

ABSTRACT
Flavivirus replication is mediated by a complex machinery that consists of viral enzymes, nonenzymatic viral proteins, and host factors. Many of the nonenzymatic viral proteins, such as NS4B, are associated with the endoplasmic reticulum membrane. How these membrane proteins function in viral replication is poorly understood. Here we report a robust method to express and purify dengue virus (DENV) and West Nile virus NS4B proteins. The NS4B proteins were expressed in Escherichia coli, reconstituted in dodecyl maltoside (DDM) detergent micelles, and purified to >95% homogeneity. The recombinant NS4B proteins dimerized in vitro, as evidenced by gel filtration, chemical cross-linking, and multiangle light scattering experiments. The dimeric form of NS4B was also detected when the protein was expressed alone in cells as well as in cells infected with DENV type 2 (DENV-2). Mutagenesis analysis showed that the cytosolic loop (amino acids 129 to 165) and the C-terminal region (amino acids 166 to 248) are responsible for NS4B dimerization. trans-Complementation experiments showed that (i) two genome-length RNAs containing distinct NS4B lethal mutations could not trans-complement each other, (ii) the replication defect of NS4B mutant RNA could be restored in cells containing DENV-2 replicons, and (iii) expression of wild-type NS4B protein alone was not sufficient to restore the replication of the NS4B mutant RNA. Collectively, the results indicate that trans-complementation of a lethal NS4B mutant RNA requires wild-type NS4B presented from a replication complex.
IMPORTANCE The reported expression and purification system has made it possible to study the biochemistry and structure of flavivirus NS4B proteins. The finding of flavivirus NS4B dimerization and the mapping of regions important for NS4B dimerization provide the possibility to inhibit viral replication through blocking NS4B dimerization. The requirement of NS4B in the context of the replication complex for successful trans-complementation enhances our understanding of NS4B in flavivirus replication.
INTRODUCTION
Many flaviviruses are significant human pathogens, including dengue virus (DENV), West Nile virus (WNV), yellow fever virus (YFV), Japanese encephalitis virus (JEV), and tick-borne encephalitis virus (TBEV). The four serotypes of DENV represent the most prevalent mosquito-borne viral pathogen for humans. DENV causes about 390 million human infections annually, leading to 96 million cases with manifest symptoms (1). No clinically approved vaccine or antiviral is currently available for DENV. A pilot study of the most advanced DENV vaccine candidate reported an overall efficacy of only 30.2% in a recent clinical trial (2). More efforts are needed to understand the biology of DENV and other flaviviruses to enable vaccine development.
The genomes of flaviviruses are single-stranded, positive-sense RNAs of 11 kb in length. The genomic RNA harbors a long open reading frame (ORF) flanked by the 5′- and 3′-untranslated regions (UTRs). The ORF encodes a polyprotein which is co- or posttranslationally processed into three structural proteins (capsid [C], premembrane [prM], and envelope [E] proteins) and seven nonstructural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5) (3). Structural proteins are the components of virions. Nonstructural proteins function in viral replication, virion assembly, and invasion against the host immune response (3–5). Among them, NS3 and NS5 harbor multiple enzymatic activities. NS3 contains protease (using NS2B as a cofactor), RNA helicase, and nucleotide triphosphatase (NTPase) activities (6–8). NS5 functions as a methyltransferase (MTase) (9–12) and an RNA-dependent RNA polymerase (RdRp) (13). Due to their high hydrophobicities, the three membrane proteins NS2A, NS4A, and NS4B remain poorly characterized. NS2A contains five transmembrane segments and is essential for viral replication and assembly (14–16). Together with other viral and host factors, NS4A induces membrane rearrangement for the formation of the replication complex (17, 18). These viral nonstructural proteins, in concert with host proteins, form replication complexes to amplify viral RNA and to facilitate assembly and release of progeny virions.
DENV type 2 (DENV-2) NS4B is an integral membrane protein consisting of 248 amino acids, among which 40% are hydrophobic residues. Using a biochemical approach, Miller and colleagues proposed a membrane topology of DENV-2 NS4B (17). In this model, the N-terminal 100 amino acids of NS4B form two predicted transmembrane domains (pTMD1 and pTMD2) and are located in the endoplasmic reticulum (ER) lumen, TMD3 (residues 101 to 129) traverses the membrane from the luminal to the cytoplasmic side, TMD4 (residues 165 to 190) spans the membrane from the cytoplasmic to the luminal side, and TMD5 (residues 217 to 244) either traverses the membrane from the luminal to the cytoplasmic side or remains in the ER lumen (17). Functional studies showed that different regions of the NS4B protein play distinct roles in viral replication and virus-host interactions, as follows. (i) The first 125 amino acids of DENV NS4B are responsible for suppression of alpha/beta interferon (IFN-α/β) signaling (19). (ii) TMD3 and TMD5 of DENV NS4B contribute to suppression of the host RNA interference (RNAi) response (20). (iii) DENV NS4B interacts with NS3 to negatively regulate the helicase function (21). (iv) WNV NS4B interacts with NS1, and such an interaction is required for viral replication (22). In addition, flavivirus NS4B has been reported to be an antiviral target for small-molecule inhibitors (23, 24); however, the molecular details of how the inhibitors suppress viral NS4B remain to be determined.
In this study, we report that NS4B dimerizes in cells infected with DENV-2. The NS4B protein forms dimers when expressed inside cells in the absence of other viral proteins. A recombinant DENV-2 NS4B protein dimerizes in vitro. Similar to that of DENV-2, a recombinant WNV NS4B protein also forms dimers. Mutagenesis analysis indicates that the cytosolic loop and the C-terminal region of DENV-2 NS4B contribute to its dimerization. Functional analysis shows that the replication defect of NS4B mutation can be partially rescued in cells harboring DENV replicons. In contrast, the replication defect of the same NS4B mutation cannot be restored in cells that express wild-type (WT) NS4B alone. The results demonstrate for the first time that (i) flavivirus NS4B dimerizes and (ii) trans-complementation of NS4B requires the wild-type NS4B protein presented in a replication complex.
MATERIALS AND METHODS
Cells, viruses, and antibodies.
BHK-21 cells and Vero cells were purchased from the American Type Culture Collection (ATCC) and maintained in high-glucose Dulbecco modified Eagle medium (DMEM) (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) (HyClone Laboratories, Logan, UT) and 1% penicillin-streptomycin (Invitrogen). 293T cells and Huh 7 cells were grown in low-glucose DMEM (Invitrogen) containing 10% FBS and 1% penicillin-streptomycin. Mosquito C6/36 cells were grown in RPMI 1640 medium (Invitrogen) with 10% FBS and 1% penicillin-streptomycin. DENV-2 (strain NGC; GenBank accession number AF038403) was generated from its corresponding infectious cDNA clone (pACYC-NGC FL) (25). The following antibodies were used in this study: mouse monoclonal antibody (MAb) against DENV-2 NS4B protein (clone 44-4-7) (26), anti-enhanced green fluorescent protein (anti-EGFP) mouse MAb (Roche), mouse MAb 4G2 (ATCC), anti-EGFP rabbit polyclonal antibody (pAb) (Abcam), anti-hemagglutinin (anti-HA) rabbit pAb (Sigma), anti-DENV-2 NS1 rabbit pAb (GeneTex), and goat anti-mouse or goat anti-rabbit antibody conjugated to horseradish peroxidase (HRP) (Sigma).
Plasmid construction.
Standard molecular biology procedures were performed for all plasmid constructions. Fragments encoding N-terminally Myc-tagged NS4A-2K-NS4B or C-terminally HA-tagged full-length (FL) or truncated NS4B were amplified from pACYC-NGC FL by PCRs using corresponding primer pairs. The PCR products were digested with the EcoRI and XhoI restriction enzymes and cloned into the pXJ expression vector (15). For constructs used for in situ fluorescence protease protection (FPP) assay, different truncations of NS4B or NS4B-MTase (including FL NS4B and the N-terminal 300 amino acids of NS5) were amplified from pACYC-NGC FL, digested with EcoRI and BamHI, and cloned into plasmid pXJ-EGFP as described previously (15), resulting in pXJ-2K-NS4B truncate-EGFP constructs. All constructs were validated by DNA sequencing. Primer sequences are available upon request.
In situ FPP assay.
