at doses ranging from 0.78 to 3.12 μM, which is clearly consistent with several antibiotics reported to prevent MDR A. baumannii infections. Colistin (polymyxin E), for example, is effective against urinary tract infections (30) and wound infections (31) caused by the bacterium. In addition, biofilm-forming A. baumannii strains are sensitive to imipenem and ciprofloxacin (32). Nonetheless, most A. baumannii strains have become resistant to both carbapenem and colistin (33, 34). For example, several groups (35–38) have shown that clinically isolated A. baumannii strains are resistant to all cephalosporins, including cefepime, ampicillin-sulbactam, ciprofloxacin, and imipenem. They are also resistant to most beta-lactams and aminoglycosides (39). It is thus apparent that many antimicrobial compounds have become considerably less effective against these biofilm-forming organisms and that alternative treatment strategies are urgently needed.
TABLE 1.
MICs of commercial antibiotics against a nonresistant Acinetobacter baumannii strain and 19 emerging nosocomial drug-resistant strains
| Strain | MIC (μM) |
|||||
|---|---|---|---|---|---|---|
| Ampicillin | Cefotaxime | Ciprofloxacin | Erythromycin | Tobramycin | Polymyxin | |
| Nonresistant | 400 | 25 | 1.56 | 12.5 | 6.25 | 0.78 |
| 1 | >400 | >400 | >400 | >400 | >400 | 1.56 |
| 2 | >400 | >400 | >400 | >400 | >400 | 1.56 |
| 3 | >400 | >400 | >400 | >400 | >400 | 1.56 |
| 4 | >400 | >400 | >400 | >400 | >400 | 3.12 |
| 5 | >400 | >400 | >400 | >400 | >400 | 1.56 |
| 6 | >400 | >400 | >400 | >400 | >400 | 0.78 |
| 7 | >400 | >400 | >400 | >400 | >400 | 3.12 |
| 8 | >400 | >400 | >400 | >400 | >400 | 3.12 |
| 9 | >400 | >400 | >400 | >400 | >400 | 1.56 |
| 10 | >400 | >400 | >400 | >400 | >400 | 0.78 |
| 11 | >400 | >400 | >400 | >400 | >400 | 3.12 |
| 12 | >400 | >400 | >400 | >400 | >400 | 3.12 |
| 13 | >400 | >400 | >400 | >400 | >400 | 1.56 |
| 14 | >400 | >400 | >400 | >400 | >400 | 3.12 |
| 15 | >400 | >400 | >400 | >400 | >400 | 0.78 |
| 16 | >400 | >400 | >400 | >400 | >400 | 1.56 |
| 17 | >400 | >400 | >400 | >400 | >400 | 3.12 |
| 18 | >400 | >400 | >400 | >400 | >400 | 1.56 |
| 19 | >400 | >400 | >400 | >400 | >400 | 0.78 |
Effects of antimicrobial peptides on MDR A. baumannii cells.
The use of AMPs is one potentially effective approach to the treatment of MDR A. baumannii. AMPs are particularly attractive as new antimicrobial agents against MDR bacteria, including A. baumannii, because their mechanism of action, which involves permeation of the bacterial membranes, reduces the likelihood of emergence of resistance (40–45). That said, other bacteria do exhibit mechanisms to resist AMPs (46–48). However, chimeric AMPs were previously shown to have bactericidal activity against colistin-resistant A. baumannii strains (10, 11, 49), and the antibacterial activities of four chimeric AMPs (CAME, HPME, HPMA, and CAMA) were tested against these 19 MDR A. baumannii strains. Notably, the results showed all 19 resistant strains to be susceptible to all four AMPs tested (MIC = 3.12 to 12.5 μM), among which CAME showed the lowest MIC (3.12 to 6.25 μM), while HPME, HPMA, and CAMA each showed a MIC of 3.12 to 12.5 μM (Table 2).
TABLE 2.
MICs of four chimeric AMPs against a nonresistant A. baumannii strain and 19 emerging nosocomial drug-resistant strains
| Strain | MIC (μM) |
|||
|---|---|---|---|---|
| HPME | HPMA | CAME | CAMA | |
| Nonresistant | 6.25 | 6.25 | 3.12 | 12.5 |
| 1 | 6.25 | 6.25 | 3.12 | 12.5 |
| 2 | 12.5 | 6.25 | 3.12 | 6.25 |
| 3 | 6.25 | 6.25 | 3.12 | 12.5 |
| 4 | 6.25 | 12.5 | 3.12 | 3.12 |
| 5 | 3.12 | 6.25 | 3.12 | 3.12 |
| 6 | 12.5 | 6.25 | 6.25 | 12.5 |
| 7 | 6.25 | 3.12 | 6.25 | 12.5 |
| 8 | 12.5 | 6.25 | 6.25 | 12.5 |
| 9 | 6.25 | 6.25 | 3.12 | 6.25 |
| 10 | 12.5 | 6.25 | 6.25 | 6.25 |
| 11 | 6.25 | 6.25 | 3.12 | 6.25 |
| 12 | 3.12 | 12.5 | 3.12 | 6.25 |
| 13 | 12.5 | 6.25 | 6.25 | 6.25 |
| 14 | 6.25 | 6.25 | 6.25 | 6.25 |
| 15 | 6.25 | 3.12 | 3.12 | 12.5 |
| 16 | 6.25 | 6.25 | 6.25 | 6.25 |
| 17 | 12.5 | 6.25 | 6.25 | 6.25 |
| 18 | 12.5 | 6.25 | 6.25 | 12.5 |
| 19 | 3.12 | 6.25 | 3.12 | 3.12 |
Evaluation of the synergistic effects of combinations of antimicrobial peptides and conventional antibiotics on MDR A. baumannii cells.
