

ABSTRACT
The HIV-1 envelope glycoprotein binds cooperatively to its cellular receptor CD4 and a coreceptor, principally CXCR4 or CCR5. We have previously improved a natural amino-acid form of a scorpion toxin-derived CD4-mimetic peptide and in parallel generated sulfopeptide mimetics of the CCR5 amino terminus. Here we show that some fusions of these CCR5- and CD4-mimetic peptides, expressed as immunoadhesins, neutralize HIV-1 more efficiently than CD4-Fc or equimolar mixtures of immunoadhesin forms of each peptide. Specifically, double-mimetic peptides with linkers of 11 amino acids or greater, and with the CCR5-mimetic component preceding the CD4-mimetic component, were more efficient than constructs with shorter linkers or in a reverse orientation. The potency of these constructs derives from (i) their ability to simultaneously and cooperatively bind the CD4- and CCR5-binding sites of a single gp120 monomer of the HIV-1 envelope glycoprotein trimer and (ii) the ability of the CCR5-mimetic component to prevent the CD4-mimetic peptide from promoting infection when cellular CD4 is limiting. Thus, there is a significant advantage to simultaneously targeting both conserved regions of the HIV-1 envelope glycoprotein.
IMPORTANCE This report describes a novel class of peptides that potently inhibit HIV-1 entry. These peptides simultaneously target the receptor- and coreceptor-binding sites of the HIV-1 envelope glycoprotein gp120. Peptides of this class overcome key limitations of inhibitors that target only one gp120 binding region and illustrate the utility of binding the sulfotyrosine-binding pockets of gp120.
INTRODUCTION
The HIV-1 envelope glycoprotein is a trimeric complex of heterodimers composed of a surface glycoprotein, gp120, and a transmembrane component, gp41. HIV-1 entry is mediated by its envelope glycoprotein and requires cellular expression of CD4 and a coreceptor, principally CCR5 or CXCR4 (1–4). When the envelope glycoprotein binds CD4, gp120 undergoes conformational changes that promote its high-affinity association with a coreceptor (e.g., 21, 37, 40, and 45). Association of gp120 with a coreceptor induces additional conformational changes in gp41, which in turn promote mixing of the membrane lipids, ultimately facilitating fusion of the viral and cellular membranes.
High-affinity binding of the HIV-1 envelope glycoprotein to the coreceptor CCR5 requires sulfation of the amino-terminal tyrosines of CCR5. In addition, a number of antibodies which bind the coreceptor-binding site of gp120 contain sulfated tyrosines in their CDRH3 regions (5). Sulfated peptides based on the amino terminus of CCR5, or on the CDRH3 loops of sulfated antibodies, can specifically bind the HIV-1 envelope glycoprotein and inhibit HIV-1 entry (6, 7). Antibody-derived sulfopeptides retain an arrangement of sulfotyrosines similar to that of the amino terminus of CCR5 and bind a broad range of R5, X4, and dual-tropic envelope glycoproteins (7, 8). These CCR5-mimetic peptides bind gp120 inefficiently but more efficiently than peptides based directly on the CCR5 amino terminus. Binding is substantially improved in the presence of CD4 or a CD4-mimetic peptide (7–9).
Small CD4-mimetic peptides, based on a scorpion toxin scaffold, also bind the envelope glycoprotein and inhibit HIV-1 entry (10–12). These peptides induce the CD4-bound conformation of gp120 and, like soluble CD4, can promote infection when cellular CD4 is limiting (9, 12, 13). The best characterized of these peptides include nonnative amino acids, preventing their expression from mammalian cells or their improvement by phage-display techniques. To address these limitations, we previously generated a native amino acid form of this peptide, which we called CD4mim1. Inspection of the structures of gp120 bound to various CD4-mimetic peptides indicated that their amino termini were held by a disulfide bond in an orientation that sterically precluded amino-terminal extensions (9, 14). To study amino-terminal fusions with CCR5-mimetic peptides, we eliminated this first disulfide bond of CD4mim1. This peptide was subsequently improved by phage display, resulting in CD4mim6, a peptide with higher affinity for gp120 (12).