The in situ FPP assay was performed using a previously reported protocol (15, 27). Briefly, BHK-21 cells were transfected with 1 μg plasmids encoding the corresponding NS4B-EGFP fusion proteins. At 24 h posttransfection (p.t.), the cells were washed with KHM buffer (110 mM potassium acetate, pH 7.2, 20 mM HEPES, and 2 mM MgCl2) and permeabilized with 50 μM digitonin (Merck) at room temperature for 3 min, followed by treatment with 50 μg/ml protease K (New England BioLabs). The fluorescence intensities were quantified immediately at 8-s intervals by using a Zeiss LSM 5 Duo laser scanning confocal microscope. The relative fluorescence intensity was calculated according to previously described methods (15).
Expression and purification of recombinant NS4B and its cytosolic loop.
The cDNA encoding the full-length NS4B protein of DENV-2 strain TSV01 (GenBank accession number AY037116) with an N-terminal decahistidine tag was amplified by fusion PCR, digested with the NcoI and NotI restriction enzymes, and inserted into pET28a, resulting in the pET28a-NS4B FL plasmid. After being confirmed by DNA sequencing, the plasmid was transformed into Escherichia coli BL21(DE3) (Stratagene). The transformed E. coli strain was grown in Difco 2× YT (BD) medium supplemented with 34 μg/ml chloramphenicol and 50 μg/ml kanamycin (Sigma) to an optical density at 600 nm (OD600) of 0.6 to 0.8. The culture was induced by the addition of 0.1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and incubation at 12°C for 16 h. Next, cells were pelleted by centrifugation at 7,000 × g for 7 min at 4°C, resuspended in lysis buffer (20 mM Tris-HCl, 300 mM NaCl, 10% glycerol, pH 8.0), and disrupted by sonication using a Digital Sonifier 450 (Branson) at 40% amplitude for 10 min. The cell lysates were clarified by centrifugation at 10,000 × g for 10 min at 4°C to remove the cell debris and unlysed cells. The supernatants were centrifuged at 35,000 rpm for 1 h at 4°C, using a Beckman Ti70 rotor to pellet membranes and their associated proteins. The membrane pellets were then resuspended thoroughly in lysis buffer supplemented with 1% n-dodecyl-β-d-maltopyranoside (DDM) (Anatrace) by stirring at 4°C for 1 h. The postsolubilized lysates were clarified by ultracentrifugation at 35,000 rpm for 1 h at 4°C to remove the insoluble fractions. Afterwards, the supernatants were purified by nickel-affinity chromatography, using a HisTrap Fast Flow column (GE Healthcare) equilibrated with buffer A (20 mM Tris-HCl, 300 mM NaCl, 20 mM imidazole, 2 mM Tris[2-carboxyethyl]phosphine [TCEP], and 0.05% DDM, pH 8.0). Proteins were eluted using a linear gradient of imidazole from 40 mM to 500 mM. The fractions containing His10-NS4B proteins were pooled, concentrated, and subjected to gel filtration (GF) using a HiLoadSuperdex 200 16/60 column (GE Healthcare) and buffer B (20 mM Tris-HCl, 300 mM NaCl, 10% glycerol, 2 mM TCEP, and 0.05% DDM, pH 8.0). Fractions containing His10-NS4B proteins were pooled and concentrated to approximately 2 to 5 mg/ml before storage at −80°C.
The expression and purification of the WNV NS4B protein followed a protocol similar to that described above. Briefly, a construct encompassing residues E2 to K254 of the NS4B protein from WNV (NY-99 strain; GenBank accession number KC407666.1) was cloned into pNIC28-Bsa4 (GenBank accession number EF198106), resulting in a construct with an N-terminal hexahistidine tag and a tobacco etch virus (TEV) protease recognition site. The protein was purified in two steps, by metal-affinity purification followed by gel filtration on a HiLoadSuperdex 200 16/60 column with a buffer containing 20 mM Tris-HCl, pH 8.0, 300 mM NaCl, 2 mM TCEP, 0.03% DDM, and 0.006% cholesterol.
The DNA sequence encoding the cytosolic loop (amino acids 121 to 171) of DENV-2 NS4B (strain TSV01) was cloned into pNIC28-Bsa4 to generate a fusion protein with an N-terminal His6 tag and a TEV cleavage site. E. coli BL21(DE3) was transformed with the expression plasmid and grown on LB plates containing 50 μg/ml kanamycin. Two or three colonies were inoculated into 50 ml of M9 medium. The overnight culture was then transferred into 1 liter of M9 medium. Recombinant protein was induced by addition of 1 mM IPTG at 25°C for 3 h and purified using Ni2+-nitrilotriacetic acid (Ni2+-NTA) chromatography. The buffer of the purified protein was exchanged with 20 mM Tris-HCl, 150 mM NaCl, 1 mM dithiothreitol (DTT; Sigma), and 2 mM CaCl2 through overnight dialysis at 4°C. The recombinant protein was then digested by TEV protease at a molar ratio of 1:0.5 (recombinant protein:TEV protease), and the cleaved His tag and uncleaved protein were removed through a Ni2+-NTA column. The protein fractions from the column were buffer exchanged with a gel filtration buffer containing 20 mM sodium phosphate, 150 mM NaCl, pH 6.5, and 1 mM DTT. Gel filtration chromatography was conducted on a Superdex 200 10/300 GL column. Protein fractions were analyzed by SDS-PAGE and mass spectroscopy.
Co-IP.
293T cells in 10-cm dishes were transfected with various constructs by use of X-tremeGENE 9 DNA transfection reagent (Roche). At 48 h p.t., cells were lysed in 1 ml immunoprecipitation (IP) buffer (20 mM Tris, pH 7.5, 100 mM NaCl, 0.5% DDM, and EDTA-free protease inhibitor cocktail [Roche]) with rotation at 4°C for 1 h. Lysates were clarified by centrifugation at 20,000 × g and 4°C for 30 min and subjected to co-IP using protein G-conjugated magnetic beads according to the manufacturer's instructions (Millipore). Briefly, immune complexes were formed at 4°C overnight by mixing 200 μl of cell lysate with mouse anti-EGFP MAb (2 μg) in a 500-μl reaction system containing 400 mM sodium chloride. Subsequently, the complexes were precipitated with protein G-conjugated magnetic beads at 4°C for 1 h with rotation, followed by washing extensively with phosphate-buffered saline (PBS) containing 0.1% Tween 20. Finally, proteins were eluted with 4× lithium dodecyl sulfate (LDS) sample buffer (Life Technologies) supplemented with 100 mM DTT, heated at 70°C for 10 min, and analyzed by Western blotting.
Protein dimerization assay.
A protein dimerization assay was performed using a previous method (28), with some modifications. 293T cells in a 10-cm dish were transfected with 10 μg plasmid encoding NS4B or its variants. At 48 h p.t., cells were lysed in HEPES lysis buffer (20 mM HEPES, pH 7.5, 100 mM NaCl, 0.5% DDM, and EDTA-free protease inhibitor cocktail) at 4°C for 1 h. The cell lysates were clarified by centrifugation at 20,000 × g and 4°C for 30 min. Supernatants were incubated with glutaraldehyde (Sigma) at a final concentration of 0.01% (wt/vol) at room temperature for the indicated times. The reaction was quenched by adding 1 M Tris-HCl (pH 8.0) to a final concentration of 100 mM. Samples were heated with 4× LDS sample buffer supplemented with 100 mM DTT, followed by SDS-PAGE and Western blotting.
Protein dimerization using MALS.
The oligomeric state of the NS4B protein from WNV was studied using a MALS-RI-UV detector system composed of a multiangle light scattering (MALS) detector (Dawn-Heleos; Wyatt Technology Corporation) for which measurements were performed at the standard 658-nm wavelength, a differential refractive index (RI) detector (Optilab rEX; Wyatt Technology Corporation), and a UV detector (Äkta Mikro GE) for which measurements were performed at 280 nm. Prior to the measurement, the system was calibrated with bovine serum albumin (BSA), using a theoretical molecular mass of 30,003 Da and an extinction coefficient of 51,450 liters mol−1cm−1. The value for dn/dc, where n is the refractive index and c is the concentration, was taken as 0.180 for the protein in a buffer containing 20 mM Tris-HCl at pH 8, 300 mM NaCl, 2 mM TCEP, 0.03% DDM, and 0.006% cholesterol. The dn/dc value for DDM was taken as 0.1435. The protein (6 mg/ml) was loaded onto a Superdex 200 5/150 GL column and eluted in the same buffer. The data from the three different detectors (MALS, RI, and UV) were collected and analyzed with Astra software (version 6.1.1). The experiment was repeated five times with consistent results.