There are several reports in the literature of cationic AMPs acting synergistically with commercial antibiotics against MDR bacterial strains (50–52). In particular, membranolytic peptides reportedly enhance the efficacy of antibiotics against MDR A. baumannii by permeabilizing the outer membrane, which likely increases access to the cytoplasmic membrane following breakdown of the peptidoglycan (9, 12). Other studies have also shown that a chimera composed of cecropin A-melittin [CA(1-8)M(1-18)] disrupts the outer and inner membranes of A. baumannii (11), and synergy was observed when CA(1-7)M(2-9)NH2, CA(1-8)M(1-18), CA(1-7)M(2-9), or CA(1-7)M(5-9) was combined with a beta-lactam antibiotic against A. baumannii (10). In the present study, all four chimeric AMPs exerted synergistic effects with antibiotics against isolates from patients with cholelithiasis caused by MDR A. baumannii. Synergy therapy is used with the aims of expanding the antimicrobial or bactericidal spectrum, minimizing cytotoxicity, preventing the emergence of resistant bacterial mutants during therapy, and obtaining synergistic antimicrobial activity. Moreover, the rationale is that cefotaxime, ciprofloxacin, or erythromycin provides susceptibility to the bacterial strains in a wound environment. Therefore, the emergence of strains susceptible to synergistic combinations (for example cefotaxime, ciprofloxacin, or erythromycin combined with an AMP) may give us another choice of drug (combination drug) for treatment.
We used the checkerboard format and assessed synergy by calculating the FICi, which is an interaction coefficient that indicates whether the combined inhibitory/bacteriostatic effects of drugs are synergistic (entirely or partially), additive, or indifferent. Using this protocol, we found that the combination of HPMA with cefotaxime, ciprofloxacin, or erythromycin gave a FICi of 0.25 to 0.375 against two strains (no. 5 and 12), which is indicative of a synergistic effect, and a FICi of 0.5 to 0.5625 against the remaining 19 strains, which is indicative of a partially synergistic effect. In contrast, HPME with cefotaxime gave a FICi of 1 or 2, which indicates additive and indifferent effects, respectively (Table 3). On the other hand, HPME with erythromycin showed partial synergy effects against three strains (no. 4, 5 and 12). Good synergy (FICi of 0.25 to 0.375) was observed when CAMA was applied in combination with cefotaxime, ciprofloxacin, or erythromycin against the 19 strains, but no entirely synergistic effect was seen when HPME or HPMA was applied with cefotaxime or erythromycin (FICi of 0.5 to 1). CAME in combination with erythromycin or cefotaxime exerted synergistic effects ((FICi of 0.312 to 0.375) against strains 4 and 19. Our finding that these AMPs act synergistically with cefotaxime, ciprofloxacin, or erythromycin against MDR A. baumannii strains suggests they act in a manner similar to other chimeric AMPs in terms of their activity toward MDR A. baumannii (10).
TABLE 3.
FICi values for the four chimeric AMPs with cefotaxime, ciprofloxacin, and erythromycin against resistant strains
| MDR strain | FICi for AMP with druga |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HPME |
HPMA |
CAME |
CAMA |
|||||||||
| Cefotaxime | Ciprofloxacin | Erythromycin | Cefotaxime | Ciprofloxacin | Erythromycin | Cefotaxime | Ciprofloxacin | Erythromycin | Cefotaxime | Ciprofloxacin | Erythromycin | |
| 4 | 1 (additive) | 0.625 (partial synergy) | 0.75 (partial synergy) | 0.5 (partial synergy) | 0.375 (synergy) | 0.3125 (synergy) | 0.75 (partial synergy) | 1 (additive) | 0.375 (synergy) | 0.75 (partial synergy) | 0.5625 (partial synergy) | 0.5 (partial synergy) |
| 5 | 2 (indifferent) | 0.75 (partial synergy) | 0.75 (partial synergy) | 0.375 (synergy) | 0.25 (synergy) | 0.375 (synergy) | 0.75 (partial synergy) | 0.75 (partial synergy) | 0.75 (partial synergy) | 0.75 (partial synergy) | 0.75 (partial synergy) | 0.375 (synergy) |
| 12 | 2 (indifferent) | 0.375 (synergy) | 0.625 (partial synergy) | 0.3125 (synergy) | 0.25 (synergy) | 0.3125 (synergy) | 0.625 (partial synergy) | 0.625 (partial synergy) | 0.625 (partial synergy) | 0.5 (partial synergy) | 0.3125 (synergy) | 0.625 (partial synergy) |
| 19 | 0.5 (partial synergy) | 0.3125 (synergy) | 1 (additive) | 0.5625 (partial synergy) | 0.375 (synergy) | 0.5 (partial synergy) | 0.3125 (synergy) | 0.5 (partial synergy) | 0.5 (partial synergy) | 0.2656 (synergy) | 0.375 (synergy) | 0.25 (synergy) |
Serial dilutions of antibiotics were tested in the presence of a constant amount of each chimeric peptide. FIC of drug A (FIC A) = MIC of drug A in combination/MIC of drug A alone; FIC of drug B (FIC B) = MIC of drug B in combination/MIC of drug B alone. The FIC index (FICi) was calculated as the sum of each FIC and was interpreted as follows: FICi of <0.5, synergy; FICi of >0.5 and <1, partial synergy; FICi of 1, additive; FICi of >2 and <4, indifferent; and FICi of >4, antagonism.