The affinity of CCR5-mimetic peptides for the HIV-1 envelope glycoprotein is low in the absence of soluble CD4 (7). CD4-mimetic peptides bind with higher affinity, but their neutralization potency is limited in part by a tendency to promote infection at subneutralizing concentrations (12). Because the binding sites of these peptides are close (separated by approximately 28 Å; Fig. 1A), we reasoned that fusions of CD4- and CCR5-mimetic peptides (“double-mimetic” peptides, represented in Fig. 1B and C) would bind gp120 cooperatively and with high avidity. We further anticipated that the CCR5-mimetic peptide would limit the ability of the CD4-mimetic peptide to promote infection. We show here that double-mimetic peptides can indeed overcome the limitations of each single-mimetic peptide and efficiently neutralize HIV-1.
FIG 1.

Receptor-mimetic constructs. (A) A ribbon diagram of HIV-1 gp120 (gray) is shown bound to a CD4-mimetic peptide (red) and to a sulfopeptide (blue, with sulfate oxygen and sulfur atoms shown in red and yellow, respectively). Sulfotyrosines were derived from the heavy chain of CD4-induced antibody 412d. The figure was generated by aligning the gp120 molecules of the gp120-CD4-412d complex (Protein Data Bank [PDB] 2QAD) with those of the gp120-F23 complex (PDB 1YYM) (10, 19). The dashed line indicates the 28-Å distance from the amino-terminal α-carbon of the CD4 mimetic to the α-carbon of 412d sulfotyrosine 100. (B) Diagram of mimetic-Fc fusion constructs studied here. CCR5mim3 is shown as a blue circle and CD4mim6 as a red circle, and the Fc domain (including the hinge, CH2, and CH3 domains of human IgG1) is indicated in gray. (C) Sequences of the single- and double-mimetic peptides used in this paper. Blue indicates CCR5mim3, and red indicates CD4mim6. Linker regions are not highlighted.
MATERIALS AND METHODS
Plasmids and protein expression.
Fc fusions were expressed using a previously described pcDM8-derived plasmid expressing the signal sequence of CD5 and the Fc region (comprising hinge and CH2 and CH3 domains) of human IgG1. Fc monomers were expressed from a similar vector in which the hinge cysteines, the CH2 N-glycosylation site, and 3 hydrophobic residues at the CH3 dimer interface had all been modified (C219A, C226N, C229G, N297D, L368R, F405H, and Y407E; numbering according to the Kabat database). Maxiprep DNA was prepared from 250 ml LB cultures of MC1061/P3 Escherichia coli using PurLink maxiprep kits (Invitrogen) according to the manufacturer's instructions. 293T cells grown to 50% confluence in 140-mm-diameter plates were transfected with 25 μg/plate using standard calcium phosphate transfection. Constructs bearing CCR5-mimetic peptides were contransfected with a plasmid expressing tyrosylprotein sulfotransferase TPST-2 at a 5:1 ratio. At 12 h posttransfection, 10% fetal calf serum-Dulbecco's modified Eagle's medium (FCS-DMEM) was replaced with serum-free 293 Freestyle medium (Invitrogen). Medium was collected 48 h later, and debris was cleared by centrifugation for 10 min at 1,500 × g and filtered in 0.45-μm-pore-size filter flasks (Millipore). Complete protease inhibitor mixture (Roche) was added to the filtered supernatants. A 500-μl bed volume of protein A-Sepharose beads (GE Healthcare) was added and agitated at 4°C overnight. The bead/medium mixture was collected by gravity flow column (Bio-Rad) and washed with 30 ml of phosphate-buffered saline (PBS) (Lonza) with the NaCl concentration increased to 0.65 M followed by 10 ml of PBS. Protein was eluted with 5 ml of 2 M arginine (pH 4) into 1 ml 1 M Tris (pH 7.5). Buffer was exchanged for PBS, and protein was concentrated to 1 mg/ml by ultrafiltration (Amicon Ultra) at 4,000 × g.
