

Abstract
Infection with the human gammaherpesviruses, Epstein-Barr virus (EBV) and Kaposi's sarcoma-associated herpesvirus (KSHV), is associated with several cancers. During lytic replication of herpesviruses, viral genes are expressed in an ordered cascade. However, the mechanism by which late gene expression is regulated has not been well characterized in gammaherpesviruses. In this study, we have investigated the cis element that mediates late gene expression during de novo lytic infection with murine gammaherpesvirus 68 (MHV-68). A reporter system was established and used to assess the activity of viral late gene promoters upon infection with MHV-68. It was found that the viral origin of lytic replication, orilyt, must be on the reporter plasmid to support activation of the late gene promoter. Furthermore, the DNA sequence required for the activation of late gene promoters was mapped to a core element containing a distinct TATT box and its neighboring sequences. The critical nucleotides of the TATT box region were determined by systematic mutagenesis in the reporter system, and the significance of these nucleotides was confirmed in the context of the viral genome. In addition, EBV and KSHV late gene core promoters could be activated by MHV-68 lytic replication, indicating that the mechanisms controlling late gene expression are conserved among gammaherpesviruses. Therefore, our results on MHV-68 establish a solid foundation for mechanistic studies of late gene regulation.
INTRODUCTION
The human gammaherpesviruses Epstein-Barr virus (EBV) and human herpesvirus 8/Kaposi's sarcoma-associated herpesvirus (HHV-8/KSHV) are linked to several diseases and cancers. Like all herpesviruses, EBV and KSHV have two distinct transcriptional programs: latency and lytic replication. During latency, the viral genome exists as a circular form in the nucleus, where only a few viral genes are expressed and no infectious viral particles are produced. In contrast, when herpesviruses undergo lytic replication, numerous viral genes are expressed in a highly ordered manner, leading to the production of viral progeny for subsequent infection of naive cells and transmission to other hosts. Both latency and lytic replication contribute to viral pathogenesis. Viral latency is the predominant form of infection in most EBV- and KSHV-associated diseases and thus is thought to be directly linked to tumorigenesis. Nevertheless, viral lytic replication is found in a small fraction of infected cells and may contribute to viral pathogenesis by increasing the transforming potential of latently infected cells in a paracrine fashion and by replenishing the pool of latently infected cells with continuous infection of naive cells. Furthermore, in some virus-associated diseases, such as with EBV in oral hairy leukoplakia (1) and KSHV in multicentric Castleman disease (2, 3), the virus is found to be primarily in the lytic replication phase.
During lytic replication, herpesviral genes are expressed in a tightly regulated cascade and can be categorized into three groups: immediate early, early, and late. Immediate early (α) genes are the first group of viral genes expressed following viral infection of cells and characterized by the fact that their RNA expression is resistant to inhibition of protein synthesis. The expression of immediate early genes is thought to be activated by incoming proteins associated with the virions and/or preexisting cellular proteins. One of the functions of immediate early genes is to activate the expression of early (β) genes. Early genes usually encode proteins involved in genome replication and nucleotide metabolism. Their RNA expression is sensitive to inhibition of protein synthesis but resistant to inhibition of viral DNA replication. Late (γ) genes refer to those genes whose expression is absolutely dependent on viral DNA synthesis. They largely encode viral structural proteins and are necessary for infectious virion formation.
Compared with the regulation of viral immediate early and early gene expression, the mechanisms controlling the expression of viral late genes are poorly characterized. Two groups have mapped several promoters of EBV late genes to minimal regions (4, 5). Despite some differences in the findings of these two groups, they both have shown that the cis element required for late gene activation comprises a short core sequence including an unconventional TATT sequence, rather than a canonical TATA box. Similarly, in KSHV, a region of only 12 bp was found to be sufficient to support activation of the K8.1 late gene promoter. This 12-bp region also contains a TATT sequence (6). Nevertheless, how such a short core sequence governs the expression of late genes remains unclear. Aside from the conclusion that a TATT sequence is required, the sequence requirement at each position of the EBV and KSHV late gene core promoters has not been well defined, and the role of individual nucleotides in controlling late gene expression remains to be demonstrated in the context of viral genomes.
Murine gammaherpesvirus 68 (MHV-68, also referred to γHV-68) is closely related to KSHV and EBV (7, 8) and has served as a tractable model system to study gammaherpesviruses, especially in regard to de novo lytic replication in vitro and virus-host interactions in vivo. Unlike KSHV and EBV, MHV-68 readily undergoes de novo lytic replication in many cell lines. The MHV-68 genome contains approximately 80 open reading frames (ORFs), 80% of which are homologous to genes of other gammaherpesviruses, including KSHV and EBV. Therefore, we have exploited MHV-68 to study the regulation of viral late gene expression during lytic replication to identify both viral trans-acting proteins and cis regulatory DNA sequences essential for this process. Toward this goal, we previously systematically identified five viral proteins (ORF18, ORF24, ORF30, ORF31, and ORF34) whose deletion results in a loss of viral late gene transcription but has no effect on viral early gene expression or genome replication (9–12). Precisely how these proteins coordinate to direct late gene expression, including whether they interact with the late gene promoter, remains under investigation. Interestingly, studies on human cytomegalovirus (HCMV), a betaherpesvirus, have shown that UL79, UL87, UL91, UL92, and UL95 (the homologues of ORF18, ORF24, ORF30, ORF31, and ORF34, respectively) are also essential for late gene transcription (13–16).
In this study, we deciphered the cis elements required for activation of late gene transcription. To facilitate the study of promoter mapping, a reporter assay was established to measure the promoter activity of late genes during de novo lytic replication of MHV-68. Using this reporter system, we found that a 15-bp sequence derived from the putative late gene promoter region that contains an atypical TATT box is both necessary and sufficient for activating reporter expression. Comprehensive mutagenesis of this 15-bp core promoter sequence revealed that the nucleotide composition of the TATT box is crucial for the viral late gene promoter activity. Finally, when critical nucleotides mapped by the reporter assay were mutated in the viral genome, the expression of the corresponding viral late gene was drastically reduced during de novo lytic replication. Taken together, our findings support the conclusion that viral late gene expression is regulated by a core promoter that contains a noncanonical TATT sequence, which has interesting mechanistic implications for the regulation of late gene expression.
MATERIALS AND METHODS
Construction of the orilyt reporter vector.
The minimal orilyt-containing fragment was cut from pMOΔ16 using the PvuII restriction enzyme (17) and then cloned into pGL3-Basic (Promega) that was linearized with PshAI. The resulting plasmid was named pGL3-Ori.
Construction of late gene promoter reporter constructs.
The regions upstream of translational initiation codons containing putative promoters were amplified from the MHV-68 genome using forward and reverse primers bearing XhoI (CTCGAG) and HindIII (AAGCTT) sites, respectively. The PCR fragment was then cloned into the XhoI and HindIII sites of the pGL3-Ori reporter vector. The sequences of the PCR primers are shown in Table 1. For all constructs containing less than 40 bp of the putative viral promoter sequences, two complementary oligonucleotides were generated with a TGCA 5′ overhang and an AGCT 3′ overhang after annealing. The double-stranded promoter insert was ligated into pGL3-orilyt digested with XhoI and HindIII. The oligonucleotide sequences used for each construct are shown in Tables 2 and 3.
TABLE 1.
Sequences of oligonucleotides used for PCR to construct reporter constructs
| Primer name | Sequencea |
|---|---|
| Luc-R | CTTCGTCCACAAACACAACTCC |
| ORF52p-250bp-F | TGCTCGAGatcacccaagaaaccacacc |
| ORF52pR-HindIII | CTAAGCTTcaggttgtctccagggcact |
| ORF57p-250bp-F | TGCTCGAGccacagaaaaatgcaaatga |
| ORF57pR-HindIII | CTAAGCTTgcacggtgcaatgtgtca |
| M7p-100bp-F | TGCTCGAGtggagtcataaacggtgccaa |
| M7p-169bp-F | TGCTCGAGtggagtcataaacggtgccaa |
| M7p-249bp-F | TGCTCGAGtatggcaaaccaggacactgc |
| M7pR-HindIII | CTAAGCTTgttgagggttttatagcgtc |
| ORF52p-40bp-F | CTCTCTCGAGctgcacccttgctcaacttg |
| ORF52p-71bp-F | CCGGCTCGAGggataatatttaagcagtgc |
| ORF52p-87bp-F | CCGGCTCGAGcctcataatgttgggaggat |
| ORF52p-100bp-F | CCGGCTCGAGcttactaccatgacctcata |
| ORF52p-133bp-F | CCGGCTCGAGgtctttttgatctacgcatg |
| ORF52p-153bp-F | CCGGCTCGAGtagtcacagtgtctagtggt |
| ORF52p-173bp-F | CCGGCTCGAGggcaataattaatgggtcta |
| ORF52p-173bp-F | CCGGCTCGAGggcaataattaatgggtcta |
| ORF52pR-HindIII | CCGGAAGCTTcgctcagggagaataacaac |
| ORF54p-100bp-F | TGCTCGAGaaacagctataaaataggtg |
| ORF54p-200bp-F | TCCTCGAGagatgagtcatcaggttttg |
| ORF54p-400bp-F | TCCTCGAGtgtctccagggcactgc |
| ORF54pR-HindIII | CGAAGCTTggcaaataattgctctgt |
The restriction enzyme sequences are in italic uppercase, and the viral sequences are in lowercase.
