

ABSTRACT
Influenza virus transcription requires functional coupling with cellular transcription for the cap-snatching process. Despite this fact, RNA polymerase II (RNAP II) is degraded during infection in a process triggered by the viral polymerase. Reassortant viruses from the A/PR/8/34 (PR8) strain that induce (hvPR8) or do not induce (lvPR8) RNAP II degradation led to the identification of PA and PB2 subunits as responsible for the degradation process. Three changes in the PB2 sequence (I105M, N456D, and I504V) and two in PA (Q193H and I550L) differentiate PA and PB2 of lvPR8 from those of hvPR8. Using recombinant viruses, we observed that changes at position 504 of PB2, together with 550 of PA, confer the ability on lvPR8 for RNAP II degradation and, conversely, abolish hvPR8 degradation capacity. Since hvPR8 is more pathogenic than lvPR8 in mice, we tested the potential contribution of RNAP II degradation in a distant viral strain, the 2009 pandemic A/California/04/09 (CAL) virus, whose PA and PB2 subunits are of avian origin. As in the hvPR8 virus, mutations at positions 504 of PB2 and 550 of PA in CAL virus abolished its RNAP II degradation capacity. Moreover, in an in vivo model, the CAL-infected mice lost more body weight, and 75% lethality was observed in this situation compared with 100% survival in mutant-CAL- or mock-infected animals. These results confirm the involvement of specific PB2 and PA residues in RNAP II degradation, which correlates with pathogenicity in mice of viruses containing human or avian polymerase PB2 and PA subunits.
IMPORTANCE The influenza virus polymerase induces the degradation of RNAP II, which probably cooperates to avoid the antiviral response. Here, we have characterized two specific residues located in the PA and PB2 polymerase subunits that mediate this degradation in different influenza viruses. Moreover, a clear correlation between RNAP II degradation and in vivo pathogenicity in mice was observed, indicating that the degradative process constitutes a viral pathogenicity factor.
INTRODUCTION
The influenza virus induces profound alterations in the host cell, and several mechanisms triggered by the influenza virus have been reported to be involved in cellular shutoff, among them cap snatching of cellular pre-mRNAs (1), inhibition of cleavage and polyadenylation of cellular pre-mRNAs (2, 3), nuclear retention of poly(A)-containing cellular mRNAs (4), degradation of cytoplasmic cellular mRNAs (5, 6), and preferential utilization of the translation machinery by virus-specific mRNAs (7, 8). More recently, a degradative process affecting RNA polymerase II (RNAP II) (9–11), as well as CHD6 chromatin remodeler (12), has been reported. Degradation and disabling of host cell factors involved in genome expression may contribute to hijacking the metabolism of the infected cell and to suppressing the establishment of the host antiviral defense against viral pathogens, contributing to viral pathogenicity.
The RNAP II and CHD6 degradation observed seems to be a general feature triggered by influenza A viruses (10, 12), but we have previously reported that degradation of RNAP II does not occur upon infection with the attenuated A/PR/8/34 (PR8) strain (9), which would contribute to the attenuated phenotype of the strain. However, a hypervirulent PR8 (hvPR8) variant, which multiplies much faster than standard PR8 (lvPR8) in infected cells and is more virulent in mice than the parental PR8 virus, efficiently induces RNAP II degradation (9). It has been reported that the inclusion of PA or PB2 segments from hvPR8 in lvPR8 recombinant viruses greatly increases their pathogenicity (13). Studies with reassortant viruses containing defined genome segments of both hvPR8 and lvPR8 indicate that PA and PB2 subunits individually contribute to the ability of the influenza virus to degrade RNAP II (9). Using recombinant hvPR8 and lvPR8 viruses with defined PA and PB2 mutations, we characterized the individual contributions of specific residues to RNAP II degradation, and moreover, we tested their contributions in a very distant viral background. A new influenza A virus from the H1N1 subtype [A(H1N1)pdm09], possessing high transmissibility but relatively low virulence, emerged in 2009, rapidly spread across the entire globe, and caused the first pandemic of the 21st century (14, 15). Infection with the new pandemic viruses produced mild symptoms in the majority of infected people, but compared with the previous seasonal H1N1, a small proportion of severe cases and deaths occurred among young and apparently healthy patients (16, 17). The new virus was a reassortant virus containing segments of avian, human, and porcine origin: for instance, PB2 and PA polymerase segments were avian, while PB1 was of human origin (15). Here, we checked the contributions of those PB2 and PA amino acids that modulate RNAP II degradation in the PR8 virus in a distant strain, the 2009 pandemic A/California/04/09 (CAL) virus. In addition, the possible importance of RNAP II degradation to the in vivo pathogenicity of the CAL strain was also studied. The same residues that abolish RNAP II degradation in the hvPR8 virus eliminate the RNAP II degradation capacity of the CAL strain. This CAL virus was less pathogenic than the wild-type (wt) CAL virus in mice, resembling the reported reduced pathogenicity of lvPR8 compared with hvPR8 in the mouse model. These results suggest that RNAP II degradation contributes to viral pathogenicity in in vivo models and that specific mutations in PB2 and PA subunits generate attenuated viruses.
MATERIALS AND METHODS
Ethics statement.
All the procedures that required the use of animals complied with Spanish and European legislation concerning vivisection and the use of genetically modified organisms, and the protocols were approved by the National Center for Biotechnology Ethics Committees on Animal Experimentation and the Consejo Superior de Investigaciones Científicas (CSIC) Bioethics Subcommittee. In particular, we followed the guidelines included in the current Spanish legislation on protection for animals used in research and other scientific aims (RD 1201/2005, 10 October, and the current European Union Directive 86/609/CEE, DOCE 12.12.86 [N.L358/1 to N.L358/28]) on protection for animals used in experimentation and other scientific aims.
Biological materials.
Human embryonic kidney 293T (HEK293T) and Madin-Darby canine kidney (MDCK) cells were grown in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal calf serum. The plasmid pHH-NS CAT expresses, under an RNA polymerase I promoter, the chloramphenicol acetyltransferase (CAT) gene in antisense orientation flanked by the untranslated region (UTR) sequences of the NS segment. It was constructed by insertion of the NS-CAT fragment from pT7NSCAT-RT (18) into the pHH plasmid.
Western blotting.
