

ABSTRACT
Bean pod mottle virus (BPMV) is a bipartite, positive-sense (+) RNA plant virus in the Secoviridae family. Its RNA1 encodes proteins required for genome replication, whereas RNA2 primarily encodes proteins needed for virion assembly and cell-to-cell movement. However, the function of a 58-kDa protein (P58) encoded by RNA2 has not been resolved. P58 and the movement protein (MP) of BPMV are two largely identical proteins differing only at their N termini, with P58 extending MP upstream by 102 amino acid residues. In this report, we unveil a unique role for P58. We show that BPMV RNA2 accumulation in infected cells was abolished when the start codon of P58 was eliminated. The role of P58 does not require the region shared by MP, as RNA2 accumulation in individual cells remained robust even when most of the MP coding sequence was removed. Importantly, the function of P58 required the P58 protein, rather than its coding RNA, as compensatory mutants could be isolated that restored RNA2 accumulation by acquiring new start codons upstream of the original one. Most strikingly, loss of P58 function could not be complemented by P58 provided in trans, suggesting that P58 functions in cis to selectively promote the accumulation of RNA2 copies that encode a functional P58 protein. Finally, we found that all RNA1-encoded proteins are cis-acting relative to RNA1. Together, our results suggest that P58 probably functions by recruiting the RNA1-encoded polyprotein to RNA2 to enable RNA2 reproduction.
IMPORTANCE Bean pod mottle virus (BPMV) is one of the most important pathogens of the crop plant soybean, yet its replication mechanism is not well understood, hindering the development of knowledge-based control measures. The current study examined the replication strategy of BPMV RNA2, one of the two genomic RNA segments of this virus, and established an essential role for P58, one of the RNA2-encoded proteins, in the process of RNA2 replication. Our study demonstrates for the first time that P58 functions preferentially with the very RNA from which it is translated, thus greatly advancing our understanding of the replication mechanisms of this and related viruses. Furthermore, this study is important because it provides a potential target for BPMV-specific control, and hence could help to mitigate soybean production losses caused by this virus.
INTRODUCTION
The replication process of positive-sense [(+)] RNA viruses involves both virus-encoded functions and host factors (1–3). Most (+) RNA plant viruses encode at least two proteins that, together with viral RNA, form the core viral replication complex (VRC). Typically, multiple copies of a more abundantly expressed viral protein, commonly referred to as auxiliary protein (AP), interact with a certain type of membrane-based organelle to induce the formation of partially enclosed spherules that house viral genomic RNA (gRNA), a virus-encoded protein with RNA-dependent RNA polymerase (RdRP) activity, and various host proteins (2, 4). The replication of the viral genome then takes place inside the protective spherules, or VRCs.
(+) RNA plant viruses with multipartite genomes have proven to be intriguing models for examining the replication mechanisms of (+) RNA viruses (5). Indeed, two of the best-studied (+) RNA viruses, Brome mosaic virus (BMV) and Red clover necrotic mosaic virus (RCNMV), partition their genetic information into multiple genomic RNA segments. This partitioning poses interesting challenges for the gRNA segments that encode none or only some of the VRC constituents. For example, both RNA3 of BMV and RNA2 of RCNMV encode neither AP nor RdRP, and thus must have the capacity to recruit these proteins for their own replication. In both of these cases, a specific RNA structural element was identified that interacts with the APs of corresponding viruses to facilitate entry into VRCs by these RNAs (6, 7).
It should be noted that for both BMV and RCNMV, functional viral proteins are translated from viral gRNAs or subgenomic RNAs (sgRNAs) as final translation products. An alternative gene expression strategy used by many other viruses, including Bean pod mottle virus (BPMV), of the genus Comovirus and family Secoviridae, is to pack all coding capacities into one large polyprotein per genome segment. These polyproteins are subsequently proteolytically processed to release functional proteins. Similar to RCNMV, BPMV is a bipartite (+) RNA virus, with its RNA1 encoding proteins required for genome replication and RNA2 encoding proteins required for virion assembly and cell-to-cell movement (8) (Fig. 1). However, unlike the case for RCNMV, functional BPMV proteins are produced through the processing of polyproteins translated from the two gRNA segments by the viral protease (Pro) (9). Given the significant difference in gene expression strategies, an as yet unresolved question is how RNA2 of BPMV and other comoviruses gains access to RNA1-encoded replication proteins.
FIG 1.

P58 is required for RNA2 accumulation. (A) Schematic representations of BPMV RNA1, RNA1m (a nonreplicating RNA1 mutant), RNA2-GFP (R2G), and the series of R2G mutants used in this study, accompanied by images of lima bean cotyledons bombarded with these constructs. The mutation contained in RNA1m (GDD → AAH) is highlighted in a gray box. Details of RNA2 mutants are provided on their respective diagrams. The images of bombarded cotyledons were taken at 48 h postbombardment, using cameras equipped with a dissecting or epifluorescence microscope. Some infection foci are highlighted with arrows. (B) Replication of BPMV in bombarded cotyledons as measured by semiquantitative RT-PCR to detect (−) RNA1 (top panel) and (−) RNA2 (middle panel). The relative accumulation levels of the mutants were calculated by using that of wild-type R2G as the reference (arbitrarily set as 1). An RT-PCR fragment of lima bean actin mRNA was used as a control.
The current study set out to resolve this question by using BPMV as a model. Like those of all comoviruses, BPMV RNA1 encodes one polyprotein which is thought to be processed into at least five proteins: a putative protease cofactor (C-Pro), a putative RNA helicase (Hel), a small virus-encoded protein (VPg) covalently linked to both genomic RNA segments at their 5′ termini, a viral protease (Pro) needed for the processing of polyproteins, and the viral RdRP (Fig. 1A) (8). Strikingly, BPMV RNA2 actually encodes two, albeit largely overlapping, polyproteins, as its translation can initiate from two separate in-frame AUG codons (at nucleotide [nt] positions 467 and 773) (Fig. 1A). These two polyproteins are hence mostly identical, differing only at their N termini (Fig. 1A). Both polyproteins are processed twice at the same sites, producing four different mature proteins: the larger N-terminal protein of 58 kDa, designated P58, with an unknown function(s); the smaller N-terminal protein, which is the movement protein (MP); and two capsid protein subunits (L-CP and S-CP) (Fig. 1A). Since BPMV RNA2 is not known to encode any replication-related protein, it depends primarily on RNA1-encoded proteins for its own replication.