TABLE 2.
Sequences of oligonucleotides used for annealing and construction of the reporter constructs for the ORF52 promotera
| Construct | Forward primer | Reverse primer |
|---|---|---|
| 52p-20bp | tcgaGGATAATATTTAAGCAGTGC | agctGCACTGCTTAAATATTATCC |
| 52p-15bp | tcgaGATAATATTTAAGCA | agctTGCTTAAATATTATC |
| 52p-15bp-1A | tcgaAATAATATTTAAGCA | agctTGCTTAAATATTATT |
| 52p-15bp-2T | tcgaGTTAATATTTAAGCA | agctTGCTTAAATATTAAC |
| 52p-15bp-3A | tcgaGAAAATATTTAAGCA | agctTGCTTAAATATTTTC |
| 52p-15bp-4T | tcgaGATTATATTTAAGCA | agctTGCTTAAATATAATC |
| 52p-15bp-5T | tcgaGATATTATTTAAGCA | agctTGCTTAAATAATATC |
| 52p-15bp-6A | tcgaGATAAAATTTAAGCA | agctTGCTTAAATTTTATC |
| 52p-15bp-7T | tcgaGATAATTTTTAAGCA | agctTGCTTAAAAATTATC |
| 52p-15bp-8A | tcgaGATAATAATTAAGCA | agctTGCTTAATTATTATC |
| 52p-15bp-9A | tcgaGATAATATATAAGCA | agctTGCTTATATATTATC |
| 52p-15bp-10A | tcgaGATAATATTAAAGCA | agctTGCTTTAATATTATC |
| 52p-15bp-11T | tcgaGATAATATTTTAGCA | agctTGCTAAAATATTATC |
| 52p-15bp-12T | tcgaGATAATATTTATGCA | agctTGCATAAATATTATC |
| 52p-15bp-13A | tcgaGATAATATTTAAACA | agctTGTTTAAATATTATC |
| 52p-15bp-14A | tcgaGATAATATTTAAGAA | agctTTCTTAAATATTATC |
| 52p-15bp-15T | tcgaGATAATATTTAAGCT | agctAGCTTAAATATTATC |
| 52p-15bp-1T | tcgaTATAATATTTAAGCA | agctTGCTTAAATATTATA |
| 52p-15bp-2G | tcgaGGTAATATTTAAGCA | agctTGCTTAAATATTACC |
| 52p-15bp-3G | tcgaGAGAATATTTAAGCA | agctTGCTTAAATATTCTC |
| 52p-15bp-4G | tcgaGATGATATTTAAGCA | agctTGCTTAAATATCATC |
| 52p-15bp-5G | tcgaGATAGTATTTAAGCA | agctTGCTTAAATACTATC |
| 52p-15bp-6G | tcgaGATAAGATTTAAGCA | agctTGCTTAAATCTTATC |
| 52p-15bp-7G | tcgaGATAATGTTTAAGCA | agctTGCTTAAACATTATC |
| 52p-15bp-8G | tcgaGATAATAGTTAAGCA | agctTGCTTAACTATTATC |
| 52p-15bp-9G | tcgaGATAATATGTAAGCA | agctTGCTTACATATTATC |
| 52p-15bp-10G | tcgaGATAATATTGAAGCA | agctTGCTTCAATATTATC |
| 52p-15bp-11G | tcgaGATAATATTTGAGCA | agctTGCTCAAATATTATC |
| 52p-15bp-12G | tcgaGATAATATTTAGGCA | agctTGCCTAAATATTATC |
| 52p-15bp-13T | tcgaGATAATATTTAATCA | agctTGATTAAATATTATC |
| 52p-15bp-14T | tcgaGATAATATTTAAGTA | agctTACTTAAATATTATC |
| 52p-15bp-15G | tcgaGATAATATTTAAGCG | agctCGCTTAAATATTATC |
| 52p-15bp-1C | tcgaCATAATATTTAAGCA | agctTGCTTAAATATTATG |
| 52p-15bp-2C | tcgaGCTAATATTTAAGCA | agctTGCTTAAATATTAGC |
| 52p-15bp-3C | tcgaGACAATATTTAAGCA | agctTGCTTAAATATTGTC |
| 52p-15bp-4C | tcgaGATCATATTTAAGCA | agctTGCTTAAATATGATC |
| 52p-15bp-5C | tcgaGATACTATTTAAGCA | agctTGCTTAAATAGTATC |
| 52p-15bp-6C | tcgaGATAACATTTAAGCA | agctTGCTTAAATGTTATC |
| 52p-15bp-7C | tcgaGATAATCTTTAAGCA | agctTGCTTAAAGATTATC |
| 52p-15bp-8C | tcgaGATAATACTTAAGCA | agctTGCTTAAGTATTATC |
| 52p-15bp-9C | tcgaGATAATATCTAAGCA | agctTGCTTAGATATTATC |
| 52p-15bp-10C | tcgaGATAATATTCAAGCA | agctTGCTTGAATATTATC |
| 52p-15bp-11C | tcgaGATAATATTTCAGCA | agctTGCTGAAATATTATC |
| 52p-15bp-12C | tcgaGATAATATTTACGCA | agctTGCGTAAATATTATC |
| 52p-15bp-13C | tcgaGATAATATTTAACCA | agctTGGTTAAATATTATC |
| 52p-15bp-14G | tcgaGATAATATTTAAGGA | agctTCCTTAAATATTATC |
| 52p-15bp-15C | tcgaGATAATATTTAAGCC | agctGGCTTAAATATTATC |
| 52p-15bp-9A10A | tcgaGATAATATAAAAGCA | agctTGCTTTTATATTATC |
| 52p-15bp-4T5T | tcgaGATTTTATTTAAGCA | agctTGCTTAAATAAAATC |
| 52p-15bp-13C14G | tcgaGATAATATTTAACGA | agctTCGTTAAATATTATC |
The viral sequences are in uppercase, and the overhangs are in lowercase.
TABLE 3.
Sequences of oligonucleotides used for annealing and construction of the reporter constructs for viral promotersa
| Construct | Forward primer | Reverse primer |
|---|---|---|
| M7p-31bp-F | tcgaTATAATGTTTATATTTAACTGAATAAAAGAA | agctTTCTTTTATTCAGTTAAATATAAACATTATA |
| M7p-20bpF | tcgaTGTTTATATTTAACTGAATAA | agctTTATTCAGTTAAATATAAACA |
| M7p-15bpF | tcgaGTTTATATTTAACTG | agctCAGTTAAATATAAAC |
| M7p-15bpF-4A | tcgaGTTAATATTTAACTG | agctCAGTTAAATATTAAC |
| M7p-15bpF-5T | tcgaGTTTTTATTTAACTG | agctCAGTTAAATAAAAAC |
| M7p-9A10A-15bpF | tcgaGTTTATATAAAACTG | agctCAGAAAAATATAAAC |
| M7p-6A-15bpF | tcgaGTTTAAATTTAACTG | agctCAGTTAAATTTAAAC |
| ORF4-15bpF | tcgaATCTCTATTAAAGGG | agctCCCTTTAATAGAGAT |
| ORF7-15bpF | tcgaAGACAATATTATAAC | agctGTTATAATATTGTCT |
| ORF8-15bpF | tcgaGAGGGTATTTAAAGA | agctTCTTTAAATACCCTC |
| ORF17-15bpF | tcgaTAGCGTATTTAATTT | agctAAATTAAATACGCTA |
| ORF19-15bpF | tcgaCCTGATATTTAAGAG | agctCTCTTAAATATCAGG |
| ORF20-15bpF | tcgaTTGTCTATTATAGGG | agctCCCTATAATAGACAA |
| ORF22-15bpF | tcgaCCGGCTATTAAAACA | agctTGTTTTAATAGCCGG |
| ORF23-15bpF | tcgaAAAAATATTAACAGG | agctCCCTATAATAGACAA |
| ORF25-15bpF | tcgaAAGTATATTAAATTG | agctCAATTTAATATACTT |
| ORF26-15bpF | tcgaAAGAGTATTAAAGAT | agctATCTTTAATACTCTT |
| ORF27-15bpF | tcgaTTCAATATTTAATCT | agctAGATTAAATATTGAA |
| ORF29-15bpF | tcgaGCTCATATTAATTGC | agctGCAATTAATATGAGC |
| ORF32-15bpF | tcgaCATGCTATTTAAATG | agctCATTTAAATAGCATG |
| ORF33-15bpF | tcgaTTTGTTATTAAAGGA | agctTCCTTTAATAACAAA |
| ORF38-15bpF | tcgaGGGTGTATATAAAAG | agctCTTTTATATACACCC |
| ORF39-15bpF | tcgaACGATTATTAAAAAG | agctCTTTTTAATAATCGT |
| ORF42-15bpF | tcgaTAGTCTATATAACTG | agctCAGTTATATAGACTA |
| ORF45-15bpF1 | tcgaACACATATTTAACGC | agctGCGTTAAATATGTGT |
| ORF45-15bpF2 | tcgaTCTTGTATTAAAGGC | agctGCCTTTAATACAAGA |
| ORF53-15bpF | tcgaTTGTCTATTATAGGG | agctCCCTATAATAGACAA |
| ORF54-15bpF | tcgaACAGCTATAAAATAG | agctCTATTTTATAGCTGT |
| ORF64-15bpF | tcgaGCAAATATTTACACA | agctTGTGTAAATATTTGC |
| ORF65-15bpF | tcgaTGCAGTATTAAGCTG | agctCAGCTTAATACTGCA |
| K8.1p15bpF | tcgaAGCAATATTAAAGGG | agctCCCTTTAATATTGCT |
| BcLF1-15bpF | tcgaAGAATTATTAAACCG | agctCGGTTTAATAATTCT |
| BdRF1-15bpF | tcgaAGTCGTATTTAAAGG | agctCCTTTAAATACGACT |
The viral sequences are in uppercase, and the overhangs are in lowercase.
