

ABSTRACT
Untreated human immunodeficiency virus (HIV) infection is characterized by depletion of CD4+ T cells, ultimately leading to the impairment of host immune defenses and death. HIV-infected CD4+ T cells die from direct virus-induced apoptosis and CD8 T-cell-mediated elimination, but a broader and more profound depletion occurs in uninfected CD4+ T cells via multiple indirect effects of infection. We fit mathematical models to data from experiments that tested an HIV eradication strategy in which five macaques with a proportion of CD4+ T cells resistant to simian-human immunodeficiency virus (SHIV) entry were challenged with SHIV89.6P, a highly pathogenic dual-tropic chimeric SIV-HIV viral strain that results in rapid loss of both SHIV-susceptible and SHIV-resistant CD4+ T cells. Our results suggest that uninfected (bystander) cell death accounts for the majority of CD4+ T-lymphocyte loss, with at least 60% and 99% of CD4+ T cell death occurring in uninfected cells during acute and established infection, respectively. Mechanisms to limit the profound indirect killing effects associated with HIV infection may be associated with immune preservation and improved long-term survival.
IMPORTANCE HIV infection induces a massive depletion of CD4+ T cells, leading to profound immunodeficiency, opportunistic infections, and eventually death. While HIV induces apoptosis (programmed cell death) by directly entering and replicating in CD4+ T cells, uninfected CD4+ T cells also undergo apoptosis due to ongoing toxic inflammation in the region of infection. In this paper, we use mathematical models in conjunction with data from simian-human immunodeficiency virus SHIV89.6P infection in macaques (a model of HIV infection in humans) to estimate the percentage of cell death that occurs in uninfected cells during the initial period of infection. We reveal that the vast majority of cell death occurs in these cells, which are not infected. The “bystander effects” that lead to enormous reductions in the number of uninfected CD4+ T cells may be a target for future interventions that aim to limit the extent of damage caused by HIV.
INTRODUCTION
Human immunodeficiency virus (HIV) infection causes a decline in CD4+ T cells that ultimately leads to loss of host cellular immune defenses and death in the absence of antiretroviral therapy. HIV replication is known to exert a direct lethal effect on CD4+ T cells (1, 2, 3, 4). Protease generation of Casp8p41 and an integrase-mediated DNA damage response are important mechanisms that initiate apoptosis of HIV-infected cells (5, 6). In addition, CD8 T cells exert contact-dependent death of HIV-infected cells. However, there are multiple other indirect mechanisms that kill both infected and uninfected, or bystander, CD4+ lymphocytes during untreated infection (7, 8, 9, 10). HIV-induced inflammation leads to massive apoptosis in gastrointestinal-associated lymphatic tissue and microbial translocation, which in turn induce more CD4+ T-cell activation and death (11, 12, 13). HIV envelope glycoprotein 120 induces expression of preapoptotic membrane-bound and -soluble factors, such as Fas, Fas ligand, and tumor necrosis factor alpha (14, 15, 16). HIV proteases cleave host proteins into cellular mediators of apoptosis, and HIV induces an accumulation of incomplete reverse transcripts, leading to a proapoptotic response (17). While less than 1% of CD4+ T cells are infected during chronic infection (18, 19), CD4+ T-cell turnover is approximately 2 to 6 times more rapid in infected than uninfected people (20, 21, 22), suggesting a high rate of CD4+ T-cell death due to bystander-mediated killing (23). Yet, the proportion of cells killed by direct viral infection versus indirect effects remains unknown.
We applied mathematical models to data from experiments testing an HIV eradication strategy in which macaques with a known proportion of CD4+ T cells resistant to simian-human immunodeficiency virus (SHIV) entry were challenged with SHIV89.6P, a highly pathogenic virus that results in rapid and irreversible depletion of CD4+ T cells (24, 25, 26, 27). Our model predicted the viral load as well as total populations of infected and uninfected CD4+ cells throughout the course of a SHIV infection. We also identified that the majority of CD4+ T-cell death during peak viremia and following establishment of viral load steady state is due to indirect viral killing, with an overwhelming majority of cell death occurring in uninfected cells.
MATERIALS AND METHODS
Gene modification protocol for SHIV eradication.
Hematopoietic stem cells (HSC) from five pigtail macaques were harvested and transduced with a lentiviral vector expressing either green fluorescent protein (GFP+) only or GFP and a SHIV fusion inhibitor, mC46. This inhibitor is a short peptide that corresponds to a 46-amino-acid sequence of gp41, which has previously been shown to potently inhibit HIV and SHIV viral entry when fused to a membrane anchor (mC46) and expressed on the surface of susceptible CD4+ T cells and other HSC-derived target cells (28). In addition, an in vivo chemoselection cassette was included in both control and mC46 coding lentiviral vectors to permit selection of gene-modified cells following infusion of genetically modified HSCs. All animals received total body radiation and were then transplanted with these modified HSCs. Two control macaques (C1 and C2) received an autologous transplant with HSCs expressing GFP but not mC46. The other three experimental macaques (E1, E2, and E3) were transplanted with HSCs that expressed GFP and mC46. Following transplant, HSCs naturally differentiated to CD4+ T cells.
All animals were engrafted with gene-modified cells, and the percentage of GFP-expressing CD4+ T cells was serially recorded. We assumed that the numbers of GFP-expressing CD4+ T cells in the experimental groups were equal to the number of mC46-expressing cells. Just before virus inoculation, 10%, 17%, and 57% of the respective CD4+ T cells in animals E1, E2, and E3 were modified. All monkeys were challenged with SHIV89.6P. Animals C1 and E2 received a challenge dose (50 50% tissue culture infective doses [TCID50]) that was five times greater than the dose the other animals (10 TCID50). See reference 27 for full details of this part of the experiment.
Figure 1A and B shows plasma viral loads and total CD4+ T-cell counts recorded over the first 10 weeks of infection. All monkeys exhibited a stereotypical viral load trajectory, with an observed peak occurring at day 10 (C1 and E2) or at day 14 (C2, E1, and E3) after infection. All monkeys had similar exponential viral growth (Fig. 1A), but this growth was delayed by 2 days in monkeys that received a lower challenge dose (C2, E1, and E3). The percentage of modified cells at the time of SHIV inoculation correlated negatively with viral load at day 60.
FIG 1.

(A) SHIV RNA trajectories in plasma for the five macaques. (B) Total CD4 T-cell trajectories in plasma for the five macaques. (C to E) Unmodified and modified populations of CD4+ T cells for the three experimental animals (E1 to E3). (C) E1 had 10% modified cells prior to infection; (D) E2 had 17% modified cells prior to infection; (E) E3 had 55% modified cells prior to infection. This research was originally published in Blood (2) and is reprinted here with permission from the publisher, the American Society of Hematology.
