

ABSTRACT
The U21 gene product from human herpesvirus 7 binds to and redirects class I major histocompatibility complex (MHC) molecules to a lysosomal compartment. The molecular mechanism by which U21 reroutes class I MHC molecules to lysosomes is not known. Here, we have reconstituted the interaction between purified soluble U21 and class I MHC molecules, suggesting that U21 does not require additional cellular proteins to interact with class I MHC molecules. Our results demonstrate that U21, itself predicted to contain an MHC class I-like protein fold, interacts tightly with class I MHC molecules as a tetramer, in a 4:2 stoichiometry. These observations have helped to elucidate a refined model describing the mechanism by which U21 escorts class I MHC molecules to the lysosomal compartment.
IMPORTANCE In this report, we show that the human herpesvirus 7 (HHV-7) immunoevasin U21, itself a class I MHC-like protein, binds with high affinity to class I MHC molecules as a tetramer and escorts them to lysosomes, where they are degraded. While many class I MHC-like molecules have been described in detail, this unusual viral class I-like protein functions as a tetramer, associating with class I MHC molecules in a 4:2 ratio, illuminating a functional significance of homooligomerization of a class I MHC-like protein.
INTRODUCTION
Human herpesvirus 6 (HHV-6) and HHV-7 are two closely related viruses that, together with human cytomegalovirus (HCMV), comprise the β subgroup of human herpesviruses. Primary infections with HHV-6 and -7 usually occur before the age of three and are often characterized by a high fever. These viruses affect over 90% of the population, and like other herpesviruses, HHV-6 and -7 remain latent or establish lifelong persistent infections in their hosts.
Lifelong interaction of herpesviruses with their hosts has resulted in the evolution of numerous strategies to evade detection by the immune system. Many viruses, including most members of the herpesvirus family, have evolved mechanisms to interfere with viral antigen presentation by class I major histocompatibility complex (MHC) molecules as a means of escaping detection by cytotoxic T lymphocytes (CTLs). We have found that HHV-6 and HHV-7 both encode one such gene product, U21 (1–3).
The host is not limited to cytotoxic T cells in its defense against viruses. Before an adaptive immune response against virus-infected cells can begin, virus infection induces the expression of natural killer (NK)-activating ligands on the surface of virus-infected cells. These virus-induced activating ligands can be recognized by NK cells and some CD8+ T cells. Many viruses have evolved means of escape from NK recognition as well. For HHV-7, U21 seems to do it all: U21 binds to and reroutes class I MHC molecules to a lysosomal compartment, presumably to escape recognition by CTLs (3). U21 also downregulates the NK-activating ligands MICA and MICB from the cell surface, which prevents NK killing of U21-expressing cells (4).
U21 is a type I integral membrane protein that binds to newly synthesized, properly folded MHC class I molecules in the endoplasmic reticulum (ER), shortly after their synthesis. In previous studies, we demonstrated that expression of U21 in U373 cells results in a dramatic redistribution of class I molecules to lysosomes and a commensurate reduction of class I molecules on the plasma membrane (2, 3). The mechanism by which U21 reroutes class I MHC molecules to lysosomes is not yet clear. Initially, we hypothesized that the lumenal domain of U21 bound to class I MHC molecules and that the cytoplasmic tail of U21 contained the lysosomal targeting information necessary to reroute the two molecules to the lysosomal compartment. We tested this hypothesis by expressing a U21 molecule lacking its cytoplasmic tail in U373 cells. We found, to our surprise, that the cytoplasmic tail of U21 was not necessary for the ability of U21 to divert class I molecules to lysosomes (2); the tailless U21 molecule could divert class I MHC molecules to lysosomes, as could a soluble version of U21, lacking both its transmembrane domain and cytoplasmic tail (2, 5). Thus, the lumenal domain of U21 is not only responsible for associating with class I molecules, but it also contains the information necessary to induce rerouting of class I MHC molecules to the lysosomal compartment.
To better understand U21's function, in the present work we have reconstituted the interaction between purified soluble forms of U21 and class I MHC molecules. We show that U21 binds to class I molecules predominantly as a tetramer, and we extend this finding to show that U21 also forms oligomers in vivo as it associates with class I molecules. These results lead us to a refined model to describe the function of U21 as it reroutes class I MHC molecules to the lysosomal compartment.
MATERIALS AND METHODS
Cell lines and antibodies.
Human embryonic kidney (HEK293T) and U373 astrocytoma cell lines were cultured in Dulbecco's modified Eagle medium (DMEM), 5% fetal bovine serum, and 5% newborn calf serum in the presence or absence of puromycin (final concentration, 375 ng/ml; Sigma-Aldrich, St. Louis, MO). Expression of U21, U21NSBPHA, and the untagged ER-lumenal domain of U21 (U21N) was carried out via lentivirus-mediated gene transfer, using the vector pHAGE-Puro-MCS (pPM), which carries the selectable marker for puromycin resistance (6, 7).
Stable expression of U21 and U21TEVHAHA-tail in U373 cells was carried out via retrovirus-mediated gene transfer, using the vector pLNCX, which carries the selectable marker for neomycin resistance. Expression of U21NSBPHA and U21TEVHAHA-tail in U373 cells was carried out via lentivirus-mediated gene transfer. Cells coexpressing wild-type U21 with a second U21-based construct were generated by introducing the mutant U21 construct into a parental, wild-type U21-expressing cell line.
The W6/32 monoclonal antibody (MAb) recognizes assembled, β2m-associated HLA molecules (8). The HC10 MAb recognizes most free MHC class I heavy chain molecules that are not associated with β2m (9). The HA11 MAb recognizes the influenza hemagglutinin (HA) epitope (Covance, San Diego, CA). The MCW62 (U21N) polyclonal antibody was raised against the putative α1α2 domain of HHV-7 U21, and the MCW50 (U21C) polyclonal antibody was raised against the cytoplasmic tail of U21 (5). Horseradish peroxidase (HRP)-conjugated goat anti-mouse and goat anti-rabbit secondary MAbs were used for immunoblotting (Bio-Rad, Hercules, CA). The SBPHA epitope tag (10) was fused to the C terminus of soluble HHV-7 U21 ER-lumenal domains.
The sequence of the SBPHA tag is as follows: ISDEKTTGWRGGHVVEGLAGELEQLRARLEHHPQGQREPIDYDIPTTASENLYFQGELKTAALAQHDEAYPYDVPDYA. Bolding indicates a streptavidin binding protein tag, underlining indicates a tobacco etch virus (TEV) protease cleavage site, and bold italic font indicates an HA epitope tag. The TEVHAHA tail tag consisted of a TEV protease cleavage site followed by two successive HA tags, inserted between amino acids D384 and G385 of the cytoplasmic tail of U21.
Class I MHC molecules.
HLA-A2:01 biotinylated monomers loaded with the LLFGYPVYV peptide were generously provided by the NIH Tetramer Core Facility (National Institute of Allergy and Infectious Disease, Emory University, Atlanta, GA). HLA-A2 molecules provided by the Tetramer Core Facility were purified from bacterial inclusion bodies and refolded in the presence of human β2m. Production and purification of recombinant sHLA B*07:02 molecules were performed as described previously (11, 12). Briefly, stable sHLA-transfected Epstein-Barr virus (EBV)-transformed B lymphoblastoid cells (721.221 cells) were used to cultivate two hollow-fiber bioreactor units of the CP-2500 Cell Pharm System (Biovest International, Minneapolis, MN). Secreted sHLA product was collected as crude harvest and affinity purified using a Sepharose 4B-W6/32 matrix. Eluted molecules were buffer exchanged with phosphate-buffered saline (PBS) at pH 7.2 and stored at 4°C until further use.
Immunoprecipitations.
For pulse label experiments, U373 cells were metabolically labeled in methionine- and cysteine-free DMEM containing 500 μCi/ml of 35S-Express label (1,175 Ci/mmol; PerkinElmer, Boston, MA) at 37°C for 20 min. Cells were lysed in 1% Nonidet P-40 lysis buffer (50 mM Tris-HCl [pH 7.4], 1% NP-40, 5 mM MgCl2). Lysates were centrifuged to pellet nuclei and debris, followed by immunoprecipitation with specific antisera and protein A agarose (RepliGen Corporation, Waltham, MA). In metabolic labeling experiments, immunoprecipitates were normalized to equal trichloroacetic acid-precipitable counts prior to immunoprecipitation. The immunoprecipitates were washed four times with NP-40 wash buffer (50 mM Tris-HCl [pH 7.4], 0.5% NP-40, 5 mM EDTA, 150 mM NaCl) and subjected to SDS-PAGE. For some immunoprecipitations, W6/32 was covalently linked to protein A agarose using a protocol modified from Pierce (Rockford, IL), in which protein A agarose was washed with PBS and incubated with purified W6/32 at room temperature for 45 min. The coupled beads were washed with PBS and then incubated in 0.225 mM disuccinimidyl suberate (DSS) in PBS for 45 min. Cross-linked beads were washed with 100 mM glycine, pH 2.8, and then equilibrated with NP-40 wash buffer. Immunoprecipitates were washed six times with Triton X-100 wash buffer (50 mM Tris-HCl [pH 7.4], 1% Triton X-100, 0.5% deoxycholate, 150 mM NaCl) and eluted with 0.5 N NaOH and 0.5 mM EDTA, with shaking at 23°C for 20 min. Eluates were flash frozen in liquid nitrogen and lyophilized. Dried eluates were resuspended in Laemmli buffer containing tris(2-carboxyethyl)phosphine (Sigma, St. Louis, MO) and subjected to SDS-PAGE, followed by silver staining.