In a previous study, we succeeded in identifying a cis-acting RNA element in BPMV RNA2 by using a particle bombardment-based experimental system (10). In the current study, we took advantage of this system to resolve whether RNA2 encodes any proteins critical to RNA2 replication. We focused our primary attention on P58 because it was the only RNA2-encoded protein with an undefined function(s). Furthermore, there is some preliminary evidence suggesting that the P58 counterpart of cowpea mosaic virus (CPMV), a relative of BPMV, might be necessary for CPMV RNA2 replication (11). Separately, RNA2 of grapevine fanleaf nepovirus (GFLV), a virus more distantly related to BPMV, encodes an N-terminally located protein (2A) that has been implicated in RNA2 replication (12). Our results showed that eliminating the P58 coding capacity rendered RNA2 undetectable in BPMV host cells and that the N-terminal 12-kDa portion of P58 was sufficient to preserve this activity. We further inferred that the translation of P58, rather than the RNA sequence coding for P58, is necessary for its function, as compensatory mutations that restored BPMV infectivity always introduced alternative start codons for P58 at various upstream positions. Most importantly, we determined that P58 functioned in a cis-acting manner relative to its coding RNA (RNA2), as P58 provided in trans was unable to complement its defect. Finally, we established that all RNA1-encoded proteins were cis-acting relative to RNA1. Together, our results strongly suggest that P58 functions to recruit the RNA1-encoded polyprotein to RNA2 to facilitate RNA2 replication.
MATERIALS AND METHODS
Constructs.
The infectious cDNA of BPMV RNA1 used in this study was derived from a Kentucky isolate (K-Ho1) of BPMV and kindly provided to us by Said Ghabrial (8). The RNA2 cDNA was originally derived from an Iowa isolate (IA-Di1) and was subsequently modified by Zhang and colleagues (13) to include a green fluorescent protein (GFP) cDNA insert between MP and L-CP. The resulting construct, designated RNA2-GFP (R2G), was a kind gift from Steve Whitham. An RNA2-mCherry (R2R) construct was created by replacing the GFP coding sequence of R2G with the coding sequence of mCherry. RNA1, R2G, and R2R cDNAs were subcloned into pRTL2 to acquire the 35S promoter and terminator (P35S and T35S) of cauliflower mosaic virus (CaMV), using previously described procedures (10, 14). RNA1m, a replication-defective mutant of RNA1 produced in a previous study (10), was also included as a negative control in most of our experiments. Constructs containing both RNA1 (and RNA1m) and R2G cassettes were also produced for some experiments.
All mutants were derivatives of RNA1 or R2G constructs. Sequences of primers used to produce these mutants are available upon request. Most mutants were generated by first producing mutation-containing PCR fragments by two-step PCR and subsequently inserting the PCR fragments into the respective viral cDNA by using appropriate restriction enzyme sites. For example, to produce R2G-mMP (Fig. 1A), two overlapping PCR fragments were first synthesized. The first fragment, encompassing nt 1 to 800 of RNA2, incorporated the desired ATG → CCT mutation at nt 773 to 775, at its 3′ end. Conversely, the second fragment, covering nt 750 to 904 of RNA2, incorporated the same mutation at its 5′ end. These two fragments were then used as templates to produce a mutation-containing fragment spanning nt 1 to 904, which also contained an XhoI site prior to nt 1 and a unique SacI site inherent to RNA2 cDNA at nt 822 to 827. This fragment was then digested with XhoI and SacI and used to replace its counterpart in wild-type R2G cDNA. Wherever possible, a new restriction enzyme site was created at or near the mutation site to facilitate convenient screening of candidate clones. The identities of all mutants were confirmed by sequencing before they were used in our experiments. A slight variation of this strategy was used to produce R2G-CPMV14K, in that three instead of two fragments were first produced and served as templates for the secondary PCR, with the middle fragment substituting the CPMV 14K protein for the BPMV 12K protein.
For all RNA1 mutants, the conserved amino acid (aa) residues in each of the proteins were first identified through multiple-sequence alignments of at least six different como- and nepoviruses (F. Qu, data not shown). These conserved aa (see Fig. 4D) were then mutated using the procedures described above.
FIG 4.
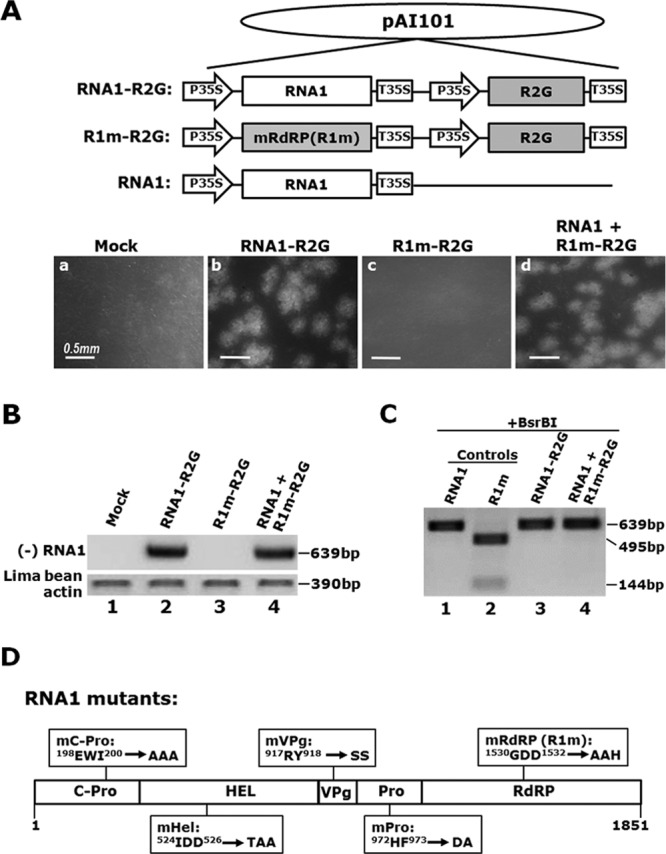
Replication-related proteins encoded by RNA1 are all cis-acting. (A) Schematic representation of the constructs used in this set of experiments (top), along with their fates in bombarded lima bean cotyledons (bottom). (B) Confirmation of results shown in panel A by (−) RNA1-specific sqRT-PCR. (C) The defect of RNA1m could not be complemented by functional RdRP provided by wild-type RNA1. Digestion with BsrBI, which cuts the PCR fragment of the R1m mutant but not that of wild-type RNA1 (lanes 1 and 2), was used to detect potential R1m-specific RT-PCR products amplified from RNA1-plus-R1m-2G-cobombarded cotyledons. (D) Schematic representation of five RNA1 mutants tested in this study. The names of the mutants, together with the specific aa changes introduced by these mutants, are highlighted in their corresponding boxes.
Particle bombardment of lima bean cotyledons.
Lima beans of the Henderson Bush variety were purchased from Earl May Seed and Nursery (Shenandoah, IA). The bombardment experiments were carried out following the procedure described by Hernandez-Garcia and colleagues (15).
Inoculation of soybean seedlings.