An initial decrease in total CD4+ T-cell counts was observed in all of the animals (Fig. 1B), although this decrease was less pronounced for the monkey with a higher proportion of modified cells (E3, with 55% modified cells). Figure 1C to E shows the unmodified and modified populations of CD4+ T cells for the three experimental animals: unmodified CD4+ T cells experienced a sharp decline during peak viral load followed by a recovery. Modified CD4+ T-cell populations also exhibited a substantial decline at peak viremia, though the monkey with the most modified cells (E3) exhibited the least substantial decrease (Fig. 1E; see also Fig. S1 in the supplemental material). The percentage of modified cells at time of SHIV inoculation correlated positively with plasma CD4+ T-cell count at day 60, and gene-modified cells predominated at day 60 in animal E3. Similar patterns of CD4+ T-cell depletion were observed in all tissues examined, including colon, duodenum, auxiliary lymph nodes, and peripheral blood (see Fig. S2 in the supplemental material) (27).
Mathematical models.
We hypothesized that these observations could represent either incomplete effectiveness of the mC46 SHIV fusion inhibitor or indirect effects of infection leading to bystander-mediated CD4+ T-cell death. We therefore constructed competing mathematical models to consider these effects. The modeling core structure of “the incomplete efficacy model” (model 1 [see the Appendix]) was adapted from previous models of viral dynamics (29, 30, 31, 32). Briefly, the model consists of 5 differential equations describing the dynamics of unmodified target cells (T), modified target cells (M), infected target cells (Iu), infected modified target cells (Im), and free virus (V). Both infected cell populations die at a rate of δ and produce k viral particles per cell. Virus is cleared at rate c (in days−1). Susceptible modified and unmodified CD4+ T cells follow logistic growth in the absence of infection, with a carrying capacity of Tmax (the maximum number of CD4+ T cells recorded pre- and postinfection) and become infected with an infectivity of β. We assumed that modified cells could still become infected but that the rate of infection would be reduced by (1 − θ), where θ is the efficacy of the SHIV fusion inhibitor. Parameter θ served as a fitting parameter for all simulations. Data from in vitro experiments and our macaque study (Fig. 2) demonstrated nearly complete elimination of SHIV/HIV entry into gene-modified cells (24, 25, 26), suggesting that the value of θ should be >99%. We considered a much wider window for this parameter and allowed it to be as low as 90%. Hence, we concluded that any model where θ is forced to take on a low value (<0.90) is unlikely to be a valid approximation of infection. It is important to note that while mathematical models compartmentalize cells into infected and uninfected, it might be challenging to measure the varying states of cell death in an in vivo model. Here, however, we fit the models to absolute numbers of CD4+ T cells concurrently with viral load, therefore avoiding variables associated with various stages of cells death.
FIG 2.
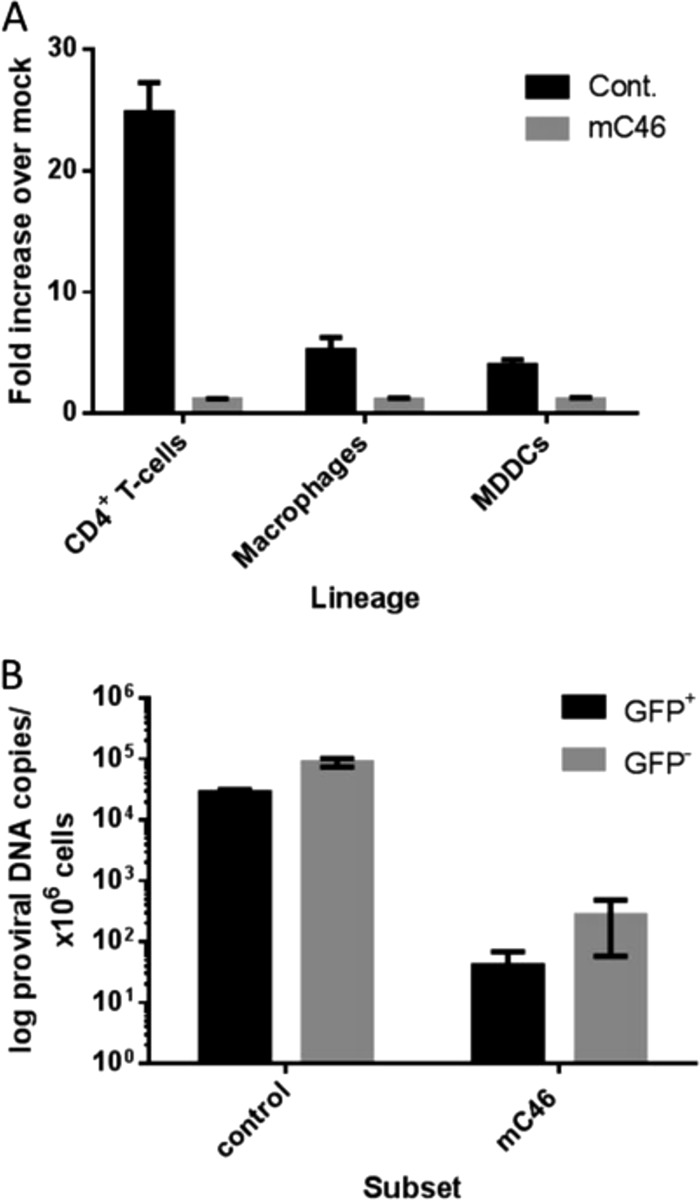
mC46 expression potently inhibits infection of primary cells both in vivo and in vitro. (A) Primary CD4+ T cells, macrophages, and monocyte-derived dendritic cells (MDDCs) were incubated for 4 h with SHIV89.6 Env-pseudotyped, replication-deficient reporter virus encoding luciferase (2 ng p24). Cells were washed and cultured for an additional 3 days in appropriate medium for each cell type. Cells were lysed in passive lysis buffer, and luciferase activity was detected using a luminometer. A minimal increase was observed in mC46-expressing CD4+ T cells (1.1-fold), macrophages (1.09-fold), and MDDCs (1.12-fold). (B) The proviral DNA copy number was determined following sorting of GFP+ and GFP− CD4+ T cells isolated from peripheral blood of SHIV89.6-infected macaques. Values were adjusted to the number of proviral DNA copies per 106 CD4+ T cells. This research was originally published in Blood (2) and is reprinted here with permission from the publisher, the American Society of Hematology.
In the “indirect effects model” (model 2 in the Appendix), we encompassed all mechanisms by which a cell might undergo programmed cell death as a result of the SHIV/HIV infection under a single new term, and we assumed that these effects are proportional to the amount of virus present in the system (with the proportionality constant a), with this effect saturating with constant n. Complete model descriptions and equations are presented in the Appendix.