Protein purification.
293T cells expressing U21NSBPHA were grown in plates in CD 293 medium (Gibco) (serum-free medium [SFM]) for 24 h before the medium was harvested and centrifuged to pellet any nonadherent cells. Harvested SFM was replaced with more SFM for additional collection. U21NSBPHA was purified from SFM by first concentrating the SFM using an Amicon stirred cell concentrator (model 8200) and an Amicon YM10-regenerated cellulose filter (molecular mass cutoff [MWCO] of 10 kDa) (Millipore, Billerica, MA). The SFM was concentrated ∼30× and diafiltered with PBS to remove biotin. Concentrated SFM was centrifuged to pellet any precipitates, and U21NSBPHA was purified from the supernatant in batch using high-capacity streptavidin agarose resin (Thermo Fisher Scientific, Waltham, MA). Streptavidin agarose resin was washed with PBS, and U21NSBPHA was eluted with PBS containing 8 mM biotin.
Immunoblotting.
Cells were washed with ice-cold PBS and lysed in 1% NP-40 lysis buffer, and nuclei and debris were pelleted. Cell lysates were assayed for protein concentration in triplicate using a Pierce BCA protein assay kit (Pierce, Rockford, IL). Equal concentrations of protein from each sample were subjected to SDS-PAGE. The separated proteins were transferred to a BA-S 85 nitrocellulose membrane (Whatman, Florham Park, NJ) (13). Nitrocellulose membranes were probed with the indicated antibodies followed by an HRP-conjugated secondary antibody (Bio-Rad, Hercules, CA). Bands were visualized using Pierce SuperSignal chemiluminescence reagents (Thermo Fisher Scientific).
Native gel shift assays.
Protein samples were incubated in native gel loading buffer (250 mM Tris [pH 8.8], 10% glycerol) for 30 min at 4°C. Samples were loaded on 8% polyacrylamide gels, at 90 V (constant voltage) at 4°C for 5.5 h in 25 mM Tris and 190 mM glycine running buffer. Following electrophoresis, proteins were visualized by silver staining or colloidal Coomassie blue staining or were transferred to a BA-S 85 nitrocellulose membrane for immunoblot analysis, as described above.
MALDI.
Purified U21 was concentrated and desalted on a C4 Zip-Tip column (Millipore), according to the manufacturer's instructions. Elution was carried out with 15 μl of 50% acetonitrile and 0.1% trifluoroacetic acid (TFA). The sample was lyophilized in a SpeedVac (Savant; Thermo Fisher Scientific), resuspended in 5 μl 0.1% TFA, mixed 1:1 with a saturated sinapinic acid solution (Sigma, St. Louis, MO), and then spotted onto a matrix-assisted laser desorption ionization (MALDI) target. The samples were then dried using vacuum crystallization. After drying, spots were briefly rinsed with ice-cold water and 0.1% TFA and then recrystallized in 50/50 acetonitrile-H2O in 0.1% TFA. Bovine serum albumin (BSA) was used as an external calibration standard for the material. MALDI spectra were acquired in linear (+) ion mode and calibrated against a linear curve from the [M+H]+ [M+2H]2+ ions of BSA (ca. 66,000, 33,000 m/z). A mass range of 20 kDa to 100 kDa m/z was monitored with an ion gate at 19 kDa. Three thousand laser shots were acquired at a frequency of 25 Hz. Spectra were baseline corrected and smoothed using a 100-m/z 2-pass Gaussian smooth (14). The molecular mass of purified U21 was estimated to be 59,222 ± 300 Da, from which we conclude it to contain approximately 10.3 kDa of carbohydrates.
Analytical ultracentrifugation experiments.
Sedimentation equilibrium (SE) and sedimentation velocity (SV) experiments were carried out as described previously (15) in Optima XL-I analytical ultracentrifuges equipped with the ProteomeLabTM XL-A/XL-I graphical user interface acquisition software version 6.0 (firmware version 5.06) (Beckman Coulter, Indianapolis, IN).
Briefly, for SV experiments, 400 μl of protein samples and reference buffer (PBS) were inserted in a cell assembly with charcoal-filled Epon double-sector centerpieces of a 3-mm or 12-mm path length and sapphire windows. Sample cells were placed in an eight-hole rotor, and temperature was equilibrated at 20°C at rest in the rotor chamber of the ultracentrifuge for 2 h, before it was accelerated from 0 to 50,000 rpm. Both Rayleigh interferometric fringe shift data and absorbance optical data with 0.003-cm radial intervals and a single acquisition per radius were collected. The maximum scan frequency was used, which produced time intervals of 3 to 4 min between scans. For experiments with U21 alone ranging from 0.04 to 0.85 mg/ml, absorbance wavelengths of 280 nm or 230 nm were used. For multisignal experiments (16), absorbance data were acquired both at 280 nm and 250 nm simultaneously with interference optical detection.
For SE experiments, 130 μl of U21 at concentrations of 4 μM, 1.25 μM, and 0.5 μM, dissolved in PBS, were loaded in double-sector centerpieces, and sedimentation equilibrium was attained sequentially at rotor speeds of 4,500 rpm, 7,500 rpm, and 13,000 rpm at a rotor temperature of 4°C. Absorbance data at 230 nm, 250 nm, and 280 nm were acquired in radial increments of 0.001 cm with 20 replicates. Attainment of equilibrium was tested by comparison of scans taken in 6-h intervals.
The protein partial specific volume of U21 was calculated as a weight average of the 48.9-kDa polypeptide chain, predicted from amino acid composition (17) in SEDFIT (https://sedfitsedphat.nibib.nih.gov/software/default.aspx), and the 10.3-kDa carbohydrate component, as determined by MALDI, using an average partial specific volume of 0.62 ml/g for carbohydrates (18), resulting in a value of 0.712 ml/g. The partial specific volume of HLA-A2 was calculated from amino acid composition as 0.722 ml/g. Buffer density and viscosity were determined in a DMA 5,000 M density meter and an AMVn automated microviscometer (both Anton Paar, Graz, Austria), respectively.
Sedimentation coefficient distribution analysis and multisignal sedimentation velocity analysis.
SV data of U21 samples were modeled with diffusion-deconvoluted sedimentation coefficient distributions [c(s)] (19) in SEDFIT, using algebraic noise decomposition (20), and with the signal-average frictional ratio and the meniscus position refined by nonlinear regression. Accurate scan time intervals were derived from the file timestamps (21), and finite acceleration of the rotor was accounted for in the evaluation of Lamm equation solutions (22, 23). Maximum entropy regularization was applied at a confidence level of P = 0.68. Final root-mean-square deviation (RMSD) typically was ∼0.005 fringes (or optical density [OD]) or better, comparable to the magnitude of noise in data acquisition.
Multisignal analysis of sedimentation velocity (MSSV) was conducted in SEDPHAT (https://sedfitsedphat.nibib.nih.gov/software/default.aspx), globally analyzing interference optical data and absorbance data at both 280 nm and 250 nm. For U21, the molar extinction coefficient at 280 nm calculated by the method of Pace et al. (24) in SEDFIT was used as a fixed reference for the signal, and signal coefficients for interference and 250-nm absorbance detection were calculated by MSSV analysis of U21 alone. For HLA-A2, the refractive index increment was calculated in SED-FIT (25) and used as a fixed reference to calculate extinction coefficients at 250 nm and 280 nm in a global MSSV analysis of HLA-A2 alone. Based on interference and absorbance data at 280 nm, Dnorm for the pair of proteins was calculated to be 0.31 (26).
Sedimentation equilibrium analysis and global sedimentation equilibrium and velocity analysis.
Global multispeed and multiwavelength analysis of SE absorbance data from all cells at suitable wavelengths was carried out in SEDPHAT. Twenty-one absorbance profiles were included in the global analysis, using a discrete species model combined with implicit conservation of mass constraints (27). The bottom position of the solution column was refined in the global analysis, constrained to be the same for data from the same cells. The global analysis of SE and SV experiments was conducted in the same model in SEDPHAT, joining the SE data with a single family of fringe profiles from an SV experiment conducted side by side with a sample from the same protein preparation. In order to exclude contaminating trace species at very low and very high sedimentation coefficients to bias the SV data set, a partial-boundary analysis was used (22) for the SV set in the global analysis, restricting the consideration to a radial range of boundary profiles corresponding to species between 3 S and 11 S. All SE and SV plots were created in GUSSI (http://biophysics.swmed.edu/MBR/software.html), kindly provided by C. Brautigam.