SDS-PAGE and Western blotting.
Proteins were separated in 12% or 15% SDS-PAGE gels and transferred onto a polyvinylidene difluoride (PVDF) membrane by using a Trans-Blot Turbo transfer system (Bio-Rad). The blots were blocked in TBST buffer (10 mM Tris-HCl, pH 7.5, 150 mM NaCl, and 0.1% Tween 20) supplemented with 5% skim milk for 1 h at room temperature, followed by probing with primary antibodies (1:1,000 dilution) for 1 h. After two washes with TBST buffer, the blots were incubated with a goat anti-mouse or goat anti-rabbit IgG conjugated to HRP (1:30,000 dilution) in TBST buffer with 5% milk for 1 h, followed by three washes with TBST buffer. The antibody-protein complexes were detected using Amersham ECL Prime Western blotting detection reagent (GE Healthcare).
In vitro deglycosylation assay.
Vero cells were infected with DENV-2 (multiplicity of infection [MOI] of 1.0) at 37°C for 48 h. Cells were lysed at 4°C for 1 h in RIPA buffer comprising 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% Nonidet P-40 (NP-40), 0.5% sodium deoxycholate, 0.1% SDS, and EDTA-free protease inhibitor cocktail. Cell lysates were centrifuged at 20,000 × g and 4°C for 30 min to remove the unlysed cells and debris. Supernatants were mixed with 4 volumes of acidified acetone-methanol (1:1) and incubated at −20°C overnight. Proteins were pelleted by centrifugation at 20,000 × g and 4°C for 30 min, dried completely under a speed vacuum (Thermo Scientific) for 5 min, and subjected to a deglycosylation assay using a protein deglycosylation cocktail (peptide-N-glycosidase F O-glycosidase, neuraminidase, β-1,4-galactosidase, and β-N-acetylglucosaminidase; NEB) according to the manufacturer's instructions. In brief, proteins were dissolved in 1× G7 buffer, heated at 95°C for 10 min, and then chilled on ice. About 10 μg proteins was digested in a 100-μl reaction system containing 1% NP-40 and 5 μl deglycosylation enzyme cocktail at 37°C for 4 h. Subsequently, the samples were mixed with 4× LDS sample buffer (Invitrogen) containing 100 mM DTT, heated at 95°C for 10 min, and examined by Western blotting.
trans-Complementation analysis.
trans-Complementation was performed as described previously (23). Briefly, DENV-2 NS4B mutations were engineered using a QuikChange II XL site-directed mutagenesis kit (Stratagene). Wild-type (WT) and mutant DENV-2 genome-length RNAs were transcribed in vitro by use of a T7 mMessage mMachine kit (Ambion, Austin, TX) from cDNA plasmids prelinearized by XbaI. RNA transcripts (10 μg) were electroporated into BHK-21 cells or BHK-21 cells containing a DENV-2 replicon (BHK-21-Rep cells) (29). At 96 h p.t., the expression of E protein was examined by an immunofluorescence assay (IFA) using the mouse MAb 4G2 (ATCC). For trans-complementation in 293T cells, 5 × 104 cells per well were seeded in an 8-well chamber (Nunc) 1 day before transfection. Cells were transfected with 1 μg pXJ-2K-NS4B plasmid by use of X-tremeGENE 9 DNA transfection reagent. At 24 h p.t., the cells were subsequently transfected with WT or NS4B K143A genome-length RNA by use of TransIT-mRNA transfection reagent (Mirus Bio LLC) according to the manufacturer's instructions. Seventy-two hours after the first transfection, the NS4B and E proteins were monitored by an IFA using MAbs 44-4-7 and 4G2, respectively. Images were processed and merged using Adobe Photoshop software.
RESULTS
Recombinant NS4B protein of DENV-2 forms dimers.
We expressed recombinant NS4B protein by using an E. coli expression system. A His10 tag was fused to the N terminus of NS4B to facilitate purification. No detectable expression of NS4B was observed by SDS-PAGE after the E. coli cells were induced by IPTG; however, NS4B could be detected by Western blotting (data not shown). To mimic the membrane environment, we reconstituted the NS4B protein by using detergent micelles. Different detergents, including decylmaltoside (DM), dodecyl maltoside (DDM), 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS), lauryldimethyl amine oxide (LDAO), and octylglucoside (OG), were used to extract NS4B from the E. coli cell membrane. Among them, DDM, LDAO, and CHAPS extracted the protein more efficiently than the other detergents (data not shown). DDM is a mild and nondenaturing nonionic detergent with a critical micelle concentration (CMC) of 0.17 mM (0.009% [wt/vol]); many membrane proteins were successfully solubilized in DDM and retained their protein functions (30, 31). Our optimized protocol used 1% DDM (wt/vol) to extract NS4B from the cellular membrane, followed by reconstituting the protein in 0.03% DDM (wt/vol). NS4B was barely detectable after the membrane fractions were solubilized by 1% DDM (data not shown). However, after purification through a His-trap column, a dominant band with a molecular mass of 28 kDa and purity of >90% was recovered (Fig. 1A); an extra band with a molecular mass of 56 kDa was also detected (lanes 1 to 6). It should be noted that the authentic NS4B protein derived from DENV-infected cells migrated at the 25-kDa position in SDS-PAGE gels (see below); the N-terminally fused His10 tag contributed to increasing the molecular mass to 28 kDa.
FIG 1.

Dimerization of recombinant DENV-2 NS4B protein. (A) SDS-PAGE analysis of NS4B fractions from His-trap column. Lanes 1 to 10, purified NS4B fractions eluted from His-trap column; lane M, protein standards (Bio-Rad). The protein molecular masses (in kDa) are shown on the left. (B) Gel filtration profile. Fractions 8 to 10 from the His-trap column (A) were pooled, concentrated, and resolved by gel filtration. The x axis represents the fractionated samples (5 ml per fraction), and the y axis shows the absorbance (milli-absorbance units [mAU]) of the flowthrough. The dimer (middle) and monomer (right) peaks are indicated by an asterisk and an arrow, respectively. The left peak represents the void volume of gel filtration. (C) SDS-PAGE analysis of gel filtration fractions. Approximately 10-μl samples from each fraction were examined by 12% SDS-PAGE. The numbers at the top of the panel indicate the fractions (fractions 9 to 17) derived from gel filtration. Lane I represents concentrated protein (∼2 mg/ml) pooled from fractions 14 to 17. (D) Western blot analysis of the sample described for lane I in panel C. (E) Cross-linking analysis. Purified NS4B protein (2.5 ng/μl) was analyzed at room temperature in a 100-μl reaction volume supplemented with 0.01% glutaraldehyde (wt/vol). The cross-linked reaction mixtures were analyzed by Western blotting using a MAb against NS4B. The cross-linking time (0 or 2 min) is indicated. The asterisk and arrow indicate bands corresponding to dimers and monomers, respectively, of the NS4B protein.
Next, we pooled the fractions containing the 28-kDa NS4B protein (Fig. 1A, lanes 8 to 10). The pooled NS4B protein was further purified through a gel filtration column. Three distinct peaks were observed in the gel filtration chromatogram (Fig. 1B). SDS-PAGE analysis under denaturing conditions (heating with DTT prior to loading of the gel) showed that fractions from the second chromatogram peak contained both the 28- and 56-kDa bands (Fig. 1C, fractions 9 to 13), whereas fractions from the third chromatogram peak contained only the 28-kDa band (fractions 14 to 17). When the 28-kDa protein pooled from gel filtration fractions 14 to 17 was concentrated to 2.5 mg/ml, about 30% of the total NS4B protein was converted to the 56-kDa form, and even under denaturing conditions (sample buffer containing LDS and DTT and heating at 95°C for 5 min), the 56-kDa band could not be disrupted completely (Fig. 1C, lane I). The identity of the 28- and 56-kDa bands was confirmed to be NS4B by Western blotting using a monoclonal antibody against DENV-2 NS4B (Fig. 1D) and mass spectrometry (data not shown). Furthermore, when the 28-kDa protein was treated with glutaraldehyde (a chemical commonly used to cross-link proteins) for 2 min, more than 80% of the NS4B protein formed dimers and larger polymers (Fig. 1E). After purification by His-trap chromatography and gel filtration, the overall yield of NS4B protein was about 1 mg per liter of E. coli culture. Taken together, the results demonstrate that recombinant NS4B of DENV-2 can be expressed and purified and that the purified NS4B protein forms dimers in vitro.