Biofilm formation and quantification.
We quantified biofilm formation based on crystal violet staining. The absorbance values obtained with all 19 strains of MDR A. baumannii clearly differed from that with the negative control (A. baumannii 2508), and most of those strains showed biofilm formation (Fig. 1), which is consistent with earlier findings showing that MDR clinical isolates of A. baumannii form biofilms (53). Moreover, there is a significant association between biofilm formation and resistance to multiple drugs, including imipenem, cefotaxime, amikacin, and ciprofloxacin (53), although A. baumannii isolates from wound patients were also resistant to amikacin, ampicillin, aztreonam, ceftriaxone, ciprofloxacin, gentamicin, imipenem, tobramycin, and vancomycin (31). Importantly in that regard, A. baumannii causes mainly nosocomial infections, including wound and surgical site infections (1, 54–57), and its ability to form a biofilm on human skin plays a key role in its persistence there. Therefore, the ability of an antibiotic to prevent biofilm formation could make it effective against A. baumannii in both the free and biofilm states when applied topically to the skin (e.g., in burn patients) (58).
FIG 1.
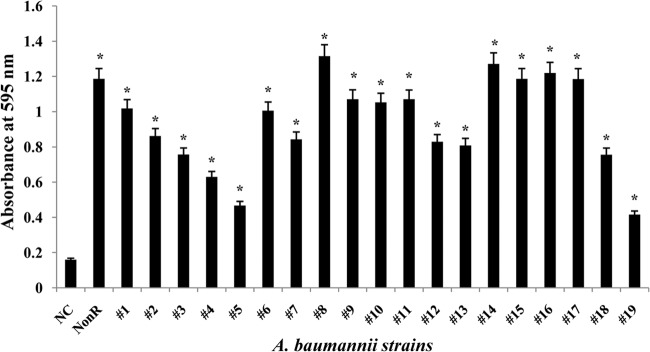
Formation and quantification of biofilms formed by clinical isolates of A. baumannii (19 strains). Bacteria were allowed to grow in 96-well microtiter plates, and the biofilms were quantified by staining with crystal violet. *, P < 0.5. NC, negative control; NonR, nonresistant strain.
Effects of antimicrobial peptides and conventional antibiotics on biofilm formation by MDR A. baumannii cells.
In the present study, none of the commercial antibiotics tested except polymyxin prevented biofilm formation by A. baumannii (Table 4). The inhibitory effect of polymyxin is likely due to its ability to induce leakage of ions and other materials from A. baumannii cells (11). However, polymyxins are controversial, as they can exhibit toxicity and have a lower success rate than other antimicrobials (59). For that reason, we next evaluated the ability of chimeric AMPs to inhibit biofilm formation by A. baumannii. All four chimeric AMPs inhibited biofilm formation by all the strains of A. baumannii isolated from patients with cholelithiasis (Table 4), with MBICs ranging from 6.25 to 50 μM. In addition to the chimeric AMPs, the human AMP LL-37 and its fragments also inhibited biofilm formation by MDR A. baumannii (60), indicating that these AMPs (CAME, HPME, HPMA, CAMA, and LL-37 and its analogs) could potentially serve as therapeutic agents against MDR A. baumannii infections in a wound environment.
TABLE 4.
Inhibitory effects of HPME, HPMA, CAME, CAMA, and antibiotics on biofilms formed by A. baumannii strains
| Strain | MBIC (μM) |
|||||||
|---|---|---|---|---|---|---|---|---|
| Cefotaxime | Ciprofloxacin | Erythromycin | Polymyxin | HPME | HPMA | CAME | CAMA | |
| Nonresistant | 100 | 6.25 | 50 | 1.56 | 12.5 | 12.5 | 6.25 | 25 |
| 1 | >400 | >400 | >400 | 3.12 | 25 | 25 | 12.5 | 50 |
| 2 | >400 | >400 | >400 | 3.12 | 25 | 12.5 | 6.25 | 12.5 |
| 3 | >400 | >400 | >400 | 1.56 | 25 | 25 | 12.5 | 50 |
| 4 | >400 | >400 | >400 | 3.12 | 12.5 | 25 | 6.25 | 6.25 |
| 5 | >400 | >400 | >400 | 1.56 | 12.5 | 25 | 12.5 | 12.5 |
| 6 | >400 | >400 | >400 | 3.12 | 25 | 12.5 | 12.5 | 25 |
| 7 | >400 | >400 | >400 | 1.56 | 12.5 | 6.25 | 12.5 | 25 |
| 8 | >400 | >400 | >400 | 6.25 | 25 | 6.25 | 12.5 | 25 |
| 9 | >400 | >400 | >400 | 1.56 | 25 | 25 | 12.5 | 25 |
| 10 | >400 | >400 | >400 | 1.56 | 25 | 12.5 | 12.5 | 12.5 |
| 11 | >400 | >400 | >400 | 6.25 | 12.5 | 12.5 | 12.5 | 12.5 |
| 12 | >400 | >400 | >400 | 6.25 | 6.25 | 25 | 12.5 | 25 |
| 13 | >400 | >400 | >400 | 3.12 | 25 | 12.5 | 12.5 | 25 |
| 14 | >400 | >400 | >400 | 3.12 | 25 | 25 | 25 | 12.5 |
| 15 | >400 | >400 | >400 | 1.56 | 6.25 | 6.25 | 6.25 | 12.5 |
| 16 | >400 | >400 | >400 | 1.56 | 12.5 | 12.5 | 12.5 | 25 |
| 17 | >400 | >400 | >400 | 6.25 | 25 | 25 | 12.5 | 12.5 |
| 18 | >400 | >400 | >400 | 3.12 | 25 | 12.5 | 12.5 | 25 |
| 19 | >400 | >400 | >400 | 0.78 | 12.5 | 25 | 12.5 | 12.5 |
Synergistic effects of combinations of HPMA and antibiotics on MDR A. baumannii biofilms.