5′ rapid amplification of cDNA ends (RACE).
Total RNA isolated from infected cells was used for reverse transcription with an ORF52 gene-specific primer (ORF52GSP1; 5′-TTA TTC ATG ATC ATG TCT GTG TCT TG-3′). After RNase H digestion, the reverse transcription product was incubated with dCTP and terminal deoxynucleotidyl transferase (TdT) to add poly(C) tails to the 3′ ends. The tailed product was then amplified with PCR using an Abridged Anchor Primer (5′-GGC CAC GCG TCG ACT AGT ACG GGI IGG GII GGG IIG-3′) and the ORF52GSP1 for 30 cycles. The PCR product was reamplified using the Abridged Universal Amplification Primer (5′-GGC CAC GCG TCG ACT AGT AC-3′) and ORF52GSP2 (5′-TTC ATA GGT TTT ATC AGG CTT-3′) for an additional 30 cycles and then analyzed by gel electrophoresis.
S1 nuclease protection assay.
A 60-bp probe was designed to contain the complementary sequence (nucleotides [nt] 71370 to 71424) upstream of the translational start site of ORF52 (nt 71364), as well as 5 nt of the noncomplementary sequence at the 3′ end (5′-AGG GAG AAT AAC AAC AAG TTG AGC AAG GGT GCA GGT TGT CTC CAG GGC AC TGC TTG GGC G-3′). The nucleotide numbers in this study are assigned on the basis of the published MHV-68 sequence (7). The probe was end labeled with 32P using T4 polynucleotide kinase. Total RNA harvested from uninfected or MHV-68-infected BHK-21 cells was purified and allowed to anneal with the radiolabeled probe. The RNA was then digested with S1 nuclease (200 or 400 U), and the remaining RNA/DNA hybrid was resolved on a denaturing polyacrylamide-urea gel.
Dual-luciferase reporter assays.
The reporter plasmids were transfected into BHK-21 cells together with an internal control plasmid, pRLSV40 (Promega), which contains Renilla luciferase under the control of the simian virus 40 (SV40) promoter, using Lipofectamine with Plus reagent (Invitrogen). At 12 h posttransfection, cells were tryspinized and reseeded into multiple wells for infection. Twelve hours later, cells were infected with MHV-68 at a multiplicity of infection (MOI) of 2, and cell lysates were harvested 24 h later for the dual-luciferase assay (Promega). Firefly luciferase activity was normalized against Renilla luciferase activity, and the fold activation was calculated by dividing the normalized values of infected samples with those obtained from uninfected samples.
Construction of M7 promoter mutant viruses.
The M7 promoter (M7p) mutant BAC plasmids were constructed by allelic exchange using a recA+ Escherichia coli strain, GS500, harboring the wild-type (WT) MHV-68 BAC plasmid and the conjugation-competent E. coli GS111 strain containing the donor suicide shuttle vector pGS284/M7p-6A or pGS284/M7p-9A10A. pGS284/M7p-6A and pGS284/M7p-9A10A were generated using site-directed mutagenesis of pGS284/M7p, which harbors 600 bp of the M7 promoter. The M7p mutant viruses were reconstituted by transfecting BHK-21 cells with the recombinant viral MHV-68 BAC plasmids using Lipofectamine 2000 (Invitrogen).
Electrophoretic mobility shift assay (EMSA).
Flp-In T-REx 293 cells (Invitrogen) engineered to inducibly express FLAG-tagged ORF24 of MHV-68 upon the addition of doxycycline (12) were used for the production of ORF24 protein. After being treated with 1 μg/ml of doxycycline for 24 h, cells were harvested in lysis buffer containing 50 mM Tris HCl (pH 7.4), 500 mM NaCl, and 1% NP-40. FLAG-tagged ORF24 was purified from whole-cell extract using FLAG resin (Sigma), washed five times in lysis buffer, and eluted from the resin with 1× FLAG peptide (Elim BioPharm). Protein purity was assessed by colloidal Coomassie staining (18). Purified protein was dialyzed against 20 mM HEPES (pH 7.9), 40% glycerol, 100 mM KCl, 0.5 mM dithiothreitol (DTT), and 0.5 mM phenylmethylsulfonyl fluoride (PMSF). Buffer (1 μg/μl of salmon sperm DNA, 0.5 mg/ml of bovine serum albumin [BSA], 0.05% Triton X-100, 10% glycerol, 10 mM HEPES [pH 7.9], 40 mM KCl, 5 mM MgCl2, and 10 μM β-mercaptoethanol [BME]) was added to binding reaction mixtures containing 50 to 200 nM protein and 1 nM radiolabeled probe. For the generation of the probe, oligonucleotides were end labeled with [γ-32P]ATP using T4 polynucleotide kinase (NEB) and purified using a spin column (Bio-Rad). Reverse-complemented oligonucleotides were annealed together to create double-stranded probes; see Fig. 8 for probe sequences used. Binding reactions were assembled and incubated at 37°C for 30 min and immediately loaded into prechilled native PAGE gels. Gels were run for 2 h at 4°C, dried, and scanned using a PharosFX Imager System (Bio-Rad).
FIG 8.
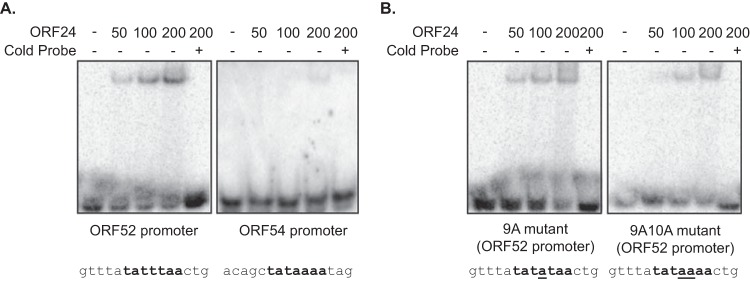
ORF24 interacts with the TATA box-containing sequences. Increasing amounts of purified FLAG-tagged ORF24 protein (50 nM to 200 nM) were incubated with a 1 nM concentration of the indicated radiolabeled probe (probe sequence listed at the bottom) with or without a 50-fold excess of cold probe (50 nM) as a competitor. The resulting complexes were resolved on 5% Tris-borate-EDTA gels.
RESULTS
MHV-68 orilyt is required in cis for activation of the late gene promoter in the reporter system.
A reporter assay was used to assess the activity of viral promoters representing different kinetic classes. Previously, we showed that late gene promoters in a reporter bearing the MHV-68 origin of lytic replication (orilyt) are activated during viral infection (11). For the present study, we constructed a modified reporter that would replicate upon infection by inserting the MHV-68 orilyt into pGL3-Basic. This modified reporter was named pGL3-Ori.
The promoter of an MHV-68 early gene, ORF57, has been previously characterized and shown to require the viral replication and transcription activator (RTA) for its transactivation (19). The RTA-responsive region of the ORF57 promoter was mapped to a 183-bp DNA sequence (nt 75599 to 75782) upstream of the transcription start site (19). We therefore cloned the 250-bp region (nt 75533 to 75782) containing the RTA-responsive ORF57 promoter into either pGL3-Basic or pGL3-Ori. Upon transfection followed by infection, the ORF57 promoter was activated 90-fold in the pGL3-Basic reporter (basic) and 80-fold in the pGL3-Ori reporter (ori) (Fig. 1A). The activation of both constructs was not reduced but increased with treatment of a DNA replication inhibitor, phosphonoacetic acid (PAA), as expected for an early viral gene promoter. The results also indicate that the presence of the orilyt does not alter the nature of early cis regulatory elements.
FIG 1.