Recombinant NS4B protein of WNV forms dimers.
To examine whether NS4B proteins of other flaviviruses could also form dimers, we prepared a recombinant NS4B protein of WNV by using a similar expression/purification protocol. SDS-PAGE analysis of various fractions from the final purification step showed a dominant band migrating at 21.5 kDa (Fig. 2A); this molecular mass is much smaller than the theoretical mass (30 kDa) of WNV NS4B. A weak band migrating at 43 kDa was also observed. The identity of the 21.5- and 43-kDa bands was confirmed to be NS4B by Western blotting (Fig. 2B) using a monoclonal antibody (MAb 44-4-7) that cross-reacts with both DENV and WNV NS4B (26). Using mass spectrometry, we confirmed the identity of both bands to be WNV NS4B by analyzing trypsin-digested peptides (data not shown); because NS4B was tightly associated with detergent in solution, measurement of the molecular mass of the complete protein was inaccurate. Overall, the results suggest that the 21.5- and 43-kDa bands may represent the monomeric and dimeric forms of the NS4B protein, respectively. The faster mobility on SDS-PAGE than that expected for the theoretical molecular mass was possibly due to the detergent buffer conditions.
FIG 2.

Dimerization of the WNV NS4B protein according to MALS–UV–RI–size-exclusion chromatography (SEC) analysis. (A) SDS-PAGE analysis of recombinant NS4B protein of WNV. Samples represent different fractions derived from gel filtration. Because the protein migrated faster than its theoretical mass of 30 kDa, its identity was confirmed by mass spectrometry (data not shown). (B) Western blot analysis of the purified WNV NS4B protein by use of MAb 44-4-7. (C) MALS-UV-RI-SEC analysis. The NS4B protein (6 mg/ml) was purified through an S200 5/150 GL column by using a buffer containing 20 mM Tris-HCl (pH 8.0), 300 mM NaCl, 2 mM TCEP, 0.03% DDM, and 0.006% cholesterol. The right y axis represents the OD280, and the left y axis represents the molecular mass. Colored traces show the measured molecular masses of the protein-detergent conjugate (red), protein (blue), and detergent (green).
We analyzed protein oligomerization by using a multiangle light scattering method. As shown in Fig. 2C, the data from three different detectors (MALS, RI, and UV) allowed us to measure the mass of the protein-detergent conjugate in the buffer containing DDM and cholesterol (with a mass of 395 kDa). Under these experimental conditions (6 mg/ml of NS4B), over 95% of the protein was found in a dimeric form (60.02 kDa), with the protein-detergent conjugate having a mass of 455 kDa. Given the theoretical mass of 30 kDa for the NS4B construct from WNV, the protein appears predominantly as a dimer, with about 5% of the protein forming larger multimers. No NS4B monomers were detected in the multiangle light scattering experiment. The results indicate that recombinant NS4B protein of WNV also forms dimers in vitro.
The above results showed that WNV NS4B was detected predominantly as a monomer by SDS-PAGE, whereas the same protein was observed as dimers and multimers by the multiangle light scattering method. This discrepancy was most likely due to the denaturing conditions used during the SDS-PAGE analysis, leading to dissociation of dimers/multimers to monomers. In contrast, the multiangle light scattering method examined the protein under nondenaturing conditions, reflecting a more authentic oligomeric status of the protein.
Validation of membrane topology of DENV-2 NS4B.
We took a mutagenesis approach to map which region of NS4B is important for dimerization. To enable the design of constructs for mutagenesis study, we first validated the membrane topology of DENV-2 NS4B (Fig. 3A) (17) by using an in situ fluorescence protease protection (FPP) assay. The FPP assay has been well established to probe the topology of membrane proteins (27). To this end, we constructed a panel of plasmids in which the 2K peptide was fused to various C-terminally truncated NS4B-EGFP constructs (Fig. 3B). BHK-21 cells were transfected with individual plasmids, followed by treatment with digitonin, which selectively permeabilizes the plasma membrane without affecting the ER membrane. Next, the transfected cells were incubated with protease K and quantified for the fluorescence intensity of EGFP. If the construct expresses EGFP in the cytoplasm, EGFP will be accessible to protease K digestion; in contrast, if the construct expresses EGFP in the lumen, EGFP will largely be protected from protease K digestion. The relative intensity of fluorescence at the end of protease digestion (5 min) and the initial rate of fluorescence degradation (Fig. 3C and D) could be used to indicate the luminal or cytosolic localization of EGFP. In cells expressing the 2K-NS4B (1–146)-EGFP or 2K-NS4B-MTase-EGFP (containing the full-length NS4B protein followed by the MTase domain of NS5) construct, only 2% and 4% of the fluorescence intensity, respectively, was retained after 5 min of digestion with protease K (Fig. 3C and D), indicating that the C termini of both constructs were located in the cytoplasm. In contrast, in cells expressing 2K-EGFP (without NS4B), 2K-NS4B (1–32)-EGFP, 2K-NS4B (1–56)-EGFP, NS4B (1–190)-EGFP, or NS4B (1–248)-EGFP (full-length NS4B without any NS5 sequence), about 53% to 68% of the fluorescence intensity was retained after the protease K digestion (Fig. 3C and D), suggesting that the EGFP portions of these constructs were located in the lumen. It should be noted that based on the topology of flavivirus polyproteins (3), the 2K-EGFP and 2K-NS4B-MTase-EGFP constructs were expected to localize the EGFP reporter to the ER lumen and cytoplasm, respectively; therefore, these two constructs served as internal controls for the FPP assay.
FIG 3.

In situ fluorescence protease protection assay. (A) Topology model of DENV NS4B on the ER membrane (17). Two predicted transmembrane domains (pTMD1 and pTMD2), three validated transmembrane domains (TMD3 to -5), and the amino acid positions in NS4B are indicated. (B) Schematic diagram of 2K-NS4B truncate-EGFP fusion constructs. Each NS4B construct was fused with the N-terminal 2K signal peptide (black boxes) and a C-terminal EGFP tag. The transmembrane domains (pTMDs and TMDs) are shown as white boxes. MTase derived from the N-terminal 300 amino acids of NS5 is indicated with an oval. (C) FPP assay. BHK-21 cells were transfected with the indicated NS4B truncate-EGFP constructs. At 24 h posttransfection, the sensitivities of EGFP were examined by FPP assay (see Materials and Methods for details). Curves of relative intensities (%) versus time (s) were plotted. The relative intensities were calculated according to previously described formulas (15). The NS4B constructs are indicated in the corresponding fluorescence panels. (D) Summary of relative fluorescence intensities and initial rates of fluorescence degradation. Average values for at least two independent experiments are presented. (E) Schematic diagram of the membrane topology of NS4B before (left) and after (right) cleavage of the MTase of NS5 by viral NS2B/NS3 protease. The cleavage of MTase from NS4B regulates the translocation of TMD5 from the cytosol to the ER lumen.
Unexpectedly, for the 2K-NS4B (1–93)-EGFP construct, the degradation of fluorescence intensity displayed a pattern significantly different from the two patterns described above (Fig. 3C and D). After 5 min of protease K treatment, 23% of the fluorescence intensity was retained, suggesting a dual possibility of luminal and cytosolic orientation of the C terminus of this construct. It is likely that the C terminus of this construct is partially buried in the membrane in situ. The exact topology needs further investigation by other biophysical approaches, such as nuclear magnetic resonance (NMR) or X-ray crystallography. Nevertheless, the overall FPP results support the previously proposed topology of DENV NS4B (17).
Notably, the FPP results showed that the 2K-NS4B (1–248)-EGFP and 2K-NS4B-MTase-EGFP constructs targeted EGFP to the ER lumen and cytoplasm, respectively (Fig. 3C and D). This result suggests that, during polyprotein translation, the uncleaved NS5 protein facilitates translocation of the C terminus of NS4B to the cytoplasm; once NS5 has been cleaved from NS4B by viral NS2B/NS3 protease, the C terminus of NS4B is probably translocated to the ER lumen (Fig. 3E). Our result is in agreement with the previous observation that TMD5 (residues 217 to 244) could either traverse the ER membrane from the lumen to the cytosol or stay in the ER lumen (17). The cleavage-modulated translocation of the NS4B C terminus may convert the protein into a replication-active form in the context of a replicase complex, leading to the switch from viral translation to RNA synthesis; experiments are ongoing to validate this speculation.