The bombarded lima bean cotyledons were ground in 1 ml inoculation buffer (0.01 M sodium phosphate, pH 7.0, 1% celite). The resultant extracts were rub inoculated onto the unifoliate leaves of 1-week-old soybean plants. Soybean plants were maintained under greenhouse conditions after inoculation.
Extraction of total RNAs from lima bean cotyledons and soybean leaves and (−) strand-specific RT-PCR.
Total RNA was extracted from lima bean cotyledons at 48 h postbombardment by using a published protocol (10, 16) and from soybean leaves by use of TRIsure reagent (Bioline, Tauton, MA). In some experiments, small lima bean cotyledon sections containing GFP-positive infection foci were pooled for RNA extraction. These GFP-positive infection foci were collected by first marking them with a Sharpie pen under an epifluorescence dissecting microscope and then carving them out with a razor blade. The quality and concentration of the isolated RNAs were verified using NanoDrop ND-1000 spectrophotometry (Thermo Fisher Scientific Inc.) and gel electrophoresis. Approximately 2 μg RNA per sample was treated with Turbo DNA-free DNase (Ambion, Austin, TX) according to the manufacturer's instructions. Negative [(−)] strand-specific reverse transcription (RT) was carried out by using a primer with a sequence identical to nt 1 to 30 of the RNA2 cDNA (sequence available upon request) and reverse transcriptase purchased from Clontech (Palo Alto, CA), with 1 μg of DNase-treated RNA per sample as the template. Subsequently, 1 μl cDNA per sample was used for semiquantitative PCR (sqPCR). PCR was carried out using EconoGreen PCR master mix (Lucigen, Middleton, WI) with appropriate primers (sequences available upon request) for 30 cycles of denaturation at 95°C for 30 s, annealing at 60°C for 30 s, and extension at 72°C for 1 min. A cDNA fragment for the lima bean actin mRNA (PlACT1) was amplified in parallel to serve as the control. The relative levels of the RT-PCR products were estimated using Quantity One 1-D analysis software (version 4.6.2; Bio-Rad, Hercules, CA) according to the manufacturer's instructions.
Fluorescence and confocal microscopy.
In most experiments, the bombarded cotyledons were monitored on a daily basis with a fluorescence dissecting microscope (model MZFLII; Leica, Heerbrugg, Switzerland) equipped with a custom-made image collection system composed of a GFP-2 filter set and a Spot-RT charge-coupled device (CCD) digital camera (Diagnostic Instruments Inc., Sterling Heights, MI). Small GFP-positive dots on some cotyledons were further examined using an epifluorescence microscope (model DM IRB; Leica, Heerbrugg, Switzerland) equipped with a cooled digital camera (model Retiga-2000; QImaging Scientific, Surrey, British Columbia, Canada) to reveal the boundaries of the infection foci. For this purpose, the upper epidermal layer of the bombarded cotyledons was manually sectioned with a scalpel and immersed in a droplet of distilled water prior to microscopic inspection. Samples for confocal microscopy (model TCS SP5II; Leica, Wetzler, Germany) were prepared in a similar manner.
RESULTS
Tracking the replication and cell-to-cell movement of BPMV in detached lima bean cotyledons bombarded with BPMV cDNA constructs.
Since a rigorous protoplast system suitable for examining BPMV replication is not yet available, we recently developed an easily tractable, alternative approach based on lima bean cotyledons for this purpose (10). Briefly, we demonstrated that cells of lima bean cotyledons supported robust BPMV multiplication upon receipt of BPMV cDNAs delivered with particle bombardment (10). We further used this experimental system to characterize stem-loop C (SLC), a cis-acting RNA element located in the 5′-untranslated region (5′ UTR) of BPMV RNA2 that is essential for RNA2 accumulation in these cells (10). This experimental system utilizes cDNAs of BPMV RNA1 and RNA2 that are flanked by the 35S promoter and terminator (P35S and T35S) (Fig. 1A) of CaMV. As a result, these constructs permit replication-independent transcription of BPMV RNAs by DNA-dependent RNA polymerase II (Pol II) of the host plant once the constructs are inside host cells. These primary BPMV RNAs then initiate autonomous BPMV replication to produce infectious viruses. To distinguish between the Pol II-driven primary transcripts and autonomously replicating BPMV RNA, we created the negative-control construct RNA1m (also referred to as mRdRP in Fig. 4D), a replication-defective mutant of RNA1, by replacing the highly conserved glycine-aspartic acid-aspartic acid (GDD) motif of viral RdRP with alanine-alanine-histidine (AAH) (Fig. 1A; also see Fig. 4D). Additionally, to visualize the progression of the BPMV infection process, we adopted RNA2-GFP (R2G), a modified RNA2 containing a GFP insert between MP and L-CP (Fig. 1A) (13). Cobombardment of RNA1 and R2G constructs into lima bean cotyledons resulted in rigorous BPMV replication and cell-to-cell movement, as indicated by brightly green fluorescent infection foci encompassing multiple cells (Fig. 1A, panels f and f′). In addition, extracts of BPMV-positive cotyledons could infect soybean plants systemically, as indicated by bright GFP fluorescence in upper uninoculated leaves (Fig. 2B, panel a′). The presence of infectious BPMV in both lima bean cotyledons and systemically infected soybean leaves was further confirmed by the detection of RNA1 and R2G-specific (−) RNA by strand-specific RT-PCR (Fig. 1B, top two panels, lane 3) (data not shown). As expected, the negative control, RNA1m plus R2G, failed to produce any green fluorescent infection foci in either bombarded lima bean cotyledons or soybean leaves (Fig. 1A, panels g and g′, and B, top two panels, lane 2) (data not shown). Together, these experiments proved that this system is suitable for examining the functions of individual BPMV-encoded proteins as well.
FIG 2.