Surface staining of envelope glycoprotein-expressing cells.
Cells expressing HIV-1 envelope glycoprotein trimers were prepared by transient transfection of plasmids expressing envelope glycoproteins truncated in the C-terminal cytoplasmic domains to facilitate surface expression (gp160-ΔCT). T75 flasks of 293T cells grown to 50% confluence were transfected with 20 μg of plasmid encoding gp160-ΔCT and 3 μg of plasmid encoding tat by the calcium phosphate technique, and media (DMEM–10% FCS) were changed at 18 h after transfection. Cells were collected 36 h posttransfection by treatment with nonenzymatic cell dissociation solution (Sigma) followed by centrifugation for 5 min at 1,200 rpm. Cells were resuspended in fluorescence-activated cell sorter (FACS) buffer (PBS–2% donor goat serum–0.1% sodium azide). Cells were incubated with Fc fusion proteins diluted to various concentrations in FACS buffer for 45 min on ice followed by two FACS buffer washes. Allophycocyanin (APC)-conjugated anti-human Fc-γ (Jackson ImmunoResearch) diluted in FACS buffer was added, and cells were incubated on ice for 30 min. Cells were then washed two times with PBS–2% goat serum and then fixed for flow cytometric analysis with 1% paraformaldehyde–PBS. Using a BD Accuri C6 flow cytometer, 10,000 events/well were collected and data were analyzed by software provided by the manufacturer.
HIV-1 neutralization and entry studies.
Pseudotyped HIV-1 was produced by coexpression of envelope glycoproteins of the indicated HIV-1 isolates, or the vesicular stomatitis virus (VSV)-G protein, with NL43ΔEnv. 293T cells grown to 50% confluence in T175 (Falcon) flasks were transfected with 25 μg of plasmid encoding envelope glycoprotein and 45 μg of NL43ΔEnv by the standard calcium phosphate technique. The 10% FCS–DMEM was changed at 12 h, and medium was collected at 48 h. Viral supernatants were cleared by centrifugation for 10 min at 1,500 × g, passed through a 0.45-μm-pore-size syringe filter (Millipore), and stored at −80°C. Neutralization by Fc fusion constructs was performed according to the protocol described by Li et al. (14). In brief, various concentrations of purified Fc fusion proteins were incubated with virus at a volume of 100 μl/well in 96-well plates for 1 h, after which 100 μl of medium containing 10,000 TZM-bl cells was added. Plates were incubated 48 h at 37°C, after which 100 μl of medium was replaced with freshly prepared Brite-Lite reagent (PerkinElmer Life Sciences), and luminescence data were collected on a Victor3V counter (PerkinElmer Life Sciences). All neutralizations studies were performed in triplicate. To study infection of CCR5-positive, CD4-negative cells, CF2Th-CCR5 cells were incubated in the presence of serial dilutions of Fc fusion proteins with pseudoviruses generated with pNL4–3.Luc.R-E bearing the indicated HIV-1 envelope glycoprotein and encoding firefly luciferase. Infection was measured as described for the neutralization studies.
Immunoprecipitation of gp120 with Fc peptides.