Our goal was to fit our models to viral load and CD4+ T-cell counts concurrently, which required considering these variables on equivalent scales. Viruses are highly diffusible particles that are likely to exist at relatively equivalent concentrations within tissues and blood during chronic infection, whereas lymphocytes traffic in and out of lymphatic tissues where they exist in higher concentrations than in blood. Hence, we assumed that a proportion, p, of SHIV measured in peripheral blood is produced by CD4+ T cells in blood, while (1 − p) of SHIV in blood is produced by CD4+ T cells in tissue. As the balance of CD4+ T cells between blood and tissue may vary between macaques, we estimated the proportionality constant p as an unknown parameter for each monkey.
RESULTS
Superiority of the indirect effects model.
We fit each monkey separately with 7 unknown parameter values (see Table S1 in the supplemental material) and compared the competing models with nonlinear least-squares and Akaike information criteria (AIC) (33) (see the Appendix). There was a better model fit with the indirect effects model than the incomplete efficacy model (see Fig. S3 and S4 and Table S2 in the supplemental material). Moreover, the indirect effects model accurately reproduced the rank of viral loads, total CD4+ T- cell count, and gene-modified CD4+ T-cell count between monkeys at day 60 (see Fig. S5 in the supplemental material). Model 1 had lower AIC scores for the control animals (see Table S2), but its oscillatory behavior makes it an unrealistic biological approximation of the data (see Fig. S4). With the indirect effects model, we identified a good overall fit to viral load and total CD4+ T-cell count as well as susceptible and modified CD4+ T cells, although the model overestimated the total CD4+ T-cell set point for one macaque, C2 (see Fig. S3). Only the indirect effects model produced sharp declines in gene-modified target cells while predicting realistic ranges for parameter θ (range, 0.993 to 0.999). In contrast, the incomplete efficacy model failed to reproduce key elements of the data (for example, viral load set point), unless the parameter θ was allowed to have unrealistic values (0.70 < θ < 0.90). Regardless of the value of θ, the incomplete efficacy model failed to reproduce dynamics of modified target cells (see Fig. S4).
With the indirect effects model, we estimated that 1.33% (range, 0.89% to 1.62%) of SHIV measured in blood is from CD4+ T cells in blood versus tissue. The mean burst size (k = 5.02 × 102 RNA copies cell−1 day−1), mean infection rate (β = 4.8 × 10−9 ml RNA copies−1 day−1), and mean infected cell death rate (δ = 2.05 × 10−1 day−1) were consistent with previous human and macaque studies (34, 35, 36, 32, 37, 38, 39, 40, 41). The value for the mean saturation rate n was found to be 5.05 × 104 RNA copies ml−1, implying a relatively high viral load threshold for the indirect effect to saturate. Parameter values were relatively preserved across the 5 monkeys (Table 1). We performed sensitivity analysis by starting the optimization routines under several (≥100) different initial conditions and selected among them those parameter sets considered viable solutions. This analysis showed that our model choice and estimates were robust, performing better than the other models considered (see Fig. S6 to S10 in the supplemental material; see also the Appendix).
TABLE 1.
Individual estimated parameters for the indirect effects modela
| Animal | Parameter estimate |
||||||
|---|---|---|---|---|---|---|---|
| p | a | β | δ | k | n | θ | |
| E1 (10% modified) | 1.62e−02 | 5.59e−01 | 5.82e−09 | 1.39e−01 | 3.61e+02 | 2.49e+04 | 9.97e−01 |
| E2 (17% modified) | 1.92e−02 | 5.55e−01 | 3.63e−09 | 1.64e−01 | 6.83e+02 | 1.06e+04 | 9.93e−01 |
| E3 (55% modified) | 9.63e−03 | 5.07e−01 | 3.40e−09 | 4.09e−01 | 5.42e+02 | 1.54e+03 | 9.96e−01 |
| C1 | 1.26e−02 | 6.29e−01 | 9.00e−09 | 2.83e−01 | 4.20e+02 | 1.88e+05 | NA |
| C2 | 8.96e−03 | 6.85e−01 | 2.15e−09 | 3.16e−02 | 5.03e+02 | 2.77e+04 | NA |
| Mean | 1.33e−02 | 5.87e−01 | 4.80e−09 | 2.05e−01 | 5.02e+02 | 5.05e+04 | 9.95e−01 |
NA, not applicable.
The indirect effects model is also predictive.
To address the small sample size of our study, we investigated the ability of the model to predict viral load and CD4+ T-cell dynamics in 4 macaques, based on parameter results from model fitting to a single experimental macaque. To this end, we fit the model to a single macaque (E1) with 7 unknown parameters. For the remaining 4 macaques, all parameter values were carried forward except for a single parameter that was most likely to vary the most between macaques (the proportion of SHIV produced by CD4+ T cells in blood versus tissue, parameter p) and was fit in all predictive simulations. The possible variability in this parameter was highlighted by different blood levels of plasma CD4+ T-cell counts in the monkeys prior to infection (Fig. 1B).
The prediction model successfully reproduced viral load, as well as modified and unmodified CD4+ T-cell trajectories (Fig. 3). It was also able to recapitulate other relevant features of the data. First, at 60 days postinoculation, the viral load set point inversely correlated with the preinfection percentage of modified cells (Fig. 4A), although this effect was more pronounced when all parameters were allowed to vary between monkeys (see Fig. S5 in the supplemental material). Second, the total CD4+ T-cell count set point correlated with the preinfection percentage of modified cells (Fig. 4B). Third, unmodified CD4+ T cells followed a sharp decline during peak viral load in all macaques (Fig. 3, second row). Fourth, modified CD4+ T cells exhibited a decline during peak viral load, and this decline was inversely correlated with the number of modified cells (Fig. 4C). Modified and unmodified cells again exhibited replenishment following initial decay (Fig. 3, second and third rows). Similar results were obtained when the model was fit to any of the five monkeys in order to predict viral load and CD4+ T-cell trajectories in the other 4 monkeys (see Fig. S11 and Table S3 in the supplemental material).
FIG 3.

Best-fit trajectories and experimentally observed data for the experimental and control animals. The indirect effects model (model 2; see the Appendix) was fit with 7 unknown parameters for experimental animal 1 (E1). All parameter values were then carried forward except for a single parameter (the proportion of SHIV produced by CD4+ T cells in blood (p). This unique parameter was fit to the data of the other 4 animals. (A to C) Data for E1, which had 10% modified cells prior to infection. (D to F) Data for E2, which had 17% modified cells prior to infection. (G to I) Data for E3, which had 55% modified cells prior to infection. (J to K) Data for control animal 1 (C1). (L to M) Data for C2.
FIG 4.

(A) Modeled viral loads for the five macaques. (B) Modeled total CD4 T-cell counts for the five macaques. (C) Modeled modified CD4+ T-cell counts for the experimental macaques. The indirect effects model (model 2; see the Appendix) was fit with 7 unknown parameters for experimental animal 1. All parameter values were then carried forward except for a single parameter (the proportion of SHIV produced by CD4+ T cells in blood, p). This unique parameter was fit to the data for the other 4 animals.