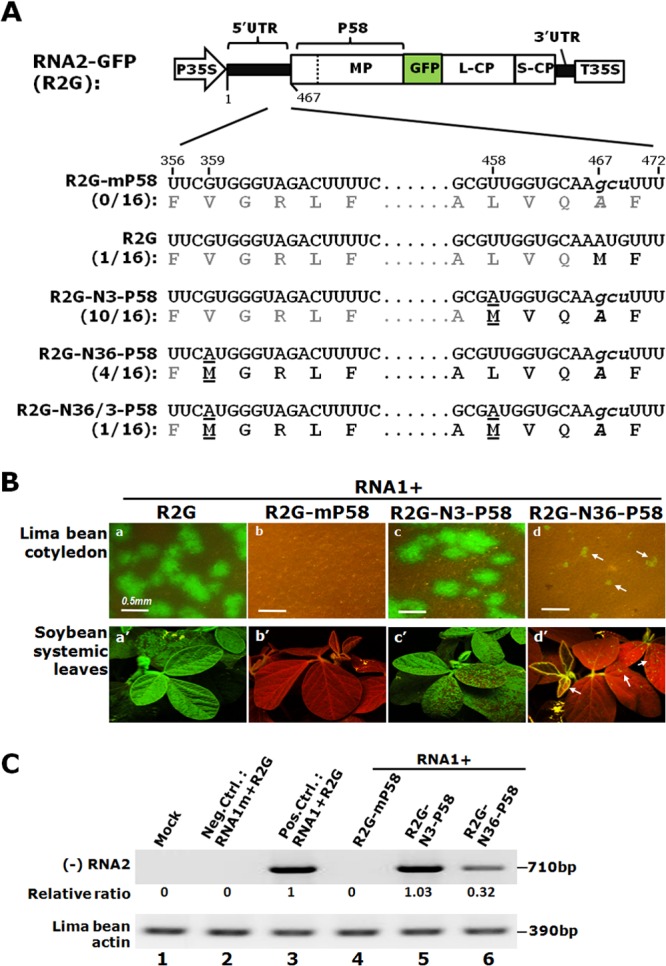
Compensatory mutants isolated from lima bean cotyledons bombarded with RNA1 plus R2G-mP58. (A) The nt sequences and deduced aa sequences of the compensatory mutants (R2G, R2G-N36-P58, R2G-N3-P58, and R2G-N3/N36-P58) were aligned against those of the original R2G-mP58 mutant to highlight the positions and identities of the compensatory mutations. Only the section of R2G containing mutations is shown. The numbers underneath the names of the mutants represent the numbers of clones with the corresponding sequences. All except the R2G revertant retained the original AUG → gcu change. Underlined letters show compensatory mutations that recreated start codons (AUG) for P58, as well as the corresponding aa (M). Nontranslatable aa are shown in gray. (B) Infectivities of the mutants in panel A, as illustrated by GFP-positive lima bean cotyledons (48 h after bombardment) (top panels) and systemically infected soybean leaves (2 weeks postinoculation) (bottom panels). (C) Verification of active replication in lima bean cotyledons by (−) RNA2-specific sqRT-PCR.
P58 is required for RNA2 accumulation in host cells of BPMV.
As described earlier, P58 is the N-terminal-most protein encoded by BPMV RNA2, and it differs from MP by harboring a 102-aa N-terminal extension (Fig. 1A). In order to assess its role in BPMV infections, we first determined if it shared any functional overlap with MP. To do this, we created the series of mutants shown in Fig. 1A to disrupt the expression of P58 and/or MP to various degrees. These mutant constructs were then bombarded into cells of lima bean cotyledons together with the wild-type RNA1 construct and observed at 48 h postbombardment with a fluorescence dissecting scope and an epifluorescence microscope. As depicted in Fig. 1A, the R2G-mMP mutant lost the AUG initiation codon of MP due to an AUG → CCU mutation (Fig. 1A). It was thus unable to produce an authentic MP but should have produced a nearly wild-type form of P58 (except for the methionine [AUG]-to-proline [CCU] change). As shown in Fig. 1A, panel a, cotyledons treated with RNA1 plus R2G-mMP developed numerous small but bright GFP foci under the dissecting scope. Compared with cotyledons treated with positive-control constructs (Fig. 1A, panel f), the dramatically reduced sizes of GFP foci strongly suggest that the cell-to-cell movement of the R2G-mMP mutant was severely debilitated. Further inspection with fluorescence microscopy showed that these foci commonly occupied two or three cells (Fig. 1A, panel a′). Thus, the P58 protein translated from this mutant probably acted as a weak MP to enable very limited cell-to-cell movement of the mutant virus. These results illustrate that a functional BPMV MP requires its translation to initiate from the second AUG codon of RNA2 and that inclusion of the N-terminal 102-aa extension compromises its function in cell-to-cell movement.
However, the replication of either RNA1 or RNA2 (R2G) in these few infected cells was not appreciably affected, as the GFP foci were sufficiently intense to permit their detection even without any magnification (Fig. 1A, panel a) (data not shown). This conclusion was further corroborated by the infection outcome of the R2G-ΔMP mutant, in which the MP coding sequence was truncated with a 516-nt (172-aa) in-frame deletion (Fig. 1A). GFP foci were again detected at large numbers with relative ease on cotyledons treated with RNA1 plus R2G-ΔMP (Fig. 1A, panel b), although these foci were now limited to single cells, indicating a complete loss of cell-to-cell movement (Fig. 1A, panel b′). Nevertheless, the high intensity of the fluorescence was suggestive of robust replication of BPMV RNAs in these individual cells. Collectively, the results with the first two mutants suggest that BPMV MP does not play a discernible role in RNA2 replication.
The next mutant we tested was R2G-mP58, in which the P58 AUG was mutated to GCU yet the MP AUG remained intact (Fig. 1A). Surprisingly, nearly all cotyledons treated with RNA1 plus R2G-mP58 failed to develop any GFP foci, even under a fluorescence microscope (Fig. 1A, panels d and d′). This result is in stark contrast with earlier results showing that both MP mutants still replicated robustly in the cells they invaded. It hence indicates that P58 plays a distinct, MP-independent role to facilitate the accumulation of BPMV RNA2. Furthermore, since the majority of the sequence shared by P58 and MP could be deleted without affecting RNA2 levels (the R2G-ΔMP mutant) (Fig. 1A, panels b and b′), this unique function of P58 is likely afforded by the first 120 aa of the protein, including the 102-aa (12-kDa) region not shared by MP (Fig. 1A).
To further delineate the minimal region required for the unique function of P58, we created R2G-ΔMP2, in which the sequence encoding the first 304 aa of MP (912 nt) was replaced with a 5-aa flexible linker (SGSGS) (Fig. 1A), leaving only a 40-aa C-terminal portion that serves to preserve the proteolytic processing site between MP and GFP. As shown in Fig. 1A, panels c and c′, R2G-ΔMP2 still replicated to easily detectable levels (also see Fig. 1B, middle panel, lane 6). These results indicated that the entire MP region of P58 could be deleted without debilitating its role in facilitating RNA2 accumulation.
Interestingly, an earlier study found that the P58 counterpart of CPMV, a close relative of BPMV, was also critical for the accumulation of RNA2 of that virus in protoplasts and that the N-terminal 14-kDa region unique to CPMV P58 was sufficient for this role (11). Therefore, even though the 14-kDa region of CPMV P58 and the 12-kDa region of BPMV P58 share little sequence similarity, it was still possible that they could complement each other. To test this possibility, we replaced the first 102-aa region of BPMV P58 with its counterpart from CPMV. The resulting construct, R2G-CPMV14K, was then examined in lima bean cotyledons. As shown in Fig. 1A, panels e and e′, no GFP foci indicative of active replication were detected on the treated cotyledons. This result suggests that this particular region of comoviral P58 is virus specific and irreplaceable for each virus.