RESULTS
The ever-increasing accumulation of known protein structural data has allowed protein modeling servers to recognize common protein folds using protein threading. Although HHV-7 U21 has no obvious primary amino acid sequence similarity to any other protein other than the U21 open reading frame (ORF) encoded in HHV-6 (HHV-6 U21 and HHV-7 U21 share 30% identity), when we submitted the HHV-6 and HHV-7 U21 sequences to four different threading or structure prediction servers, all servers predicted the existence of a class I MHC-like fold in the full lumenal domain of HHV-6 U21, two of the four servers predicted an MHC-I fold in the full lumenal domain of HHV-7 U21, and all servers predicted class I-like folds when we omitted the putative immunoglobulin domain (α3) from the query sequence of either HHV-6 or HHV-7 U21 (data not shown) (28–32). A hypothetical schematic of a class I MHC molecule and U21, with cartoon reference to their overall domain structure, is illustrated in Fig. 1a.
FIG 1.
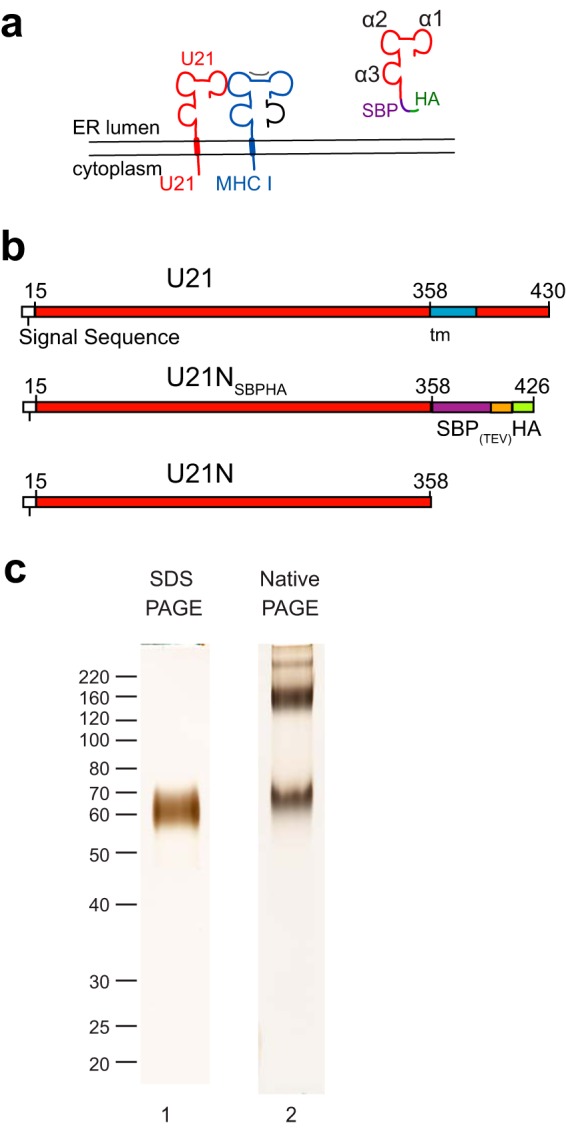
Purified U21 migrates as multiple species in native gels. (a) Topological schematic of the predicted similarity in structure between U21(red) and class I MHC, in which a class I MHC heavy chain (blue) is depicted bound to peptide (gray) and β2-microglobulin (black). Putative α1, α2, and α3 domains are noted in the cartoon depiction of U21NSBPHA. (b) Schematic of U21 constructs used for purification: top, full-length U21, with amino acid numbers, signal sequence (white), and transmembrane domain (blue); middle, soluble U21NSBPHA, with SBP tag (purple), TEV protease cleavage site (orange), and HA tag (green); bottom, U21N (or U21 lumenal domain) alone. (c) Lane 1, purified U21NSBPHA subjected to SDS-polyacrylamide gel electrophoresis and silver stained. Molecular weight size markers are noted. Lane 2, purified U21NSBPHA subjected to native gel electrophoresis and silver stained.
Production and purification of soluble U21.
We have shown previously that U21 binds to and reroutes class I MHC molecules to lysosomes, but neither the requirements for binding of U21 to class I MHC molecules nor the mechanism of U21-mediated lysosomal trafficking are well understood (3, 5). It is unclear, for example, whether the binding of U21 to class I MHC molecules is direct or whether their association might rely upon a third, cellular protein. To better understand the structural requirements for U21 binding to class I MHC molecules, we set out to determine whether U21 and class I MHC molecules could interact in vitro. To purify soluble U21 for in vitro binding experiments, we utilized a streptavidin-binding protein-HA tandem tag (SBPHA), consisting of a 37-amino-acid streptavidin binding protein and a 9-amino-acid HA epitope tag, separated by a tobacco etch virus (TEV) site-specific cleavage site (10), fusing it to the C terminus of the soluble ER-lumenal domain of U21 (see schematics, Fig. 1b). This truncated soluble SBPHA-tagged U21 molecule, U21NSBPHA, retained its functionality when expressed in U373 astrocytoma cells; soluble U21NSBPHA could bind to and reroute class I MHC molecules to lysosomes (5). The U21NSBPHA that did not traffic to lysosomes was secreted, which may suggest the existence of a saturable binding partner for U21; soluble U21 expressed in excess of such a binding partner is secreted.
To produce larger quantities of soluble U21, we utilized lentivirus-mediated gene transfer to stably express soluble U21NSBPHA in 293T cells. Secreted U21NSBPHA molecules could be purified easily and cleanly on streptavidin agarose resin. U21NSBPHA migrated as a single 59-kDa polypeptide in SDS-PAGE, and when silver staining was allowed to develop until long after the purified U21 became apparent, we observed no other copurifying polypeptides, suggesting relative purity of the soluble U21 (Fig. 1c, lane 1). Surprisingly, when subjected to native gel electrophoresis, purified U21NSBPHA migrated as multiple stable nonreducible species, demonstrating the existence of different oligomeric states (Fig. 1c, lane 2).
Native gel analysis of U21 binding to class I MHC molecules.
To assess the ability of purified soluble U21NSBPHA to associate with class I MHC molecules, we mixed U21NSBPHA with purified soluble class I MHC molecules and analyzed the resulting complexes on a native gel. We tested binding of U21 to class I MHC molecules (human leukocyte antigen allele A*02:01 [HLA-A*02:01]) refolded from bacterial inclusion bodies (Fig. 2a, lane 2) and to a human cell-produced soluble HLA-B*07:02 molecule, which possesses a single N-linked glycan that exhibits heterogeneous sialylation (Fig. 2a, lane 5). In the native gel shift assay, we mixed purified U21NSBPHA (3 μM) with purified HLA-A*02:01 (11 μM) or purified HLA-B*07:02 (3.5 μM, with 2 μM U21) and examined whether the addition of a molar excess of HLA-A or -B resulted in an electrophoretic mobility shift of the purified U21NSBPHA. Interestingly, only the slower-migrating oligomers of U21NSBPHA shifted upward, demonstrating that, within the sensitivity of this assay, only the larger oligomeric U21 species can associate with class I MHC molecules (Fig. 2a, lanes 3 and 6). These data suggest that oligomeric U21NSBPHA associates with class I MHC molecules in vitro and that the N-linked glycans on class I MHC molecules do not play a critical role in their association. We also note that higher-order U21 complexes also shift with the addition of class I MHC molecules (Fig. 2a, lanes 3 and 6).
FIG 2.
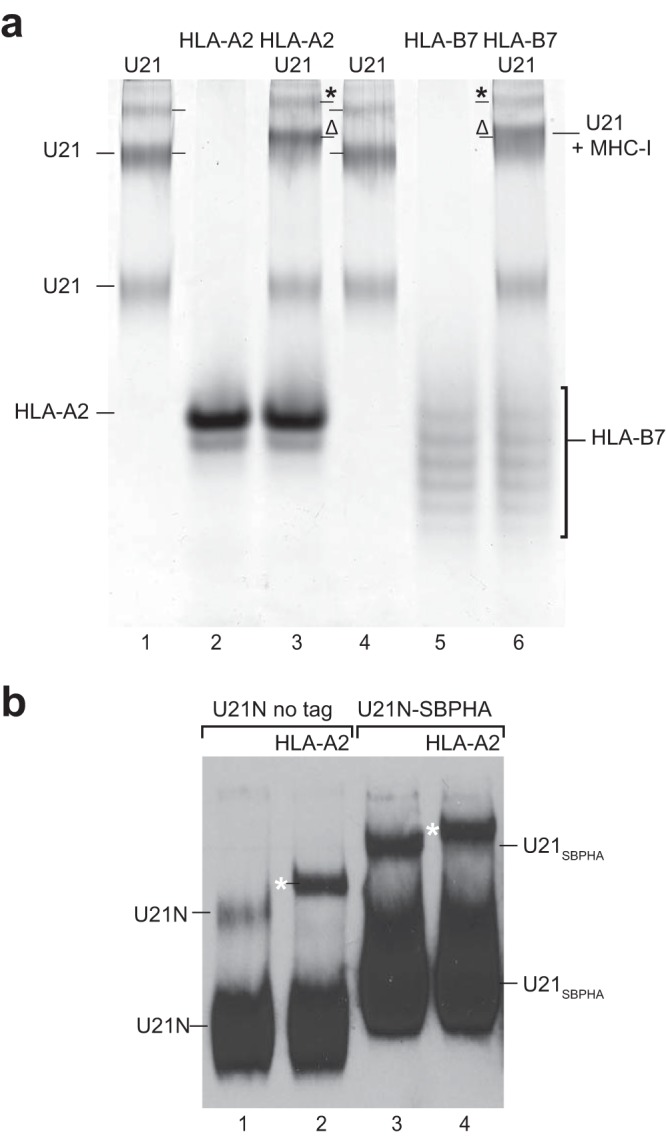
The slower of the U21 bands associates with purified HLA-A and -B molecules. (a) Purified U21 (lanes 1 and 4), HLA-A2 (lane 2), HLA-B7 (lane 5), or a mixture of these molecules, as noted (lanes 3 and 6), were separated on native gels and silver stained. U21 and HLA molecules are noted. Shifted U21 and class I MHC molecules are denoted by the Δ symbols, and higher-order-shifted U21 and HLA molecules are denoted with asterisks. (b) Equal volumes of serum-free 293T culture medium containing secreted U21NSBPHA or untagged U21N were incubated alone (lanes 1 and 3) or with purified HLA-A2 (lanes 2 and 4) for 30 min at 4°C, subjected to native-PAGE, and immunoblotted with anti-U21N. Migration positions of U21N and U21NSBPHA are noted. The migration positions of shifted U21N and U21NSBPHA in the presence of HLA-A2 are denoted with white asterisks.