The cytosolic loop and the C-terminal region of NS4B are important for dimerization.
To map the region(s) responsible for the dimerization of DENV-2 NS4B, we performed a chemical cross-linking experiment using a set of NS4B deletion mutants (Fig. 4A). All NS4B variants contained a C-terminal HA tag and were transiently expressed in 293T cells; the cell lysates were cross-linked using glutaraldehyde (0.01% [wt/vol]) at 22°C. As expected, full-length 2K-NS4B was efficiently cross-linked to form dimers after 2 min of glutaraldehyde treatment; after 10 min of cross-linking, more dimers were formed (Fig. 4B). The 2K-NS4B (1–165)-HA mutant, with a deletion of both TMD4 and -5, did not significantly affect dimer formation (Fig. 4B). Further deletion of the cytosolic loop, represented by the 2K-NS4B (1–129)-HA mutant, almost abolished dimer formation (Fig. 4B). The results suggest that the cytosolic loop is important for the dimerization of NS4B.
FIG 4.

Mapping the regions responsible for NS4B dimerization by chemical cross-linking. (A) Schematic diagram of 2K-NS4B truncate-HA fusion constructs. (B) Dimerization analysis of truncated or full-length NS4B by cross-linking. The cross-linking time (min) is indicated at the top. Schematic diagrams of HA-tagged NS4B constructs are shown at the bottom. N.C., negative control. (C) Transient expression of NS4B (166–248) construct. 293T cells were transfected with a plasmid expressing the C-terminal fragment (amino acids 166 to 248) of NS4B fused with an HA tag [construct NS4B (166–248)-HA; WT] or transfected with the NS4B (166–248)-HA construct with two Cys mutations (C177A and C191A). At 48 h posttransfection, the samples were lysed in RIPA buffer, mixed with 4× LDS sample buffer, and incubated at room temperature. The samples were resolved by SDS-PAGE and detected by Western blotting using a rabbit anti-HA IgG. (D) Gel filtration profile of NS4B (121–171). The peaks with retention volumes of 17 ml and 21 ml are labeled peaks I and II, respectively. (E) Molecular mass determination of peaks I and II by mass spectrometry. The numbers in each panel represents molecular masses.
Although C-terminal deletion of NS4B did not affect dimerization [as indicated by the 2K-NS4B (1–165)-HA construct], the C-terminal region may be able to dimerize by itself. To test this possibility, we transiently expressed the C-terminal region (amino acids 166 to 248, spanning TMD4 and TMD5) in 293T cells. As shown in Fig. 4C, in the absence of cross-linking agent, the C-terminal fragment alone could form dimers under conditions of no heating or DTT treatment. Since there are two Cys residues, at positions 177 and 191, it is possible that dimers are formed through Cys-mediated intermolecular disulfide bond formation. To exclude this possibility, we replaced Cys177 and Cys191 with Ala. SDS-PAGE analysis showed that Ala substitutions of both Cys residues did not affect dimer formation of the C-terminal fragment (Fig. 4C), indicating that the dimerization of NS4B (166–248) is mediated by the intrinsic property of the fragment rather than by intermolecular disulfide bonds. These data demonstrate that the C-terminal region of NS4B has a redundant role in mediating protein dimerization.
To test whether the cytosolic loop alone can form dimers, we expressed and purified the loop of DENV-2 NS4B (residues 121 to 171) as a soluble protein. Gel filtration analysis of the loop protein showed several peaks (Fig. 4D). Mass spectrometry showed that peaks with retention volumes of <16 ml represented impurities (data not shown), whereas peaks I and II represented intact NS4B loops (Fig. 4E). Peak I corresponded to a molecular mass of about 12 kDa, suggesting that it represents the dimeric form of the loop (the theoretical molecular mass of the NS4B loop is about 6 kDa). Peak II corresponded to a molecular mass of about 6 kDa, suggesting that peak II represents the monomeric form of the loop. Collectively, the results suggest that the cytosolic loop of NS4B can form dimers under our experimental conditions.
NS4B forms dimers when expressed alone in cells.
We transiently expressed DENV-2 NS4B alone and examined its ability to form dimers by co-IP. Cells (293T) were cotransfected with two plasmids: one plasmid expressing C-terminally HA-tagged 2K-NS4B (construct 2K-NS4B-HA) and one plasmid expressing C-terminally EGFP-tagged 2K-NS4B (construct 2K-NS4B-EGFP). Co-IP experiments showed that anti-EGFP MAb was able to pull down 2K-NS4B-EGFP together with 2K-NS4B-HA (Fig. 5A, right lane). In contrast, when EGFP alone was coexpressed with 2K-NS4B-HA, anti-EGFP MAb could pull down only EGFP, but not together with 2K-NS4B-HA (Fig. 5A, middle lane). As a negative control, 2K-NS4B-HA alone did not bind to beads or antibodies under our co-IP conditions (Fig. 5A, left lane). Notably, when 2K-NS4B-EGFP and its variants were expressed in 293T cells, a band migrating at a position identical to that of EGFP was observed (Fig. 5A and B); this band most likely represents EGFP derived from an unknown cleavage of the 2K-NS4B-EGFP protein. Nevertheless, these data indicate that NS4B proteins can interact directly with each other when expressed in mammalian cells.
FIG 5.

Co-IP analysis of NS4B dimerization. (A) Dimerization of NS4B expressed in 293T cells. Cells were transfected with one plasmid, encoding 2K-NS4B-HA (left lane), or with two plasmids, encoding 2K-NS4B-HA and EGFP (middle lane) or 2K-NS4B-HA and 2K-NS4B-EGFP (right lane). At 48 h posttransfection, the cells were subjected to co-IP as described in Materials and Methods. A mouse anti-EGFP MAb was used to perform IP analysis. Rabbit anti-HA and anti-EGFP IgGs were used to detect NS4B protein in cell lysates and IP eluates, respectively. (B) Mapping of dimerization determinants of NS4B protein. Cells were cotransfected with two plasmids, encoding 2K-NS4B-HA and 2K-NS4B truncate-EGFP. The various 2K-NS4B truncate-EGFP constructs are shown in Fig. 3B. The co-IP experiments were performed as described for panel A.
Next, we used the co-IP method to confirm the regions that are important for NS4B dimerization. Cells were cotransfected with two plasmids: one plasmid expressing a full-length NS4B protein with an HA tag (construct 2K-NS4B-HA) and one plasmid expressing a C-terminally truncated NS4B protein with an EGFP tag (as depicted in Fig. 3B). The co-IP experiments showed that the expression levels of 2K-NS4B-HA protein were comparable among different co-IP samples (Fig. 5B, bottom panel). Anti-EGFP MAb efficiently pulled down various EGFP-tagged NS4B constructs (Fig. 5B, top panel); however, different EGFP-tagged NS4B constructs pulled down the 2K-NS4B-HA protein with various efficiencies. Compared with EGFP-tagged full-length NS4B (construct 1–248), the 1–56, 1–93, 1–146, and 1–190 NS4B constructs reduced the pull-down efficiency of 2K-NS4B-HA to 1%, 10%, 35%, and 80%, respectively; 2K and the 1–32 NS4B construct did not pull down the 2K-NS4B-HA protein (Fig. 5B, middle panel). These data indicate that the cytosolic loop as well as its downstream C-terminal region is important for NS4B dimerization.
Dimerization of NS4B in DENV-2-infected cells.
To examine the biological relevance of NS4B dimerization, we performed Western blotting of DENV-2-infected Vero cells by using a monoclonal antibody against NS4B (26). At 48 h postinfection, cell lysates were prepared in buffer containing no DTT and resolved by SDS-PAGE at 4°C (to avoid dissociation of protein complexes due to the heat generated during electrophoresis). Western blotting revealed four bands, corresponding to molecular masses of 25, 27, 37, and 50 kDa (Fig. 6A, lane 2). Based on the calculated molecular mass, the strong 25-kDa band and the weak 50-kDa band could represent the monomeric and dimeric forms of NS4B, respectively. The 27-kDa band was most likely the uncleaved 2K-NS4B construct; glycosylation analysis showed that the 27-kDa band was not a glycosylated form of NS4B (Fig. 6B). Specifically, treatment of the DENV-2-infected cell lysates with a mixture of deglycosylation enzymes did not affect the intensity of the 27-kDa band; as a positive control, the same deglycosylation treatment shifted the migration of the NS1 protein in the SDS-PAGE gel (Fig. 6B). The 37-kDa band represents uncleaved NS4A-2K-NS4B, because transient expression of a plasmid encoding NS4A-2K-NS4B produced a band that migrated at the identical position (Fig. 6A, lane 4). Similar results were obtained using DENV-2-infected mosquito C6/36 cells (Fig. 6C, lane 1) and human Huh 7 cells (Fig. 6C, lane 3).