Finally, we evaluated the reproducibility of these five mutants by detecting (−) BPMV RNA in the treated cotyledons by a strand-specific RT-PCR procedure. By focusing solely on (−) RNA, we could capture only the replicating mutants that produced (−) RNA replication intermediates, while avoiding the interference by replication-independent transcription of (+) RNA driven by P35S (see Fig. 1A for construct details). As shown in Fig. 1B, top panel, (−) RNA1 was abundantly detected in the positive-control (RNA1 plus R2G; lane 3) cotyledons but absent in the negative-control (RNA1m plus R2G; lane 2) cotyledons, as expected. Surprisingly, low levels of (−) RNA1 could be detected in all five infections in which RNA1 was mixed with various RNA2 mutants, suggesting that our approach was sufficiently sensitive to detect infections that were limited to individual cells (lanes 5 and 6) and that RNA2 accumulation is not needed for RNA1 replication (lanes 7 and 8). Similarly, low levels of (−) RNA2 were also detected for the three MP mutants (Fig. 1B, middle panel, lanes 4 to 6). In contrast, (−) RNA2 was undetectable in cotyledons containing either R2G-m58K or R2G-CPMV14K (lanes 7 and 8). These results are highly consistent with the differences in the number, size, and intensity of GFP foci, thus further confirming a critical role of P58 in BPMV RNA2 accumulation.
We hasten to note that since the start codon of BPMV P58 lies only 2 nt downstream of the cis-acting RNA stem-loop C (SLC; nt 415 to 464) characterized in our previous study (10), we needed to rule out the possibility that altering the P58 start codon (as in mP58) might have inadvertently disrupted the integrity of SLC, thus abrogating RNA2 replication. However, our in silico analysis of the first 500 nt of BPMV RNA2 (including the 5′ UTR and the first 34 nt of the P58 coding sequence) by use of the Mfold algorithm (http://mfold.rna.albany.edu/?q=mfold/rna-folding-form) showed that SLC was part of the three most favorable RNA secondary structures predicted for both wild-type BR2G and mP58, with ΔG values of about −119.5 and −120.8 kcal/mol, respectively (Qu, data not shown). More importantly, as we show in the next section, we were able to isolate several compensatory mutants that preserved the original mutation at the P58 start site (AUG → GCU) but restored the translatability of P58 by introducing new start codons upstream, thus ruling out the possibility of a serious disruption of SLC by the mutation.
P58 functionality is restored by spontaneous second-site mutations that extend the N terminus of P58 by various lengths.
We described earlier that most of the lima bean cotyledons bombarded with RNA1 plus R2G-mP58 were devoid of GFP foci, thus suggesting a lack of RNA2 replication. However, upon prolonged incubation (7 days after bombardment), a few bombarded cotyledons developed one or two isolated GFP foci that slowly expanded. To determine the underlying reason for the sporadic and delayed development of GFP foci, we physically separated the focus-containing areas from the rest of the cotyledons and pooled them for RNA extraction. This dissecting step was necessary to enrich potential compensatory mutants, as we needed to minimize the interference by the nonreplicating mP58 RNA2 present in other bombardment foci on the same cotyledon(s), whose transcription was driven by P35S. cDNA corresponding to the first 900 nt of BPMV RNA2 was then obtained by subjecting the extracted RNA to RT-PCR and subsequently cloning the product into a plasmid vector. The sequences of the inserts were then determined for 16 recombinant plasmids. As summarized in Fig. 2A, none of the clones contained solely the original mP58 mutation (AUG → GCU). Similarly noteworthy is that only 1 of the 16 clones completely reverted to the wild-type sequence (R2G). Rather, 15 of 16 clones retained the AUG → GCU mutation but also possessed additional novel changes upstream. Among them, 10 clones, designated R2G-N3-P58, contained a U → A change (at nt 458) 9 nt upstream of the original mutation, thus creating a new AUG codon in-frame with P58 and adding three additional aa to its N terminus. Similarly, the R2G-N36-P58 variant, accounting for 4 of the 16 clones, recreated a new start codon for P58, located 36 aa upstream of the original mutation, through a G → A change at nt 359. Finally, 1 of the 16 clones (R2G-N36/3-P58) contained both of the new start codons.
To demonstrate the viability of these spontaneous mutants, we recreated full-length cDNA constructs of R2G-N3-P58 and R2G-N36-P58 and tested their infectivity (together with RNA1) in lima bean cotyledons and, subsequently, soybean plants. As shown in Fig. 2B, panels c and c′, the R2G-N3-P58 mutant caused robust infections in both lima bean cotyledons and soybean plants that were indistinguishable from those with wild-type R2G (Fig. 2B, panels a and a′). In contrast, the R2G-N36-P58 construct, with a 36-aa N-terminal extension, induced much smaller infection foci in bombarded lima bean cotyledons (Fig. 2B, panel d) yet was still able to infect soybean plants systemically (Fig. 2B, panel d′). Finally, the results of infection assays were confirmed with strand-specific RT-PCR that detected (−) RNA2 (Fig. 2C). Note that the compensatory mutations were not expected to affect the structure of the SLC RNA element: the U → A mutation at nt 458 of R2G-N3-P58 changed a C-U mismatch to a C-A mismatch in the lower part of the SLC stem, whereas the G → A mutation at nt 359 of R2G-N36-P58 is 56 nt upstream of SLC (nt 415 to 464), within a region dispensable for RNA2 accumulation (SLB in reference 10). Together, these data demonstrated that it is the P58 protein, not the RNA sequence/structure coding for P58, that is needed for BPMV RNA2 accumulation.
P58 is a cis-acting protein of BPMV RNA2.
To further elucidate the functional mechanism of P58, we sought to complement the loss of P58 function in the R2G-mP58 mutant with intact P58 expressed from a different RNA2 construct. For this purpose, we created modified versions of R2G-mMP and R2G-mP58 that replaced the GFP coding sequence with that of red-fluorescing mCherry. These new mutants, designated R2R-mMP and R2R-mP58 (Fig. 3A, rows b and e), were expected to produce functional P58 and MP, respectively, and could potentially complement the GFP-expressing R2G-mP58 and R2G-mMP constructs. Similar to their GFP-tagged counterparts, R2R-mMP but not R2R-mP58 replicated to levels detectable by confocal microscopy in foci that spanned one to three cells (Fig. 3A, compare confocal images in rows b and e).
FIG 3.

P58 is a cis-acting protein. (A) Complementation between RNA2 mutants with defects in P58 and MP, as revealed by confocal microscopy of the bombarded cotyledons. The diagrams of the mutants used, as well as those included in each of the complementation sets, are shown on the left, with the outcomes of infections/complementations shown on the right. mCh, red fluorescent mCherry protein. (B) Results of (−) RNA2-specific sqRT-PCR confirming the complementation results shown in panel A. Note that the relative accumulation levels were calculated by using R2G-mMP (top panel) or R2R-mMP (middle panel) as a reference (arbitrarily set as 1).