To dispel concerns that the 80-amino-acid SBPHA tag might contribute to anomalous oligomerization of our purified soluble U21NSBPHA molecules, we generated 293T cells secreting soluble nontagged U21 molecules (U21N; see schematic, Fig. 1a). Although the lack of an epitope tag precluded the possibility of affinity purification, we could track the nontagged U21N molecule in immunoblots following native gel electrophoresis, using a polyclonal antibody directed against the N terminus of U21 (Fig. 2b, lanes 1 and 2). To confirm that nontagged U21N oligomerized similarly to U21NSBPHA, we examined the migration of nontagged and SBPHA-tagged U21 on the same native gel. Both U21NSBPHA and U21N migrated similarly (Fig. 2b, lanes 1 and 3). To assess whether the nontagged U21 behaved similarly in its association with class I MHC molecules, we added purified HLA-A2 molecules to the U21N or U21NSBPHA-containing 293T culture supernatant and subjected the mixture to native gel electrophoresis. Both nontagged U21N and U21NSBPHA shifted in the presence of HLA-A2, suggesting that the SBPHA tag neither induces artificial U21 oligomerization nor interferes with binding to class I MHC molecules (Fig. 2b, lanes 2 and 4, white asterisks).
Analysis of the oligomeric state of U21 by SV and SE analytical ultracentrifugation.
We next employed sedimentation velocity (SV) and sedimentation equilibrium (SE) analytical ultracentrifugation (AUC) to determine the oligomeric state of U21. MALDI analysis showed the monomer molecular mass of U21NSBPHA to be 59.3 ± 0.3 kDa (data not shown), indicating a substantial degree of glycosylation, since the molecular mass of the nonglycosylated protein is predicted to be 48.7 kDa (33). To establish the oligomeric state of purified U21NSBPHA in solution, we carried out side-by-side SE and SV experiments at a range of loading concentrations, from 0.04 to 0.85 mg/ml. In the SV experiment, two separate sedimentation boundaries could be clearly discerned (Fig. 3a), corresponding to two major peaks in the sedimentation coefficient distribution [c(s)] at s20,w values of 5.67 S and 8.42 S (Fig. 3b). We also observed trace populations of 11-S material (Fig. 3b). The invariance of the peak positions over the 20-fold concentration range indicated that these were not reaction boundaries but instead species independently sedimenting and not interconverting on the time scale of the SV experiment (Fig. 3b).
FIG 3.

Sedimentation velocity analysis of U21NSBPHA. (a) Representative radial absorbance profiles at 280 nm as a function of time, from an experiment at a loading concentration of 0.26 mg/ml U21NSBPHA in 12-mm path length centerpieces recorded at 280 nm (symbols, with times indicated by the color temperature); best-fit profiles from the c(s) distribution (solid lines). For clarity, only every 3rd data point of every 3rd scan is shown. Residuals of the fit, with an RMSD of 0.0051 OD, are shown in the lower panel. (b) c(s) distribution from the model shown in panel a (purple). Also shown are equivalent distributions obtained at a 2-fold-higher concentration in a 3-mm path length centerpiece recorded at 280 nm (blue) and a 10-fold-lower concentration in a 12-mm centerpiece recorded at 230 nm (green).
The molecular mass values implied by the average boundary spread in the sedimentation coefficient distribution c(s) are 114 kDa and 204 kDa, which, within the typical error of the c(s)/c(M) method, demonstrate that the two peaks represent dimers and tetramers of U21. Given the observed s values, the dimer and tetramer would have frictional coefficients of 1.65 and 1.76, respectively. These hydrated frictional ratios are high but common for heavily glycosylated proteins such as U21NSBPHA (34). The contribution of the high degree of glycosylation to the hydrodynamic friction values prevented sensible interpretation of the frictional ratios in terms of hydrodynamically equivalent smooth ellipsoids.
Interestingly, the dimer and tetramer remained similar in abundance over the entire concentration range studied (with weighted average sedimentation coefficient varying by less than 1.3%). This would in principle be consistent with the dimer and tetramer not being in a reversible equilibrium and instead the result of persistent structural or chemical differences, even though both the dimer and tetramer are functional in binding HLA-A2 (see below). Alternatively, the hydrodynamic separation of species at virtually concentration-independent sedimentation coefficients and populations would be consistent with species that are in a very slow equilibrium on the time scale of days (15). Assuming the latter, the approximately equal abundance of dimers and tetramers in samples equilibrating for several days at low micromolar concentrations would suggest in a back-of-the-envelope estimate an equilibrium dissociation constant (KD) for the dimer-tetramer association in the low micromolar range.
As an independent confirmation of the oligomeric states, we conducted SE experiments at multiple loading concentrations, sequentially attaining equilibrium at three rotor speeds, leading to characteristic low-speed and meniscus depletion profiles (Fig. 4). The best global two-species fit was obtained with species of 2.1- and 4.3-fold the monomer mass, leading to a relative reduced global χ2 of 0.996. While a model accounting for two species corresponding exactly to the dimer and tetramer led to a slightly higher χ2 of 1.057, the inclusion of hexamer (or, similarly, octamers) in the model led to a significantly lower global χ2 of 0.725, by far the best fit (Fig. 4). Constraining the analysis to a dimer/tetramer model in chemical equilibrium resulted in a global χ2 of 2.231. When hexameric species were allowed for, the resulting global χ2 was 1.694. These calculations suggest that the dimer and tetramer have not come to chemical equilibrium at 4°C on the time scale of several days in SE. With regard to the higher-order species larger than the tetramer, from the analytical ultracentrifugation analysis it is unclear whether these represent functional higher-order states in slow exchange that are detectable only at the higher concentrations in the SE gradient and invisible at the loading concentration accessible in SV or if they represent small aggregates of degradation products that form during the extended time of SE experiments. With this model, an excellent global fit of both SE and SV data was achieved, the latter constrained in a partial boundary model to radial ranges corresponding to 3 S to 11 S (data not shown), which effectively excluded hexamers from consideration in SV. Together, the SV and SE experiments clearly show that the predominant species of U21NSBPHA in solution are dimers and tetramers in very slow exchange.
FIG 4.

Sedimentation equilibrium analysis of U21NSBPHA. Representative data from a global analysis of 0.9 μM, 2.2 μM, and 7.1 μM U21NSBPHA in sedimentation equilibrium sequentially at rotor speeds of 4,500 rpm, 7,500 rpm, and 13,000 rpm, scanned at wavelengths of 230 nm, 250 nm, and 280 nm. Shown are absorbance data at 250 nm at a loading concentration of 7.1 μM at the three rotor speeds (symbols), best-fit distributions (solid lines) with a model of dimer, tetramer, and hexamer at total equivalent loading concentrations of 43%, 37%, 20%, respectively. Residuals, amounting to an RMSD of 0.0039 OD, are shown in the lower panel.
Analysis of the binding stoichiometry of U21 and HLA-A2 by MSSV.
To determine the binding stoichiometry of U21 and HLA-A2, we performed multisignal sedimentation velocity (MSSV) experiments to characterize the mode of association between U21NSBPHA and class I MHC molecules. Potentially, both the U21NSBPHA dimer and tetramer could be functional units for class I MHC binding, at unknown stoichiometry. The method of choice for unraveling potentially multiple coexisting mixed complexes in multicomponent mixtures in solution is MSSV (16, 26, 35), which exploits spectral differences in proteins to extend diffusion-deconvoluted sedimentation coefficient distributions [c(s)] into molar multicomponent sedimentation coefficient distributions [ck(s)] by direct global modeling of SV data acquired at different signals.