Cells were infected at a multiplicity of infection (MOI) of 3 PFU/cell. At different times postinfection (p.i.), cells were collected in Laemmli sample buffer. Western blotting was carried out as described previously (10). The following primary antibodies were used: for RNA polymerase II, monoclonal antibody 8WG16 (1:500) from Covance; for PA, monoclonal antibodies 2 and 9 (1:250 each) (19); for PB1, a rabbit polyclonal antibody (1:1,000) (20); for NP, a rabbit polyclonal antibody (1:5,000) (21); for DDX5, a goat anti-human antibody (1:1,000) from Abcam; for β-tubulin, a mouse monoclonal antibody (1:15,000) from Sigma. RNAP II quantification was done by quantifying the Western blots by densitometry using the ImageJ program and normalizing by the β-tubulin levels.
Generation of recombinant viruses.
For generation of the different lvPR8 and hvPR8 recombinant viruses, mutations were introduced into the corresponding ambisense expression vector pDZ, which contains a human RNA polymerase I promoter and a chicken β-actin promoter, using the QuikChange Site-Directed mutagenesis kit (Stratagene), and rescue of lvPR8 and hvPR8 recombinant viruses was done as previously described (13).
Rescue of the recombinant A/California/04/2009 viruses was done as previously described (22). The mutations in the PA and PB2 segments were introduced into the corresponding pPolI plasmids, using the QuikChange Site-Directed mutagenesis kit (Stratagene).
In vitro infection.
Cultured MDCK cells were infected at 5 × 10−3 PFU/cell (low MOI), or HEK293T cells were infected at 3 PFU/cell (high MOI). After 1 h, the unbound virus was rinsed off, and at different times postinfection, cell supernatants (MDCK) or cell extracts (HEK293T) were collected and used for virus titration by plaque assay or Western blotting, respectively.
CAT assays.
HEK293T cells were transfected with pHH-NS CAT plasmid. Twenty-four hours later, the cells were infected with the different recombinant influenza viruses at 3 PFU/cell. At 10 h p.i., cell extracts were collected and CAT accumulation was assayed by enzyme-linked immunosorbent assay (ELISA).
In vivo infection.
Female BALB/c mice (6 to 8 weeks old) were infected intranasally with 5 × 106 PFU of the corresponding viruses or were mock infected. The animals were monitored daily for body weight. On days 2, 4, and 6 postinfection, mice were euthanized and necropsied. Clinical specimens from lungs were homogenized in phosphate-buffered saline–0.3% bovine serum albumin in a homogenizer and used for determination of viral titers by plaque assay.
Virus titration.
Tissue samples from mice were homogenized (10% [wt/vol]) in phosphate-buffered saline (PBS), and debris was pelleted by centrifugation (2,000 × g; 5 min). Virus titers of triplicate tissue samples or cell culture supernatants were determined by standard plaque assay on MDCK cells.
RESULTS
Characterization of PA and PB2 residues involved in RNAP II degradation.
Three changes in the PB2 sequence (amino acids [aa] 105, 456, and 504) and two in PA (aa 193 and 550) differentiate PB2 and PA of lvPR8 from those of hvPR8. To characterize the individual contributions of these amino acids to the RNAP II degradation process, recombinant viruses were constructed containing the corresponding residues from lvPR8 in the hvPR8 context and the opposite, the residues present in hvPR8 in the context of lvPR8. Table 1 shows the nomenclature of these PB2 and PA residues, depending on the amino acid that is present in the lvPR8 or hvPR8 virus. Recombinant viruses containing each of the individual residues, as well as the double mutations, were constructed and confirmed by sequencing. The rescued viruses were amplified and used to infect HEK293T cells at a high multiplicity of infection to examine their ability to degrade RNAP II. In parallel, the accumulation levels of DDX5, a host factor that interacts with influenza virus polymerase (23); of PB1 and PA as markers for the progression of the infection; and of β-tubulin as a loading indicator were also examined. As reported, the global change of individual PB2 or PA subunits of lvPR8 in the hvPR8 virus decreases the ability of hvPR8 to degrade RNAP II, producing an intermediate degradation phenotype compared with the original virus (9). Examination of the changes introduced in the PB2 subunit of lvPR8 corresponding to hvPR8 allowed the identification of residue 504 (rlvPB23, rlvPB21,3, and rlvPB22,3, where subscript numbers refer to the subscripts in the column “Mutation name” in Table 1 [i.e., rlvPB21,3 refers to a virus with the mutations I105M and I504V in PB2 protein]) as the main element responsible for increased RNAP II degradation, while residues 105 (rlvPB21) and 456 (rlvPB22 and rlvPB21,2) did not seem to contribute to the degradation process (Fig. 1A). Regarding the PA subunit, whereas residue 193 (rlvPA3) did not contribute to RNAP II degradation, residue 550 (rlvPA2) did cause increased degradation (Fig. 1B). Furthermore, study of the corresponding changes in the hvPR8 virus containing the PB2 and PA residues present in lvPR8 confirmed that residues 504 (rhvPB26, rhvPB24,6, and rhvPB25,6) of PB2 and 550 (rlvPA4) of PA were responsible for the decreased capacity of the corresponding viruses to degrade RNAP II (Fig. 2). With this information, two additional recombinant viruses were constructed, containing aa 504 of PB2 and 550 of PA from lvPR8 or hvPR8 in the opposite background (rlvPB23-PA2 and rhvPB26-PA4), and their capacities for RNAP II degradation were examined (Fig. 3). As expected, lvPR8 with these mutations acquires degradation capacity, and conversely, hvPR8 with these changes was unable to degrade the protein (here, rlvPB23-PA2 and rhvPB26-PA4 are referred to as rlvPR8 DM and rhvPR8 DM, indicating the presence of the double mutations in PB2 and PA that produce the change in RNAP II degradation). In all cases (Fig. 1 to 3), the experiments were repeated 3 to 5 times, and the quantification of RNAP II levels from these experiments is shown at the bottom of the corresponding RNAP II Western blots. In summary, amino acids 504 of PB2 and 550 of PA play a major role in the ability of the PR8 influenza virus to degrade RNAP II.
TABLE 1.
Mutations in PB2 and PA of hvPR8 and lvPR8 implicated in RNAP II degradation
| Virus | Mutation name | Mutation |
|
|---|---|---|---|
| PB2 | PA | ||
| lvPR8 | PB21 | I105M | |
| PB22 | N456D | ||
| PB23 | I504V | ||
| PA1 | Q193H | ||
| PA2 | I550L | ||
| hvPR8 | PB24 | M105I | |
| PB25 | D456N | ||
| PB26 | V504I | ||
| PA3 | H193Q | ||
| PA4 | L550I | ||
FIG 1.