FIG 6.
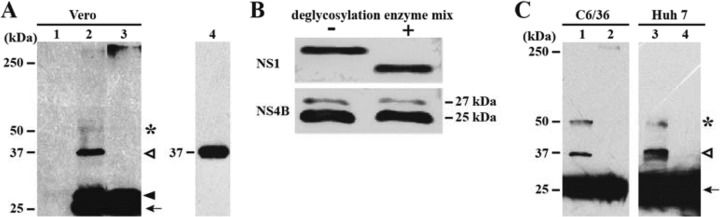
Dimerization and glycosylation analyses of NS4B from DENV-2-infected cells. (A) Dimerization of NS4B in cells infected with DENV-2. Vero cells were infected (MOI = 1.0) with DENV-2 (lanes 2 and 3) or left uninfected (as a negative control; lane 1). At 48 h postinfection, cell lysates were incubated in sample buffer without DTT at room temperature (lane 2) or with DTT at 95°C for 10 min (lane 3). Transiently expressed Myc-NS4A-2K-NS4B was incubated at room temperature without DTT (lane 4). Protein samples were resolved by SDS-PAGE and analyzed by Western blotting using a mouse MAb against DENV-2 NS4B (clone 44-4-7). The black arrow, black arrowhead, white arrowhead, and asterisk indicate bands at 25, 27, 37, and 50 kDa, respectively. (B) Glycosylation analysis of NS4B. Lysates from the DENV-2-infected cells were treated with a deglycosylation enzyme cocktail and detected with an NS4B monoclonal antibody by Western blotting (see Materials and Methods). NS1 protein was included as a positive control. (C) Dimerization of DENV-2 NS4B in mosquito C6/36 cells and human Huh 7 cells. C6/36 cells and Huh 7 cells were infected (MOI = 10) with DENV-2 for 48 h. Cell lysates were incubated in sample buffer without DTT at room temperature (lanes 1 and 3) or with DTT at 95°C for 10 min (lanes 2 and 4). Detection of NS4B was performed as described for panel A.
In contrast to the above results, when the infected cell lysates were heated at 95°C for 10 min in buffer containing DTT, the 37- and 50-kDa bands disappeared (Fig. 6A, lane 3, and C, lanes 2 and 4), suggesting that these proteins are sensitive to reducing reagents and high temperature. Collectively, the results indicate that the dimeric form of NS4B could be detected in cells infected with DENV-2.
Two genome-length RNAs containing distinct lethal mutations in NS4B cannot trans-complement each other.
We attempted to demonstrate the biological relevance of NS4B dimerization by using a trans-complementation approach. We hypothesized that if two distinct NS4B lethal mutant viruses could trans-complement each other and rescue viral replication, this could be strong evidence for the biological relevance of NS4B dimerization. To perform this experiment, we performed a mutagenesis analysis of NS4B in the context of DENV-2 genome-length RNA. Ala scanning mutagenesis of charged amino acids that are conserved among the cytosolic loops (residues 129 to 165) of flavivirus NS4B proteins identified a K143A mutation that is lethal for viral replication (Fig. 7A). Specifically, after electroporation of equal amounts of WT and K143A mutant genome-length RNAs into BHK-21 cells, the WT RNA generated E protein IFA-positive cells. No IFA-positive cells were detected in the K143A RNA-transfected cells; no revertants were obtained (as indicated by IFA, reverse transcription-PCR [RT-PCR], and plaque assay) after 5 rounds of passaging of the transfected cell supernatant in Vero cells (data not shown).
FIG 7.

trans-Complementation of NS4B. BHK-21 cells (A) or BHK-21-Rep cells (B) were electroporated with genome-length RNAs encoding WT NS4B, NS4B K143A, NS4B P104R, or NS4B K143A plus NS4B P104R (as indicated). At 96 h posttransfection, the expression of viral E protein was detected by an IFA using mouse MAb 4G2 and Alexa Fluor 488-conjugated goat anti-mouse IgG as primary and secondary antibodies, respectively. Nuclei were stained by 4′,6-diamidino-2-phenylindole (DAPI) and are shown in blue. (C) 293T cells were first transfected with a plasmid encoding 2K-NS4B protein. At 24 h posttransfection, cells were further transfected with wild-type or NS4B K143A genome-length RNA. Seventy-two hours after the first transfection, the NS4B and E proteins were monitored by IFAs using NS4B 44-4-7 MAb and E 4G2 MAb, respectively. (D) Co-IP analysis of dimerization of NS4B K143A. 293T cells were cotransfected with two plasmids, encoding 2K-NS4B-HA (WT or K143A mutation) and EGFP or 2K-NS4B-EGFP (WT or K143A mutation). Co-IP was performed as described in Materials and Methods.
We previously reported another NS4B mutant, the P104R mutant, which is lethal for viral replication (23). As expected, cells transfected with the NS4B P104R genome-length RNA did not yield any IFA-positive cells (Fig. 7A). To test whether two distinct NS4B lethal mutant viruses could trans-complement each other, we cotransfected P104R and K143A genome-length RNAs into BHK-21 cells; no IFA-positive cells were observed. The result indicates that two distinct NS4B lethal viruses could not trans-complement each other and restore viral replication.
trans-Complementation of lethal NS4B mutant requires wild-type NS4B in the context of a replication complex.
We performed trans-complementation analysis of NS4B K143A in BHK-21 cells harboring a DENV-2 replicon (BHK-21-Rep). Since BHK-21-Rep cells did not express any E protein, IFA targeting the viral E protein was used to monitor the trans-complementation of genome-length RNA. Upon transfection into BHK-21-Rep cells, WT genome-length RNA yielded E protein IFA-positive cells (Fig. 7B, left panel); however, the number of E-positive cells was smaller than that after transfection into naive BHK-21 cells (compare Fig. 7A and B), most likely due to replication exclusion between the replicon and the WT genome-length RNA (32). Compared with the WT, genome-length RNA containing the K143A or P104R mutation generated fewer but clearly detectable IFA-positive signals in the BHK-21-Rep cells (Fig. 7B, middle and right panels). This result confirms our previous observation that the replication defect of NS4B can be trans-complemented in BHK-21-Rep cells (23).
Next, we asked whether trans-complementation could be achieved when NS4B is expressed in the absence of other viral proteins. To this end, we transfected 293T cells with a plasmid encoding the WT 2K-NS4B protein. At 24 h posttransfection, the cells were further transfected with the WT or NS4B K143A genome-length RNA. Seventy-two hours after the first transfection, IFA was performed to examine the expression of NS4B or E protein. As shown in Fig. 7C, after the double transfections, up to 80% of the cells were NS4B positive, and up to 20% of the cells transfected with the WT genome-length RNA were E positive; in contrast, no E-positive cells were detected in the cells transfected with the NS4B K143A genome-length RNA. These results demonstrate that expression of NS4B protein alone is not able to trans-complement the replication defects of NS4B mutants. Taken together, our results indicate that trans-complementation of lethal NS4B mutants requires WT NS4B presented in the replication complex.
Furthermore, we examined whether the K143A mutation affects the dimerization of NS4B protein. Co-IP experiments showed that the pull-down efficiencies were similar among the four pairs of 2K-NS4B-EGFP plus 2K-NS4B-HA molecules (Fig. 7D), i.e., 2K-NS4B-EGFP plus 2K-NS4B-HA, 2K-NS4B (K143A)-EGFP plus 2K-NS4B-HA, 2K-NS4B-EGFP plus 2K-NS4B (K143A)-HA, and 2K-NS4B (K143A)-EGFP plus 2K-NS4B (K143A)-HA. The results indicate that (i) the K143A mutation alone does not abolish NS4B dimerization and (ii) the replication defect of K143A genome-length RNA is not due to a defect in NS4B dimerization.