The synergistic effects of HPMA administered in combination with conventional antibiotics on inhibition of biofilm formation by A. baumannii were evaluated using FICi with the checkerboard method (Table 5 and Fig. 2). We found that the inhibitory effects of HPMA on biofilm formation by A. baumannii were significantly potentiated by coadministration with ciprofloxacin. The MBICs for HPMA plus ciprofloxacin were thus lower than those obtained with HPMA alone or HPMA plus cefotaxime or erythromycin. This finding is consistent with other studies reporting that the biofilm-inhibiting activity of an AMP against A. baumannii could be enhanced by combination with a commercial antibiotic (27). The synergistic effects of HPMA in combination with ciprofloxacin were observed against biofilms formed by A. baumannii strains 4, 5, and 12 (Table 5), and a partial synergistic interaction was observed against biofilms formed by strain 19. Other combinational activities of HPMA and antibiotics were found to be partial or additive synergistic interactions.
TABLE 5.
Activities of HPMA in combination with different antibiotics
| MDR strain | FICia |
||
|---|---|---|---|
| HPMA + cefotaxime | HPMA + ciprofloxacin | HPMA + erythromycin | |
| 4 | 1 (additive) | 0.375 (synergy) | 0.75 (partial synergy) |
| 5 | 0.625 (partial synergy) | 0.3125 (synergy) | 0.5 (partial synergy) |
| 12 | 1 (additive) | 0.375 (synergy) | 1 (additive) |
| 19 | 0.5 (partial synergy) | 0.5 (partial synergy) | 0.5 (partial synergy) |
The FICi was calculated using the following formula: FICi = (MICDrug A in combination/MICDrug A alone) + (MICDrug B in combination/MICDrug B alone).
FIG 2.
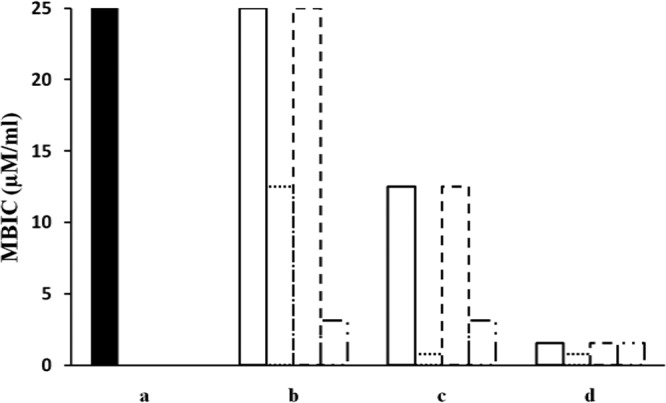
Synergistic effects of HPMA against A. baumannii biofilm when administered in combination with a commercial antibiotic. a, MBIC values for HPMA against A. baumannii strains 4, 5, 12, and 19. b to d, A. baumannii biofilms were exposed to HPMA (25 μM) plus cefotaxime (400 μM) (b), HPMA (25 μM) plus erythromycin (400 μM) (c), or HPMA (25 μM) plus ciprofloxacin (400 μM) (d). Bars with solid lines depict the MBIC values for HPMA-antibiotic combinations against strain 4; dotted lines, strain 5; dashed lines, strain 12; and long-dashed and dotted lines, strain 19.
Effects of AMPs on HaCaT human skin keratinocytes.
The cytotoxicity of the four AMPs against HaCaT human skin keratinocytes was assessed in order evaluate their potential to serve as antimicrobial agents against bacterial skin infections. The survival rate among HaCaT cells was nearly always 100%, although it was ≥92% in the presence of 100 μM CAME (Fig. 3). The AMPs were therefore considered to be slightly cytotoxic or noncytotoxic. Indeed, earlier cytotoxicity studies showed that these peptides most likely do not have toxic effects on HaCaT cells. So far, however, in vivo studies of AMPs in clinical settings have been hampered in part by the fact that AMPs can become highly toxic in vivo (61). At present, therefore, most therapeutic peptides are being developed for topical application against bacterial infections of the skin. Moreover, our observation that HPMA and a commercial antibiotic act synergistically to inhibit biofilm formation by A. baumannii suggests that chimeric AMPs should be considered a preferred class of antimicrobial substances for future investigations of the treatment of A. baumannii biofilms in a wound environment, either alone or in combination with other antibiotics.
FIG 3.