Identification of amino acids of PB2 and PA lvPR8 involved in loss of RNAP II degradation. HEK293T cells were mock infected or infected with the recombinant lvPR8 viruses listed in panel B, right, and at 15 h p.i., RNAP II and the indicated proteins were monitored by Western blotting of total cell extracts. Quantification of the amount of RNAP II during the virus infection normalized to the β-tubulin levels is shown below the RNAP II blots (means ± standard deviations). (A) Recombinant lvPB2 viruses. (B) Recombinant lvPA viruses. The asterisks indicate affected mutants. Three to five independent experiments were carried out, and a representative experiment is shown.
FIG 2.
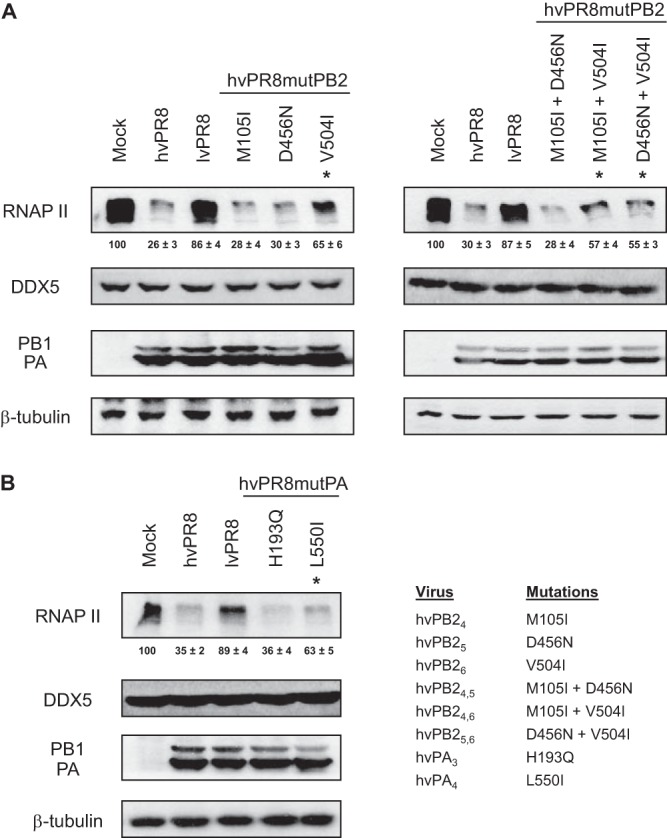
Identification of amino acids of PB2 and PA hvPR8 implicated in RNAP II degradation. HEK293T cells were mock infected or infected with the recombinant hvPR8 viruses indicated in panel B, right, and at 15 h p.i., RNAP II and the indicated proteins were monitored in Western blots of total cell extracts. Quantification of the amount of RNAP II during the virus infection normalized to the β-tubulin levels is shown below the RNAP II blots (means ± standard deviations). (A) Recombinant hvPB2 viruses. (B) Recombinant hvPA viruses. The asterisks indicate affected mutants. Three to five independent experiments were carried out, and a representative experiment is shown.
FIG 3.
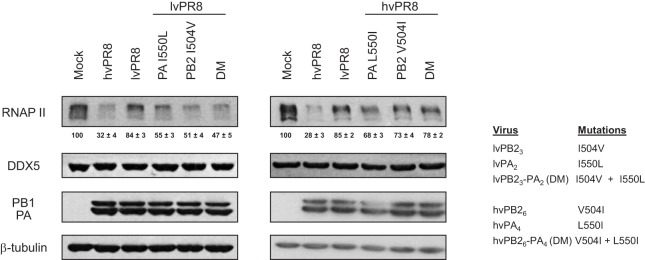
Analysis of RNAP II degradation induced by double-mutant lvPR8 and hvPR8 viruses. HEK293T cells were mock infected or infected with the recombinant lvPR8 (left) or hvPR8 (center) viruses indicated on the right. At 15 h p.i., RNAP II and the indicated proteins were monitored in Western blots of total cell extracts. Quantification of the amount of RNAP II during the virus infection normalized to the β-tubulin levels is shown below of the RNAP II blots (means ± standard deviations). Three to five independent experiments were carried out, and a representative experiment is shown.
Previous reports have indicated the individual contributions of aa 504 of PB2 and 550 of PA to modulating the influenza virus polymerase activity of lvPR8. It has been shown that an isoleucine-to-valine change at position 504 in PB2 (PB23) and an isoleucine-to-leucine change at position 550 in PA (PA2) enhanced the activity of the polymerase complex of the virus in a minireplicon system, although the combined contributions of the two residues were not examined (24). Then, the pooled contributions of these changes in hvPR8 and lvPR8 recombinant viruses were assayed. With that aim, a transfection-infection system was used in which HEK293T cells were first transfected with plasmid pHH-NSCAT, expressing a negative-sense virus-like CAT RNA under the polymerase I promoter, and then infected with different recombinant viruses. After 10 h p.i., the CAT accumulation and protein content were measured and used to quantify the replication/transcription activity of the viral polymerase. Insertion of mutations in PB2 and PA in the lvPR8 virus (rlvPR8 DM) that increased the ability of the strain to degrade RNAP II caused an important increase in the activity of the viral polymerase (Fig. 4A). The inclusion of the corresponding PB2 and PA mutations in the hvPR8 strain, which decreased the degradation capacity (rhvPR8 DM), caused only a moderate decrease in polymerase activity (Fig. 4B). Therefore, the insertion of these mutations in the viral polymerase has an impact on viral polymerase activity, although a clear correlation between RNAP II degradation and polymerase activity cannot be established.
FIG 4.

Polymerase activities of recombinant lvPR8 and hvPR8 double mutants. HEK293T cells were transfected with a plasmid encoding a CAT viral minigenome construct. Twenty-four hours later, the cells were infected at an MOI of 3 with the recombinant wt and double-mutant (DM) lvPR8 (A) or hvPR8 (B) virus. The cultures were harvested at 10 h p.i., and the amount of CAT protein present in the total cell extract was analyzed by ELISA. On the right, aliquots of the same samples were analyzed for the presence of PB1 and β-tubulin by Western blotting. Three independent experiments were carried out, and a representative experiment is shown. The error bars indicate the standard errors of the mean. Student's t test was performed to determine the P values. *, P < 0.05.
Effects of PB2 504 and PA 550 residues on virus pathogenicity.