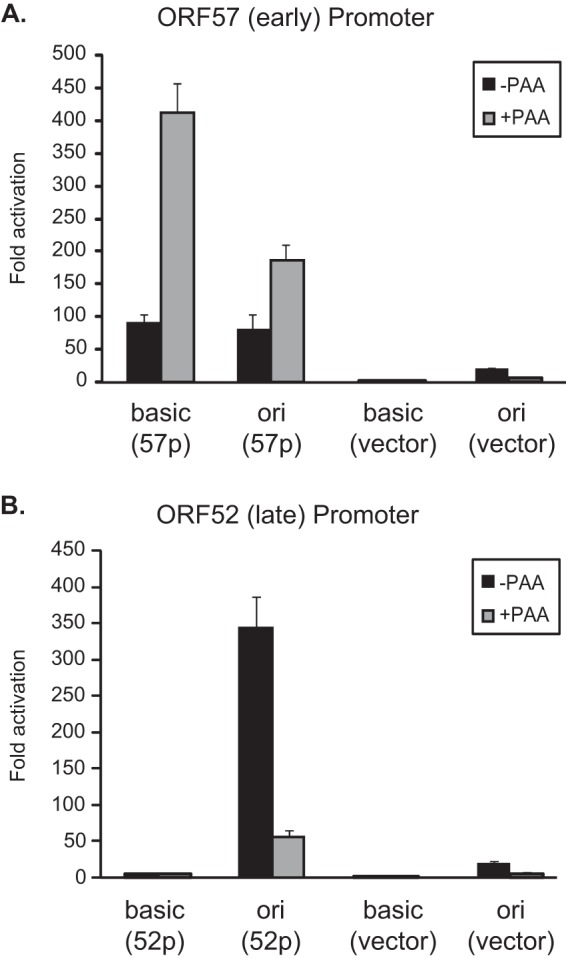
Activation of a late gene promoter requires the MHV-68 origin of lytic replication. The 250-bp fragment of the ORF57 (A) or ORF52 (B) promoter was cloned into the pGL3-Basic or pGL3-Ori vector. Each reporter was transfected and tested for activation induced by MHV-68 infection. Fold activation was calculated by comparing the promoter activity in the infected cells to that in the uninfected cells.
Next, we examined the promoter of a late gene, ORF52. ORF52 is highly expressed and its transcription is sensitive to PAA treatment (20). We cloned a 250-bp region (nt 71401 to 71650) upstream of the ORF52 translational start site into the two reporter vectors, pGL3-Ori and pGL3-Basic. After transfection of the reporters and infection, the putative ORF52 promoter was activated only 4-fold in the pGL3-Basic reporter, and this activation was not inhibited by PAA treatment (Fig. 1B). In contrast, over 300-fold activation was observed for the same DNA sequence cloned in the pGL3-Ori reporter, and this activation was largely abolished by PAA (Fig. 1B). Therefore, full activation of the ORF52 promoter in the reporter requires the presence of the orilyt in cis and depends upon viral DNA replication, recapitulating the defined features of late gene expression. Collectively, these results confirm the utility of the pGL3-Ori system for characterizing the late gene promoters of MHV-68.
Promoter mapping reveals distinctions in the activation of early and late gene promoters.
Next, we constructed a series of reporters for 5′ deletion analysis of the 250-bp ORF52 late gene promoter (Fig. 2). A 5′ deletion up to nt 71436 had no significant effect on the induction of the reporter. However, a further 5′ truncation of the 71-bp region to a 40-bp region (nt 71404 to 71365) greatly diminished reporter activation, resulting in only 3% activity compared to the other reporters, an activation level similar to that with the empty pGL3-Ori vector. We then carried out 3′ deletion mapping of the 71-bp promoter region. To our surprise, both of the resultant 20-bp and 15-bp DNA fragments remained capable of supporting reporter activation upon infection. Close examination of the 15-bp sequence (nt 71434 to 71420) revealed a potential TATA box in the middle. This TATA box has a sequence of TATTTA, with a single deviation at the fourth position from the consensus, TATAT/AA. Our mapping analysis of the ORF52 promoter indicates that the TATT-containing 15-bp sequence is sufficient to support activation of the pGL3-Ori reporter during MHV-68 infection.
FIG 2.
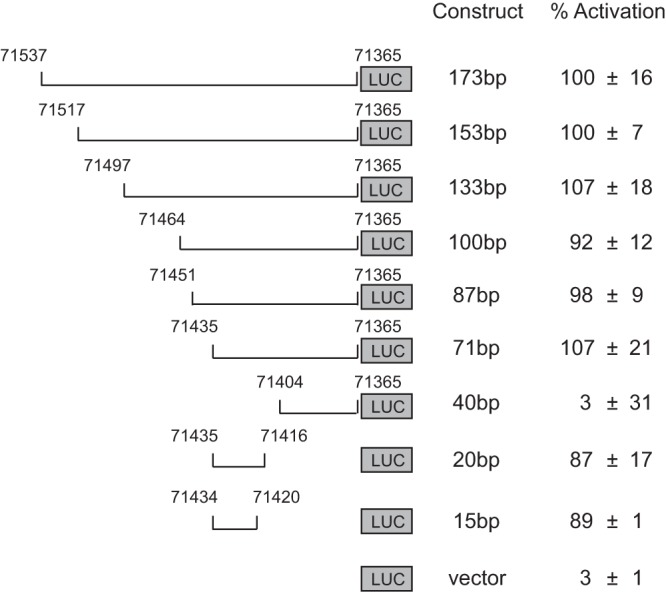
The ORF52 late gene promoter maps to a 15-bp region. The 173-bp region upstream of the ORF52 translational start site was cloned into pGL3-Ori. Successive deletions from the 5′ and 3′ ends were generated, and their boundaries (indicated by nucleotide positions corresponding to the viral genome) are shown. Each construct was transfected and tested for activation induced by MHV-68 infection without PAA treatment. Fold activation was derived from comparing promoter activity in the infected cells to that in the uninfected cells. The percent activation was calculated based on the fold activation for the largest promoter construct (173 bp) being set as 100%.
We next conducted a similar study for the promoter of an early gene, ORF54 (20–22). The 400-bp, 200-bp, and 100-bp regions upstream of the ORF54 translational start codon were cloned into pGL3-Ori. A potential TATA box with a canonical sequence, TATAAA, is located between 89 and 93 bp upstream of the translation start site. The 400-bp and 200-bp constructs were activated in our reporter assay, but the 100-bp construct (which still contains the TATA box) was not induced above the background level (pGL3-Ori vector) (Fig. 3). The finding that the TATA-containing 100-bp sequence failed to activate the pGL3-Ori reporter is in sharp contrast to the ORF52 promoter mapping result. The ORF52 late promoter therefore consists of a 15-bp sequence with a TATT core promoter motif, whereas activation of the ORF54 early promoter depends on additional upstream cis elements within the region between 100 and 200 bp upstream of the translation initiation codon. This distinction between the early and late promoters is also observed for other herpesviruses (4–6, 23, 24) and thus appears to be a conserved feature of herpesviral gene regulation.
FIG 3.
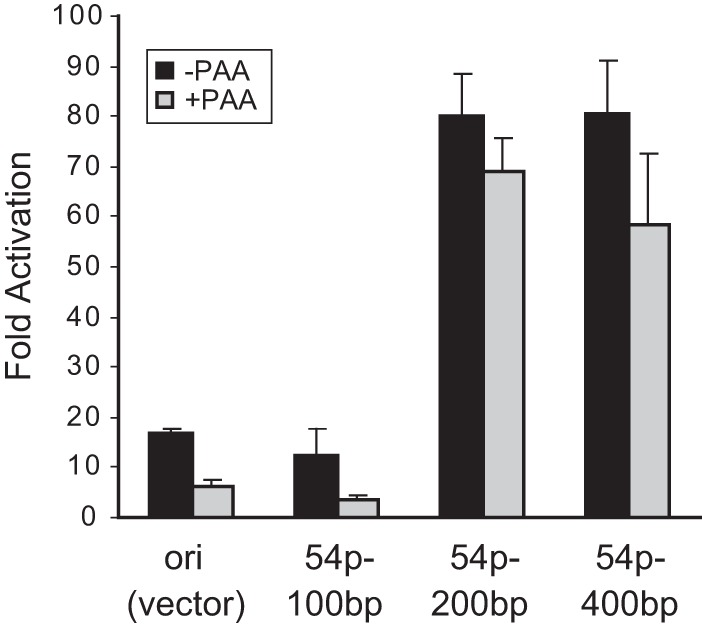
Activation of the ORF54 early gene promoter requires additional regulatory sequences upstream of the TATA box. The 400-bp, 200-bp, and 100-bp regions upstream of the translational start site of ORF54 were individually cloned into pGL3-Ori. Each construct was transfected and tested for activation induced by MHV-68 infection. Fold activation was derived from comparing the promoter activity in the infected cells to that in the uninfected cells.
Mapping of the transcriptional start site of ORF52.