DISCUSSION
During the past decade, significant progress has been made toward understanding flavivirus structural proteins and nonstructural proteins with enzymatic activities (33, 34). These breakthroughs have set the foundation for modern drug discovery (35). In contrast, how nonstructural proteins with no enzymatic activities function in flavivirus replication remains poorly characterized. In this study, we developed a robust protocol to express and purify the flavivirus NS4B protein. To our knowledge, this is the first report of successful production of a recombinant NS4B protein of a flavivirus in large quantity. The ability to obtain recombinant NS4B protein makes it possible to use it for future biochemical and structural studies.
Three lines of evidence support the conclusion that flavivirus NS4B dimerizes in vitro and in vivo. (i) Recombinant NS4B proteins of both DENV-2 and WNV form dimers, as indicated by gel filtration, chemical cross-linking, and multiangle light scattering. Among the three methods, chemical cross-linking generated predominantly NS4B dimers as well as some higher-order multimers (Fig. 1E); such dimers were also observed in the gel filtration (Fig. 1B and C and 2A) and multiangle light scattering (Fig. 2C) experiments. The larger amount of multimers observed in Fig. 1E could be due to the irreversible nature of the chemical cross-linking method: multiple NS4B molecules could be covalently cross-linked into larger complexes over the incubation time. In contrast, the formation of NS4B dimers and multimers is dynamically reversible under gel filtration and multiangle light scattering conditions. Therefore, caution should be taken in interpreting the higher-order multimers observed in the cross-linking experiment. In addition, oligomerization is concentration dependent, so variations in protein concentration could lead to different distributions of the various multimeric statuses. (ii) When DENV-2 NS4B protein alone was expressed in cells, proteins tagged with two distinct tags (EGFP and HA) could be pulled down with each other, indicating that NS4B molecules interact with each other in the context of the cellular environment. (iii) A monoclonal antibody against DENV-2 NS4B could detect both monomeric and dimeric forms of NS4B in cells infected with DENV. Similar to flavivirus NS4B, hepatitis C virus (HCV) NS4B multimerizes, which is important for inducing the membrane structure during viral replication (28). However, during flavivirus infection, membrane remodeling is mediated by the NS4A protein, which has also been shown to form oligomers (36, 37).
Two complementary approaches (cross-linking and pull-down methods) were used to map the regions that are responsible for DENV-2 NS4B dimerization. Cross-linking of C-terminally truncated NS4B suggested that the cytosolic loop (amino acids 129 to 165) is critical for dimerization (Fig. 4). The C-terminal fragment of NS4B (amino acids 166 to 248; downstream of the cytosolic loop) alone could form dimers without cross-linking treatment. These results suggest that either the cytosolic loop or the C-terminal region of NS4B is sufficient to mediate dimerization. This conclusion was further supported by a pulldown experiment showing that full-length NS4B was coimmunoprecipitated with different efficiencies by a set of C-terminally truncated NS4B variants (Fig. 5B). Using the cross-linking approach, we attempted to further map the determinants for dimerization within the cytosolic loop or within the C-terminal region. Unfortunately, those results were inconclusive due to the following technical limitations: (i) different NS4B constructs drastically affected the protein expression levels and (ii) the cross-linking efficiency varied significantly depending on the expression levels of NS4B constructs (data not shown).
One straightforward approach to demonstrate the biological relevance of NS4B dimerization is to identify a mutation that could abolish or significantly ablate NS4B dimerization. Analysis of such a mutation in the context of viral replication should indicate the function of NS4B dimerization. This approach could not be applied to this study because (i) either the cytosolic loop or the C-terminal region of NS4B was sufficient to mediate dimerization and (ii) multiple mutations within NS4B could ablate functions of this protein other than dimerization. As an alternative approach, we examined whether two distinct lethal point mutants (P104R and K143A) of NS4B could complement each other; no complementation could be observed to restore viral replication. However, the replication defect of individual NS4B mutants could be rescued in replicon-containing cells. These data confirmed our previous result showing that NS4B mutants can be trans-complemented in the presence of a wild-type replication complex (23). It should be noted that trans-complementation in replicon cells does not prove that NS4B dimerization is essential for viral replication. To further explore the requirement of NS4B trans-complementation, we tested whether wild-type NS4B alone was sufficient to restore the replication defect of the NS4B mutant. Under these conditions, no trans-complementation could be achieved for the NS4B lethal genome-length RNA. This result demonstrates that trans-complementation of NS4B requires wild-type NS4B provided in the context of a viral replication complex.
In summary, we have developed a robust method to generate recombinant NS4B proteins of flaviviruses. Recombinant NS4B protein as well as the NS4B protein from infected cells forms dimers. The cytoplasmic loop and its downstream C-terminal region are critical for NS4B dimerization. DENV-2 NS4B is not glycosylated. trans-Complementation of NS4B requires wild-type NS4B presented in the replication complex. Since NS4B inhibitors have been reported for YFV (24) and DENV (23, 38), it will be interesting to examine whether these inhibitors suppress viral replication through blocking NS4B dimerization.
ACKNOWLEDGMENTS
We thank our colleagues at the Novartis Institute for Tropical Diseases (NITD) for helpful discussions during the course of this study.
This project was partially supported by the TCR flagship STOP Dengue program from the National Medical Research Council in Singapore to NITD and by grant CRP2008 from the National Research Foundation to the J.L. lab.
Footnotes
Published ahead of print 3 January 2014
REFERENCES
- 1.Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, Myers MF, George DB, Jaenisch T, Wint GR, Simmons CP, Scott TW, Farrar JJ, Hay SI. 2013. The global distribution and burden of dengue. Nature 496:504–507. 10.1038/nature12060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sabchareon A, Wallace D, Sirivichayakul C, Limkittikul K, Chanthavanich P, Suvannadabba S, Jiwariyavej V, Dulyachai W, Pengsaa K, Wartel TA, Moureau A, Saville M, Bouckenooghe A, Viviani S, Tornieporth NG, Lang J. 2012. Protective efficacy of the recombinant, live-attenuated, CYD tetravalent dengue vaccine in Thai schoolchildren: a randomised, controlled phase 2b trial. Lancet 380:1559–1567. 10.1016/S0140-6736(12)61428-7 [DOI] [PubMed] [Google Scholar]
- 3.Lindenbach BD, Thiel H-J, Rice CM. 2007. Flaviviridae: the viruses and their replication, p 1101–1152 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 4.Munoz-Jordan JL, Sanchez-Burgos GG, Laurent-Rolle M, Garcia-Sastre A. 2003. Inhibition of interferon signaling by dengue virus. Proc. Natl. Acad. Sci. U. S. A. 100:14333–14338. 10.1073/pnas.2335168100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morrison J, Aguirre S, Fernandez-Sesma A. 2012. Innate immunity evasion by dengue virus. Viruses 4:397–413. 10.3390/v4030397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Warrener P, Tamura JK, Collett MS. 1993. RNA-stimulated NTPase activity associated with yellow fever virus NS3 protein expressed in bacteria. J. Virol. 67:989–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wengler G, Czaya G, Farber PM, Hegemann JH. 1991. In vitro synthesis of West Nile virus proteins indicates that the amino-terminal segment of the NS3 protein contains the active centre of the protease which cleaves the viral polyprotein after multiple basic amino acids. J. Gen. Virol. 72:851–858. 10.1099/0022-1317-72-4-851 [DOI] [PubMed] [Google Scholar]