HIV-1 gp120 constructs were transfected into 6-well plates at 5 μg/well. At 12 h after transfection, medium was replaced with cysteine-methionine-negative DMEM containing ExpressLabel (Cole Parmer) and 5% dialyzed calf serum (Invitrogen). Supernatants were collected 36 h posttransfection and were cleared by centrifugation at 3,000 rpm for 2 min followed by 1 min at 10,000 rpm, after which Complete protease inhibitor mixture was added. Immunoprecipitations were conducted by rocking 1 μg of Fc fusion protein with protein A Sepharose beads for 2 h at 4°C. The bound beads were washed with PBS and then incubated for 20 min with 20 μl of supernatant containing radiolabeled gp120. Beads were washed three times with PBS-Tween (0.01%), and protein was eluted by heating 10 min at 70°C in NuPage sample buffer. Samples were run on NuPage bis-tris gels, treated with methanol/acetic acid fixative, and dried on Whatman paper. Radiolabeled protein was quantified with a PhosphorImager (Fuji).
RESULTS
Double-mimetic peptides efficiently neutralize HIV-1 isolates.
For these studies, we combined CD4mim6 (12) with CCR5mim3, a shortened form of the previously described CCR5-mimetic peptide CCR5mim2 (8). We produced a series of CCR5mim3/CD4mim6 fusion proteins separated by linkers of 1, 6, 11, and 16 amino acids (1-Fc, 6-Fc, 11-Fc, 16-Fc; Fig. 1B and C). We also generated a reverse CD4mim6/CCR5mim3 fusion protein with a linker of 11 amino acids (Rev-Fc). The fusion protein with the shortest linker, 1-Fc, was consistently the least efficient at neutralizing HIV-1 and was in most cases less efficient than CD4-Fc (Fig. 2A). Fusion proteins with linker lengths of 11 and 16 neutralized all HIV-1 isolates with comparable efficiencies and with markedly greater efficiencies than CD4-Fc. With some isolates (HXB2, SG3), 6-Fc, with a six-amino-acid linker, neutralized as efficiently as 11-Fc and 16-Fc. With others (89.6, BaL), 6-Fc was clearly less efficient than 11-Fc. Isolate-specific differences in the region between the CD4- and CCR5-binding sites may account for these differences in 6-Fc neutralization. Together, these data suggest that with linkers of 11 amino acids or greater, both mimetic peptides are engaged, likely to the same gp120 monomer. Consistent with this inference, mixtures of both mimetic peptides alone, each at the same total concentration as 16-Fc, neutralized HIV-1 much less efficiently than 16-Fc itself (Fig. 2B). Similarly, Rev-Fc, a construct in which the order of mimetic peptides was reversed and the peptides were separated by an 11-amino-acid linker, also neutralized less efficiently than 16-Fc (Fig. 2B). This observation is consistent with the structure of a CD4-mimetic peptide bound to gp120 (see Fig. 1A), which indicates that a linker considerably longer than 11 amino acids would be required to bridge the distance between the peptide carboxy terminus and the nearest gp120 sulfopeptide-binding pocket. We conclude that 11-Fc and 16-Fc potently neutralize HIV-1, in part because they simultaneously engage the CD4- and coreceptor-binding sites of gp120.
FIG 2.

Neutralization assays of single- and double-mimetic peptides. (A) HIV-1 pseudotyped with the envelope glycoproteins of the indicated HIV-1 isolates was incubated with TZM-bl cells and with CD4-Fc or the indicated double-mimetic Fc fusions. Infection was determined by luciferase activity 48 h later and is shown as the percentage of luciferase units in the absence of inhibitor. (B) Assay similar to that described for panel A except that CD4mim6-Fc, CCR5mim3-Fc, a mixture of these constructs, Rev-Fc, 16-Fc, and CD4-Fc were assayed. All neutralization studies were performed in triplicate, with bars representing standard errors. Experiments are representative of two with similar results.
A double-mimetic peptide does not promote infection of CD4-negative cells.