We first successfully complemented the cell-to-cell movement defects of R2G-mMP and -ΔMP2 with R2R-mP58, as evidenced by the observation of large clusters of green fluorescent cells in the cobombarded cotyledons (Fig. 3A, compare confocal images in rows f and g with those in rows a and c). These results suggested that the R2R-mP58 mRNA encoded functional MP, which in turn enabled the cell-to-cell spread of R2G-mMP and -ΔMP2 mutants. Surprisingly, the accumulation defect of R2R-mP58 itself was not complemented by cobombarded constructs (R2G-mMP and R2G-ΔMP2) that should provide P58 or its functional equivalent, as evidenced by a lack of mCherry-borne red fluorescence in the cobombarded cotyledons (rows f and g, mCherry panels). These results raised the intriguing possibility that the R2R-mP58 mutant could not utilize the P58 protein (or its functional equivalent in R2G-ΔMP2) provided by its complementation partners. This in turn suggested that P58 is a cis-acting protein encoded by RNA2 to facilitate its own accumulation.
To further confirm this interesting observation, we carried out reciprocal complementation experiments by using R2R-mMP to provide P58 in trans to complement R2G-mP58 (Fig. 3A, row h). Consistent with the above-described complementation attempts, no GFP foci was detected even at the single-cell level, indicating that P58 provided by R2R-mMP was unable to complement the loss of P58 function in R2G-mP58 (Fig. 3A, row h). Instead, R2R-mMP itself spread to multiple cells thanks to the functional MP provided by R2G-mP58 in trans. Therefore, mutants that do not encode functional P58 (R2G-mP58 and R2R-mP58) are unable to use P58 provided in trans to enhance their own accumulation, even though their P35S-driven transcripts could readily translate functional MP. Together, these results strongly suggest that P58 is a cis-acting protein that functions only with the RNA2 molecules from which it is translated.
We also verified these findings by using strand-specific RT-PCR (Fig. 3B). Note here that we used the levels of R2G-mMP/R2R-mMP, rather than wild-type R2G, as references to calculate the relative accumulation levels of other mutants, as a wild-type R2R construct was not included in this set of experiments. In cobombarded cotyledons in which the movement defect of R2G-mMP or R2G-ΔMP2 was complemented by R2R-mP58, the RT-PCR assay detected an approximately 2-fold increase of GFP-specific (−) RNA2 levels (Fig. 3B, top panel, compare lanes 5 and 7 with lanes 6 and 8, respectively), which is probably an underestimate of the complementation outcomes, judging by the number of cells in the green fluorescent foci (Fig. 3A, rows f and g). In contrast, mCherry-specific (−) RNA was not detected in any of these samples, confirming the inability of the mCherry-tagged, P58-defective R2R-mP58 construct to produce (−) RNA to detectable levels (Fig. 3B, middle panel, lanes 6 and 8). Similarly, the GFP-tagged, P58-defective R2G-mP58 construct was likewise unable to generate a detectable level of GFP-specific (−) RNA, despite its ability to complement R2R-mMP (Fig. 3B, two upper panels, lanes 10 and 11). In summary, the data so far implicate P58 as a cis-acting protein that facilitates RNA2 accumulation in BPMV-infected cells.
Replication-related proteins encoded by BPMV RNA1 are all cis-acting relative to RNA1.
Having established that P58 is required in cis for RNA2 accumulation, we wondered if the cis-acting mode is unique to P58 or shared by other BPMV proteins functioning in the genome replication process. Since all other replication-related proteins are encoded by RNA1 but needed for RNA2 replication as well, it seemed counterintuitive to expect these proteins to be cis-acting. Nevertheless, we reasoned that some of the RNA1-encoded proteins could still be cis-acting relative to RNA1, and hence must be recruited to RNA2 through RNA2-specific mechanisms, as previously established for other multipartite viruses (6, 7, 17). To test this possibility, we first focused on BPMV RdRP, as we had available an RNA1 mutant in which the highly conserved GDD motif of RdRP was changed to AAH, rendering RdRP nonfunctional. This mutant, designated RNA1m or mRdRP (Fig. 1A and 4A), was used in many of our previous experiments as a negative control.
To determine if the loss of RdRP function in this mutant could be complemented by a functional RdRP provided in trans, we first created R1m-R2G, a new construct that combined the expression cassettes of both R1m and R2G into one single plasmid to ensure the maximal coexistence of both RNA segments in the same cells (Fig. 4A). A similarly configured positive-control construct, RNA1-R2G, was also created (Fig. 1A). As shown in Fig. 4A, panel c, when bombarded into lima bean cotyledons alone, R1m-R2G failed to give rise to any GFP foci, thus verifying that the mutation in R1m abolished the replication of RNA1. When R1m-R2G was cobombarded into cotyledons with a wild-type RNA1 construct, the replication of RNA2 was restored to levels similar to those of RNA1-R2G (Fig. 4A, panel d), suggesting that wild-type RNA1 provided through a different construct readily supported RNA2 replication. Indeed, this was also confirmed by the detection of a (−) RNA1-specific RT-PCR fragment in these cotyledons (Fig. 4B, lane 4).
However, this complementation assay did not allow us to assess whether the defect in R1m was complemented by RdRP provided by wild-type RNA1 in trans. To do this, we took advantage of a BsrBI restriction enzyme site introduced into R1m at the time of mutant creation. As shown in Fig. 4C, lanes 1 and 2, digestion of control PCR products derived from RNA1- and R1m-containing plasmids failed to fragmentize the wild-type RNA1 product but cut the R1m product into two smaller fragments, confirming the presence of the BsrBI site, as well as the effectiveness of this enzyme. However, the equivalent, (−) RNA1-specific PCR products derived from cotyledons infected with RNA1-R2G or RNA1 plus R1m-R2G were both resistant to BsrBI digestion (Fig. 4C, lanes 3 and 4), indicating that despite its presence in the same cells with R2G, the inability of R1m to replicate was not reversed by wild-type RNA1 that facilitated R2G replication. These results revealed two important insights: (i) the mutated RdRP of R1m, if produced, did not repress wild-type RdRP trans-dominantly; and (ii) conversely, wild-type RdRP provided in trans was also incapable of complementing the R1m defect. In conclusion, RdRP of BPMV is cis-acting relative to RNA1.
We went on to make four additional RNA1 mutants, mC-Pro, mHel, mVPg, and mPro, by replacing two or three conserved amino acids within each of the four corresponding RNA1 proteins. The detailed changes are summarized in Fig. 4D. These mutants were then analyzed by procedures similar to those used to examine R1m. None of these mutants could infect host cells when cointroduced into lima bean cotyledons together with R2G (data not shown). Furthermore, despite repeated attempts, we were unable to complement the defects in any of these mutants with wild-type RNA1. Thus, we concluded that all RNA1-encoded proteins are cis-acting proteins relative to RNA1.