In preliminary MSSV experiments with both U21NSBPHA and HLA-A2 molecules separately (data not shown), molar signal coefficients were determined for refractive index-based Rayleigh interference detection and UV absorbance at 280 nm and 250 nm. Due to the differences in aromatic amino acids, and exacerbated by the glycosylation of U21NSBPHA contributing to molecular refractive index increment but not UV absorbance, U21NSBPHA and HLA-A2 were spectrally very well distinguishable with a Dnorm value (26) of 0.31, which permitted their straightforward discrimination in MSSV without attachment of any extrinsic chromophoric labels. SV experiments on HLA-A2 alone were carried out at loading concentrations of 39 μM and 0.39 μM, both resulting in single peaks in the sedimentation coefficient distribution at virtually identical s values (data not shown), implying a monomeric molecule with a best-fit molecular mass of ∼42 kDa, consistent with expectations for a class I MHC molecule.
A first mixture experiment was conducted at a high concentration, with 39.4 μM HLA-A2 and 11.8 μM U21NSBPHA, in order to determine the maximal stoichiometry of the U21NSBPHA/HLA-A2 complex in large molar excess of HLA-A2 (data not shown). As shown in Fig. 5a, little free dimer of U21NSBPHA is present (5.9 S), and no free tetramer (9 S) can be discerned. Instead, a new species at ∼10.75 S emerged, with a best-fit molar ratio of 2.33 U21NSBPHA/HLA-A2, statistically equivalent to a 2:1 molar ratio of U21NSBPHA/HLA-A2. Similar results with a best-fit molar ratio of 1.87:1 were obtained at half the concentrations of both components (data not shown). In a segmented c(s) analysis, the best-fit frictional ratio associated solely with the 10.75-S peak implies a buoyant molecular mass of 96.7 kDa, which is within typical experimental error consistent with the buoyant molecular mass of 89.4 kDa of a 4:2 complex. Using the compositional molecular weight of a 4:2 complex and the measured s20,w value, this implies a complex with a frictional ratio of 1.60, suggesting an extended hydrodynamic shape similar to that of U21NSBPHA. At the ∼10-fold-lower concentrations of 4.9 μM HLA-A2 and 3.3 μM U21NSBPHA, a significant population of free U21NSBPHA dimer (6 S) was found to coexist with the same ∼4:2 complex of U21NSBPHA/HLA-A2 (Fig. 5B). At these low concentrations, the absence of U21 tetramers and the coexistence of U21NSBPHA dimers with HLA-A2 molecules indicates that U21 dimers possess lower affinity for HLA-A2 molecules than do the tetramers and confirms a thermodynamic barrier in the formation of U21 tetramers from U21 dimers (consistent with the slow dissociation of tetramers observed by SV, described above).
FIG 5.

Multisignal sedimentation velocity (MSSV) analysis based on a global fit of data recorded by Rayleigh interference, absorbance at 280 nm and 250 nm, and analyzed in terms of a spectrally and diffusionally deconvoluted component sedimentation coefficient distribution [ck(s)] of HLA-A2 (blue) and U21NSBPHA (red). For ease of comparison, vertical bars denote the sedimentation coefficients of U21 dimers (gray) and tetramers (green), as shown in Fig. 3. (a) Multisignal sedimentation velocity ck(s) analysis of a loading mixture of 39.4 μM HLA-A2 and of 11.8 μM U21NSBPHA. Raw boundary data and global fits are available from the author. (b) ck(s) analysis of a loading mixture of 4.9 μM HLA-A2 and 3.3 μM U21NSBPHA.
While the apparent kinetically limited equilibration of species does not allow us to estimate equilibrium binding constants, in experiments performed using a molar excess of U21NSBPHA over HLA-A2, we could discern no free HLA-A2; thus, all of the HLA-A2 molecules appeared to be absorbed in complexes with U21NSBPHA, suggesting KD values of tetramer/HLA-A2 in the nanomolar range or lower (data not shown). This is consistent with our previous observation that the two proteins coimmunoprecipitate under fairly stringent conditions (3).
In vivo analysis of U21 oligomers.
Since our AUC experiments and native gel analyses were performed in solution with purified soluble U21, we next sought to determine whether U21 oligomers could be the operative binding unit in vivo as well. We have often observed a higher ratio of U21 to class I MHC molecules in immunoprecipitations of class I MHC molecules, regardless of whether immunoprecipitations were performed from metabolically labeled lysates of HHV-7-infected cells or U21-expressing cells (3, 5). Until now, however, we were hesitant to interpret these data as a reflection of the true in vivo ratio of U21: class I MHC molecules in a complex, since the efficiency of metabolic labeling could have differed between the two proteins, and since the immunoprecipitations contained a mixture of radiolabeled and nonradiolabeled class I molecules. To circumvent these limitations of metabolic labeling, we performed similar immunoprecipitations of class I molecules after covalently linking the W6/32 antibody to agarose beads. Strikingly, just as for metabolically labeled immunoprecipitations with W6/32, silver-stained SDS-PAGE of these immunoprecipitates show a similarly high ratio of U21/class I MHC molecules (Fig. 6, lane 3). Quantitation of the U21/class I heavy chain bands using ImageJ revealed that the W6/32 antibody recovered 2.05-fold more U21 than class I MHC, consistent with an in vivo association ratio of 2:1 or 4:2 (36). We note that the amount of class I heavy chain molecules recovered at steady state in this immunoprecipitation is 5.6-fold reduced in U21-expressing cells, reflecting the degradation of class I molecules that occurs in lysosomes (6).
FIG 6.
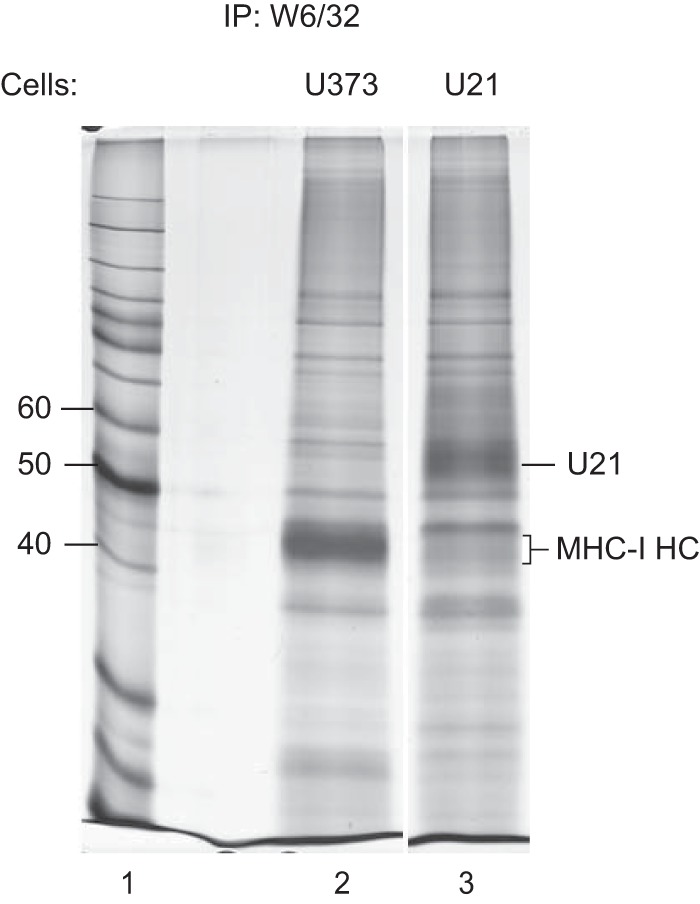
In vivo stoichiometry of U21 and class I MHC molecules. Lysates from U373 or U21-expressing U373 cells were immunoprecipitated using W6/32 covalently linked to protein A agarose. The migration positions of U21 and class I MHC heavy chain molecules are indicated. Molecular weight standards are indicated to the left (lane 1). Quantitation of the U21 and class I heavy chain bands was performed using NIH ImageJ (36).
To more specifically assess whether U21 forms oligomers in vivo, we performed coimmunoprecipitation experiments from U373 cells coexpressing both U21 and an HA-tagged U21 possessing a TEV protease cleavage site followed by tandem HA tags engineered into the junction between the transmembrane domain and its cytoplasmic tail (see schematic, Fig. 7a). The TEVHAHA-tagged U21 migrated more slowly in an SDS-polyacrylamide gel and thus allowed us to distinguish between the nontagged and TEVHAHA-tagged molecules. We metabolically labeled cells expressing U21 or both U21 and HA-tagged U21 and immunoprecipitated U21 with antibodies directed against either U21 or HA. When we recovered U21 from U21-expressing cells, we observed U21 and coprecipitating class I MHC molecules (Fig. 7b, lane 1). From cells expressing both U21 and U21TEVHAHA-tail, with the anti-U21 antibody, we recovered both U21 molecules and class I MHC molecules (Fig. 7b, lane 2). When we immunoprecipitated the TEVHAHA-tagged U21 with an anti-HA antibody, we recovered nothing from cells expressing U21, and we recovered both tagged and nontagged U21 molecules, as well as class I MHC molecules, from cells expressing the TEVHAHA-tagged U21 (Fig. 7b, lanes 3 and 4). The only way for nontagged U21 to have coimmunoprecipitated with the anti-HA antibody is if U21 associated in vivo with the HA-tagged U21; thus, these results suggest that U21 forms oligomers within the cell as well as in vitro. Of note, in these experiments, we recovered more TEVHAHA-tagged U21 than nontagged U21 in U21 immunoprecipitations. We found similar ratios of tagged/nontagged U21 also reflected in the relative expression levels of these two constructs in this cell line (Fig. 7c, lanes 2 and 3).