Most CD4+ T-cell death during SHIV infection occurs in uninfected cells.
We next investigated the differential contributions in cell death of indirect versus direct effects. In our model simulations, any susceptible (modified or unmodified) CD4+ T cell could theoretically be removed from its respective compartment by becoming infected or by dying from indirect effects (bystander death). Once infected, a cell could either die from indirect effects or direct infection. Hence, we compared two measures to quantify cell death: the proportion of cell death attributable to indirect effects versus direct infection and the proportion of cell death occurring in uninfected versus infected cells.
Figure 5A shows modeled death rates for indirect effects (in both uninfected and infected cells) versus direct effects (infected cells only). The direct effect of viral infection follows the trajectory of viral load, with a rise during acute infection and a sharp decline afterwards. Our model suggests that the maximum percentage of cells killed from direct effects varied from 2% (E3) to 10% (C1) during acute infection. By 20 days postinfection, the indirect effects accounted for over 99% of the cell death, with limited difference across animals (Fig. 5B). A similar pattern was observed even among infected cells, where over 80% of cell death in this group was due to indirect effects (see Fig. S12A in the supplemental material). These results suggest that the indirect effects, once established during acute infection, are overwhelming and persist throughout with little variability across animals.
FIG 5.

Kinetics of cell death rates during SHIV infection (the predictive model fits were used for these plots). (A) Indirect (solid lines) versus direct (dashed lines) death rates as functions of time. The indirect cell death rate encompasses the indirect terms affecting all target cells in the model (susceptible, modified, infected, and modified infected), while the direct effect rate accounts for the rate at which infected cells die from direct virus-induced apoptosis in the model. (B) Percentage of total cell death due to indirect effects (solid lines) and to direct virus-induced apoptosis of infected cells (dashed lines) during infection. (C) Percentage of total cell death in uninfected (bystander) cells (solid lines) versus infected (dashed) lines. (D) Rates of indirect (solid) versus direct (dashed) removal per cell for susceptible cells.
Furthermore, our results suggest that uninfected (bystander) cell death accounts for the majority of CD4+ T-lymphocyte loss during SHIV infection. During peak acute infection, between 18% and 44% of total cell death occurred in infected cells, with the lowest percent occurring in the animal with the highest percentage of gene-modified cells (E3). However, for all the animals, less than 1% of total cell death occurred in infected cells during the chronic phase of infection (Fig. 5C). Finally, we evaluated the fate of susceptible unmodified target cells on a per cell basis and identified that the transition rate from the susceptible to infected compartment was less than the transition rate from susceptible to death from indirect effects (Fig. 5D). Moreover, the transition rate from the susceptible to the infected compartment reached a sharp peak during acute infection but then declined considerably. Our model simulations predict that during acute infection, around 30% of total CD4+ T cells will be infected, but less than 1% of these cells are infected during the chronic phase (see Fig. S12B and C in the supplemental material). This is in agreement with previous work showing similar percentages of infected cells during acute infection (42) and showing that during chronic infection very few infected cells are detectable in the periphery (3, 18, 19).
Models with immunity.
HIV and SHIV dynamics are extremely complex, based on the fact that a bidirectional relationship occurs between the virus and its target cells, which are directly involved in the immune response against viral infections. Indeed, viral loads decreased after day 60 in the macaques in this study, with the highest CD4+ T-cell counts (27). In the above models, in order to avoid overparameterization of the model, we excluded analysis of data beyond day 60 and did not consider the effect of the immune response. To ascertain whether inclusion of immunity might improve model fit to data from the first 60 days of infection, we tested the indirect effects model against several models that incorporated immunity. The first immunity model assumed a delayed immune response, phasing in gradually between days 14 and 18 postinfection, when an immune response to HIV-infected cells is typically evident (43, 44, 45). We allowed the immune response to act by decreasing the infectivity, decreasing the burst size of the virus, or increasing the death rate of infected cells. This model was tested against the data both with (model 3 [see Appendix]) and without (model 4 [see Appendix]) the indirect effects.
Inclusion of the immunity term in the absence of the indirect effects term resulted in poor model fit in all 5 macaques (Fig S13). When the indirect effects term was included, the fit approximated that of the indirect effects model without immunity (Fig S14). However, AIC scores were superior in the model without immunity (Table S2), owing to the added parameters and complexity of the immune model. These results imply that indirect effect are critical for explaining viral and CD4+ T-cell dynamics early during infection, and that immune mediated protection might have a role in determining viral and CD4+ T-cell set point as well.
Finally, we considered a different immunity model based on information reported in reference 36; this model added a term to the infected cell equation to account for the removal of infected cells in a density-dependent fashion (model 5 [see Appendix]), but the fit to the data was poor and failed to demonstrate the sharp decline of modified cells after peak viremia (see Fig. S15 in the supplemental material).
DISCUSSION
Mathematical models have provided critical estimates of rapid replication and clearance of HIV in the body, resulting in important implications for therapy and drug resistance (29, 46, 47, 32, 48, 49). We propose that a key omission in models to date is the indirect killing effect that SHIV or HIV is likely to have on CD4+ lymphocytes. While these effects were impossible to quantify based on lack of available technology to follow a population of SHIV- or HIV-resistant target cells, our current data set indicates that CD4+ T cells, which are rendered resistant to SHIV infection, undergo a sharp decline a few days after viral load peaks during primary infection.
We constructed a mechanistic, mathematical model that was able to recapitulate the viral load and CD4+ T-cell trajectories concurrently in any of the five animals and then predict analogous trajectories in the other animals. Our results suggest that the rapid decline in both susceptible and resistant CD4+ T cells during peak viremia is due to indirect effects of infection. Furthermore, the majority of cell death occurring during steady-state SHIV viremia is due to these indirect effects, with over 99% of cell death occurring in uninfected cells.
Previous work analyzing lymph nodes in HIV-infected children and SIV-infected macaques showed that apoptosis occurred primarily in uninfected CD4+ T cells (50). Recent cross-sectional evaluations suggest that 95% of the dying cells in lymphoid tissue are uninfected and that their death results from abortive HIV infection, where the virus enters the cell but is not able to replicate (17). Our results are in alignment with an increasing body of literature suggesting that the role of bystander cell death during HIV infection is much larger than the loss of CD4+ T cells due to direct infection (12, 51, 52, 53, 54, 55, 56, 57, 58, 59). Furthermore, our model suggests that over 50% of CD4+ T cells die during acute infection. This is consistent with previous studies, which demonstrated that profound CD4+ depletion occurs around peak viral load (19, 42). However, this is the first time that a quantification of bystander death has been performed based on modeling of viral and target cell dynamics rather than single cross-sectional observations. The model also demonstrates why genetically modified cells are enriched relative to unmodified cells. Because direct effects exert a differential effect on unmodified cells, their depletion is more profound during peak viremia, a trend that continues upon CD4+ T-cell replenishment.