DISCUSSION
BPMV is a bipartite (+) RNA virus of the genus Comovirus and the family Secoviridae (18). Secoviruses share substantial similarities with animal viruses of the Picornaviridae family in terms of genome organization, mode of gene expression, and virion symmetry (19). One notable difference between these two virus families is that while picornaviruses typically have monopartite genomes, many secoviruses, including all comoviruses, have bipartite genomes. Like other comoviruses, the larger RNA1 segment of BPMV encodes all viral proteins required for its own replication and replicates in single cells in the absence of RNA2 (J. Lin and F. Qu, unpublished data) (Fig. 1B). In contrast, RNA2 encodes mostly functions needed for virion assembly and cell-to-cell trafficking, and hence has to rely on RNA1-encoded proteins for replication. Exactly how RNA2 recruits RNA1-encoded proteins to replicate itself is not well understood for BPMV or comoviruses in general. Earlier investigations using CPMV, another comovirus, showed that mutations disrupting the coding region of the N-terminally located P58 open reading frame (ORF) abolished RNA2 accumulation in protoplast cells, although it was not further examined whether the P58 protein itself or its coding RNA played a more critical role or how this genome region facilitated RNA2 accumulation (11, 20, 21). It was also found that RNA2 of GFLV, a virus of the genus Nepovirus of the Secoviridae family, encodes a 5′-proximal 2A protein that is needed for RNA2 accumulation and colocalizes with some of the RNA1-encoded proteins (12). These earlier studies suggested that the recruitment of RNA1-encoded proteins by RNA2 of bipartite secoviruses might invoke a shared mechanism that requires an RNA2-encoded protein.
In the current study, we carried out an extensive investigation of P58 encoded by BPMV RNA2 with a newly developed system that allowed us to monitor BPMV RNA accumulation as well as viral cell-to-cell movement. With this new system, we were able to show that the P58 protein, rather than the sequence/structure of its coding RNA, is needed to ensure the accumulation of BPMV RNA2. We also determined that the function of P58 does not overlap that of MP, as most of the MP region could be deleted without compromising RNA2 accumulation in single cells. Most importantly, we found that P58 acts in cis, or at least cis-preferentially, to enable the accumulation of the same RNA2 molecule from which it is translated. Our results advance the current understanding of the 5′-proximally encoded P58 protein of BPMV RNA2 and lay the foundation for further in-depth examination of the replication mechanism of BPMV RNA2.
The importance of P58 in facilitating BPMV RNA2 accumulation is further highlighted by the low-frequency occurrence of compensatory mutants of R2G-mP58 that restored RNA2 accumulation by recreating new start codons upstream of the original one. The original mutation that eliminated P58 translatability was not expected to affect the integrity of the nearby RNA element SLC. Indeed, the original mutation was present in all three compensatory mutants isolated (Fig. 2). How, then, could these compensatory mutants arise if the P58-defective mutant (R2G-mP58) could not replicate? While we cannot rule out the remote possibility that this mutant could replicate at extremely low levels undetectable by our procedure, we consider it possible that our experimental system, in which the replication-independent transcription of mutant RNA is driven by the strong P35S of CaMV, could give rise to the occurrence of compensatory mutations. Furthermore, high levels of mutant RNA2 could provide the chance for occasional translational initiation from upstream non-AUG codons, producing low levels of P58 equivalents. Indeed, it has long been known that non-AUG codons such as GUG and UUG, both present in the R2G 3′ UTR in-frame with the P58 ORF, are often used by viruses to initiate protein translation (reviewed in reference 22). P58 equivalents translated from these non-AUG codons could then support suboptimal replication of mutant RNA2 in a fraction of cells, facilitating the emergence of compensatory mutations in these cells.
How does P58 ensure the accumulation of RNA2 specifically? While this was not thoroughly investigated in the current study, several lines of evidence led us to speculate that P58 functions to recruit the RNA1-encoded polyprotein to RNA2 for the purpose of replicating RNA2. First, in cotyledons treated with wild-type RNA1 and R2G-mP58 constructs, (−) RNA2 was undetectable, despite the availability of P35S-driven (+) RNA2 in these cells, and despite the fact that (−) RNA1 was readily detectable in the same cells. Second, the cis-acting mode of P58 function is commonly associated with viral replication proteins (7, 17, 23, 24). Indeed, most of the replication-related proteins encoded by poliovirus, a member of the Picornaviridae family, were found to be cis-acting (25). Our result showing that all BPMV RNA1-encoded proteins are cis-acting further extends the list of cis-acting replication proteins and, conversely, supports a replicational role for P58. Finally, the reported intracellular colocalization of the GFLV 2A protein with some of the RNA1-encoded proteins is also consistent with the involvement of one of the RNA2-encoded proteins in the replication of RNA2 itself (12). In addition, structural modeling suggests that the region of BPMV P58 not shared by MP contains a very strong transmembrane domain, which could conceivably allow it to associate with the membrane-based VRCs. Our next step will be to investigate whether P58 indeed localizes to membrane-based VRCs and whether it colocalizes with RNA1-encoded proteins.
We recently reported the identification of SLC, an RNA stem-loop structure within the 5′ UTR of BPMV RNA2 that is essential for RNA2 accumulation in infected cells (10). The characterization of P58 in the current study adds to the list of cis-acting elements needed to ensure efficient reproduction of BPMV RNA2. Our next goals are to understand how these cis-acting elements function to ensure RNA2 accumulation in infected cells and whether they coordinate with each other to achieve the optimal outcome for the virus.
ACKNOWLEDGMENTS
We greatly appreciate the generosity of Steve Whitham and John Hill at Iowa State University in providing us with the RNA2-GFP construct. We thank members of the Qu and Finer labs for stimulating discussions and technical assistance and Lucy Stewart and other members of the USDA-ARS Corn and Soybean Research Unit for sharing equipment.
This study was supported in part by an OARDC Graduate Seed award to J.L. Soybean virus-related research in the Qu lab has been supported in part by grants from the North Central Soybean Research Program and the Ohio Soybean Council.