FIG 7.
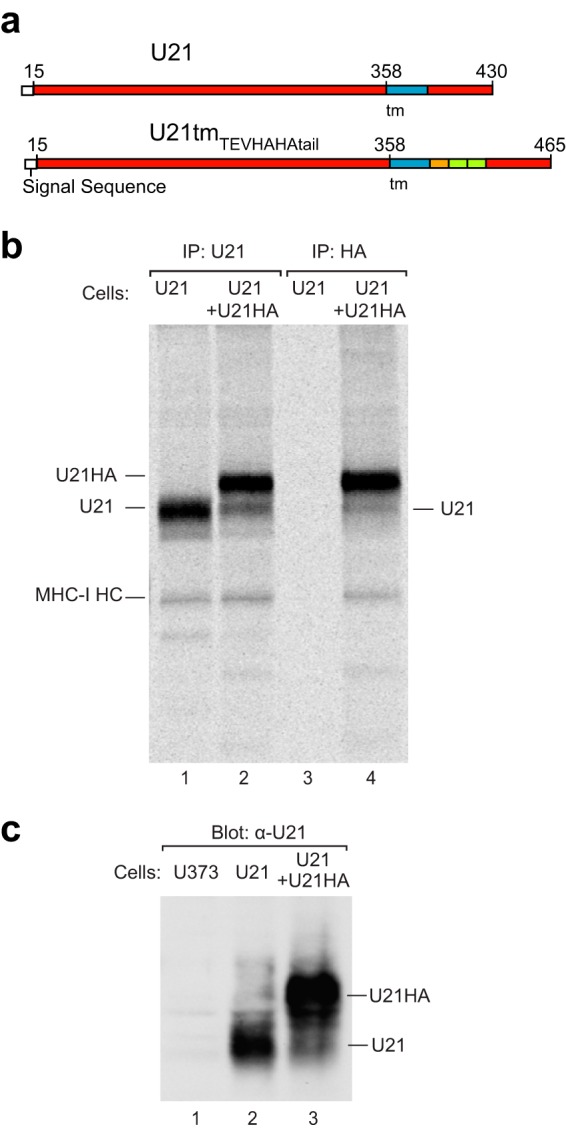
U21 multimerizes in vivo. (a) Schematic of the U21tmTEVHAHA-tail construct, with amino acid numbers, signal sequence (white), transmembrane domain (blue), TEV cleavage site (orange), and the tandem HA tags (green). (b) Immunoprecipitations of metabolically labeled U21- or U21tmTEVHAHA-tail-expressing U373 cells with anti-U21C or anti-HA, as noted. Positions of U21, class I MHC heavy chains (MHCI HC), and U21tmTEVHAHA-tail (U21HA) are noted. (c) Immunoblot of U373 or U21- or U21tmTEVHAHA-tail-expressing U373 lysates with an anti-U21 C-terminal antibody. Migration positions of U21tmTEVHAHA-tail and U21 are noted.
DISCUSSION
HHV-7 U21 is a type I integral membrane protein that binds to class I MHC molecules and reroutes them to the lysosomal compartment. Here, we provide in vitro evidence for a direct and high-affinity interaction between stable homotetramers of U21 with two class I MHC molecules, and we demonstrate that U21 oligomers may also be the functional unit of U21 in vivo.
Threading servers predict that U21 possesses an MHC-I-like protein fold. The existence of a viral protein with a class I-like protein fold is not unusual among betaherpesviruses; human cytomegalovirus (HCMV) encodes one such protein, UL18, and murine cytomegalovirus (MCMV) encodes 10 gene products with predicted class I folds (for a review, see reference 37). HCMV UL18 and MCMV m144 are class I homologs that associate with β2m and act as inhibitory NK ligands (38–41). MCMV m157 binds to inhibitory and activating NK receptors Ly49H and Ly49I (42, 43), MCMV m152 is a class I homolog that reroutes class I molecules to lysosomes and also downregulates NKG2D ligand Rae-1 family members (44), MCMV m138, m145, and m155 interfere with NK ligands or costimulatory molecules (45–48), and the functions of m17, m150, m151, m153, and m158 are not yet known (49).
Homooligomerization of class I MHC or class I-like molecules is unusual. Noncovalent homooligomerization of class I MHC molecules has, to our knowledge, not been described. Recently, however, noncovalent homodimerization of another viral MHC-I fold-containing protein, MCMV m153, was shown to occur both in vitro and in vivo (49).
In contrast, association of class I-like molecules with other MHC-I fold-containing molecules is quite common; several of the cytomegalovirus-encoded MHC-I fold-containing proteins have been shown to affect the cell surface expression of class I MHC molecules or of the class I-like NK-activating ligands. Direct binding has been demonstrated between m152, an MCMV-encoded MHC-I fold-containing protein, and Rae-1, a murine NK-activating ligand (50). Interestingly, like U21, m152 is the only other immunoevasin that has been shown to serve a dual function in downregulating both class I MHC molecules and NK-activating ligands (3, 4, 44, 50, 51). Unlike U21, however, m152 does not oligomerize, suggesting that the two virally encoded MHC fold-containing proteins utilize dissimilar mechanisms (49).
We demonstrate here that U21 is secreted as homodimers and homotetramers. We have seen no evidence for the existence of monomeric U21, even at low concentrations, suggesting that U21 possesses a high propensity for dimer formation and/or very slow dimer dissociation. Because U21 dimers and U21 tetramers remain in similar abundance over a broad concentration range, and because both oligomers are functional in binding class I molecules, we believe the tetramers may be in a very slow equilibrium with the U21 dimers, in which case an equilibrium dissociation constant for U21 tetramers would be in the low micromolar range. This indicates a very small on-rate constant and suggests the possibility of structural rearrangements accompanying tetramer formation. However, other factors leading to the stabilization of dimer or tetramer configuration, such as additional small ligands or small chemical differences, cannot be excluded at present and may become clear in future structural work. Interestingly, it is the U21 tetramers that best associate with class I molecules; while we did not directly detect a stable complex formed by U21 dimers bound to class I MHC molecules, it is possible that U21 dimers transiently form 2:1 complexes with class I molecules and that these 2:1 complexes quickly dimerize to form 4:2 complexes. In experiments performed using a molar excess of U21 over HLA-A2, both in the low micromolar concentration range, all of the HLA-A2 was found in association with U21, suggesting that the dissociation constant for U21 and HLA-A2 molecules is in the mid- to low nanomolar range.
We were initially concerned that the soluble secreted oligomers of U21 may have been an artifact of engineering of the soluble SBPHA-tagged U21 molecule. We note, however, that soluble secreted U21NSBPHA, when expressed in human cells, functions to reroute class I MHC molecules to lysosomes (5), suggesting that the soluble U21 molecules retain their function.
Our finding that U21 binds to class I (and presumably NKG2D ligands, as well) in a 4:2 ratio suggests that in a virus-infected cell, U21 molecules may need to outnumber all HLA alleles and NKG2D ligands to effect a significant change upon surface expression of these molecules. In the context of HHV-7 infection of T cells, we estimate that U21 is indeed expressed at levels that affect class I MHC trafficking (3). A 4:2 ratio of U21 to class I MHC molecules is also consistent with our previously documented correlation between increasing U21 expression levels, relocalization of NKG2D ligands to lysosomes, and reduced steady-state expression of NKG2D ligands and class I MHC molecules (4).
U21 binds to class I MHC molecules in the ER and escorts them through the Golgi en route to the lysosomal compartment. The process of trafficking of integral membrane proteins to the lysosomal compartment is generally mediated by proteins that recognize various cytoplasmic sorting signals. The lamp-1 and lamp-2 proteins, for example, which comprise perhaps as much as 70% of the total lysosomal membrane protein content, contain a tyrosine-based sorting signal in their cytoplasmic tails that is recognized by the clathrin adaptor proteins AP-1 and AP-3 (52–54). These adaptor protein complexes mediate sorting to the endolysosomal compartment.
In constructing a model for U21-mediated trafficking of class I MHC molecules to lysosomes, the simplest is one exemplified by the MCMV m06 immunoevasin (55). MCMV m06 contains a di-leucine sorting signal in its cytoplasmic tail that requires functional AP-1 and AP-3 clathrin adaptor complexes. These adaptor complexes mediate sorting of the m06-class I MHC complex to the lysosomal compartment (56). We initially postulated a similar mechanism for U21, but instead we found that U21 can affect the trafficking of class I molecules even when its cytoplasmic tail is deleted (3) and even when the U21 molecule is in a soluble secreted form (5). Thus, if the lysosomal sorting signal utilized by U21 is cytoplasmic, we can envision two possible models: either a third, cellular protein (or more than one protein) associates with U21 and class I molecules that contain the information necessary to reroute the class I MHC-U21 complexes (Fig. 8) or a high local concentration of MHC-I fold-containing proteins (octamers?) somehow induces the secretory apparatus to segregate and reroute this aggregate of integral membrane proteins to the lysosomal compartment. This aggregation model was suggested in 1997 by Wolins et al. to explain the trafficking of furin to the lysosomal compartment (57). Still, it would seem that this model must also involve a cellular protein to detect the presence of the aggregate and initiate the lysosomal trafficking process.