The TATT-containing 15-bp sequence mapped in Fig. 2 is between −71 and −56 relative to the translational initiation site (+1) of ORF52 and should be located approximately 30 bp upstream of the transcriptional start site if it functions as a core promoter. However, the transcriptional start site of ORF52 has not been previously determined. We therefore used 5′ RACE to map the approximate 5′ end of the ORF52 transcript. A PCR product of less than 400 bp was amplified using a gene-specific primer 354 bp downstream of the translational start site, indicating that the transcriptional start site is less than 50 bp from the translation initiation codon. To further determine the exact start site of the ORF52 transcript, an S1 nuclease protection assay was used. A 60-bp radiolabeled probe complementary to the region from −6 to −60 (nt 71370 to 71424) relative to the translational initiation site (+1) with an additional 5 nt of nonviral sequence was annealed to RNA harvested from infected BHK-21 cells. After S1 nuclease digestion, the products were separated on a denaturing polyacrylamide gel. In addition to the undigested probe, three protected products were detected (Fig. 4A, lanes 1 and 2). The largest one is most likely the protected product from read-through transcripts of ORF53, while the other two bands of 31 and 32 nt are products from protection of the ORF52 transcripts. Based on these results, the transcriptional start sites of ORF52 were mapped to nt 71401 and 71400 (Fig. 4B). Accordingly, the TATT sequence (nt 71429 to 71424) of the mapped 15-bp promoter sequence is located at the expected position, 25 to 30 bp upstream of the transcriptional start site.
FIG 4.

Mapping of the 5′ end of the ORF52 transcript by S1 nuclease protection assay. (A) S1 nuclease protection assay. Total RNA from uninfected (lane 3) and infected (lanes 1 and 2) BHK-21 cells was hybridized with a labeled probe and digested with 200 U (lane 1) or 400 U (lanes 2 and 3) of S1 nuclease, and the products were separated on a denaturing gel along with markers of 29 nt (lane 4) and 31 nt (lane 5) as well as the undigested probe (lane 6). (B) Nucleotide sequence of the probe and surrounding region. The translational start codon of ORF52 is indicated by underlined bold uppercase letters. A gray box represents the 15-bp ORF52 promoter region defined by the reporter studies (Fig. 2), with the TATA box highlighted by underlined italic bold letters. The probe used in the S1 nuclease protection assay is indicated by a dashed line, and the two triangles show the two mapped transcription sites.
Systematic mutagenesis of the ORF52 promoter.
To define the sequence requirement at each position of the 15-bp ORF52 promoter, systematic mutagenesis of one nucleotide at a time to the other three possible nucleotides was carried out. A total of 45 promoter mutants were generated, and two independent clones of each construct were tested for induction in our reporter assay. Notably, in the middle of the 15-bp promoter there is a distinct region of remarkable stringency which coincides with the TATT sequence, TATTTAA (Fig. 5A). Nineteen out of 21 possible mutations in this 7-bp TATT sequence greatly reduced the reporter activity, by more than 80%. Despite the variety of TATA boxes that can be found in eukaryotic promoters, the ORF52 core promoter appears to have a strict specificity for the TATTTAA sequence. Interestingly, one of the two less affected mutations, a T-to-A change at the 9th position (4th of the 7-bp TATT box), while making the sequence more similar to the consensus (TATATA), decreased promoter activation by more than 50%. This finding is consistent with previous studies on other herpesviruses (4, 25) and indicates that T at the fourth position of the 7-bp TATT sequence, though much rarer (<10%) than A (>90%) in TATA box-containing eukaryotic promoters (26), is important for the herpesviral late gene promoter activity. For the bases outside the 7-bp TATT sequence, most of the mutations were tolerated, with the exception of those adjacent to it.
FIG 5.

Mutational analysis of the 15-bp ORF52 promoter reveals a stringent sequence requirement for the TATT box. (A) Systematic single mutations. The 15-bp ORF52 promoter was mutated by individually changing each nucleotide of the 15 bp to the other three possible nucleotides. Fold activation was derived from comparing the promoter activity in the infected cells to that in the uninfected cells. The percentage of WT activation was calculated based on the fold activation for the WT reporter, set as 100%. The WT sequence of the 15-bp promoter is shown under the graph, and a gray box represents the region least tolerant to mutations. (B) Targeted double mutations. Three double mutations were tested, and the results are presented together with corresponding single mutations. The sequence of each 15-bp promoter is shown with WT nucleotides in lowercase and the specific mutations in underlined uppercase letters. Activation was calculated as described for panel A and is based on comparisons to fold activation for the WT 15-bp ORF52 promoter.
In addition, we also made three double mutants of the 15-bp promoter constructs. One double mutant construct, 9A10A, was designed to convert the TATT sequence, TATTTA, to the consensus TATA box, TATAAA. Surprisingly, the 9A10A double mutant was activated to only 10% of the WT level, indicating that the consensus TATA box sequence is unable to support ORF52 promoter activation during viral infection. Another double mutant that lost most of its activity was the 4T5T construct, in which the two A residues immediately upstream of the TATT sequence were changed to T's. The single A-to-T mutation at either position caused an ∼60% reduction, and the combination double mutant further decreased the promoter activation, by 90%. This synergistic decrease was not seen for the third double mutant, 13C14G.
A variety of 15-bp sequences from putative MHV-68 late gene promoter regions are capable of supporting reporter activation.
To test whether other MHV-68 late gene promoters function in a similar manner to the ORF52 promoter, we set out to study the promoter of another viral late gene, M7. A series of sequences upstream of the translational start codon (nt 69466) of M7 were cloned into pGL3-Ori and tested for the ability to support reporter activation upon viral infection (Fig. 6). All of the promoter constructs were activated to comparable levels, including the one driven by a 15-bp sequence (nt 69419 to 69433), which contains the TATT box, TATTTAA, the same one found in the 15-bp ORF52 promoter. These data support the hypothesis that a core promoter with a specific TATT sequence is sufficient to mediate replication-dependent gene transcription.
FIG 6.
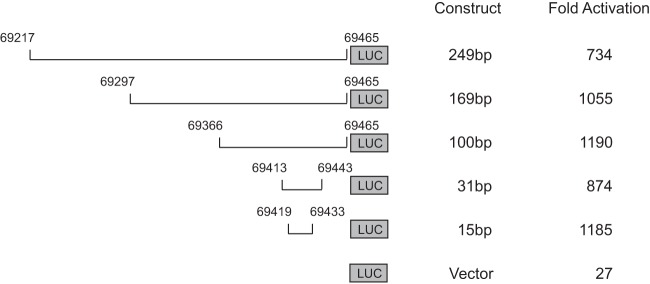
M7 late gene promoter is mapped to a 15-bp region. The schematic diagram illustrates the M7 reporter constructs used to define the M7 promoter. Shown at the right side of each construct are the promoter size and the boundaries indicated by nucleotide positions corresponding to the viral genome. The reporter constructs were individually transfected and tested for activation induced by MHV-68 infection. Fold activation was derived from comparing the promoter activity in the infected cells to that in the uninfected cells.
To extend the scope of the analysis, we constructed a collection of pGL3-Ori reporters with various 15-bp sequences derived from the putative promoter regions of viral late genes that are classified on the basis of several previously published MHV-68 transcriptional profiles (20–22). The viral genome was scanned for potential core promoters that have the distinct TATT sequence located within 200 bp upstream of the translational start site of late genes. As a control, we also included the 15-bp sequence from the putative promoter region of a viral early gene, ORF54, with a canonical TATA box, TATAAAA. In total, we cloned 20 promoters in our pGL3-Ori reporter, and their 15-bp sequences are listed in Table 4. Eleven of these 20 reporters were activated over 200-fold by MHV-68 infection (more than 20-fold higher than the background pGL3-Ori vector), and their activation was greatly reduced by PAA treatment. Ten of these 11 highly active promoters, including the 15-bp promoters of M7 and ORF52, share a TATT sequence, TATTA/TAA, while one has G at the last position. For the other nine reporters, seven were weakly activated and two were inactive. Four of the seven weakly active sequences bear either T at the sixth or C at the seventh position of the consensus, TATTA/TAA, indicating that pyrimidines at these two positions are not tolerated as well as G. The 15-bp sequence derived from the upstream region of ORF29 is inactive and contains the TATTAAT sequence. Since T is never found at the last position of the 7-bp TATT sequence in any of the active promoters, we suggest that the A-to-T variation at this position is not tolerated. The other inactive sequence is derived from the putative ORF54 promoter region and contains a canonical TATA box, TATAAAA, with A at both the fourth and fifth positions of the 7-bp sequence. This inability to support reporter activation is consistent with the phenotype of the 9A10A ORF52 promoter mutant (Fig. 5B).
TABLE 4.
Activities of gammaherpesvirus late gene promotersa

The fold activation was calculated by dividing the normalized luciferase value of the infected cells by that of the uninfected cells. The location of T (the first T of TATA box) is relative to the translation start site (+1).
In addition to the above-mentioned potential core promoters with TATT sequence, we also tested three 15-bp sequences derived from upstream regions of viral late genes, ORF38, ORF42, and ORF53, containing TATATAA with A at the fourth position. Two of the three were highly active and the third was moderately activated, demonstrating that T at the fourth position, while overrepresented among the late gene core promoters, is not absolutely required for the promoter activity. This result was not surprising since our mutagenesis study (Fig. 5A) showed that the T-to-A mutation at the fourth position of the TATA box (9th position of the 15-bp sequence) caused a 60% reduction but did not completely abolish the reporter activity. Taken together, our findings demonstrate that replication-dependent late gene transcription during viral infection is primarily controlled by a core promoter consisting of a specific TATA-like sequence, either TATTA/T or TATAT, but surprisingly not the canonical highly active sequence, TATAA (27).