Footnotes
Published ahead of print 3 January 2014
REFERENCES
- 1.den Boon JA, Ahlquist P. 2010. Organelle-like membrane compartmentalization of positive-strand RNA virus replication factories. Annu. Rev. Microbiol. 64:241–256. 10.1146/annurev.micro.112408.134012 [DOI] [PubMed] [Google Scholar]
- 2.Nagy PD, Pogany J. 2010. Global genomics and proteomics approaches to identify host factors as targets to induce resistance against Tomato bushy stunt virus. Adv. Virus Res. 76:123–177. 10.1016/S0065-3527(10)76004-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nagy PD, Barajas D, Pogany J. 2012. Host factors with regulatory roles in tombusvirus replication. Curr. Opin. Virol. 2:691–698. 10.1016/j.coviro.2012.10.004 [DOI] [PubMed] [Google Scholar]
- 4.Schwartz M, Chen J, Janda M, Sullivan M, den Boon J, Ahlquist P. 2002. A positive-strand RNA virus replication complex parallels form and function of retrovirus capsids. Mol. Cell 9:505–514. 10.1016/S1097-2765(02)00474-4 [DOI] [PubMed] [Google Scholar]
- 5.Mine A, Okuno T. 2012. Composition of plant virus RNA replicase complexes. Curr. Opin. Virol. 2:669–675. 10.1016/j.coviro.2012.09.014 [DOI] [PubMed] [Google Scholar]
- 6.Baumstark T, Ahlquist P. 2001. The brome mosaic virus RNA3 intergenic replication enhancer folds to mimic a tRNA TpsiC-stem loop and is modified in vivo. RNA 7:1652–1670 [PMC free article] [PubMed] [Google Scholar]
- 7.Iwakawa HO, Mine A, Hyodo K, An M, Kaido M, Mise K, Okuno T. 2011. Template recognition mechanisms by replicase proteins differ between bipartite positive-strand genomic RNAs of a plant virus. J. Virol. 85:497–509. 10.1128/JVI.01754-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gu H, Ghabrial SA. 2005. The Bean pod mottle virus proteinase cofactor and putative helicase are symptom severity determinants. Virology 333:271–283. 10.1016/j.virol.2005.01.020 [DOI] [PubMed] [Google Scholar]
- 9.MacFarlane SA, Shanks M, Davies JW, Zlotnick A, Lomonossoff GP. 1991. Analysis of the nucleotide sequence of bean pod mottle virus middle component RNA. Virology 183:405–409. 10.1016/0042-6822(91)90155-5 [DOI] [PubMed] [Google Scholar]
- 10.Lin J, Ali AK, Chen P, Ghabrial S, Finer J, Dorrance A, Redinbaugh P, Qu F. 2013. A stem-loop structure in the 5′ untranslated region of Bean pod mottle virus RNA2 is specifically required for RNA2 accumulation. J. Gen. Virol. 94:1415–1420. 10.1099/vir.0.051755-0 [DOI] [PubMed] [Google Scholar]
- 11.Van Bokhoven H, Le Gall O, Kasteel D, Verver J, Wellink J, Van Kammen AB. 1993. Cis- and trans-acting elements in cowpea mosaic virus RNA replication. Virology 195:377–386. 10.1006/viro.1993.1387 [DOI] [PubMed] [Google Scholar]
- 12.Gaire F, Schmitt C, Stussi-Garaud C, Pinck L, Ritzenthaler C. 1999. Protein 2A of grapevine fanleaf nepovirus is implicated in RNA2 replication and colocalizes to the replication site. Virology 264:25–36. 10.1006/viro.1999.9927 [DOI] [PubMed] [Google Scholar]
- 13.Zhang C, Bradshaw JD, Whitham SA, Hill JH. 2010. The development of an efficient multipurpose bean pod mottle virus viral vector set for foreign gene expression and RNA silencing. Plant Physiol. 153:52–65. 10.1104/pp.109.151639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qu F, Ren T, Morris TJ. 2003. The coat protein of turnip crinkle virus suppresses posttranscriptional gene silencing at an early initiation step. J. Virol. 77:511–522. 10.1128/JVI.77.1.511-522.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hernandez-Garcia CM, Bouchard RA, Rushton PJ, Jones ML, Chen X, Timko MP, Finer JJ. 2010. High level transgenic expression of soybean (Glycine max) GmERF and Gmubi gene promoters isolated by a novel promoter analysis pipeline. BMC Plant Biol. 10:237. 10.1186/1471-2229-10-237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Louime C, Vasanthaiah HKN, Jittayasothorn Y, Jiang L, Basha SM, Thipyapong P, Boonkerd N. 2008. A simple and efficient protocol for high quality RNA extraction and cloning of chalcone synthase partial CDS from muscadine grape cultivars (Vitis Rotundifolia Michx.). Eur. J. Sci. Res. 22:232–240 [Google Scholar]
- 17.Yi G, Kao C. 2008. cis- and trans-acting functions of brome mosaic virus protein 1a in genomic RNA1 replication. J. Virol. 82:3045–3053. 10.1128/JVI.02390-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sanfacon H, Wellink J, Le Gall O, Karasev A, van der Vlugt R, Wetzel T. 2009. Secoviridae: a proposed family of plant viruses within the order Picornavirales that combines the families Sequiviridae and Comoviridae, the unassigned genera Cheravirus and Sadwavirus, and the proposed genus Torradovirus. Arch. Virol. 154:899–907. 10.1007/s00705-009-0367-z [DOI] [PubMed] [Google Scholar]
- 19.Le Gall O, Christian P, Fauquet CM, King AM, Knowles NJ, Nakashima N, Stanway G, Gorbalenya AE. 2008. Picornavirales, a proposed order of positive-sense single-stranded RNA viruses with a pseudo-T=3 virion architecture. Arch. Virol. 153:715–727. 10.1007/s00705-008-0041-x [DOI] [PubMed] [Google Scholar]
- 20.Carette JE, Guhl K, Wellink J, Van Kammen A. 2002. Coalescence of the sites of cowpea mosaic virus RNA replication into a cytopathic structure. J. Virol. 76:6235–6243. 10.1128/JVI.76.12.6235-6243.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sainsbury F, Cañizares MC, Lomonossoff GP. 2010. Cowpea mosaic virus: the plant virus-based biotechnology workhorse. Annu. Rev. Phytopathol. 48:437–455. 10.1146/annurev-phyto-073009-114242 [DOI] [PubMed] [Google Scholar]
- 22.Futterer J, Hohn T. 1996. Translation in plants—rules and exceptions. Plant Mol. Biol. 32:159–189. 10.1007/BF00039382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.White KA, Skuzeski JM, Li W, Wei N, Morris TJ. 1995. Immunodetection, expression strategy and complementation of turnip crinkle virus p28 and p88 replication components. Virology 211:525–534. 10.1006/viro.1995.1434 [DOI] [PubMed] [Google Scholar]
- 24.Wang J, Yeh HH, Falk BW. 2009. cis preferential replication of Lettuce infectious yellows virus (LIYV) RNA 1: the initial step in the asynchronous replication of the LIYV genomic RNAs. Virology 386:217–223. 10.1016/j.virol.2009.01.004 [DOI] [PubMed] [Google Scholar]
- 25.Novak JE, Kirkegaard K. 1994. Coupling between genome translation and replication in an RNA virus. Genes Dev. 8:1726–1737. 10.1101/gad.8.14.1726 [DOI] [PubMed] [Google Scholar]