The neutralization activity of soluble CD4 and CD4-mimetic peptides can be attenuated by their parallel tendency to enhance infection, possibly by promoting interaction of the HIV-1 envelope glycoprotein with the coreceptor (14, 15). This tendency is most pronounced when cellular CD4 is limiting or absent. We sought to determine if CCR5mim3 prevented the enhancement of infection observed with CD4mim6 alone, explaining in part the potency of 11-Fc relative to CD4mim6-Fc. To do so, we incubated CCR5-positive, CD4-negative CF2Th cells with various concentrations of CD4-Fc, CD4mim6-Fc, or 11-Fc and with HIV-1 pseudotyped with the BaL or ADA envelope glycoproteins. Expectedly, both CD4-Fc and CD4mim-Fc promoted HIV-1 infection in these cells, whereas 11-Fc did not (Fig. 3). Thus, the CCR5mim3 domain of 11-Fc inhibits the tendency of CD4mim6 to enhance infection.
FIG 3.
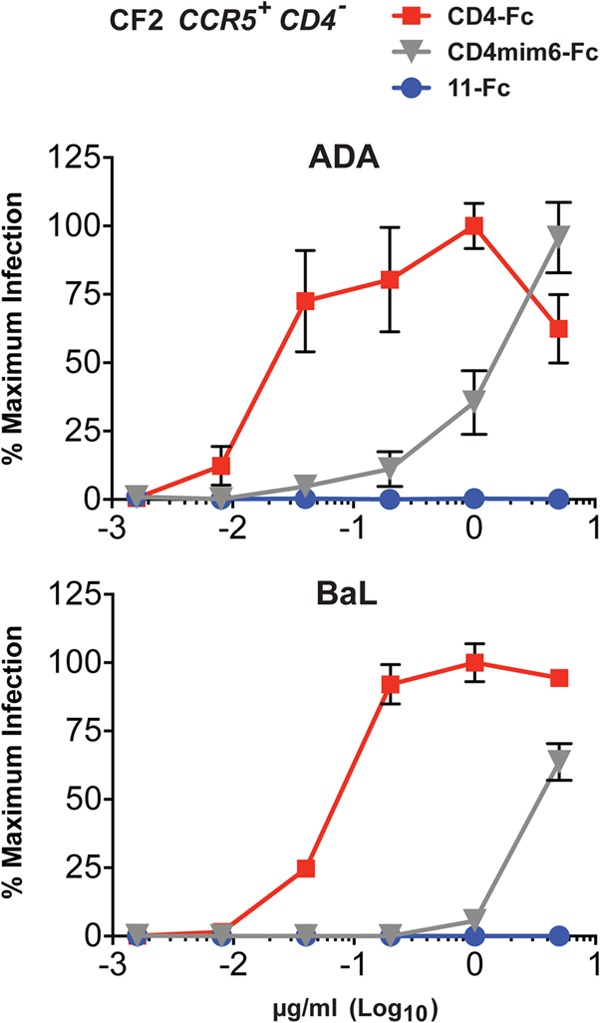
Infection of CCR5-positive, CD4-negative cells. CCR5-high, CD4-negative CF2Th cells were incubated with the indicated pseudoviruses encoding firefly luciferase in the presence of the indicated concentrations of CD4-Fc, CD4mim6-Fc, or 11-Fc. Infection was measured as luciferase activity as described for Fig. 2. Numbers represent relative luciferase activity levels normalized to the largest observed value. The experiment was performed in triplicate, with error bars representing standard errors.
A double-mimetic peptide binds both receptor-binding sites of gp120.