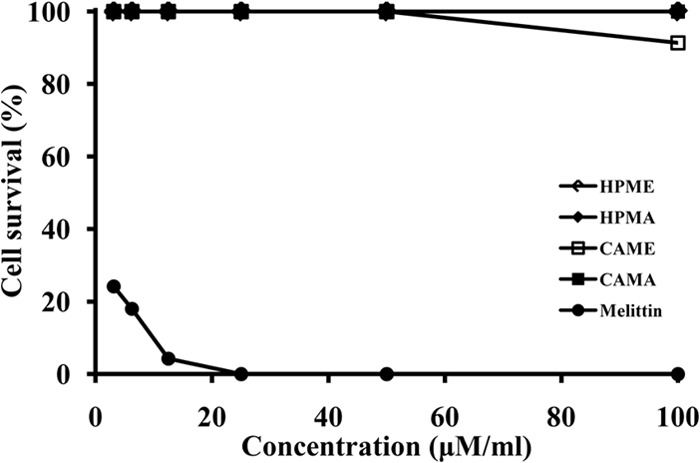
Cytotoxic effects of the four chimeric AMPs tested. The cytotoxicity of the four peptides against HaCaT human keratinocyte cells was evaluated using melittin as a positive control.
In summary, the work described here shows that the four chimeric AMPs tested are highly effective against MDR A. baumannii, exhibiting both antimicrobial and biofilm-inhibiting activities. In addition, they all showed synergistic antibacterial effects with commercial antibiotics but minimal cytotoxicity against human keratinocytes, and HPMA also showed a synergistic effect against biofilm formation by MDR A. baumannii when applied in combination with ciprofloxacin. Our findings indicate that these four AMPs possess considerable potential for use in combination with other antibiotics to treat infections of the skin caused by MDR A. baumannii pathogens.
ACKNOWLEDGMENTS
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MEST) (no. 2011-0017532) and the Human Resource Training Project for Regional Innovation (NRF-2013H1B8A2032054).
Footnotes
Published ahead of print 23 December 2013
REFERENCES
- 1.Bergogne-Berezin E, Towner KJ. 1996. Acinetobacter spp. as nosocomial pathogens: microbiological, clinical, and epidemiological features. Clin. Microbiol. Rev. 9:148–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peleg AY, Seifert H, Paterson DL. 2008. Acinetobacter baumannii: emergence of a successful pathogen. Clin. Microbiol. Rev. 21:538–582. 10.1128/CMR.00058-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anstey NM, Currie BJ, Hassell M, Palmer D, Dwyer B, Seifert H. 2002. Community-acquired bacteremic Acinetobacter pneumonia in tropical Australia is caused by diverse strains of Acinetobacter baumannii, with carriage in the throat in at-risk groups. J. Clin. Microbiol. 40:685–686. 10.1128/JCM.40.2.685-686.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Giamarellou H, Antoniadou A. 2001. Antipseudomonal antibiotics. Med. Clin. N. Am. 85:19–41. 10.1016/S0025-7125(05)70303-5 [DOI] [PubMed] [Google Scholar]
- 5.Bassetti M, Righi E, Esposito S, Petrosillo N, Nicolini L. 2008. Drug treatment for multidrug-resistant Acinetobacter baumannii infections. Future Microbiol. 3:649–660. 10.2217/17460913.3.6.649 [DOI] [PubMed] [Google Scholar]
- 6.Lockhart SR, Abramson MA, Beekmann SE, Gallagher G, Riedel S, Diekema DJ, Quinn JP, Doern GV. 2007. Antimicrobial resistance among Gram-negative bacilli causing infections in intensive care unit patients in the United States between 1993 and 2004. J. Clin. Microbiol. 45:3352–3359. 10.1128/JCM.01284-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fjell CD, Hiss JA, Hancock RE, Schneider G. 2012. Designing antimicrobial peptides: form follows function. Nat. Rev. Drug Discov. 11:37–51. 10.1038/nrd3591 [DOI] [PubMed] [Google Scholar]
- 8.de la Fuente-Núñez C, Korolik V, Bains M, Nguyen U, Breidenstein EB, Horsman S, Lewenza S, Burrows L, Hancock RE. 2012. Inhibition of bacterial biofilm formation and swarming motility by a small synthetic cationic peptide. Antimicrob. Agents Chemother. 56:2696–2704. 10.1128/AAC.00064-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rodriguez-Hernandez MJ, Saugar J, Docobo-Perez F, de la Torre BG, Pachon-Ibanez ME, Garcia-Curiel A, Fernandez-Cuenca F, Andreu D, Rivas L, Pachon J. 2006. Studies on the antimicrobial activity of cecropin A-melittin hybrid peptides in colistin-resistant clinical isolates of Acinetobacter baumannii. J. Antimicrob. Chemother. 58:95–100. 10.1093/jac/dkl145 [DOI] [PubMed] [Google Scholar]
- 10.Giacometti A, Cirioni O, Kamysz W, D'Amato G, Silvestri C, Del Prete MS, Lukasiak J, Scalise G. 2003. Comparative activities of cecropin A, melittin, and cecropin A-melittin peptide CA(1-7)M(2-9)NH2 against multidrug-resistant nosocomial isolates of Acinetobacter baumannii. Peptides 24:1315–1318. 10.1016/j.peptides.2003.08.003 [DOI] [PubMed] [Google Scholar]