We acknowledge that other phenomena could contribute to our observations. In particular, the decrease in the number of modified CD4+ T cells in peripheral blood a few days after virus inoculation could result in part from CD4+ T cells trafficking from lymph nodes via blood to sites of infection. Our model assumes that the proportion of CD4+ T cells present in blood is static. In reality, this proportion may be somewhat dynamic. Variability in the degree of T-cell trafficking between monkeys could partly explain why our model was not able to perfectly predict the viral load and CD4+ T-cell dynamics of each macaque. We demonstrated massive CD4+ T-cell depletion in all observed tissues, suggesting that peripheral CD4+ T-cell decline is due mostly to CD4+ T-cell death rather than trafficking. Our model may slightly overestimate the true death rate of infected cells, because cellular RNA, DNA, and proteins are degraded during late apoptosis, and these cells may therefore be overlooked with current technologies. Finally, the number of animals used to fit the model was small, making our conclusions difficult to generalize. We attempted to circumvent this issue by using results from a single monkey to predict outcomes in other monkeys.
Previous work has suggested that survival of CD4+ T cells following acute infection is ultimately critical in containing the virus, leading to a lower steady-state viral load and better disease outcomes (60). Our model effectively reproduces key components of the data with or without early immunity. However, a model that includes concepts of an immune response would be necessary for description of infection beyond the early time points studied in the present work. The steady decline in viral load in macaque E3 after day 30 is also likely to be a result of this response.
Taken together, these observations call for experiments using this new technology to specifically understand the role of bystander-mediated apoptosis during HIV infection. This could have considerable implications for new therapies and vaccines. Because CD4+ T cells are central in immunologic control of HIV, any mechanism to decrease their turnover may provide an important benefit (61, 62, 63). Even macaques with a very small percentage of modified CD4+ T cells followed a lower set point than controls and were able to replenish both compartments of CD4+ T cells. A therapy targeting bystander-mediated apoptosis could help preserve CD4+ T cells, an effect that could augment other curative approaches being considered for HIV infection.
APPENDIX
Mathematical models.
We performed our analysis with several competing models. All of them are described by a set of ordinary differential equations, where T represents the number of susceptible unmodified CD4+ T cells, M represents the number of susceptible modified CD4+ T cells, Iu and Im the number of unmodified and modified infected CD4+ T cells, and V represents free virus. Because all monkeys underwent full-body irradiation followed by a stem cell transplant and SHIV challenge, we considered that CD4+ T-cell proliferation would be the main mechanism for CD4+ T-cell replenishment, and we modeled it with a logistic growth term, with growth rate λ and carrying capacity Tmax, given by the maximum number of CD4+ T cells recorded. We also considered models without a logistic term but with a source term instead, but the fits were very poor. In all the models, we fixed the clearance rate c at 23, according to previous estimates (48). We set λ at 0.5 and the initial number of virus V0 to 10−3. This value, however, didn't affect the results: modifying V0 by 3 orders of magnitude gave similar results. We delayed by 2 days the start of infection (i.e., Vt = 0 for the first 2 days) for the animals that received a lower challenge dose (control 2, experiment 1, and experiment 3 [C2, E1, and E3, respectively]). This was done based on the observation that the monkeys receiving a lower dose had a delay of 2 days in the exponential growth phase compared with animals control 1 and experiment 2. For all the models considered, we allowed gene-modified cells to become infected, but the rate of infection was reduced by (1 − θ), where θ is the efficacy of the SHIV fusion inhibitor. The parameter θ was fit to the data.
Incomplete efficacy model.
We wished to investigate if a partially efficacious SHIV fusion inhibitor could be the unique reason responsible for the experimental results observed. We developed a basic viral dynamics model based on a target cell limitation model published previously (29, 47, 32), and we obtained the following system of equations (model 1):
| (1) |
Indirect effects model.
We next developed a model to take into account bystander-mediated apoptosis due to the indirect effects of infection. We encompassed all the different mechanisms (change of growth factors, increase in the death ligands, release of toxins, etc.) by which a cell would undergo programmed cell death as a result of the SHIV/HIV infection under a single term. To this end, we modified the above model to incorporate a term that accounted for the indirect effects of infection by killing of all types of cells—modified and unmodified cells, both susceptible and infected—and assumed that this effect is proportional to the amount of virus present in the system. The equations were modified as follows (model 2):
| (2) |
where bys = α[V/(V + n)].
Models with immunity.
We next compared model 2 with three models that incorporated an immune response.
(i) Incomplete efficacy with phasing immunity model (model 3).
This model is identical to the incomplete efficacy model (model 1), with the addition of a delayed immune response phasing in gradually between days 14 and days 18 postinfection, when an immune response to HIV-infected cells is typically evident (43, 44, 45). Since it is not clear how immunity would affect the infection process, and hence how to include immunity in our model, we took an agnostic view and allowed the immune response to act by either decreasing the infectivity (β), decreasing the burst size of the virus (k), or increasing the death rate of infected cells (δ) in a linear fashion. Namely, for each possible parameter, we assumed a value Par1 from time of infection up to day 14, a value Par2 from day 18 on, and we linearly interpolated between Par1and Par2 between days 14 and 18. This produced the following system (model 3)
| (3) |
for Par = β, δ, or k.
(ii) Indirect effects model with phasing immunity (model 4).
Next, we considered the indirect effects model (model 2) with a phasing immunity as described above, resulting in the following model (model 4):
| (4) |
for Par = β, δ, or k.
(iii) Incomplete efficacy with density-dependent immunity model (model 5).
We adapted a model proposed in reference 36 to incorporate an immune response in the model that was density dependent. To do this, we added a saturation function that accounted for the loss of infected cells (both modified and unmodified). That is, the model assumes that the immune response depends on the number of infected cells present in the system, but it saturates at a threshold (m). The equations describing this model are the following (model 5):
| 5 |
where
Parameter fitting.
For each model, we fit concurrently to viral load unmodified CD4+ T cells and modified CD4+ T cells (experimental animals only) on a log10 scale. We attributed equal weights to each of the quantities fitted. We used nonlinear least-squares optimization routines in Matlab (The Mathworks). That is, we minimized the following function:
where yi represents the data at time i and ymodel represents the numerical solution to the model at time i. Table S1 in the supplemental material provides those parameters that were fitted to the data and those that were fixed. For each model, we started the optimization routines at several starting points (>100) to ensure a wide search in the parameter space, and we collected all the solutions. We assessed the quality of model fits based mainly on three criteria.
(i) First, we selected solutions that gave the minimum of the least-squares sum (LSS).