We were also interested in testing whether the short sequences derived from upstream regions of KSHV or EBV late genes function as promoters in our reporter assays. It was previously demonstrated that a sequence of only 12 bp is sufficient for activation of the KSHV K8.1 promoter (6). The EBV BdRF1 promoter was narrowed down to the region from −49 to +30, with the transcriptional start site at +1 (5), and the BcLF1 promoter was mapped to a 35-bp sequence (4). Based on the promoter regions defined by others, we cloned the 15-bp sequences spanning the TATT sequences of KSHV K8.1, EBV BdRF1, and BcLF1 late gene promoters into the pGL3-Ori vector and tested for activation upon MHV-68 infection. All three sequences were activated in our reporter assay (Table 4). This is not unexpected, since these three promoters all contain the TATTA/T sequence, a consensus shared by a majority of the active 15-bp promoter sequences that we tested. Importantly, more than 70% of known EBV late gene promoters have the TATT sequence (4, 28). Thus, the distinct TATT box, shared by many viral late gene promoters of different gammaherpesviruses, is the major determinant for late gene expression, and the mechanism underlying its recognition is likely conserved among MHV-68, EBV, and KSHV.
Targeted mutations on the M7 promoter in MHV-68.
Using our reporter assay, we determined that in addition to DNA replication mediated by the MHV-68 orilyt, the TATT sequence plays an essential role in the activation of late gene promoters during viral infection. To validate our results in the context of the viral genome, we sought to generate mutant viruses that contain site-specific nucleotide changes in the essential promoter sequence defined by the reporter assay. We chose to study the M7 promoter because M7 is dispensable for viral replication in tissue culture (9, 29) and because its promoter region is located downstream of ORF50 without overlapping the polyadenylation signal of ORF50.
The 15-bp M7 promoter defined in this study (Fig. 6) has the same TATT sequence, TATTTAA, as the ORF52 promoter. Before constructing mutant viruses, we used reporter assays to identify the critical nucleotides within this 15-bp sequence. Moreover, we wanted to determine whether the results from the systematic mutagenesis study of the 15-bp ORF52 promoter (Fig. 5) can be applied to other late gene promotes, such as the M7 promoter. The ORF52 promoter contains an A at the fourth and fifth positions of the 15-bp sequence, immediately upstream of the TATT sequence; a mutation of either A to T in the ORF52 promoter caused >50% reduction in the reporter activation. Thus, we would predict that a T-to-A mutation at the fourth position of the M7 promoter sequence (4A mutation) enhances reporter activation, while an A-to-T mutation at the fifth position (5T mutation) attenuates activation. Additionally, mutations were also introduced at the 6th position to change the TATT sequence from TATTTAA to AATTTAA (6A mutation) or at both the 9th and 10th positions to TATAAAA (9A10A mutation) (mutations underlined). Both of the 6A and 9A10A mutations should reduce reporter activation. The constructs containing these four types of mutations in the 15-bp M7 promoter were individually generated and tested (Fig. 7A). Indeed, their actual activation relative to that of the WT reporter was as predicted. The 4A mutation moderately enhanced promoter activation. The 5T, 6A, and 9A10A mutations significantly reduced activation, to 16, 7, and 14% of the WT level, respectively, just above the vector activation (4%).
FIG 7.

Mutations at the M7 promoter TATA box diminish reporter activation and M7 gene expression from the viral genome. (A) Reporter activation. The 15-bp M7 promoter sequence of each construct, either WT or mutant, is presented along with the predicted activity based on the mutagenesis study on the ORF52 promoter (Fig. 2). The WT sequence is in lowercase, with the specific mutations in underlined uppercase letters. Each construct was tested for activation induced by MHV-68 infection, and the data shown in the right panel are percentages of WT activation based on comparisons to fold activation for the WT reporter. (B) Northern blot analysis. BHK-21 cells were infected at an MOI of 1 with WT (lane 4), mutant M7p-6A (6A, lane 1), or M7p-9A10A (9A10A, lane 2). At 24 h postinfection, total RNAs harvested from infected cells (lanes 1, 2, and 4) along with RNA from uninfected cells (UI, lane 3) were separated on an agarose gel, transferred to a membrane, and hybridized with a probe detecting either M7 (upper image) or ORF54 (lower image) transcripts. (C) Western blot analysis. NIH 3T3 cells were infected as described for panel B, and at 24 h postinfection, the total proteins from cells infected with the virus indicated above the image (lanes 2 to 4) and from uninfected cells (lane 1) were separated on a polyacrylamide gel, transferred to a membrane, and probed with antibodies detecting M7 and another two late gene products, ORF65 and ORF26.
Based on the reporter results, we constructed two recombinant viruses with mutations in the TATT sequence of the M7 promoter, the M7p-6A virus, which has a 6A mutation (AATTTAA), and the M7p-9A10A virus, which has a mutation converting the distinct late gene TATT sequence, TATTTAA, to a canonical sequence, TATAAAA. To examine the effects of the M7 promoter mutations on the M7 RNA expression, BHK-21 cells were infected with M7p-6A or M7p-9A10A mutant virus, along with the WT virus, at an MOI of 1. At 24 h postinfection, total RNA was harvested for analysis of the M7 transcript by Northern blotting. While the M7 transcript was readily detectable in the WT-infected sample, there was no obvious band of a similar size seen in the sample prepared from cells infected with M7p-6A or M7p-9A10A virus. However, it was noted that there was a barely detectable band with a slightly larger size in the M7p-9A10A sample and an even much larger RNA species in the M7p-6A sample. Hybridization with a probe detecting the ORF54 transcript demonstrated that both mutant viruses expressed ORF54, an early viral gene, without any defect (Fig. 7B). Western blotting confirmed that M7 protein expression was completely abolished in cells infected with either M7p-6A or M7p-9A10A virus (Fig. 7C), while expression of another two viral late gene products, ORF65 and ORF26, was comparable to that in the WT-infected cells. Taken together, the results demonstrate that the adverse mutations at the TATT sequence identified in the reporter assay also have detrimental effects on M7 RNA expression when introduced into the viral genome. This confirms that the TATT sequence plays a pivotal role in activating viral late gene transcription and importantly, a canonical TATA sequence, TATAAAA, is not a functional core element for the viral late gene promoter.
Impact of promoter mutations on ORF24 binding.
One of the five gammaherpesviral proteins required to mediate late gene expression, ORF24, has been previously shown to contain a domain with putative structural similarity to cellular TATA binding protein (TBP) (28), suggesting that it may directly associate with late gene promoters. Indeed, the ORF24 homologue in EBV (BcRF1) was recently shown to bind EBV late gene promoters through its TBP-like domain (30). We therefore sought to determine whether the MHV-68 ORF24 protein similarly bound the core late gene promoter sequence and whether the mutations we identified in this study as being adverse for promoter activation impacted ORF24 binding. To this end, we analyzed the promoter binding activity of purified MHV-68 ORF24 in DNA electrophoretic mobility shift assays (EMSAs) using probes for the early promoter of ORF54, as well as WT and mutant versions of the ORF52 promoter. As predicted, ORF24 associated specifically with the ORF52 promoter, which contains a TATTTAA sequence, but failed to bind the early ORF54 promoter, which contains a canonical TATAAAA sequence (Fig. 8A). This binding specificity is consistent with our reporter study (Table 4). Moreover, while the T-to-A mutation at position 9 (9A) of the ORF52 promoter did not dramatically impact ORF24 binding, ORF24 binding to the 9A10A double mutant (T-to-A mutation at positions 9 and 10) was reduced (Fig. 8B). These results are in general agreement with our mutagenesis study, in which the activity of the 9A10A promoter mutant was decreased to ∼11% of the WT and the 9A promoter activity was partially impaired. We therefore conclude that the specific sequence requirements of the late gene promoters help direct binding by the viral ORF24 protein, a known regulator of late gene expression.