- 11.Saugar JM, Alarcón T, López-Hernández S, López-Brea M, Andreu D, Rivas L. 2002. Activities of polymyxin B and cecropin A-melittin peptide CA(1-8)M(1-18) against a multiresistant strain of Acinetobacter baumannii. Antimicrob. Agents Chemother. 46:875–878. 10.1128/AAC.46.3.875-878.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saugar JM, Rodriguez-Hernandez MJ, de la Torre BG, Pachon-Ibanez ME, Fernandez-Reyes M, Andreu D, Pachon J, Rivas L. 2006. Activity of cecropin A-melittin hybrid peptides against colistin-resistant clinical strains of Acinetobacter baumannii: molecular basis for the differential mechanisms of action. Antimicrob. Agents Chemother. 50:1251–1256. 10.1128/AAC.50.4.1251-1256.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Steiner H, Hultmark D, Engstrom A, Bennich H, Boman HG. 1981. Sequence and specificity of two antibacterial proteins involved in insect immunity. Nature 292:246–248. 10.1038/292246a0 [DOI] [PubMed] [Google Scholar]
- 14.Macias EA, Rana F, Blazyk J, Modrzakowski MC. 1990. Bactericidal activity of magainin 2: use of lipopolysaccharide mutants. Can. J. Microbiol. 36:582–584. 10.1139/m90-102 [DOI] [PubMed] [Google Scholar]
- 15.Cuervo JH, Rodriguez B, Houghten RA. 1988. The magainins: sequence factors relevant to increased antimicrobial activity and decreased hemolytic activity. Pept. Res. 1:81–86 [PubMed] [Google Scholar]
- 16.Silvestro L, Axelsen PH. 2000. Membrane-induced folding of cecropin A. Biophys. J. 79:1465–1477. 10.1016/S0006-3495(00)76398-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boman HG. 1991. Antibacterial peptides: key components needed in immunity. Cell 65:205–207. 10.1016/0092-8674(91)90154-Q [DOI] [PubMed] [Google Scholar]
- 18.Boman HG. 1995. Peptide antibiotics and their role in innate immunity. Annu. Rev. Immunol. 13:61–92. 10.1146/annurev.iy.13.040195.000425 [DOI] [PubMed] [Google Scholar]
- 19.Zasloff M. 1987. Magainins, a class of antimicrobial peptides from Xenopus skin: isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc. Natl. Acad. Sci. U. S. A. 84:5449–5453. 10.1073/pnas.84.15.5449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Putsep K, Brändén CI, Boman HG, Normark S. 1999. Antibacterial peptide from H. pylori. Nature 398:671–672. 10.1038/19439 [DOI] [PubMed] [Google Scholar]
- 21.Tosteson MT, Holmes SJ, Razin M, Tosteson DC. 1985. Melittin of a red cells. J. Membr. Biol. 87:35–44. 10.1007/BF01870697 [DOI] [PubMed] [Google Scholar]
- 22.Selsted ME. 1997. HPLC methods for purification of antimicrobial peptides. Methods Mol. Biol. 78:17–33 [DOI] [PubMed] [Google Scholar]
- 23.Clinical and Laboratory Standards Institute 2006. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically, 7th ed. Approved standard M7-A7. Clinical and Laboratory Standards Institute, Wayne. PA [Google Scholar]
- 24.Jeong N, Kim JY, Park SC, Lee JK, Gopal R, Yoo S, Son BK, Hahm JS, Park Y, Hahm KS. 2010. Antibiotic and synergistic effect of Leu-Lys rich peptide against antibiotic resistant microorganisms isolated from patients with cholelithiasis. Biochem. Biophys. Res. Commun. 399:581–586. 10.1016/j.bbrc.2010.07.118 [DOI] [PubMed] [Google Scholar]
- 25.Dawis MA, Isenberg HD, France KA, Jenkins SG. 2003. In vitro activity of gatifloxacin alone in combination with cefepime, meropenem, piperacillin and gentamicin against multidrug-resistant organisms. J. Antimicrob. Chemother. 51:1203–1211. 10.1093/jac/dkg238 [DOI] [PubMed] [Google Scholar]
- 26.Christensen GD, Simpson WA, Younger JJ, Baddour LM, Barrett FF, Melton DM, Beachey EH. 1985. Adherence of coagulase-negative staphylococci to plastic tissue culture plate: a quantitative model for the adherence of staphylococci to medical devices. J. Clin. Microbiol. 22:996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hwang IS, Hwang JS, Hwang JH, Choi H, Lee E, Kim Y, Lee DG. 2013. Synergistic effect and antibiofilm activity between the antimicrobial peptide coprisin and conventional antibiotics against opportunistic bacteria. Curr. Microbiol. 66:56–60. 10.1007/s00284-012-0239-8 [DOI] [PubMed] [Google Scholar]
- 28.Wei GX, Campagna AN, Bobek LA. 2006. Effect of MUC7 peptides on the growth of bacteria and on Streptococcus mutans biofilm. J. Antimicrob. Chemother. 57:1100–1109. 10.1093/jac/dkl120 [DOI] [PubMed] [Google Scholar]
- 29.Gopal R, Lee JH, Kim YG, Kim MS, Seo CH, Park Y. 2013. Anti-microbial, anti-biofilm activities and cell selectivity of the NRC-16 peptide derived from witch flounder, Glyptocephalus cynoglossus. Mar. Drugs 11:1836–1852. 10.3390/md11061836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pour NK, Dusane DH, Dhakephalkar PK, Zamin FR, Zinjarde SS, Chopade BA. 2011. Biofilm formation by Acinetobacter baumannii strains isolated from urinary tract infection and urinary catheters. FEMS Immunol. Med. Microbiol. 62:328–338. 10.1111/j.1574-695X.2011.00818.x [DOI] [PubMed] [Google Scholar]