The above-mentioned data indicate the roles of specific PB2 and PA residues in the degradation of RNAP II triggered by influenza virus infection. Subsequently, we examined whether the contributions of these specific amino acids could also take place in a different viral background. We have previously reported that influenza viruses of avian origin belonging to different subtypes (H5N1 and H9N2) also trigger RNAP II degradation in infected cells (9). Taking into account the recent first pandemic of the 21st century caused by a reassortant influenza A virus containing porcine, avian (PB2 and PA), and human segments, we chose a representative strain of the A(H1N1)pdm09 viruses, the CAL strain, to evaluate the contributions of PB2 and PA residues in a very different polymerase context than that previously characterized, the laboratory-passaged human origin PR8 strain.
Sequence comparison of the PB2 and PA subunits of the CAL virus showed that PB2 residue 105 was different than that present in lvPR8 and hvPR8; PB2 amino acid 456 was the same in lvPR8; and PB2 residue 504, which modulates RNAP II degradation, is a valine, as in hvPR8 (Table 2). Regarding PA, a glutamine is present at position 193, as in lvPR8, while PA 550 is a leucine, as in hvPR8. Therefore, the CAL strain contains the same amino acids as hvPR8 at PB2 504 and PA 550. With this information, we checked whether these positions modulate RNAP II degradation, as well as other functions of the CAL virus, as they do in the PR8 strain. To do so, recombinant viruses containing all the genes from the CAL strain but introducing the individual PB2 or PA residues of lvPR8, or the two amino acids together, were constructed. The recombinant wild type (rCAL wt) and the mutant CAL viruses with the inserted mutations were used for HEK293T infection at 3 PFU/cell, and RNAP II, DDX5, PB1, and NP viral protein and β-tubulin contents were evaluated by Western blotting. Quantitative data on RNAP II accumulation are presented at the bottom of the RNAP II Western blots. The results (Fig. 5A) indicated that the rCAL wt virus degrades RNAP II, the viruses containing one of the PB2 or PA amino acids of lvPR8 showed decreased RNAP II degradation, and the double mutant (rCAL DM) did not cause RNAP II degradation. Thus, as in hvPR8, a valine at position 504 of PB2, together with a leucine at PA 550, is required to induce RNAP II degradation by the pandemic CAL strain, which has avian PB2 and PA segments. On the other hand, the accumulation levels of viral proteins were similar in the rCAL wt virus and recombinant mutant CAL viruses. Next, we evaluated the activity of the viral polymerase in the rCAL wt and the rCAL DM recombinant viruses by using the transfection-infection protocol described above. In agreement with the small reduction observed in the polymerase activity of the rhvPR8 DM virus, the introduction of these mutations in the CAL strain also produced a similar weak reduction in its polymerase activity (Fig. 5B).
TABLE 2.
Amino acid differences between lvPR8, hvPR8, and CAL virusesa

Residues not implicated in RNAP II degradation are in black. Amino acids that confer the ability for RNAP II degradation are shown in red, whereas residues involved in loss of RNAP II degradation are in green.
FIG 5.
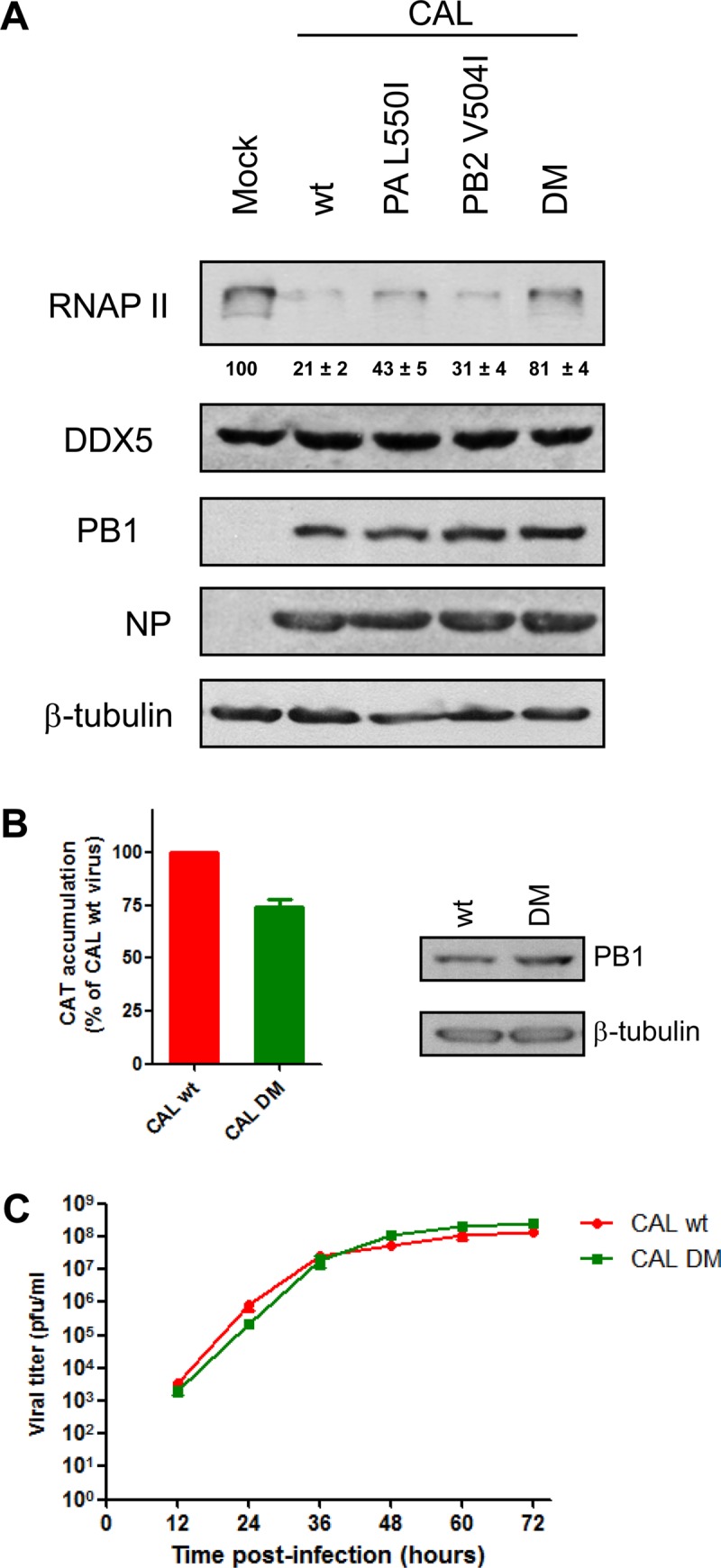
A recombinant CAL double mutant is affected in RNAP II degradation. (A) HEK293T cells were mock infected or infected with the indicated recombinant CAL viruses, and at 15 h p.i., RNAP II and the indicated proteins were monitored in Western blots of total cell extracts. Quantification of the amount of RNAP II during the virus infection normalized to the β-tubulin levels (means ± standard deviations) is shown below the RNAP II blot. (B) HEK293T cells were transfected with a plasmid encoding a CAT viral minigenome construct. Twenty-four hours later, the cells were infected with 3 PFU/cell of either CAL wt or CAL DM. The cultures were harvested at 10 h p.i., and the amount of CAT protein present in the total cell extract was analyzed by ELISA. (Right) Aliquots of the same samples were also analyzed for the presence of PB1 and β-tubulin by Western blotting. (C) Cultured MDCK cells were infected at 10−3 PFU/cell with the CAL wt or CAL DM influenza virus. At the indicated times p.i., cell supernatants were collected, and the virus titer was determined by plaque assay. In all cases, three independent experiments were carried out, and a representative experiment is shown. The error bars indicate the standard errors of the mean.