DISCUSSION
The genes of herpesviruses are generally divided into early and late genes separated by their expression before and after viral DNA replication, respectively. Studies have shown that unlike viral early gene promoters, the promoters of late genes map to the regions near the transcription start sites without the requirement for regulatory sequences upstream of the TATA box (4–6, 23, 24). In addition, several lines of evidence have indicated a crucial and regulatory role of the TATA box in controlling herpesviral late gene expression (4, 23, 25). However, the sequence that defines a functional TATA box for late gene promoters had not been thoroughly examined. In this study, we investigated the regulatory sequences of MHV-68 late gene promoters. To facilitate the analysis of the promoter, we utilized a luciferase reporter containing the MHV-68 origin of lytic replication (orilyt), which is essential for the activation of viral late gene promoters (Fig. 1). We showed that the late gene promoter of ORF52 was contained in a 15-bp sequence that has an uncoventional TATT box, TATTTAA. A detailed analysis of the 15-bp ORF52 late gene promoter by systematic point mutations revealed limited tolerance of sequence variations in the TATT box. The only two acceptable point mutations in the 7-bp sequence, TATTTAA, are T-to-A changes at fourth and fifth positions. This is in contrast to TATA-containing eukaryotic promoters in which there are diverse A/T-rich TATA-like sequences that can function as a TATA box (27, 31, 32). More surprisingly, the doubly mutated 15-bp ORF52 late gene promoter with a widely accepted consensus TATA box sequence, TATAAAA (mutations underlined), failed to support the induction of the reporter activity. The same double mutation also had detrimental effects on the 15-bp M7 late gene promoter activity in the reporter assay (Fig. 7A). Moreover, when this double T-to-A mutation was introduced to the TATT box (TATTTAA) of the M7 promoter on the viral genome, the mutated promoter with the TATAAA sequence was unable to support M7 transcription, resulting in no expression of M7 protein (Fig. 7B and C). This result indicates that transcription of viral late genes is governed by the TATT sequence. Consistent with the observed stringent sequence requirement for the TATT box of late gene ORF52 and M7 promoters, we found that a majority of the known MHV-68 late genes have TATTA/TA sequences within 200 bp upstream of the translation start codon. Furthermore, we demonstrated that various 15-bp sequences containing these potential late gene-specific TATT boxes were able to drive transcription in our reporter assay. Thus, our study supports a model where promoters governing transcription of late genes are uniquely concise and comprised of a TATT box with specific sequence requirement.
Use of an unconventional TATT box in the herpesviral late gene promoter has been reported in several studies (4, 25). The most striking characteristic of this atypical TATT sequence is a T at the fourth position, which is rarely found in the TATA box-containing promoters of eukaryotic genes (<10%) (26). The functional significance of this fourth T has been examined for other herpesviral late gene promoters. The UL44 late promoter of HCMV has a noncanonical TATT sequence, TATTAA, and mutation of the fourth T, yielding TATAAA (mutation underlined), a canonical sequence, greatly reduces the level of the UL44 late transcript (25). The importance of a similar T-to-A point mutation has been examined for the two late gene promoters of EBV. The mutation of the fourth T in the TATA box of the BcLF1 promoter (TATTAA to TATAAA) results in a loss of induction (4, 5), while the same mutation in the BdRF1 promoter (TATTTA to TATATA) does not cause similar impairment (5). Since only a single mutant was generated for each EBV promoter in these studies and the two promoters do not have the same TATT sequences, the discrepancy in the role of the fourth T in the TATA box may be due to the neighboring nucleotides. Indeed, our mutagenesis study on the ORF52 promoter indicates that both TATATA and TATTA/TA support the late gene promoter activity, but TATAAA does not. We also made attempts to examine the TATA sequences among the promoter regions of the late genes of HCMV and the closely related virus murine CMV (MCMV). Using different bioinformatic programs (e.g., the Network Promoter Prediction online server from Lawrence Berkeley National Laboratories), the sequences of TATTA/TA and TATA/TTA can been found in the promoter regions of many late genes, such as UL14, UL18, UL31, UL32, and UL48 of HCMV and M25, M31, M34, M69, M97, M100, and M102 of MCMV. Therefore, supported by several examples in HCMV, EBV, and MHV-68, although TATAAA is often considered to be the consensus sequence for a TATA box, it appears unable to mediate transcription of a herpesviral late gene promoter. These observations also suggest that the mode of regulation of late gene transcription may be conserved among betaherpesviruses and gammaherpesviruses.
The inability of a TATA box consensus sequence, TATAAA, to function as a core element for the late gene promoter is an important piece of information in understanding the regulation of late gene expression. Transcription initiation requires the assembly of the RNA polymerase II (Pol II) preinitiation complex (PIC) at the promoter, and the PIC component TFIID is responsible for recognizing and binding to the TATA box for accurate TATA-dependent transcription initiation. TFIID is a multisubunit complex that consists of the TATA binding protein (TBP) and more than 10 TBP-associated factors (TAFs), which receive and coordinate signals from activators (33). As previously mentioned, a variety of functional TATA elements are present in cellular and viral early gene promoters, indicating degenerate sequence recognition by TFIID. One possibility to explain the failure of TATAAA to function in the late gene promoter is that recognition and binding to the TATA box of late gene promoters are mediated by a distinct type of TBP that has different sequence specificity. Although a TBP-like protein (TLP) was found in humans and mice, TLP does not bind a TATA box (34). A bioinformatic analysis of BcRF1 of EBV and UL87 of HCMV, homologues of MHV-68 ORF24, revealed a possible TBP saddle-like fold in the central part of these proteins (28). Although the overall sequence homology between the viral homologues and cellular TBP is low, 6 out of the 15 residues involved in TBP-DNA binding are conserved, including the crucial phenylalanines that insert into the TATA box to bend the DNA. Moreover, the EBV BcRF1 protein was recently shown to bind to the TATT-containing sequence (30).
In this study, we observed selective binding of the MHV-68 ORF24 protein to the late gene promoter sequence (Fig. 8). Furthermore, the binding of MHV-68 ORF24 to the ORF52 late gene promoter sequence was consistently reduced upon the double mutation of the critical promoter residues at positions 9 and 10 (9A10A). These results indicate that the TATT sequence is important for the DNA-ORF24 interaction, in general agreement with our reporter study. The fact that the 9A10A double mutation, while reducing the promoter activity by 90%, did not completely abrogate the binding of ORF24 suggests additional regulatory aspects beyond ORF24 binding in late gene expression. Alternatively, it is possible that these sequences direct additional ORF24-related activities, such as conformational changes, that may be important for the late gene promoter activity but cannot be easily monitored using the in vitro binding assays. In either scenario, our results support the notion that the binding of ORF24 to the promoter is an important component of late gene expression.
Another important aspect of this study is the requirement of orilyt on the reporter to support the activation of the late promoter during viral infection. Although a higher level of empty reporter activation was obtained in the presence of orilyt, it accounts for less than 5% of the reporter containing the late gene promoter and is likely due to the increased copy number as a result of reporter replication during viral infection. In addition, we estimate on the basis of Southern analysis combined with the use of DpnI restriction enzyme that only ∼5% of the total amount of the reporter represented the replicated product (data not shown). Therefore, activation of the late gene promoter in the orilyt-containing reporter is unlikely caused by the copy number effect. Furthermore, inhibition of reporter replication abolishes the late gene promoter activity, indicating that the major role of orilyt sequence is to support DNA replication of the reporter rather than function as a DNA binding site for activators. Several hypotheses have been previously proposed to explain this replication-dependent transcription. Taking into account the unusually minimal structure of the late gene promoter, which is confined to a 15-bp sequence that contains only the TATT core element, we favor the hypothesis that the transcription complex is recruited by the viral DNA replication complex to the late gene core promoter, bypassing the requirement for conventional activators. Such a mechanism would be very effective to link viral DNA replication to the activation of late gene transcription. For example, ICP8 of herpes simplex virus 1 (HSV-1), a component of the viral DNA replication complex, interacts with the Pol II complex and can potentially assist in recruiting it to the replicating viral DNA, thereby stimulating late gene expression (35, 36).
We report here that a minimal core promoter is sufficient to mediate replication-dependent gene transcription during MHV-68 infection and that the activity of this core promoter requires a selective type of TATA box with a striking exclusion of the consensus sequence, TATAAA. Our study has also prompted several interesting directions for future investigations. The EMSA analysis (Fig. 8) supports the hypothesis that recognition and binding to the late gene core promoter are carried out by ORF24, a member of the UL87 TBP-like protein family, but the mechanism by which the viral TBP-like protein mediates the nucleation of PIC is unclear. Another important question is how a core promoter drives replication-dependent transcription beyond the basal level without involvement of conventional activators. We speculate that there are sequence-independent specific molecular interactions with the transcription complex at the sites of viral DNA replication to increase the transcription efficiency from the viral template. Future studies addressing these remaining questions will help clarify the mechanisms underlying the control of late gene expression.
ACKNOWLEDGMENTS
We thank Ronika Sitapara Leang and Harding Luan for editing the manuscript and for helpful comments. We thank Stephen Y. Chen for excellent technical assistance with bioinformatics analysis of the HCMV and MCMV promoter regions.
This work was supported by the National Basic Research Program of China (973 program, no. 2011CB504800), NSFC 2011CB504803 and 2011CB504300, NIH grants DE15752, CA91791, and AI078133, and funding from the Stop Cancer Foundation to R.S. and NIH grants CA136367 and CA160556 and a W. M. Keck Foundation Distinguished Young Scholars Award to B.G.