Double-mimetic peptides with suboptimal linkers or orientations still neutralize better than mixtures of CCR5mim3-Fc and CD4mim6-Fc (Fig. 2B), perhaps because gp120 can bind CCR5mim3 from one arm of the Fc dimer and CD4mim6 from the opposite arm. Avidity effects may also be obtained by binding of two different gp120 monomers of a single envelope glycoprotein trimer, or by cross-linking two trimers. Indeed, we have previously shown that an Fc-dimerized CCR5-mimetic peptide, but not CD4-Fc, can bind two gp120 molecules of one trimer (9). To reduce the complexity associated with the binding of a dimeric immunoadhesin to a trimeric envelope glycoprotein, we generated monomeric Fc forms of 16-Fc and 1-Fc (mono 16-Fc, represented in Fig. 1A, and mono 1-Fc). These variants were compared by flow cytometry with dimerized forms of the same constructs for their ability to bind cells expressing HIV-1 envelope glycoprotein trimers (Fig. 4A). They were also compared for their abilities to neutralize HIV-1 (Fig. 4B) and precipitate radiolabeled gp120 (Fig. 4C). In each assay, the dimer form of 16-Fc modestly outperformed the monomer form of 16-Fc and dimer form of 1-Fc. All three of these constructs bound envelope glycoprotein trimers, neutralized HIV-1, and precipitated free gp120 markedly more efficiently than monomeric 1-Fc. Figure 4D diagrams our interpretation of the gp120 precipitation studies of Fig. 4C. Specifically, we suggest that monomeric and dimeric 16-Fc and dimeric 1-Fc can bind both receptor binding sites on gp120 but that monomeric 1-Fc can bind only one. We also note the possibility that in trimer-binding studies (Fig. 4A) and in neutralization studies (Fig. 4B), one or more of the mimetic peptides could recognize a different gp120 monomer of the trimer (9) or bridge two envelope glycoprotein trimers. However, because these figures replicate the pattern observed in Fig. 4C, which represents an experiment that used only monomeric gp120, we infer that any such cross-linking effects are relatively modest. We conclude that 16-Fc, but not 1-Fc, can associate with both receptor-binding domains of monomeric gp120.
FIG 4.

Comparison of monomer and dimer forms of double-mimetic peptides. (A to C) Monomeric and dimeric Fc fusions of the indicated double-mimetic peptides were assayed by cell-surface binding to 293T cells expressing the envelope glycoprotein of the ADA isolate, as measured by flow cytometry (A); neutralization studies, as described for Fig. 2 (B); and precipitation of [35S]cysteine-labeled gp120 by Fc fusions bound to protein A-Sepharose beads (C). Results shown in panel C are represented as the percentage of maximal binding. (D) A model explaining the precipitation results corresponding to panel C. Monomeric and dimeric 16-Fc and dimeric 1-Fc can engage both receptor sites on free gp120, whereas monomeric 1-Fc can engage only one site. The experiments represented in panels A to C were performed in triplicate, with errors bars indicating standard errors, and are representative of at least two experiments with similar results.
DISCUSSION
Collectively, our data describe a novel class of peptide inhibitors of HIV-1 entry, namely, those that can simultaneously bind both the receptor- and coreceptor-binding sites of gp120. These peptides are more potent than any single-mimetic peptide that targets gp120 and, indeed, more potent than CD4-Fc against several clade B and clade C isolates. Interestingly, each component complements a weakness of the other. CCR5-mimetic peptides such as CCR5mim3 bind gp120 inefficiently, but their affinity increases in the presence of CD4-mimetic peptides. CD4-mimetic peptides such as CD4mim6 bind more efficiently, but they tend to promote infection when cell-expressed CD4 is limiting. CCR5mim3 prevents this, presumably by blocking access to cell-expressed CCR5. With appropriate linkers, double-mimetic peptides also benefit from increased avidity by binding two discrete and conserved sites of gp120.
Despite these advantages, these peptides have several limitations as therapeutics. First, the CD4-mimetic component is considerably less broad than soluble CD4 or the best CD4-binding site antibodies (12). CD4mim6 was developed by combinatorial methods against a clade B gp120 molecule and then further improved by phage display, panning against both clade B and clade C gp120 molecules (11, 12). While the CD4-binding site is well conserved among isolates, CD4mim6 appears to bind gp120 in a manner sufficiently different from that of CD4 so as to limit breadth against more-divergent envelopes (12). The CD4 mimetic might be further broadened through additional phage-display improvements. However, given the structural constraints enforced by its remaining disulfide bonds, it may prove necessary to remove an additional cysteine pair before this peptide can be further broadened. In contrast, the CCR5-mimetic peptide described here is strikingly broad and can enhance neutralization of an immunoadhesin form of CD4-Fc with a very wide range of HIV-1 and simian immunodeficiency (SIV) isolates (in preparation).