(ii) Second, we selected solutions that were biologically relevant. Based on previous studies (24, 25, 26), we considered that only models where θ ≥ 0.9 are biologically plausible. Hence, only parameter sets with θ ≥ 0.9 were taken into account.
Since we were interested in capturing the deplenishment and further replenishment of modified CD4+ T-cell populations, we selected, for models 2 and model 1 among all the solutions found, those that satisfied the following condition:
For each monkey, y0 is given by the minimum number of modified CD4+ T cells recorded over the first 60 days of infection, and ymodel is the respective numerical value of the model for the modified CD4+ T cells for that particular day (experimental animals only).
Out of these solutions, we finally selected the one with the minimum LSS value. We tried to proceed in an analogous fashion for models 3, 4, and 5, but there were no solutions that satisfied this condition, implying that these models miss the dynamics of modified CD4+ T cells during acute infection. Hence, for models 3 to 5, we simply selected the solution with the minimum LSS value.
(iii) Finally, we also computed the Akaike information criterion (AIC) for each model used. We used the corrected AIC to take into account the small number of data points (33). Table S2 in the supplemental material gives the AIC for each of the five models compared when we fitted the data to each individual monkey.
Sensitivity analysis.
We performed a sensitivity analysis by starting the optimization routines under several (≥100) different initial conditions. We then considered viable “solutions” as those with LSS of ≤10 (solutions with LSS of ≥10 were very far off from the data and we considered them as being failed results). Figures S7 to S10 in the supplemental material show the plots for all the sets of parameters found for the indirect effects model when each monkey was fit individually, together with the median parameter set. These figures show that the solution is robust with respect to the fitting, in the sense that most of the solutions behaved similarly to the median and to the “optimal” solution reported in the main text.
Supplementary Material
ACKNOWLEDGMENTS
This work was partially supported by the NIH-supported Martin Delaney Collaboratory defeatHIV Program (grants U19 AI 096111 and NIH K23 AI087206), and NIH grants R01CA114218, R01AI080326, and R01HL098489. H.-P.K. is a Markey Molecular Medicine Investigator and the recipient of the Jos Carreras/E. Donnall Thomas Endowed Chair for Cancer Research.
We thank Keith Jerome and Bryan Mayer for assistance in developing ideas for the manuscript.
Footnotes
Published ahead of print 3 January 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.03428-13.
REFERENCES
- 1.Grossman Z, Meier-Schellersheim M, Sousa A, Victorino R, Paul W. 2002. CD4+ T-cell depletion in HIV infection: are we closer to understanding the cause? Nat. Med. 8:319–323. 10.1038/nm0402-319 [DOI] [PubMed] [Google Scholar]
- 2.Hazenberg M, Hamann D, Schuitemaker H, Miedema F. 2000. T cell depletion in HIV-I infection: how CD4+ T cells go out of stock. Nat. Immunol. 1:285–289. 10.1038/79724 [DOI] [PubMed] [Google Scholar]
- 3.Haase AT. 1999. Population biology of HIV-1 infection: viral and CD4+ T cell demographics and dynamics in lymphatic tissues. Annu. Rev. Immunol. 17:625–656 [DOI] [PubMed] [Google Scholar]
- 4.McCune J. 2001. The dynamics of CD4+ T-cell depletion in HIV disease. Nature 410:974–979. 10.1038/35073648 [DOI] [PubMed] [Google Scholar]
- 5.Sainski AM, Natesampillai S, Cummins NW, Bren GD, Taylor J, Saenz DT, Poeschla EM, Badley AD. 2011. The HIV-1-specific protein Casp8p41 induces death of infected cells through Bax/Bak. J. Virol. 85:7965–7975. 10.1128/JVI.02515-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cooper A, Garcia M, Petrovas C, Yamamoto T, Koup RA, Nabel GJ. 2013. HIV-1 causes CD4 cell death through DNA-dependent protein kinase during viral integration. Nature 498:376–379. 10.1038/nature12274 [DOI] [PubMed] [Google Scholar]
- 7.Badley AD, Pilon AA, Landay A, Lynch DH. 2000. Mechanisms of HIV-associated lymphocyte apoptosis. Blood 96:2951–2964 http://bloodjournal.hematologylibrary.org/content/96/9/2951.long [PubMed] [Google Scholar]
- 8.Chase A, Zhou Y, Siliciano RF. 2006. HIV-1-induced depletion of CD4+ T cells in the gut: mechanism and therapeutic implications. Trends Pharmacol. Sci. 27:4–7. 10.1016/j.tips.2005.11.005 [DOI] [PubMed] [Google Scholar]
- 9.Cummins NW, Badley AD. 2010. Mechanisms of HIV-associated lymphocyte apoptosis: 2010. Cell Death Dis. 1:e99. 10.1038/cddis.2010.77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gougeon ML. 2003. Apoptosis as an HIV strategy to escape immune attack. Nat. Rev. Immunol. 3:392–404. 10.1038/nri1087 [DOI] [PubMed] [Google Scholar]
- 11.Gougeon ML, Piacentini M. 2009. New insights on the role of apoptosis and autophagy in HIV pathogenesis. Apoptosis 14:501–508. 10.1007/s10495-009-0314-1 [DOI] [PubMed] [Google Scholar]
- 12.Ipp H, Zemlin A. 2013. The paradox of the immune response in HIV infection: when inflammation becomes harmful. Clin. Chim. Acta 416:96–99. 10.1016/j.cca.2012.11.025 [DOI] [PubMed] [Google Scholar]
- 13.Mehandru S, Poles MA, Tenner-Racz K, Horowitz A, Hurley A, Hogan C, Boden D, Racz P, Markowitz M. 2004. Primary HIV-1 infection is associated with preferential depletion of CD4+ T lymphocytes from effector sites in the gastrointestinal tract. J. Exp. Med. 200:761–770. 10.1084/jem.20041196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fluur C, De Milito A, Fry TJ, Vivar N, Eidsmo L, Atlas A, Federici C, Matarrese P, Logozzi M, Rajnavolgyi E, Mackall CL, Fais S, Chiodi F, Rethi B. 2007. Potential role for IL-7 in Fas-mediated T cell apoptosis during HIV infection. J. Immunol. 178:5340–5350 http://www.jimmunol.org/content/178/8/5340.long [DOI] [PubMed] [Google Scholar]