FIG 8.
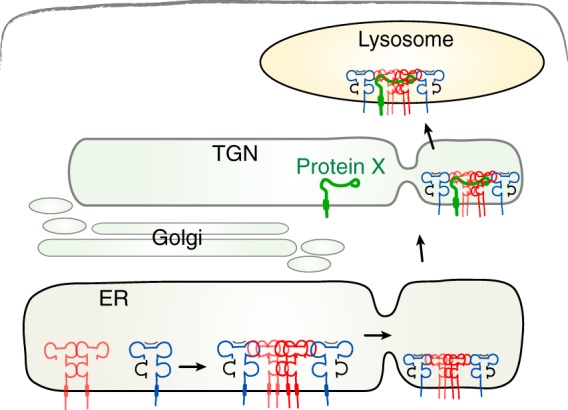
Model depicting U21-mediated rerouting of class I MHC molecules to the lysosomal compartment. U21 (red) associates with and reroutes class I MHC molecules (blue) to a lysosomal compartment through association with a cellular protein, protein X (green), which contains lysosomal targeting information in its cytoplasmic tail. The association of protein X with the 4:2 U21-MHC I complex could occur in the ER (not depicted) or later in the secretory pathway (here shown in the TGN).
Our finding that U21 interacts with class I MHC molecules in a 4:2 ratio contributes to our understanding of these models. Based on our proposed model, interruption of U21 oligomerization should reduce the efficiency of U21 molecules to reroute class I MHC molecules to the lysosomal compartment. Since U21 can traffic to lysosomes, even in cells depleted of class I MHC molecules (5), an as-yet-unknown cellular trafficking protein must be able to influence U21 without the requirement of associated class I MHC molecules. It will be informative to determine whether oligomerization of U21 is necessary for lysosomal trafficking of U21 molecules, whether the as-yet-unknown cellular protein requires tetrameric U21 for recognition, and whether a mutant of U21 can be obtained that maintains the ability to associate with class I MHC molecules but can no longer reroute them to lysosomes. More detailed structural analysis, as might be accomplished by crystallography, may reveal oligomerization sites and specify the residues that participate in oligomerization.
ACKNOWLEDGMENTS
We thank Matthew Champion (University of Notre Dame Mass Spectrometry and Proteomics Facility) for MALDI analysis and David Margulies, Ben Gewurz, Rachelle Gaudet, and Jung-Ja Kim for valuable discussions.
This work was supported by Public Health Service Grant R01-AI069099 from National Institute for Allergy and Infectious Disease (A.W.H.) and the Intramural Research Program of the National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health.
Footnotes
Published ahead of print 3 January 2014
REFERENCES
- 1.Glosson NL, Hudson AW. 2007. Human herpesvirus-6A and -6B encode viral immunoevasins that downregulate class I MHC molecules. Virology 365:125–135. 10.1016/j.virol.2007.03.048 [DOI] [PubMed] [Google Scholar]
- 2.Hudson AW, Blom D, Howley PM, Ploegh HL. 2003. The ER-lumenal domain of the HHV-7 immunoevasin U21 directs class I MHC molecules to lysosomes. Traffic 4:824–837. 10.1046/j.1398-9219.2003.0137.x [DOI] [PubMed] [Google Scholar]
- 3.Hudson AW, Howley PM, Ploegh HL. 2001. A human herpesvirus 7 glycoprotein, U21, diverts major histocompatibility complex class I molecules to lysosomes. J. Virol. 75:12347–12358. 10.1128/JVI.75.24.12347-12358.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schneider CL, Hudson A. 2011. The human herpesvirus-7 (HHV-7) U21 immunoevasin subverts NK-mediated cytoxicity through modulation of MICA and MICB. PLoS Pathog. 7:e1002362. 10.1371/journal.ppat.1002362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glosson NL, Gonyo P, May NA, Schneider CL, Ristow LC, Wang Q, Hudson AW. 2010. Insight into the mechanism of human herpesvirus 7 U21-mediated diversion of class I MHC molecules to lysosomes. J. Biol. Chem. 285:37016–37029. 10.1074/jbc.M110.125849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.May NA, Glosson NL, Hudson AW. 2010. HHV-7 U21 downregulates classical and nonclassical class I MHC molecules from the cell surface. J. Virol. 84:3738–3751. 10.1128/JVI.01782-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mostoslavsky G, Fabian AJ, Rooney S, Alt FW, Mulligan RC. 2006. Complete correction of murine Artemis immunodeficiency by lentiviral vector-mediated gene transfer. Proc. Natl. Acad. Sci. U. S. A. 103:16406–16411. 10.1073/pnas.0608130103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barnstable CJ, Bodmer WF, Brown G, Galfre G, Milstein C, Williams AF, Ziegler A. 1978. Production of monoclonal antibodies to group A erythrocytes, HLA and other human cell surface antigens-new tools for genetic analysis. Cell 14:9–20. 10.1016/0092-8674(78)90296-9 [DOI] [PubMed] [Google Scholar]
- 9.Stam NJ, Spits H, Ploegh HL. 1986. Monoclonal antibodies raised against denatured HLA-B locus heavy chains permit biochemical characterization of certain HLA-C locus products. J. Immunol. 137:2299–2306 [PubMed] [Google Scholar]
- 10.Call ME, Pyrdol J, Wiedmann M, Wucherpfennig KW. 2002. The organizing principle in the formation of the T cell receptor-CD3 complex. Cell 111:967–979. 10.1016/S0092-8674(02)01194-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buchli R, VanGundy RS, Hickman-Miller HD, Giberson CF, Bardet W, Hildebrand WH. 2004. Real-time measurement of in vitro peptide binding to soluble HLA-A*0201 by fluorescence polarization. Biochemistry 43:14852–14863. 10.1021/bi048580q [DOI] [PubMed] [Google Scholar]
- 12.Prilliman K, Lindsey M, Zuo Y, Jackson KW, Zhang Y, Hildebrand W. 1997. Large-scale production of class I bound peptides: assigning a signature to HLA-B*1501. Immunogenetics 45:379–385. 10.1007/s002510050219 [DOI] [PubMed] [Google Scholar]
- 13.Renart J, Reiser J, Stark GR. 1979. Transfer of proteins from gels to diazobenzyloxymethyl-paper and detection with antisera: a method for studying antibody specificity and antigen structure. Proc. Natl. Acad. Sci. U. S. A. 76:3116–3120. 10.1073/pnas.76.7.3116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Y, Wojcik R, Dovichi NJ, Champion MM. 2012. Quantitative multiple reaction monitoring of peptide abundance introduced via a capillary zone electrophoresis-electrospray interface. Anal. Chem. 84:6116–6121. 10.1021/ac300926h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao H, Brautigam CA, Ghirlando R, Schuck P. 2013. Overview of current methods in sedimentation velocity and sedimentation equilibrium analytical ultracentrifugation. Curr. Protoc. Protein Sci. Chapter 20:Unit20.12. 10.1002/0471140864.ps2012s71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Balbo A, Minor KH, Velikovsky CA, Mariuzza RA, Peterson CB, Schuck P. 2005. Studying multiprotein complexes by multisignal sedimentation velocity analytical ultracentrifugation. Proc. Natl. Acad. Sci. U. S. A. 102:81–86. 10.1073/pnas.0408399102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cohn E, Edsall J. 1943. Density and apparent specific volume of proteins, p 370–381 In Cohn EJ, Edsall JT. (ed), Proteins, amino acids, and peptides. Van Nostrand-Reinhold, Princeton, NJ [Google Scholar]
- 18.Lewis MS, Junghans RP. 2000. Ultracentrifugal analysis of molecular mass of glycoproteins of unknown or ill-defined carbohydrate composition. Methods Enzymol. 321:136–149 [DOI] [PubMed] [Google Scholar]
- 19.Schuck P. 2000. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 78:1606–1619. 10.1016/S0006-3495(00)76713-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schuck P, Demeler B. 1999. Direct sedimentation analysis of interference optical data in analytical ultracentrifugation. Biophys. J. 76:2288–2296. 10.1016/S0006-3495(99)77384-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao H, Ghirlando R, Piszczek G, Curth U, Brautigam CA, Schuck P. 2013. Recorded scan times can limit the accuracy of sedimentation coefficients in analytical ultracentrifugation. Anal. Biochem. 437:104–108. 10.1016/j.ab.2013.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brown PH, Balbo A, Schuck P. 2009. On the analysis of sedimentation velocity in the study of protein complexes. Eur. Biophys. J. 38:1079–1099. 10.1007/s00249-009-0514-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schuck P, Taraporewala Z, McPhie P, Patton JT. 2001. Rotavirus nonstructural protein NSP2 self-assembles into octamers that undergo ligand-induced conformational changes. J. Biol. Chem. 276:9679–9687. 10.1074/jbc.M009398200 [DOI] [PubMed] [Google Scholar]