These results, the correct accumulation of viral proteins in rCAL DM infection compared with recombinant rCAL wt at a high MOI (Fig. 5A) and the small reduction in polymerase activity in the transfection-infection studies (Fig. 5B), indicated that the introduction of the specific PB2 and PA amino acids that abolish RNAP II degradation capacity does not profoundly alter its viral functions in cultured cells. To further examine this possibility, we studied the rCAL wt and rCAL DM kinetics in cell cultures. MDCK cells were infected with each virus at a low multiplicity of infection, and viral titers were determined at different times postinfection. The two viruses presented similar kinetics and reached similar titers at later times (Fig. 5C), confirming that the introduction of these mutations does not produce an important impairment in virus fitness in cultured cells, thus allowing the examination of its virulence in vivo in animal models.
Previous reports indicated that hvPR8 presents higher pathogenicity than lvPR8 in mice and that the dissimilarity can be attributed to viral polymerase differences (11, 20). We examined whether the rCAL wt virus presents differences in pathogenicity compared to rCAL DM in the mouse model. BALB/c mice were intranasally infected with 5 × 106 PFU of rCAL wt or rCAL DM virus or were mock infected. Survival and body weight were monitored daily for 2 weeks. No loss of body weight was observed in mock-infected mice, whereas mice infected with rCAL wt and rCAL DM viruses had a peak of weight loss 5 to 7 days after inoculation. However, in the rCAL wt virus-infected mice, the weight loss was more pronounced (Fig. 6A). Importantly, 75% lethality in rCAL wt virus-infected mice was observed in comparison with 100% survival in rCAL DM virus- or mock-infected animals (Fig. 6B). Since influenza virus primarily infects the lungs of mice, lung samples of the infected animals were used to determine the viral titers at different days postinfection (Fig. 6C). At 2 days p.i., the virus titer reached its maximum value. The presence of virus was gradually reduced in the lungs of both groups of infected animals, and by 6 days p.i., infectious virus was still detectable, with higher viral titers in wt-infected mice, although this observation was not statistically significant. Collectively, these results indicate that the same specific residues of PB2 and PA modulate the capacities of the PR8 and CAL viruses to induce RNAP II degradation, which correlates with viral pathogenicity in mice.
FIG 6.

Double-mutant A/California/04/2009 virus is attenuated in vivo. Mice were intranasally inoculated with 5 × 106 PFU (50 μl) of either CAL wt or CAL DM influenza virus or were mock infected with 50 μl of PBS. (A) Body weights were determined daily for 14 days and are depicted as the percentage of body weight at the time of inoculation. The data are the mean body weights of the mice (n = 4). The error bars represent standard errors of the mean. (B) The mice were monitored daily for survival for 14 days. Animals that lost 25% of their body weight were euthanized and counted as dead animals. (C) Three mice from each group were euthanized on days 2, 4, and 6 postinfection for lung virus titration. The error bars show standard errors of the mean. For body weights, Student's t test was performed to determine the P value. *, P < 0.05. For survival, statistical significance was assessed by a log rank (Mantel-Cox) test. *, P < 0.05. For panels A and B, two independent experiments were carried out, and a representative experiment is shown.
DISCUSSION
Viruses do not possess the full equipment required for genome expression. They are thus obliged to utilize host cell factors and therefore to compete for and manipulate the host cell to their own benefit. They have developed different strategies to interfere with host cell expression, which is described as cellular shutoff, by trying to hijack the cellular expression machinery while at the same time avoiding the antiviral response and increasing viral pathogenicity. Understanding the pathogenicity of influenza viruses is critical for predicting several aspects of viral infection, such as the severity of the infection or even the pandemic potential of influenza viruses. Viral genetic determinants for high virulence have been extensively studied, especially for highly virulent viruses, such as the 1918 virus that produced the most devastating human pandemic; the avian H5N1 virus that was transmitted to humans, causing a high rate of mortality; or the recent new and mild H1N1 2009 pandemic strain. Despite extensive studies, no clear common pathogenicity traits have been determined, although there is general agreement as to the prominent contribution of viral polymerase, hemagglutinin, neuraminidase, and the NS1 protein (25–28).
Viral polymerase plays a crucial role in controlling the expression of the viral and host genomes, as well as viral pathogenicity. Studies on highly pathogenic avian influenza H5N1 viruses have shown that virulence in mammals is directly influenced by mutations in PB1 and PB2 polymerase subunits (29). Moreover, experiments with hybrid polymerase constructs suggested that the PA, and to a lesser extent the PB2, subunit of the polymerase was responsible for increased polymerase activity of a highly pathogenic duck H5N1 strain (30) and that the specific replacement of 2 amino acids, S224P and N383D, in PA contributed to the highly virulent phenotype of H5N1 infection in domestic ducks (31). In addition, specific mutations of PB2 (A221T) and PA (D529N) contributed to the high pathogenicity of a 2009 pandemic strain isolated from a fatal case (32). Accordingly, specific mutations in polymerase subunits produce attenuated temperature-sensitive viruses (33) that are commonly used for live-vaccine production (34–36). Influenza virus polymerase also has a role in the RNAP II degradative process caused by infection, since its expression from its cloned cDNAs is sufficient to cause RNAP II degradation (10). As we previously reported, ubiquitin-mediated proteasomal degradation is not involved in the RNAP II degradation caused by influenza virus infection, since degradation occurs similarly in the presence of the proteasome inhibitor MG132 (10). In agreement with that, we have not observed differences in RNAP II ubiquitination with the different viruses used in this study (data not shown), indicating that there is no correlation between RNAP II degradation and ubiquitination. RNAP II degradation appears to contribute to host cell shutoff, since inhibition of cellular transcription correlates with the degree of degradation generated by different influenza virus strains (9, 10). New polymerase-related viral strategies controlling host cell shutoff have been recently described. A new influenza virus-encoded protein named PA-X that represses cellular gene expression and modulates viral pathogenicity is produced through ribosomal frameshifting and contains the endonuclease domain of the PA polymerase subunit, followed by a new sequence translated from a different reading frame (37). In addition, new influenza virus proteins translated from the 11th and 13th in-frame AUG codons in PA mRNA have also been described (38). These new PA-encoded proteins are N-terminally truncated forms of PA, named PA-N155 and PA-N182, and mutant viruses lacking these proteins replicate more slowly in cell culture and have lower pathogenicity in mice (38). The PB1-F2 protein, encoded by an open reading frame overlapping the PB1 open reading frame, needs to be added to the new polymerase gene-encoded proteins that contribute to viral pathogenesis (39).