- 15.Joshi A, Nyakeriga AM, Ravi R, Garg H. 2011. HIV Env glycoprotein-mediated bystander apoptosis depends on expression of the CCR5 co-receptor at the cell surface and Env fusogenic activity. J. Biol. Chem. 286:36404–36413. 10.1074/jbc.M111.281659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wan ZT, Chen XL. 2010. Mechanisms of HIV envelope-induced T lymphocyte apoptosis. Virol. Sin. 25:307–315. 10.1007/s12250-010-3148-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doitsh G, Cavrois M, Lassen KG, Zepeda O, Yang Z, Santiago ML, Hebbeler AM, Greene WC. 2010. Abortive HIV infection mediates CD4 T cell depletion and inflammation in human lymphoid tissue. Cell 143:789-801. 10.1016/j.cell.2010.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chun TW, Carruth L, Finzi D, Shen X, DiGiuseppe JA, Taylor H, Hermankova M, Chadwick K, Margolick J, Quinn TC, Kuo YH, Brookmeyer R, Zeiger MA, Barditch-Crovo P, Siliciano RF. 1997. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 387:183–188 [DOI] [PubMed] [Google Scholar]
- 19.Davenport MP, Zhang L, Shiver JW, Casmiro DR, Ribeiro RM, Perelson AS. 2006. Influence of peak viral load on the extent of CD4+ T-cell depletion in simian HIV infection. J. Acquir. Immune Defic. Syndr. 41:259–265. 10.1097/01.qai.0000199232.31340.d3 [DOI] [PubMed] [Google Scholar]
- 20.McCune JM, Hanley MB, Cesar D, Halvorsen R, Hoh R, Schmidt D, Wieder E, Deeks S, Siler S, Neese R, Hellerstein M. 2000. Factors influencing T-cell turnover in HIV-1-seropositive patients. J. Clin. Invest. 105:R1–R8. 10.1172/JCI8647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mohri H, Perelson AS, Tung K, Ribeiro RM, Ramratnam B, Markowitz M, Kost R, Hurley A, Weinberger L, Cesar D, Hellerstein MK, Ho DD. 2001. Increased turnover of T lymphocytes in HIV-1 infection and its reduction by antiretroviral therapy. J. Exp. Med. 194:1277–1287. 10.1084/jem.194.9.1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sachsenberg N, Perelson AS, Yerly S, Schockmel GA, Leduc D, Hirschel B, Perrin L. 1998. Turnover of CD4+ and CD8+ T lymphocytes in HIV-1 infection as measured by Ki-67 antigen. J. Exp. Med. 187:1295–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meyaard L, Otto SA, Jonker RR, Mijnster MJ, Keet RP, Miedema F. 1992. Programmed death of T cells in HIV-1 infection. Science 257:217–219 [DOI] [PubMed] [Google Scholar]
- 24.Kiem HP, Wu RA, Sun G, von Laer D, Rossi JJ, Trobridge GD. 2010. Foamy combinatorial anti-HIV vectors with MGMTP140K potently inhibit HIV-1 and SHIV replication and mediate selection in vivo. Gene Ther. 17:37–4910. 10.1038/gt.2009.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trobridge GD, Beard BC, Gooch C, Wohlfahrt M, Olsen P, Fletcher J, Malik P, Kiem HP. 2008. Efficient transduction of pigtailed macaque hematopoietic repopulating cells with HIV-based lentiviral vectors. Blood 111:5537–5543. 10.1182/blood-2007-09-115022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trobridge GD, Wu RA, Beard BC, Chiu SY, Munoz NM, von Laer D, Rossi JJ, Kiem HP. 2009. Protection of stem cell-derived lymphocytes in a primate AIDS gene therapy model after in vivo selection. PLoS One 4(11):e7693. 10.1371/journal.pone.0007693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Younan PM, Polacino P, Kowalski JP, Peterson CW, Maurice NJ, Williams NP, Ho O, Trobridge GD, Von Laer D, Prlic M, Beard BC, Derosa S, Hu SL, Kiem HP. 2013. Positive selection of mC46-expressing CD4+ T cells and maintenance of virus specific immunity in a primate AIDS model. Blood 122:179–187. 10.1182/blood-2013-01-482224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Egelhofer M, Brandenburg G, Martinius H, Schult-Dietrich P, Melikyan G, Kunert R, Baum C, Choi I, Alexandrov A, von Laer D. 2004. Inhibition of human immunodeficiency virus type 1 entry in cells expressing gp41-derived peptides. J. Virol. 78:568–575. 10.1128/JVI.78.2.568-575.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M. 1995. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 373:123–126 [DOI] [PubMed] [Google Scholar]
- 30.Krakauer DC, Nowak M. 1999. T-cell induced pathogenesis in HIV: bystander effects and latent infection. Proc. Biol. Sci. 266:1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Perelson AS, Nelson PW. 1998. Mathematical analysis of HIV-1 dynamics in vivo. SIAM Rev. 41:3–44. 10.2478/v100006-010-0045-z [DOI] [Google Scholar]
- 32.Nowak MA, Lloyd AL, Vasquez GM, Wiltrout TA, Wahl LM, Bischofberger N, Williams J, Kinter A, Fauci AS, Hirsch VM, Lifson JD. 1997. Viral dynamics of primary viremia and antiretroviral therapy in simian immunodeficiency virus infection. J. Virol. 71:7518–7525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burnham KP, Anderson DR. 2002. Model selection and multimodel inference: a practical information-theoretic approach, 2nd ed. Springer, New York, NY [Google Scholar]
- 34.Chen HY, Di Mascio M, Perelson AS, Ho DD, Zhang L. 2007. Determination of virus burst size in vivo using a single-cycle SIV in rhesus macaques. Proc. Natl. Acad. Sci. U. S. A. 104:19079–19084. 10.1073/pnas.0707449104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bonhoeffer S, Coffin JM, Nowak MA. 1997. Human immunodeficiency virus drug therapy and virus load. J. Virol. 71:3275–3278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burg D, Rong L, Neumann AU, Dahari H. 2009. Mathematical modeling of viral kinetics under immune control during primary HIV-1 infection. J. Theor. Biol. 259:751–759. 10.1016/j.jtbi.2009.04.010 [DOI] [PubMed] [Google Scholar]
- 37.Markowitz M, Louie M, Hurley A, Sun E, Di Mascio M, Perelson AS, Ho DD. 2003. A novel antiviral intervention results in more accurate assessment of human immunodeficiency virus type 1 replication dynamics and T-cell decay in vivo. J. Virol. 77:5037–5038. 10.1128/JVI.77.8.5037-5038.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stafford MA, Corey L, Cao Y, Daar ES, Ho DD, Perelson AS. 2000. Modeling plasma virus concentration during primary HIV infection. J. Theor. Biol. 203:285–301. 10.1006/jtbi.2000.1076 [DOI] [PubMed] [Google Scholar]
- 39.Bonhoeffer S, Funk GA, Gunthard HF, Fischer M, Muller V. 2003. Glancing behind virus load variation in HIV-1 infection. Trends Microbiol. 11:499–504. 10.1016/j.tim.2003.09.002 [DOI] [PubMed] [Google Scholar]