- 31.Crane DP, Gromov K, Li D, Søballe K, Wahnes C, Büchner H, Hilton MJ, O'Keefe RJ, Murray CK, Schwarz EM. 2009. Efficacy of colistin-impregnated beads to prevent multidrug-resistant A. baumannii implant-associated osteomyelitis. J. Orthop. Res. 27:1008–1015. 10.1002/jor.20847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodríguez-Baño J, Martí S, Soto S, Fernández-Cuenca F, Cisneros JM, Pachón J, Pascual A, Martínez-Martínez L, McQueary C, Actis LA, Vila J. 2008. Biofilm formation in Acinetobacter baumannii: associated features and clinical implications. Clin. Microbiol. Infect. 14:276–278. 10.1111/j.1469-0691.2007.01916.x [DOI] [PubMed] [Google Scholar]
- 33.Kempf M, Rolain JM. 2012. Emergence of resistance to carbapenems in Acinetobacter baumannii in Europe: clinical impact and therapeutic options. Int. J. Antimicrob. Agents 39:105–114. 10.1016/j.ijantimicag.2011.10.004 [DOI] [PubMed] [Google Scholar]
- 34.Abbott I, Cerqueira GM, Bhuiyan S, Peleg AY. 2013. Carbapenem resistance in Acinetobacter baumannii: laboratory challenges, mechanistic insights and therapeutic strategies. Expert Rev. Anti Infect. Ther. 11:395–409. 10.1586/eri.13.21 [DOI] [PubMed] [Google Scholar]
- 35.Lim YM, Shin KS, Kim J. 2007. Distinct antimicrobial resistance patterns and antimicrobial resistance-harboring genes according to genomic species of Acinetobacter isolates. J. Clin. Microbiol. 45:902–905. 10.1128/JCM.01573-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simor AE, Lee M, Vearncombe M, Jones-Paul L, Barry C, Gomez M, Fish JS, Cartotto RC, Palmer R, Louie M. 2002. An outbreak due to multiresistant Acinetobacter baumannii in a burn unit: risk factors for acquisition and management. Infect. Control Hosp. Epidemiol. 23:261–267. 10.1086/502046 [DOI] [PubMed] [Google Scholar]
- 37.Vahaboglu H, Ozturk R, Aygun G, Coskunkan F, Yaman F, Kaygusuz A, Leblebicioglu H, Balik I, Aydin K, Otkun M. 1997. Widespread detection of PER-1-type extended-spectrum β-lactamases among nosocomial Acinetobacter and Pseudomonas aeruginosa isolates in Turkey: a nationwide multicenter study. Antimicrob. Agents Chemother. 41:2265–2269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yong D, Shin JH, Kim S, Lim Y, Yum JH, Lee K, Chong Y, Bauernfeind 2003. High prevalence of PER-1 extended-spectrum β-lactamase-producing Acinetobacter spp. in Korea. Antimicrob. Agents. Chemother. 47:1749–1751. 10.1128/AAC.47.5.1749-1751.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li J, Nation RL, Owen RJ, Wong S, Spelman D, Franklin C. 2007. Antibiograms of multidrug-resistant clinical Acinetobacter baumannii: promising therapeutic options for treatment of infection with colistin-resistant strains. Clin. Infect. Dis. 45:594–598. 10.1086/520658 [DOI] [PubMed] [Google Scholar]
- 40.Calandra T. 2001. Pathogenesis of septic shock: implications for prevention and treatment. J. Chemother. 13:173–180 [DOI] [PubMed] [Google Scholar]
- 41.David SA, Awasthi SK, Balaram P. 2000. The role of polar and facial amphipathic character in determining lipopolysaccharide-binding properties in synthetic cationic peptides. J. Endotoxin Res. 6:249–256. 10.1177/09680519000060030601 [DOI] [PubMed] [Google Scholar]
- 42.Guerra AN, Fisette PL, Pfeiffer ZA, Quinchia-Rios BH, Prabhu U, Aga M, Denlinger LC, Guadarrama AG, Abozeid S, Sommer JA, Proctor RA, Bertics PJ. 2003. Purinergic receptor regulation of LPS-induced signaling and pathophysiology. J. Endotoxin Res. 9:256–263. 10.1179/096805103225001468 [DOI] [PubMed] [Google Scholar]
- 43.Hancock REW, Lehrer R. 1998. Cationic peptides: a new source of antibiotics. Trends Biotechnol. 16:82–88. 10.1016/S0167-7799(97)01156-6 [DOI] [PubMed] [Google Scholar]
- 44.Shin SY, Lee MK, Kim KL, Hahm KS. 1997. Structure-antitumor and hemolytic activity relationships of synthetic peptides derived from cecropin A-magainin 2 and cecropin A-melittin hybrid peptides. J. Pept. Res. 50:279–285 [DOI] [PubMed] [Google Scholar]
- 45.Park Y, Lee DG, Hahm KS. 2004. HP(2-9)-magainin 2(1-12), a synthetic hybrid peptide, exerts its antifungal effect on Candida albicans by damaging the plasma membrane. J. Pept. Sci. 10:204–209. 10.1002/psc.489 [DOI] [PubMed] [Google Scholar]
- 46.Shi Y, Cromie MJ, Hsu FF, Turk J, Groisman EA. 2004. PhoP-regulated Salmonella resistance to the antimicrobial peptides magainin 2 and polymyxin B. Mol. Microbiol. 53:229–241. 10.1111/j.1365-2958.2004.04107.x [DOI] [PubMed] [Google Scholar]