Using recombinant viruses containing PA and PB2 residues, which distinguish viral strains that degrade (hvPR8) or do not degrade (lvPR8) RNAP II, we have characterized the specific residues involved in RNAP II degradation and demonstrated that the combination of a valine at position 504 in the PB2 subunit and a leucine at position 550 in the PA subunit confers the ability to degrade RNAP II (Fig. 3). This combination also determines the ability to degrade RNAP II in a completely different viral background, such as the CAL 2009 pandemic strain (Fig. 5).
Searches in the databases indicate that these residues are highly conserved in influenza A viruses. Among more than 9,000 influenza viruses of human origin, the total calculated frequencies of appearance of PB2 V504 and PA L550 are 99.77% and 99.88%, respectively (Table 3). Sixteen of these sequences contain isoleucine at PA 550 and 11 at PB2 504, but 9 of the downloaded sequences belong to PR8 viruses sequenced in different laboratories, indicating that the total frequency of PB2 V504 and PA L550 is even higher. Moreover, PB2 V504 and PA L550 are also highly conserved in influenza A viruses of avian and swine origins (Table 3), suggesting an important role of these amino acids in the polymerase context. On the other hand, the contribution of PA polymerase subunits to RNAP II degradation does not seem to be mediated by the newly PA-X-expressed protein. The UCCUUUCGU (positions 567 to 576 of the PA coding sequence) motif that promotes the frameshifting is located just before the PA triplet encoding aa 193 of PA, which is one of the two residues that distinguishes PAs from hvPR8 and lvPR8. As we have shown, recombinant rlvPA1, which contains all the lvPR8 segments with a Q193H mutation in the PA gene that restores hvPR8 aa 193, does not increase the ability of the rescued virus to degrade RNAP II (Fig. 1). This recombinant virus would express a PA-X identical to that of hvPR8 but an entire PA containing aa 550 from lvPR8. In addition, CAL wt and CAL DM recombinant viruses would express identical PA-X proteins, although a different point mutant PA, and the latter virus does not degrade RNAP II. Therefore, the PA-X protein does not mediate the RNAP II degradation process. Contributions by the other newly described PA-N155 and PA-N182 N-truncated proteins, to RNAP II degradation cannot be excluded, since these proteins would contain two amino acid differences between lvPR8 and hvPR8, aa 193 and 550 of PA, and one difference between CAL and CAL DM (aa 550), and this residue is relevant to RNAP II degradation. Interaction between the N-terminal 100 aa of the PA and PB2 subunits has been described (40), but the new N-terminally truncated PA proteins do not interact with PB2, since they do not contain the PB2-interacting domain. Taking into account that two residues located in different polymerase subunits (aa 550 in PA and 504 in PB2) are required for full RNAP II degradation activity and the absence of PB2 interaction, the contribution of PA-truncated forms to the degradation process seems unlikely.
TABLE 3.
Frequencies of different residues at PA 550 and PB2 504 positions in human, avian, and swine virusesc
Nine of the sequences belong to A/Puerto Rico/8/1934.
Nine of the sequences belong to A/Puerto Rico/8/1934.
Residues not implicated in RNAP II degradation are in black. Amino acids that confer the ability for RNAP II degradation are shown in red, whereas residues involved in loss of RNAP II degradation are in green.
The specific V504PB2-L550PA combination restores the ability of lvPR8 DM for RNAP II degradation and, conversely, I504PB2-I550PA combination inhibits the degradation capacity of CAL and hvPR8 double mutants. In agreement with previous publications, induction of RNAP II degradation in infected cells clearly correlates with viral pathogenicity in vivo, as has been observed in three different viral systems using both H1N1 and H3N2 viruses. The three systems include the higher pathogenicity of hvPR8 (H1N1) than of lvPR8 (13), the decreased pathogenicity of a recombinant virus of A/Victoria/3/75 (H3N2) virus that expresses a mutant PA protein with decreased proteolytic activity (10, 41), and the decreased pathogenicity of the CAL DM (H1N1) virus described here. Together, these data suggest that impairment of RNAP II could contribute to diminishing the establishment of the host antiviral response, thus contributing to viral pathogenicity. In addition, it is important to emphasize that isoleucine at positions 504 of PB2 and 550 of PB2 gives rise to attenuated viruses, and therefore, incorporation of these mutations could be useful in designing attenuated virus for vaccine strategies.
ACKNOWLEDGMENTS
We appreciate the cooperation of G. Kochs and Y. Kawaoka in providing plasmids expressing the segments of the hvPR8 and lvPR8 viruses and the A/California/04/09 viruses, respectively. We are indebted to P. Gastaminza, A. Falcón, and J. Ortín for their criticisms of the manuscript. The invaluable contribution of C. Dunn in correcting the manuscript is recognized. The technical assistance of Y. Fernández and N. Zamarreño is also gratefully acknowledged.
This work was supported by the Ministerio de Ciencia e Innovación, Plan Nacional de Investigacion Científica, Desarrollo e Innovacion Tecnologica (BFU2011-26175), Ciber de Enfermedades Respiratorias, and Programa de Investigación sobre la Gripe Pándemica GR09/0023 (ISCIII).