- 24.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. 1995. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 4:2411–2423. 10.1002/pro.5560041120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao H, Brown PH, Schuck P. 2011. On the distribution of protein refractive index increments. Biophys. J. 100:2309–2317. 10.1016/j.bpj.2011.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Padrick SB, Brautigam CA. 2011. Evaluating the stoichiometry of macromolecular complexes using multisignal sedimentation velocity. Methods 54:39–55. 10.1016/j.ymeth.2011.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vistica J, Dam J, Balbo A, Yikilmaz E, Mariuzza RA, Rouault TA, Schuck P. 2004. Sedimentation equilibrium analysis of protein interactions with global implicit mass conservation constraints and systematic noise decomposition. Anal. Biochem. 326:234–256. 10.1016/j.ab.2003.12.014 [DOI] [PubMed] [Google Scholar]
- 28.Kelley LA, Sternberg MJE. 2009. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4:363–371. 10.1038/nprot.2009.2 [DOI] [PubMed] [Google Scholar]
- 29.Roy A, Kucukural A, Zhang Y. 2010. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5:725–738. 10.1038/nprot.2010.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi J, Blundell TL, Mizuguchi K. 2001. FUGUE: sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J. Mol. Biol. 310:243–257. 10.1006/jmbi.2001.4762 [DOI] [PubMed] [Google Scholar]
- 31.Källberg M, Wang H, Wang S, Peng J, Wang Z, Lu H, Xu J. 2012. Template-based protein structure modeling using the RaptorX Web server. Nat. Protoc. 7:1511–1522. 10.1038/nprot.2012.085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu S, Zhang Y. 2007. LOMETS: a local meta-threading-server for protein structure prediction. Nucleic Acids Res. 35:3375–3382. 10.1093/nar/gkm251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. 2005. Protein identification and analysis tools on the ExPASy server, p 571–607 In Walker JM. (ed), The proteomics protocols handbook. Humana Press, New York, NY [Google Scholar]
- 34.Dam J, Schuck P. 2004. Calculating sedimentation coefficient distributions by direct modeling of sedimentation velocity concentration profiles, vol 384, p 185–212 Elsevier, Amsterdam, Netherlands: [DOI] [PubMed] [Google Scholar]
- 35.Padrick SB, Deka RK, Chuang JL, Wynn RM, Chuang DT, Norgard MV, Rosen MK, Brautigam CA. 2010. Determination of protein complex stoichiometry through multisignal sedimentation velocity experiments. Anal. Biochem. 407:89–103. 10.1016/j.ab.2010.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rasband WS. 1997–2012. ImageJ. National Institutes of Health, Bethesda, MD: http://imagej.nih.gov/ij/ [Google Scholar]
- 37.Revilleza MJ, Wang R, Mans J, Hong M, Natarajan K, Margulies DH. 2011. How the virus outsmarts the host: function and structure of cytomegalovirus MHC-I-like molecules in the evasion of natural killer cell surveillance. J. Biomed. Biotechnol. 2011:724607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reyburn HT, Mandelboim O, Vales-Gomez M, Davis DM, Pazmany L, Strominger JL. 1997. The class I MHC homologue of human cytomegalovirus inhibits attack by natural killer cells. Nature 386:514–517. 10.1038/386514a0 [DOI] [PubMed] [Google Scholar]
- 39.Farrell HE, Vally H, Lynch DM, Fleming P, Shellam GR, Scalzo AA, Davis-Poynter NJ. 1997. Inhibition of natural killer cells by a cytomegalovirus MHC class I homologue in vivo. Nature 386:510–514. 10.1038/386510a0 [DOI] [PubMed] [Google Scholar]
- 40.Chapman TL, Bjorkman PJ. 1998. Characterization of a murine cytomegalovirus class I major histocompatibility complex (MHC) homolog: comparison to MHC molecules and to the human cytomegalovirus MHC homolog. J. Virol. 72:460–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fahnestock ML, Johnson JL, Feldman RM, Neveu JM, Lane WS, Bjorkman PJ. 1995. The MHC class I homolog encoded by human cytomegalovirus binds endogenous peptides. Immunity 3:583–590. 10.1016/1074-7613(95)90129-9 [DOI] [PubMed] [Google Scholar]
- 42.Arase H, Mocarski ES, Campbell AE, Hill AB, Lanier LL. 2002. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science 296:1323–1326. 10.1126/science.1070884 [DOI] [PubMed] [Google Scholar]
- 43.Smith HRC, Heusel JW, Mehta IK, Kim S, Dorner BG, Naidenko OV, Iizuka K, Furukawa H, Beckman DL, Pingel JT, Scalzo AA, Fremont DH, Yokoyama WM. 2002. Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc. Natl. Acad. Sci. U. S. A. 99:8826–8831. 10.1073/pnas.092258599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lodoen M, Ogasawara K, Hamerman JA, Arase H, Houchins JP, Mocarski ES, Lanier LL. 2003. NKG2D-mediated natural killer cell protection against cytomegalovirus is impaired by viral gp40 modulation of retinoic acid early inducible 1 gene molecules. J. Exp. Med. 197:1245–1253. 10.1084/jem.20021973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arapović J, Lenac Rovis T, Reddy AB, Krmpotić A, Jonjić S. 2009. Promiscuity of MCMV immunoevasin of NKG2D: m138/fcr-1 down-modulates RAE-1epsilon in addition to MULT-1 and H60. Mol. Immunol. 47:114–122. 10.1016/j.molimm.2009.02.010 [DOI] [PubMed] [Google Scholar]
- 46.Lenac T, Budt M, Arapović J, Hasan M, Zimmermann A, Simic H, Krmpotić A, Messerle M, Ruzsics Z, Koszinowski UH, Hengel H, Jonjić S. 2006. The herpesviral Fc receptor fcr-1 down-regulates the NKG2D ligands MULT-1 and H60. J. Exp. Med. 203:1843–1850. 10.1084/jem.20060514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mintern JD, Klemm EJ, Wagner M, Paquet ME, Napier MD, Kim YM, Koszinowski UH, Ploegh HL. 2006. Viral interference with B7-1 costimulation: a new role for murine cytomegalovirus Fc receptor-1. J. Immunol. 177:8422–8431 [DOI] [PubMed] [Google Scholar]
- 48.Loewendorf AI, Steinbrueck L, Peter C, Busche A, Benedict CA, Kay-Jackson PC. 2011. The mouse cytomegalovirus glycoprotein m155 inhibits CD40 expression and restricts CD4 T cell responses. J. Virol. 85:5208–5212. 10.1128/JVI.02178-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mans J, Natarajan K, Balbo A, Schuck P, Eikel D, Hess S, Robinson H, Simic H, Jonjić S, Tiemessen CT, Margulies DH. 2007. Cellular expression and crystal structure of the murine cytomegalovirus major histocompatibility complex class I-like glycoprotein, m153. J. Biol. Chem. 282:35247–35258. 10.1074/jbc.M706782200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhi L, Mans J, Paskow MJ, Brown PH, Schuck P, Jonjić S, Natarajan K, Margulies DH. 2010. Direct interaction of the mouse cytomegalovirus m152/gp40 immunoevasin with RAE-1 isoforms. Biochemistry 49:2443–2453. 10.1021/bi902130j [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krmpotić A, Busch DH, Bubić I, Gebhardt F, Hengel H, Hasan M, Scalzo AA, Koszinowski UH, Jonjić S. 2002. MCMV glycoprotein gp40 confers virus resistance to CD8+ T cells and NK cells in vivo. Nat. Immunol. 3:529–535. 10.1038/ni799 [DOI] [PubMed] [Google Scholar]
- 52.Höning S, Griffith J, Geuze HJ, Hunziker W. 1996. The tyrosine-based lysosomal targeting signal in lamp-1 mediates sorting into Golgi-derived clathrin-coated vesicles. EMBO J. 15:5230–5239 [PMC free article] [PubMed] [Google Scholar]
- 53.Williams MA, Fukuda M. 1990. Accumulation of membrane glycoproteins in lysosomes requires a tyrosine residue at a particular position in the cytoplasmic tail. J. Cell Biol. 111:955–966. 10.1083/jcb.111.3.955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Höning S, Hunziker W. 1995. Cytoplasmic determinants involved in direct lysosomal sorting, endocytosis, and basolateral targeting of rat lgp120 (lamp-I) in MDCK cells. J. Cell Biol. 128:321–332. 10.1083/jcb.128.3.321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reusch U, Muranyi W, Lucin P, Burgert HG, Hengel H, Koszinowski UH. 1999. A cytomegalovirus glycoprotein re-routes MHC class I complexes to lysosomes for degradation. EMBO J. 18:1081–1091. 10.1093/emboj/18.4.1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reusch U, Bernhard O, Koszinowski U, Schu P. 2002. AP-1A and AP-3A lysosomal sorting functions. Traffic 3:752–761. 10.1034/j.1600-0854.2002.31007.x [DOI] [PubMed] [Google Scholar]
- 57.Wolins N, Bosshart H, Küster H, Bonifacino JS. 1997. Aggregation as a determinant of protein fate in post-Golgi compartments: role of the luminal domain of furin in lysosomal targeting. J. Cell Biol. 139:1735–1745. 10.1083/jcb.139.7.1735 [DOI] [PMC free article] [PubMed] [Google Scholar]