A second limitation to the therapeutic use of this class of peptides is the presence of multiple sulfotyrosines, which complicate its chemical synthesis (16). However, bacterial expression systems have been developed which can incorporate sulfotyrosines during translation, and these might be used to express double-mimetic peptides on a large scale (17, 18). Finally, like any peptide, those of this class cannot be administered orally, and given the wide array of oral therapeutics now available, these peptides might be best used as part of a microbicide rather than as a complement or replacement for peptides such as enfuvirtide/T20. However, as this report illustrates, these peptides can be useful as probes of the structure and function of the HIV-1 envelope glycoprotein, and they highlight the utility of simultaneously targeting both receptor-binding sites.
ACKNOWLEDGMENT
This work was supported by the National Institutes of Health (R01 AI080324, R01 AI0914767, and P01 AI100263).
Footnotes
Published ahead of print 3 January 2013
REFERENCES
- 1.Choe H, Farzan M, Sun Y, Sullivan N, Rollins B, Ponath PD, Wu L, Mackay CR, LaRosa G, Newman W, Gerard N, Gerard C, Sodroski J. 1996. The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell 85:1135–1148. 10.1016/S0092-8674(00)81313-6 [DOI] [PubMed] [Google Scholar]
- 2.Dalgleish AG, Beverley PC, Clapham PR, Crawford DH, Greaves MF, Weiss RA. 1984. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature 312:763–767. 10.1038/312763a0 [DOI] [PubMed] [Google Scholar]
- 3.Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Nagashima KA, Cayanan C, Maddon PJ, Koup RA, Moore JP, Paxton WA. 1996. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5 [see comments]. Nature 381:667–673. 10.1038/381667a0 [DOI] [PubMed] [Google Scholar]
- 4.Feng Y, Broder CC, Kennedy PE, Berger EA. 1996. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 272:872–877. 10.1126/science.272.5263.872 [DOI] [PubMed] [Google Scholar]
- 5.Choe H, Li W, Wright PL, Vasilieva N, Venturi M, Huang CC, Grundner C, Dorfman T, Zwick MB, Wang L, Rosenberg ES, Kwong PD, Burton DR, Robinson JE, Sodroski JG, Farzan M. 2003. Tyrosine sulfation of human antibodies contributes to recognition of the CCR5 binding region of HIV-1 gp120. Cell 114:161–170. 10.1016/S0092-8674(03)00508-7 [DOI] [PubMed] [Google Scholar]
- 6.Farzan M, Vasilieva N, Schnitzler CE, Chung S, Robinson J, Gerard NP, Gerard C, Choe H, Sodroski J. 2000. A tyrosine-sulfated peptide based on the N terminus of CCR5 interacts with a CD4-enhanced epitope of the HIV-1 gp120 envelope glycoprotein and inhibits HIV-1 entry. J. Biol. Chem. 275:33516–33521. 10.1074/jbc.M007228200 [DOI] [PubMed] [Google Scholar]
- 7.Dorfman T, Moore MJ, Guth AC, Choe H, Farzan M. 2006. A tyrosine-sulfated peptide derived from the heavy-chain CDR3 region of an HIV-1-neutralizing antibody binds gp120 and inhibits HIV-1 infection. J. Biol. Chem. 281:28529–28535. 10.1074/jbc.M602732200 [DOI] [PubMed] [Google Scholar]