Footnotes
Published ahead of print 8 January 2014
REFERENCES
- 1.Krug RM, Broni BA, Bouloy M. 1979. Are the 5′-ends of influenza viral mRNAs synthesized in vivo donated by host mRNAs? Cell 18:329–334. 10.1016/0092-8674(79)90052-7 [DOI] [PubMed] [Google Scholar]
- 2.Chen Z, Li Y, Krug RM. 1999. Influenza A virus NS1 protein targets poly(A)-binding protein II of the cellular 3′-end processing machinery. EMBO J. 18:2273–2283. 10.1093/emboj/18.8.2273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nemeroff ME, Barabino SML, Li Y, Keller W, Krug RM. 1998. Influenza virus NS1 protein interacts with the cellular 30 kDa subunit of CPSF and inhibits 3′ end formation of cellular pre-mRNAs. Mol. Cell 1:991–1000. 10.1016/S1097-2765(00)80099-4 [DOI] [PubMed] [Google Scholar]
- 4.Fortes P, Beloso A, Ortín J. 1994. Influenza virus NS1 protein inhibits pre-mRNA splicing and blocks RNA nucleocytoplasmic transport. EMBO J. 13:704–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beloso A, Martínez C, Valcárcel J, Fernández-Santarén J, Ortín J. 1992. Degradation of cellular mRNA during influenza virus infection: its possible role in protein synthesis shutoff. J. Gen. Virol. 73:575–581. 10.1099/0022-1317-73-3-575 [DOI] [PubMed] [Google Scholar]
- 6.Inglis SC. 1982. Inhibition of host protein synthesis and degradation of cellular mRNAs during infection by influenza and herpes simplex virus. Mol. Cell. Biol. 2:1644–1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burgui I, Yángüez E, Sonenber N, Nieto A. 2007. Influenza mRNA translation revisited: is the eIF4E cap-binding factor required for viral mRNA translation? J. Virol. 81:12427–12438. 10.1128/JVI.01105-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Katze MG, DeCorato D, Krug RM. 1986. Cellular mRNA translation is blocked at both initiation and elongation after infection by influenza virus or adenovirus. J. Virol. 60:1027–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rodriguez A, Perez-Gonzalez A, Hossain MJ, Chen LM, Rolling T, Perez-Brena P, Donis R, Kochs G, Nieto A. 2009. Attenuated strains of influenza A viruses do not induce degradation of RNA polymerase II. J. Virol. 83:11166–11174. 10.1128/JVI.01439-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rodriguez A, Pérez-Gonzalez A, Nieto A. 2007. Influenza virus infection causes specific degradation of the largest subunit of cellular RNA polymerase II. J. Virol. 81:5315–5324. 10.1128/JVI.02129-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vreede FT, Chan AY, Sharps J, Fodor E. 2010. Mechanisms and functional implications of the degradation of host RNA polymerase II in influenza virus infected cells. Virology 396:125–134. 10.1016/j.virol.2009.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alfonso R, Rodriguez A, Rodriguez P, Lutz T, Nieto A. 2013. CHD6, a cellular repressor of influenza virus replication is degraded in human alveolar epithelial cells and mice lungs during infection. J. Virol. 87:4534–4544. 10.1128/JVI.00554-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grimm D, Staeheli P, Hufbauer M, Koerner I, Martinez-Sobrido L, Solorzano A, Garcia-Sastre A, Haller O, Kochs G. 2007. Replication fitness determines high virulence of influenza A virus in mice carrying functional Mx1 resistance gene. Proc. Natl. Acad. Sci. U. S. A. 104:6806–6811. 10.1073/pnas.0701849104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garten RJ, Davis CT, Russell CA, Shu B, Lindstrom S, Balish A, Sessions WM, Xu X, Skepner E, Deyde V, Okomo-Adhiambo M, Gubareva L, Barnes J, Smith CB, Emery SL, Hillman MJ, Rivailler P, Smagala J, de Graaf M, Burke DF, Fouchier RA, Pappas C, Alpuche-Aranda CM, Lopez-Gatell H, Olivera H, Lopez I, Myers CA, Faix D, Blair PJ, Yu C, Keene KM, Dotson PD, Jr, Boxrud D, Sambol AR, Abid SH, St George K, Bannerman T, Moore AL, Stringer DJ, Blevins P, Demmler-Harrison GJ, Ginsberg M, Kriner P, Waterman S, Smole S, Guevara HF, Belongia EA, Clark PA, Beatrice ST, Donis R, Katz J, Finelli L, Bridges CB, Shaw M, Jernigan DB, Uyeki TM, Smith DJ, Klimov AI, Cox NJ. 2009. Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science 325:197–201. 10.1126/science.1176225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neumann G, Noda T, Kawaoka Y. 2009. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 459:931–939. 10.1038/nature08157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Louie JK, Acosta M, Winter K, Jean C, Gavali S, Schechter R, Vugia D, Harriman K, Matyas B, Glaser CA, Samuel MC, Rosenberg J, Talarico J, Hatch D. 2009. Factors associated with death or hospitalization due to pandemic 2009 influenza A(H1N1) infection in California. JAMA 302:1896–1902. 10.1001/jama.2009.1583 [DOI] [PubMed] [Google Scholar]
- 17.Perez-Padilla R, de la Rosa-Zamboni D, Ponce de Leon S, Hernandez M, Quinones-Falconi F, Bautista E, Ramirez-Venegas A, Rojas-Serrano J, Ormsby CE, Corrales A, Higuera A, Mondragon E, Cordova-Villalobos JA. 2009. Pneumonia and respiratory failure from swine-origin influenza A (H1N1) in Mexico. N. Engl. J. Med. 361:680–689. 10.1056/NEJMoa0904252 [DOI] [PubMed] [Google Scholar]
- 18.Perales B, Sanz-Ezquerro JJ, Gastaminza P, Ortega J, Fernandez-Santarén J, Ortín J, Nieto A. 2000. The replication activity of influenza virus polymerase is linked to the capacity of the PA subunit to induce proteolysis. J. Virol. 74:1307–1312. 10.1128/JVI.74.3.1307-1312.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bárcena J, Ochoa M, de la Luna S, Melero JA, Nieto A, Ortín J, Portela A. 1994. Monoclonal antibodies against the influenza virus PB2 and NP polypeptides interfere with the initiation step of viral mRNA synthesis in vitro. J. Virol. 68:6900–6909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gonzalez S, Ortín J. 1999. Distinct regions of influenza virus PB1 polymerase subunit recognize vRNA and cRNA templates. EMBO J. 18:3767–3775. 10.1093/emboj/18.13.3767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coloma R, Valpuesta JM, Arranz R, Carrascosa JL, Ortin J, Martin-Benito J. 2009. The structure of a biologically active influenza virus ribonucleoprotein complex. PLoS Pathog. 5:e1000491. 10.1371/journal.ppat.1000491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neumann G, Watanabe T, Ito H, Watanabe S, Goto H, Gao P, Hugues M, Perea DR, Donis R, Hoffmann E, Hobom G, Kawaoka Y. 1999. Generation of influenza A viruses entirely from cloned cDNAs. Proc. Natl. Acad. Sci. U. S. A. 96:9345–9350. 10.1073/pnas.96.16.9345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jorba N, Juarez S, Torreira E, Gastaminza P, Zamarreno N, Albar JP, Ortin J. 2008. Analysis of the interaction of influenza virus polymerase complex with human cell factors. Proteomics 8:2077–2088. 10.1002/pmic.200700508 [DOI] [PubMed] [Google Scholar]