- 40.Ribeiro RM, Qin L, Chavez LL, Li D, Self SG, Perelson AS. 2010. Estimation of the initial viral growth rate and basic reproductive number during acute HIV-1 infection. J. Virol. 84:6096–6102. 10.1128/JVI.00127-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vaidya NK, Rong L, Marconi VC, Kuritzkes DR, Deeks SG, Perelson AS. 2010. Treatment-mediated alterations in HIV fitness preserve CD4+ T cell counts but have minimal effects on viral load. PLoS Comput. Biol. 6(11):e1001012. 10.1371/journal.pcbi.1001012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mattapallil JJ, Douek DC, Hill B, Nishimura Y, Martin M, Roederer M. 2005. Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature 434:1093–1097. 10.1038/nature03501 [DOI] [PubMed] [Google Scholar]
- 43.Davenport MP, Ribeiro RM, Perelson AS. 2004. Kinetics of virus-specific CD8+ T cells and the control of human immunodeficiency virus infection. J. Virol. 78:10096–10103. 10.1128/JVI.78.18.10096-10103.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kaur A, Hale CL, Ramanujan S, Jain RK, Johnson RP. 2000. Differential dynamics of CD4(+) and CD8(+) T-lymphocyte proliferation and activation in acute simian immunodeficiency virus infection. J. Virol. 74:8413–8424. 10.1128/JVI.74.18.8413-8424.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu H, Wang X, Lackner AA, Veazey RS. 2013. CD8 down-regulation and functional impairment of SIV-specific cytotoxic T lymphocytes in lymphoid and mucosal tissues during SIV infection. J. Leukoc. Biol. 93:943–950. 10.1189/jlb.1112580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Perelson AS, Kirschner DE, De Boer R. 1993. Dynamics of HIV infection of CD4+ T cells. Math. Biosci. 114:81–125 [DOI] [PubMed] [Google Scholar]
- 47.Perelson AS, Neumann AU, Markowitz M, Leonard JM, Ho DD. 1996. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science 271:1582–1586 [DOI] [PubMed] [Google Scholar]
- 48.Ramratnam B, Bonhoeffer S, Binley J, Hurley A, Zhang L, Mittler JE, Markowitz M, Moore JP, Perelson AS, Ho DD. 1999. Rapid production and clearance of HIV-1 and hepatitis C virus assessed by large volume plasma apheresis. Lancet 354:1782–1785 [DOI] [PubMed] [Google Scholar]
- 49.Wei X, Ghosh SK, Taylor ME, Johnson VA, Emini EA, Deutsch P, Lifson JD, Bonhoeffer S, Nowak MA, Hahn BH. 1995. Viral dynamics in human immunodeficiency virus type 1 infection. Nature 373:117–122 [DOI] [PubMed] [Google Scholar]
- 50.Finkel TH, Tudor-Williams G, Banda NK, Cotton MF, Curiel T, Monks C, Baba TW, Ruprecht RM, Kupfer A. 1995. Apoptosis occurs predominantly in bystander cells and not in productively infected cells of HIV- and SIV-infected lymph nodes. Nat. Med. 1:129–134 [DOI] [PubMed] [Google Scholar]
- 51.Ameisen JC, Capron A. 1991. Cell dysfunction and depletion in AIDS: the programmed cell death hypothesis. Immunol. Today 12:102–105 [DOI] [PubMed] [Google Scholar]
- 52.Alimonti JB, Ball TB, Fowke KR. 2003. Mechanisms of CD4+ T lymphocyte cell death in human immunodeficiency virus infection and AIDS. J. Gen. Virol. 84:1649–1661. 10.1099/vir.0.19110-0 [DOI] [PubMed] [Google Scholar]
- 53.Embretson J, Zupancic M, Ribas JL, Burke A, Racz P, Tenner-Racz K, Haase AT. 1993. Massive covert infection of helper T lymphocytes and macrophages by HIV during the incubation period of AIDS. Nature 362:359–362 [DOI] [PubMed] [Google Scholar]
- 54.Esser MT, Bess JWJ, Suryanarayana K, Chertova E, Marti D, Carrington M, Arthur LO, Lifson JD. 2001. Partial activation and induction of apoptosis in CD4(+) and CD8(+) T lymphocytes by conformationally authentic noninfectious human immunodeficiency virus type 1. J. Virol. 75:1152–1164. 10.1128/JVI.75.3.1152-1164.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Garg H, Mohl J, Joshi A. 2012. HIV-1 induced bystander apoptosis. Viruses 4:3020–3043. 10.3390/v4113020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Holm GH, Zhang C, Gorry PR, Peden K, Schols D, De Clercq E, Gabuzda D. 2004. Apoptosis of bystander T cells induced by human immunodeficiency virus type 1 with increased envelope/receptor affinity and coreceptor binding site exposure. J. Virol. 78:4541–4551. 10.1128/JVI.78.9.4541-4551.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Holm GH, Gabuzda D. 2005. Distinct mechanisms of CD4+ and CD8+ T-cell activation and bystander apoptosis induced by human immunodeficiency virus type 1 virions. J. Virol. 79:6299–6311. 10.1128/JVI.79.10.6299-6311.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jekle A, Keppler OT, De Clercq E, Schols D, Weinstein M, Goldsmith MA. 2003. In vivo evolution of human immunodeficiency virus type 1 toward increased pathogenicity through CXCR4-mediated killing of uninfected CD4 T cells. J. Virol. 77:5846–5854. 10.1128/JVI.77.10.5846-5854.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Silvestri G, Sodora DL, Koup RA, Paiardini M, O'Neil SP, McClure HM, Staprans SI, Feinberg MB. 2003. Nonpathogenic SIV infection of sooty mangabeys is characterized by limited bystander immunopathology despite chronic high-level viremia. Immunity 18:441–452. 10.1016/S1074-7613(03)00060-8 [DOI] [PubMed] [Google Scholar]
- 60.Sun Y, Permar SR, Buzby AP, Letvin NL. 2007. Memory CD4+ T-lymphocyte loss and dysfunction during primary simian immunodeficiency virus infection. J. Virol. 81:8009–8015. 10.1128/JVI.00482-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Petravic J, Davenport MP. 2011. Simian-human immunodeficiency infection: is the course set in the acute phase? PLoS One 6(2):e17180. 10.1371/journal.pone.0017180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roger PM, Breittmayer JP, Durant J, Sanderson F, Ceppi C, Brignone C, Cua E, Clevenbergh P, Fuzibet JG, Pesce A, Bernard A, Dellamonica P. 2002. Early CD4(+) T cell recovery in human immunodeficiency virus-infected patients receiving effective therapy is related to a down- regulation of apoptosis and not to proliferation. J. Infect. Dis. 185:463–470. 10.1086/338573 [DOI] [PubMed] [Google Scholar]
- 63.Kalams SA, Walker BD. 1998. The critical need for CD4 help in maintaining effective cytotoxic T lymphocyte responses. J. Exp. Med. 188:2199–2204 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.