- 8.Li H, Clum S, You S, Ebner KE, Padmanabhan R. 1999. The serine protease and RNA-stimulated nucleoside triphosphatase and RNA helicase functional domains of dengue virus type 2 NS3 converge within a region of 20 amino acids. J. Virol. 73:3108–3116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Egloff MP, Benarroch D, Selisko B, Romette JL, Canard B. 2002. An RNA cap (nucleoside-2′-O-)-methyltransferase in the flavivirus RNA polymerase NS5: crystal structure and functional characterization. EMBO J. 21:2757–2768. 10.1093/emboj/21.11.2757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ray D, Shah A, Tilgner M, Guo Y, Zhao Y, Dong H, Deas T, Zhou Y, Li H, Shi P-Y. 2006. West Nile virus 5′-cap structure is formed by sequential guanine N-7 and ribose 2′-O methylations by nonstructural protein 5. J. Virol. 80:8362–8370. 10.1128/JVI.00814-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou Y, Ray D, Zhao Y, Dong H, Ren S, Li Z, Guo Y, Bernard K, Shi P-Y, Li H. 2007. Structure and function of flavivirus NS5 methyltransferase. J. Virol. 81:3891–3903. 10.1128/JVI.02704-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong H, Chang DC, Hua MH, Lim SP, Chionh YH, Hia F, Lee YH, Kukkaro P, Lok SM, Dedon PC, Shi PY. 2012. 2′-O methylation of internal adenosine by flavivirus NS5 methyltransferase. PLoS Pathog. 8:e1002642. 10.1371/journal.ppat.1002642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ackermann M, Padmanabhan R. 2001. De novo synthesis of RNA by the dengue virus RNA-dependent RNA polymerase exhibits temperature dependence at the initiation but not elongation phase. J. Biol. Chem. 276:39926–39937. 10.1074/jbc.M104248200 [DOI] [PubMed] [Google Scholar]
- 14.Leung JY, Pijlman GP, Kondratieva N, Hyde J, Mackenzie JM, Khromykh AA. 2008. Role of nonstructural protein NS2A in flavivirus assembly. J. Virol. 82:4731–4741. 10.1128/JVI.00002-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie X, Gayen S, Kang C, Yuan Z, Shi PY. 2013. Membrane topology and function of dengue virus NS2A protein. J. Virol. 87:4609–4622. 10.1128/JVI.02424-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kummerer BM, Rice CM. 2002. Mutations in the yellow fever virus nonstructural protein NS2A selectively block production of infectious particles. J. Virol. 76:4773–4784. 10.1128/JVI.76.10.4773-4784.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miller S, Sparacio S, Bartenschlager R. 2006. Subcellular localization and membrane topology of the dengue virus type 2 non-structural protein 4B. J. Biol. Chem. 281:8854–8863. 10.1074/jbc.M512697200 [DOI] [PubMed] [Google Scholar]
- 18.Roosendaal J, Westaway EG, Khromykh A, Mackenzie JM. 2006. Regulated cleavages at the West Nile virus NS4A-2K-NS4B junctions play a major role in rearranging cytoplasmic membranes and Golgi trafficking of the NS4A protein. J. Virol. 80:4623–4632. 10.1128/JVI.80.9.4623-4632.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Munoz-Jordan JL, Laurent-Rolle M, Ashour J, Martinez-Sobrido L, Ashok M, Lipkin WI, Garcia-Sastre A. 2005. Inhibition of alpha/beta interferon signaling by the NS4B protein of flaviviruses. J. Virol. 79:8004–8013. 10.1128/JVI.79.13.8004-8013.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kakumani PK, Ponia SS, S RK, Sood V, Chinnappan M, Banerjea AC, Medigeshi GR, Malhotra P, Mukherjee SK, Bhatnagar RK. 2013. Role of RNA interference (RNAi) in dengue virus replication and identification of NS4B as an RNAi suppressor. J. Virol. 87:8870–8883. 10.1128/JVI.02774-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Umareddy I, Chao A, Sampath A, Gu F, Vasudevan SG. 2006. Dengue virus NS4B interacts with NS3 and dissociates it from single-stranded RNA. J. Gen. Virol. 87:2605–2614. 10.1099/vir.0.81844-0 [DOI] [PubMed] [Google Scholar]
- 22.Youn S, Li T, McCune BT, Edeling MA, Fremont DH, Cristea IM, Diamond MS. 2012. Evidence for a genetic and physical interaction between nonstructural proteins NS1 and NS4B that modulates replication of West Nile virus. J. Virol. 86:7360–7371. 10.1128/JVI.00157-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xie X, Wang QY, Xu HY, Qing M, Kramer L, Yuan Z, Shi PY. 2011. Inhibition of dengue virus by targeting viral NS4B protein. J. Virol. 85:11183–11195. 10.1128/JVI.05468-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Patkar CG, Larsen M, Owston M, Smith JL, Kuhn RJ. 2009. Identification of inhibitors of yellow fever virus replication using a replicon-based high-throughput assay. Antimicrob. Agents Chemother. 53:4103–4114. 10.1128/AAC.00074-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zou G, Chen YL, Dong H, Lim CC, Yap LJ, Yau YH, Shochat SG, Lescar J, Shi PY. 2011. Functional analysis of two cavities in flavivirus NS5 polymerase. J. Biol. Chem. 286:14362–14372. 10.1074/jbc.M110.214189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie X, Zou J, Wang Q-Y, Noble CG, Lescar J, Shi P-Y. 2014. Generation and characterization of mouse monoclonal antibodies against NS4B protein of dengue virus. Virology 450–451:250–257. 10.1016/j.virol.2013.12.025 [DOI] [PubMed] [Google Scholar]
- 27.Lorenz H, Hailey DW, Wunder C, Lippincott-Schwartz J. 2006. The fluorescence protease protection (FPP) assay to determine protein localization and membrane topology. Nat. Protoc. 1:276–279. 10.1038/nprot.2006.42 [DOI] [PubMed] [Google Scholar]
- 28.Yu GY, Lee KJ, Gao L, Lai MM. 2006. Palmitoylation and polymerization of hepatitis C virus NS4B protein. J. Virol. 80:6013–6023. 10.1128/JVI.00053-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ng CY, Gu F, Phong WY, Chen YL, Lim SP, Davidson A, Vasudevan SG. 2007. Construction and characterization of a stable subgenomic dengue virus type 2 replicon system for antiviral compound and siRNA testing. Antiviral Res. 76:222–231. 10.1016/j.antiviral.2007.06.007 [DOI] [PubMed] [Google Scholar]
- 30.Lund S, Orlowski S, de Foresta B, Champeil P, le Maire M, Moller JV. 1989. Detergent structure and associated lipid as determinants in the stabilization of solubilized Ca2+-ATPase from sarcoplasmic reticulum. J. Biol. Chem. 264:4907–4915 [PubMed] [Google Scholar]
- 31.Berger BW, Garcia RY, Lenhoff AM, Kaler EW, Robinson CR. 2005. Relating surfactant properties to activity and solubilization of the human adenosine a3 receptor. Biophys. J. 89:452–464. 10.1529/biophysj.104.051417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zou G, Zhang B, Lim PY, Yuan Z, Bernard KA, Shi PY. 2009. Exclusion of West Nile virus superinfection through RNA replication. J. Virol. 83:11765–11776. 10.1128/JVI.01205-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mukhopadhyay S, Kuhn RJ, Rossmann MG. 2005. A structural perspective of the flavivirus life cycle. Nat. Rev. Microbiol. 3:13–22. 10.1038/nrmicro1067 [DOI] [PubMed] [Google Scholar]
- 34.Lescar J, Canard B. 2009. RNA-dependent RNA polymerases from flaviviruses and Picornaviridae. Curr. Opin. Struct. Biol. 19:759–767. 10.1016/j.sbi.2009.10.011 [DOI] [PubMed] [Google Scholar]
- 35.Noble CG, Shi PY. 2012. Structural biology of dengue virus enzymes: towards rational design of therapeutics. Antiviral Res. 96:115–126. 10.1016/j.antiviral.2012.09.007 [DOI] [PubMed] [Google Scholar]
- 36.Mackenzie JM, Khromykh AA, Jones MK, Westaway EG. 1998. Subcellular localization and some biochemical properties of the flavivirus Kunjin nonstructural proteins NS2A and NS4A. Virology 245:203–215. 10.1006/viro.1998.9156 [DOI] [PubMed] [Google Scholar]
- 37.Miller S, Kastner S, Krijnse-Locker J, Buhler S, Bartenschlager R. 2007. The non-structural protein 4A of dengue virus is an integral membrane protein inducing membrane alterations in a 2K-regulated manner. J. Biol. Chem. 282:8873–8882. 10.1074/jbc.M609919200 [DOI] [PubMed] [Google Scholar]
- 38.van Cleef KW, Overheul GJ, Thomassen MC, Kaptein SJ, Davidson AD, Jacobs M, Neyts J, van Kuppeveld FJ, van Rij RP. 2013. Identification of a new dengue virus inhibitor that targets the viral NS4B protein and restricts genomic RNA replication. Antiviral Res. 99:165–171. 10.1016/j.antiviral.2013.05.011 [DOI] [PubMed] [Google Scholar]