- 24.Rolling T, Koerner I, Zimmermann P, Holz K, Haller O, Staeheli P, Kochs G. 2009. Adaptive mutations resulting in enhanced polymerase activity contribute to high virulence of influenza A virus in mice. J. Virol. 83:6673–6678. 10.1128/JVI.00212-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Wit E, Kawaoka Y, de Jong MD, Fouchier RA. 2008. Pathogenicity of highly pathogenic avian influenza virus in mammals. Vaccine 26(Suppl 4):D54–D58. 10.1016/j.vaccine.2008.07.072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Imai H, Shinya K, Takano R, Kiso M, Muramoto Y, Sakabe S, Murakami S, Ito M, Yamada S, Le MT, Nidom CA, Sakai-Tagawa Y, Takahashi K, Omori Y, Noda T, Shimojima M, Kakugawa S, Goto H, Iwatsuki-Horimoto K, Horimoto T, Kawaoka Y. 2010. The HA and NS genes of human H5N1 influenza A virus contribute to high virulence in ferrets. PLoS Pathog. 6:e1001106. 10.1371/journal.ppat.1001106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tscherne DM, Garcia-Sastre A. 2011. Virulence determinants of pandemic influenza viruses. J. Clin. Invest. 121:6–13. 10.1172/JCI44947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watanabe T, Tisoncik-Go J, Tchitchek N, Watanabe S, Benecke AG, Katze MG, Kawaoka Y. 2013. 1918 influenza virus hemagglutinin (HA) and the viral RNA polymerase complex enhance viral pathogenicity, but only HA induces aberrant host responses in mice. J. Virol. 87:5239–5254. 10.1128/JVI.02753-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lycett SJ, Ward MJ, Lewis FI, Poon AF, Kosakovsky Pond SL, Brown AJ. 2009. Detection of mammalian virulence determinants in highly pathogenic avian influenza H5N1 viruses: multivariate analysis of published data. J. Virol. 83:9901–9910. 10.1128/JVI.00608-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leung BW, Chen H, Brownlee GG. 2010. Correlation between polymerase activity and pathogenicity in two duck H5N1 influenza viruses suggests that the polymerase contributes to pathogenicity. Virology 401:96–106. 10.1016/j.virol.2010.01.036 [DOI] [PubMed] [Google Scholar]
- 31.Song J, Feng H, Xu J, Zhao D, Shi J, Li Y, Deng G, Jiang Y, Li X, Zhu P, Guan Y, Bu Z, Kawaoka Y, Chen H. 2011. The PA protein directly contributes to the virulence of H5N1 avian influenza viruses in domestic ducks. J. Virol. 85:2180–2188. 10.1128/JVI.01975-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodriguez A, Falcon A, Cuevas MT, Pozo F, Guerra S, Garcia-Barreno B, Martinez-Orellana P, Perez-Brena P, Montoya M, Melero JA, Pizarro M, Ortin J, Casas I, Nieto A. 2013. Characterization in vitro and in vivo of a pandemic H1N1 influenza virus from a fatal case. PLoS One 8:e53515. 10.1371/journal.pone.0053515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Snyder MH, Betts RF, DeBorde D, Tierney EL, Clements ML, Herrington D, Sears SD, Dolin R, Maassab HF, Murphy BR. 1988. Four viral genes independently contribute to attenuation of live influenza A/Ann Arbor/6/60 (H2N2) cold-adapted reassortant virus vaccines. J. Virol. 62:488–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.He W, Wang W, Han H, Wang L, Zhang G, Gao B. 2013. Molecular basis of live-attenuated influenza virus. PLoS One 8:e60413. 10.1371/journal.pone.0060413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Solorzano A, Ye J, Perez DR. 2010. Alternative live-attenuated influenza vaccines based on modifications in the polymerase genes protect against epidemic and pandemic flu. J. Virol. 84:4587–4596. 10.1128/JVI.00101-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Song H, Nieto GR, Perez DR. 2007. A new generation of modified live-attenuated avian influenza viruses using a two-strategy combination as potential vaccine candidates. J. Virol. 81:9238–9248. 10.1128/JVI.00893-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jagger BW, Wise HM, Kash JC, Walters KA, Wills NM, Xiao YL, Dunfee RL, Schwartzman LM, Ozinsky A, Bell GL, Dalton RM, Lo A, Efstathiou S, Atkins JF, Firth AE, Taubenberger JK, Digard P. 2012. An overlapping protein-coding region in influenza A virus segment 3 modulates the host response. Science 337:199–204. 10.1126/science.1222213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muramoto Y, Noda T, Kawakami E, Akkina R, Kawaoka Y. 2013. Identification of novel influenza A virus proteins translated from PA mRNA. J. Virol. 87:2455–2462. 10.1128/JVI.02656-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schmolke M, Manicassamy B, Pena L, Sutton T, Hai R, Varga ZT, Hale BG, Steel J, Perez DR, Garcia-Sastre A. 2011. Differential contribution of PB1-F2 to the virulence of highly pathogenic H5N1 influenza A virus in mammalian and avian species. PLoS Pathog. 7:e1002186. 10.1371/journal.ppat.1002186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hemerka JN, Wang D, Weng Y, Lu W, Kaushik RS, Jin J, Harmon AF, Li F. 2009. Detection and characterization of influenza A virus PA-PB2 interaction through a bimolecular fluorescence complementation assay. J. Virol. 83:3944–3955. 10.1128/JVI.02300-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huarte M, Falcón A, Nakaya Y, Ortín J, García-Sastre A, Nieto A. 2003. Threonine 157 of influenza virus PA polymerase subunit modulates RNA replication in infectious viruses. J. Virol. 77:6007–6013. 10.1128/JVI.77.10.6007-6013.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]