- 8.Chiang JJ, Gardner MR, Quinlan BD, Dorfman T, Choe H, Farzan M. 2012. Enhanced recognition and neutralization of HIV-1 by antibody-derived CCR5-mimetic peptide variants. J. Virol. 86:12417–12421. 10.1128/JVI.00967-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kwong JA, Dorfman T, Quinlan BD, Chiang JJ, Ahmed AA, Choe H, Farzan M. 2011. A tyrosine-sulfated CCR5-mimetic peptide promotes conformational transitions in the HIV-1 envelope glycoprotein. J. Virol. 85:7563–7571. 10.1128/JVI.00630-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang CC, Stricher F, Martin L, Decker JM, Majeed S, Barthe P, Hendrickson WA, Robinson J, Roumestand C, Sodroski J, Wyatt R, Shaw GM, Vita C, Kwong PD. 2005. Scorpion-toxin mimics of CD4 in complex with human immunodeficiency virus gp120 crystal structures, molecular mimicry, and neutralization breadth. Structure 13:755–768. 10.1016/j.str.2005.03.006 [DOI] [PubMed] [Google Scholar]
- 11.Martin L, Stricher F, Misse D, Sironi F, Pugniere M, Barthe P, Prado-Gotor R, Freulon I, Magne X, Roumestand C, Menez A, Lusso P, Veas F, Vita C. 2003. Rational design of a CD4 mimic that inhibits HIV-1 entry and exposes cryptic neutralization epitopes. Nat. Biotechnol. 21:71–76. 10.1038/nbt768 [DOI] [PubMed] [Google Scholar]
- 12.Quinlan BD, Gardner MR, Joshi VR, Chiang JJ, Farzan M. 2013. Direct expression and validation of phage-selected peptide variants in mammalian cells. J. Biol. Chem. 288:18803–18810. 10.1074/jbc.M113.452839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sullivan N, Sun Y, Sattentau Q, Thali M, Wu D, Denisova G, Gershoni J, Robinson J, Moore J, Sodroski J. 1998. CD4-Induced conformational changes in the human immunodeficiency virus type 1 gp120 glycoprotein: consequences for virus entry and neutralization. J. Virol. 72:4694–4703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sullivan N, Sun Y, Li J, Hofmann W, Sodroski J. 1995. Replicative function and neutralization sensitivity of envelope glycoproteins from primary and T-cell line-passaged human immunodeficiency virus type 1 isolates. J. Virol. 69:4413–4422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu L, Gerard NP, Wyatt R, Choe H, Paralin C, Ruffing N, Borsetti A, Cardosa AA, Desjardin E, Newman W, Gerard C, Sodroski J. 1996. CD4-induced interactions of primary HIV-1 gp120 glycoproteins with the chemokine receptor CCR-5. Nature 384:179–183. 10.1038/384179a0 [DOI] [PubMed] [Google Scholar]
- 16.Choe H, Farzan M. 2009. Chapter 7. Tyrosine sulfation of HIV-1 coreceptors and other chemokine receptors. Methods Enzymol. 461:147–170. 10.1016/S0076-6879(09)05407-X [DOI] [PubMed] [Google Scholar]
- 17.Choe H, Farzan M. 2006. Tyrosine sulfate trapped by amber. Nat. Biotechnol. 24:1361–1362. 10.1038/nbt1106-1361 [DOI] [PubMed] [Google Scholar]
- 18.Liu CC, Schultz PG. 2006. Recombinant expression of selectively sulfated proteins in Escherichia coli. Nat. Biotechnol. 24:1436–1440. 10.1038/nbt1254 [DOI] [PubMed] [Google Scholar]
- 19.Huang CC, Venturi M, Majeed S, Moore MJ, Phogat S, Zhang MY, Dimitrov DS, Hendrickson WA, Robinson J, Sodroski J, Wyatt R, Choe H, Farzan M, Kwong PD. 2004. Structural basis of tyrosine sulfation and VH-gene usage in antibodies that recognize the HIV type 1 coreceptor-binding site on gp120. Proc. Natl. Acad. Sci. U. S. A. 101:2706–2711. 10.1073/pnas.0308527100 [DOI] [PMC free article] [PubMed] [Google Scholar]