- 47.Guina T, Yi EC, Wang H, Hackett M, Miller SI. 2000. A PhoP-regulated outer membrane protease of Salmonella enterica serovar typhimurium promotes resistance to alpha-helical antimicrobial peptides. J. Bacteriol. 182:4077–4086. 10.1128/JB.182.14.4077-4086.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abi Khattar Z, Rejasse A, Destoumieux-Garzon D, Escoubas JM, Sanchis V, Lereclus D, Givaudan A, Kallassy M, Nielsen-Leroux C, Gaudriault S. 2009. The dlt operon of Bacillus cereus is required for resistance to cationic antimicrobial peptides and for virulence in insects. J. Bacteriol. 191:7063–7073. 10.1128/JB.00892-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Alarcón T, López-Hernández S, Andreu D, Saugar JM, Rivas L, López-Brea M. 2001. In vitro activity of CA(1-8)M(1-18), a synthetic cecropin A-melittin hybrid peptide, against multiresistant Acinetobacter baumannii strains. Rev. Esp. Quimioter. 14:184–190 [PubMed] [Google Scholar]
- 50.Sands M, McCarter Y, Sanchez W. 2007. Synergy testing of multidrug resistant Acinetobacter baumanii against tigecycline and polymyxin using an E test methodology. Eur. J. Clin. Microbiol. Infect. Dis. 26:521–522. 10.1007/s10096-007-0330-4 [DOI] [PubMed] [Google Scholar]
- 51.Tan TY, Ng LSY, Tan E, Huang G. 2007. In vitro effect of minocycline and colistin combinations on imipenem-resistant Acinetobacter baumanii clinical isolates. J. Antimicrob. Chemother. 60:421–423. 10.1093/jac/dkm178 [DOI] [PubMed] [Google Scholar]
- 52.Yoon J, Urban C, Terzian N, Mariano N, Rahal J. 2004. In vitro double and triple synergistic activities of polymyxin B, imipenem, and rifampin against multidrug resistant Acinetobacter baumannii. Antimicrob. Agents Chemother. 48:753–757. 10.1128/AAC.48.3.753-757.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rao RS, Karthika RU, Singh SP, Shashikala P, Kanungo R, Jayachandran S, Prashanth K. 2008. Correlation between biofilm production and multiple drug resistance in imipenem resistant clinical isolates of Acinetobacter baumannii. Indian J. Med. Microbiol. 26:333–337. 10.4103/0255-0857.43566 [DOI] [PubMed] [Google Scholar]
- 54.de Breij A, Haisma EM, Rietveld M, El Ghalbzouri A, van den Broek PJ, Dijkshoorn L, Nibbering PH. 2012. Three-dimensional human skin equivalent as a tool to study Acinetobacter baumannii colonization. Antimicrob. Agents Chemother. 56:2459–2464. 10.1128/AAC.05975-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ehrenstein B, Bernards AT, Dijkshoorn L, Gerner-Smidt P, Towner KJ, Bouvet PJ, Daschner FD, Grundmann H. 1996. Acinetobacter species identification by using tRNA spacer fingerprinting. J. Clin. Microbiol. 34:2414–2420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Joly-Guillou ML. 2005. Clinical impact and pathogenicity of Acinetobacter. Clin. Microbiol. Infect. 11:868–873. 10.1111/j.1469-0691.2005.01227.x [DOI] [PubMed] [Google Scholar]
- 57.Rodríguez-Baño J, Cisneros JM, Fernández-Cuenca F, Ribera A, Vila J, Pascual A, Martínez-Martínez L, Bou G, Pachón J. 2004. Clinical features and epidemiology of Acinetobacter baumannii colonization and infection in Spanish hospitals. Infect. Control Hosp. Epidemiol. 25:819–824. 10.1086/502302 [DOI] [PubMed] [Google Scholar]
- 58.Huang XQ, Xiang J, Song F, Huan JN. 2012. Effects of topical agents for burns on Acinetobacter baumannii within biofilm. Zhonghua ShaoShang ZaZhi 28:106–110 [PubMed] [Google Scholar]
- 59.Falagas ME, Kasiakou SK. 2005. Colistin: the revival of polymyxins for management of multidrug resistant gram negative bacterial infections. Clin. Infect. Dis. 40:1333–1341. 10.1086/429323 [DOI] [PubMed] [Google Scholar]
- 60.Feng X, Sambanthamoorthy K, Palys T, Paranavitana C. 2013. The human antimicrobial peptide LL-37 and its fragments possess both antimicrobial and antibiofilm activities against multidrug-resistant Acinetobacter baumannii. Peptides 49:131–137. 10.1016/j.peptides.2013.09.007 [DOI] [PubMed] [Google Scholar]
- 61.López-Rojas R, Docobo-Pérez F, Pachón-Ibáñez ME, de la Torre BG, Fernández-Reyes M, March C, Bengoechea JA, Andreu D, Rivas L, Pachón J. 2011. Efficacy of cecropin A-melittin peptides on a sepsis model of infection by pan-resistant Acinetobacter baumannii. Eur. J. Clin. Microbiol. Infect. Dis. 30:1391–1398. 10.1007/s10096-011-1233-y [DOI] [PubMed] [Google Scholar]