Footnotes
Published ahead of print 8 January 2014
REFERENCES
- 1.Lau R, Middeldorp J, Farrell PJ. 1993. Epstein-Barr virus gene expression in oral hairy leukoplakia. Virology 195:463–474. 10.1006/viro.1993.1397 [DOI] [PubMed] [Google Scholar]
- 2.Katano H, Sato Y, Kurata T, Mori S, Sata T. 2000. Expression and localization of human herpesvirus 8-encoded proteins in primary effusion lymphoma, Kaposi's sarcoma, and multicentric Castleman's disease. Virology 269:335–344. 10.1006/viro.2000.0196 [DOI] [PubMed] [Google Scholar]
- 3.Staskus KA, Sun R, Miller G, Racz P, Jaslowski A, Metroka C, Brett-Smith H, Haase AT. 1999. Cellular tropism and viral interleukin-6 expression distinguish human herpesvirus 8 involvement in Kaposi's sarcoma, primary effusion lymphoma, and multicentric Castleman's disease. J. Virol. 73:4181–4187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Serio TR, Cahill N, Prout ME, Miller G. 1998. A functionally distinct TATA box required for late progression through the Epstein-Barr virus life cycle. J. Virol. 72:8338–8343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amon W, Binne UK, Bryant H, Jenkins PJ, Karstegl CE, Farrell PJ. 2004. Lytic cycle gene regulation of Epstein-Barr virus. J. Virol. 78:13460–13469. 10.1128/JVI.78.24.13460-13469.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tang S, Yamanegi K, Zheng ZM. 2004. Requirement of a 12-base-pair TATT-containing sequence and viral lytic DNA replication in activation of the Kaposi's sarcoma-associated herpesvirus K8.1 late promoter. J. Virol. 78:2609–2614. 10.1128/JVI.78.5.2609-2614.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Virgin HW, IV, Latreille P, Wamsley P, Hallsworth K, Weck KE, Dal Canto AJ, Speck SH. 1997. Complete sequence and genomic analysis of murine gammaherpesvirus 68. J. Virol. 71:5894–5904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Efstathiou S, Ho YM, Hall S, Styles CJ, Scott SD, Gompels UA. 1990. Murine herpesvirus 68 is genetically related to the gammaherpesviruses Epstein-Barr virus and herpesvirus saimiri. J. Gen. Virol. 71:1365–1372. 10.1099/0022-1317-71-6-1365 [DOI] [PubMed] [Google Scholar]
- 9.Song MJ, Hwang S, Wong WH, Wu TT, Lee S, Liao HI, Sun R. 2005. Identification of viral genes essential for replication of murine gamma-herpesvirus 68 using signature-tagged mutagenesis. Proc. Natl. Acad. Sci. U. S. A. 102:3805–3810. 10.1073/pnas.0404521102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arumugaswami V, Wu TT, Martinez-Guzman D, Jia Q, Deng H, Reyes N, Sun R. 2006. ORF18 is a transfactor that is essential for late gene transcription of a gammaherpesvirus. J. Virol. 80:9730–9740. 10.1128/JVI.00246-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu TT, Park T, Kim H, Tran T, Tong L, Martinez-Guzman D, Reyes N, Deng H, Sun R. 2009. ORF30 and ORF34 are essential for expression of late genes in murine gammaherpesvirus 68. J. Virol. 83:2265–2273. 10.1128/JVI.01785-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wong E, Wu TT, Reyes N, Deng H, Sun R. 2007. Murine gammaherpesvirus 68 open reading frame 24 is required for late gene expression after DNA replication. J. Virol. 81:6761–6764. 10.1128/JVI.02726-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Isomura H, Stinski MF, Murata T, Yamashita Y, Kanda T, Toyokuni S, Tsurumi T. 2011. The human cytomegalovirus gene products essential for late viral gene expression assemble into prereplication complexes before viral DNA replication. J. Virol. 85:6629–6644. 10.1128/JVI.00384-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Omoto S, Mocarski ES. 2014. transcription of true late (γ2) cytomegalovirus genes requires ul92 function that is conserved among beta- and gammaherpesviruses. J. Virol. 88:120–130. 10.1128/JVI.02983-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Omoto S, Mocarski ES. 2013. Cytomegalovirus UL91 is essential for transcription of viral true late (gamma2) genes. J. Virol. 87:8651–8664. 10.1128/JVI.01052-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perng YC, Qian Z, Fehr AR, Xuan B, Yu D. 2011. The human cytomegalovirus gene UL79 is required for the accumulation of late viral transcripts. J. Virol. 85:4841–4852. 10.1128/JVI.02344-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deng H, Chu JT, Park NH, Sun R. 2004. Identification of cis sequences required for lytic DNA replication and packaging of murine gammaherpesvirus 68. J. Virol. 78:9123–9131. 10.1128/JVI.78.17.9123-9131.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dyballa N, Metzger S. 2009. Fast and sensitive colloidal Coomassie G-250 staining for proteins in polyacrylamide gels. J. Vis Exp. 2009:1430. 10.3791/1431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pavlova I, Lin CY, Speck SH. 2005. Murine gammaherpesvirus 68 Rta-dependent activation of the gene 57 promoter. Virology 333:169–179. 10.1016/j.virol.2004.12.021 [DOI] [PubMed] [Google Scholar]
- 20.Martinez-Guzman D, Rickabaugh T, Wu TT, Brown H, Cole S, Song MJ, Tong L, Sun R. 2003. Transcription program of murine gammaherpesvirus 68. J. Virol. 77:10488–10503. 10.1128/JVI.77.19.10488-10503.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ebrahimi B, Dutia BM, Roberts KL, Garcia-Ramirez JJ, Dickinson P, Stewart JP, Ghazal P, Roy DJ, Nash AA. 2003. Transcriptome profile of murine gammaherpesvirus-68 lytic infection. J. Gen. Virol. 84:99–109. 10.1099/vir.0.18639-0 [DOI] [PubMed] [Google Scholar]
- 22.Ahn JW, Powell KL, Kellam P, Alber DG. 2002. Gammaherpesvirus lytic gene expression as characterized by DNA array. J. Virol. 76:6244–6256. 10.1128/JVI.76.12.6244-6256.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Homa FL, Glorioso JC, Levine M. 1988. A specific 15-bp TATA box promoter element is required for expression of a herpes simplex virus type 1 late gene. Genes Dev. 2:40–53. 10.1101/gad.2.1.40 [DOI] [PubMed] [Google Scholar]
- 24.Johnson PA, Everett RD. 1986. The control of herpes simplex virus type-1 late gene transcription: a ‘TATA-box'/cap site region is sufficient for fully efficient regulated activity. Nucleic Acids Res. 14:8247–8264. 10.1093/nar/14.21.8247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Isomura H, Stinski MF, Kudoh A, Murata T, Nakayama S, Sato Y, Iwahori S, Tsurumi T. 2008. Noncanonical TATA sequence in the UL44 late promoter of human cytomegalovirus is required for the accumulation of late viral transcripts. J. Virol. 82:1638–1646. 10.1128/JVI.01917-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bucher P. 1990. Weight matrix descriptions of four eukaryotic RNA polymerase II promoter elements derived from 502 unrelated promoter sequences. J. Mol. Biol. 212:563–578. 10.1016/0022-2836(90)90223-9 [DOI] [PubMed] [Google Scholar]
- 27.Wobbe CR, Struhl K. 1990. Yeast and human TATA-binding proteins have nearly identical DNA sequence requirements for transcription in vitro. Mol. Cell. Biol. 10:3859–3867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wyrwicz LS, Rychlewski L. 2007. Identification of herpes TATT-binding protein. Antiviral Res. 75:167–172. 10.1016/j.antiviral.2007.03.002 [DOI] [PubMed] [Google Scholar]
- 29.de Lima BD, May JS, Stevenson PG. 2004. Murine gammaherpesvirus 68 lacking gp150 shows defective virion release but establishes normal latency in vivo. J. Virol. 78:5103–5112. 10.1128/JVI.78.10.5103-5112.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gruffat H, Kadjouf F, Mariame B, Manet E. 2012. The Epstein-Barr virus BcRF1 gene product is a TBP-like protein with an essential role in late gene expression. J. Virol. 86:6023–6032. 10.1128/JVI.00159-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hahn S, Buratowski S, Sharp PA, Guarente L. 1989. Isolation of the gene encoding the yeast TATA binding protein TFIID: a gene identical to the SPT15 suppressor of Ty element insertions. Cell 58:1173–1181. 10.1016/0092-8674(89)90515-1 [DOI] [PubMed] [Google Scholar]
- 32.Singer VL, Wobbe CR, Struhl K. 1990. A wide variety of DNA sequences can functionally replace a yeast TATA element for transcriptional activation. Genes Dev. 4:636–645. 10.1101/gad.4.4.636 [DOI] [PubMed] [Google Scholar]
- 33.Lemon B, Tjian R. 2000. Orchestrated response: a symphony of transcription factors for gene control. Genes Dev. 14:2551–2569. 10.1101/gad.831000 [DOI] [PubMed] [Google Scholar]
- 34.Moore PA, Ozer J, Salunek M, Jan G, Zerby D, Campbell S, Lieberman PM. 1999. A human TATA binding protein-related protein with altered DNA binding specificity inhibits transcription from multiple promoters and activators. Mol. Cell. Biol. 19:7610–7620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gao M, Knipe DM. 1991. Potential role for herpes simplex virus ICP8 DNA replication protein in stimulation of late gene expression. J. Virol. 65:2666–2675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou C, Knipe DM. 2002. Association of herpes simplex virus type 1 ICP8 and ICP27 proteins with cellular RNA polymerase II holoenzyme. J. Virol. 76:5893–5904. 10.1128/JVI.76.12.5893-5904.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]