

Abstract
Listeria monocytogenes is responsible for the life-threatening food-borne disease listeriosis. This disease mainly affects elderly and immunocompromised individuals, causing bacteremia and meningoencephalitis. In pregnant women, L. monocytogenes infection leads to abortion and severe infection of the fetus or newborn. The L. monocytogenes intracellular life cycle is critical for pathogenesis. Previous studies have established that the major virulence factor of L. monocytogenes, the pore-forming toxin listeriolysin O (LLO), is sufficient to induce L. monocytogenes internalization into human epithelial cell lines. This internalization pathway strictly requires the formation of LLO pores in the plasma membrane and can be stimulated by the heterologous pore-forming toxin pneumolysin, suggesting that LLO acts nonspecifically by forming transmembrane pores. The present work tested the hypothesis that Ca2+ and K+ fluxes subsequent to perforation by LLO control L. monocytogenes internalization. We report that L. monocytogenes perforates the host cell plasma membrane in an LLO-dependent fashion at the early stage of invasion. In response to perforation, host cells undergo Ca2+-dependent but K+-independent resealing of their plasma membrane. In contrast to the plasma membrane resealing process, LLO-induced L. monocytogenes internalization requires both Ca2+ and K+ fluxes. Further linking ion fluxes to bacterial internalization, treating cells with a combination of Ca2+ and K+ ionophores but not with individual ionophores is sufficient to induce efficient internalization of large cargoes, such as 1-μm polystyrene beads and bacteria. We propose that LLO-induced L. monocytogenes internalization requires a Ca2+- and K+-dependent internalization pathway that is mechanistically distinct from the process of plasma membrane resealing.
INTRODUCTION
Numerous pathogenic microorganisms adopt an intracellular lifestyle to infect their host. Bacterial pathogens can induce their internalization into host cells by one of two major mechanisms (1). The zipper mechanism involves bacterial surface molecules that specifically activate a host receptor-mediated signaling cascade (1). In the trigger mechanism, Gram-negative bacteria use a secretion apparatus that injects effectors into host cells to directly activate their internalization machinery (2). More recently, a third invasion mechanism used by the bacterium Listeria monocytogenes and the parasite Trypanosoma cruzi was reported (3, 4). This novel mechanism of pathogen internalization is stimulated upon perforation of the host cell plasma membrane. In the case of L. monocytogenes, membrane perforation is caused by the pore-forming toxin listeriolysin O (LLO) that belongs to the cholesterol-dependent cytolysin (CDC) family (3). This invasion mechanism is not activated only by LLO, since the heterologous CDC pneumolysin can also stimulate L. monocytogenes internalization (3). T. cruzi is believed to perforate host cells via an unidentified pore-forming protein or mechanical disruption (4). Thus, damaging the host cell plasma membrane emerges as an invasion strategy shared by bacteria and parasites. This invasion process is not the result of passive entry of the pathogen through a breach in the plasma membrane but is due to the activation of the host cell endocytic machinery (3, 4). Importantly, this invasion mechanism is relevant to numerous pathogens, because pore-forming proteins are common virulence factors of viruses, bacteria, and eukaryotic intracellular pathogens (5–11).
The food-borne pathogen Listeria monocytogenes infects a large variety of host cells, including cells that are normally nonphagocytic, such as epithelial, endothelial, and fibroblastic cells (12). L. monocytogenes can use several pathways to infect nonphagocytic cells (13, 14). The surface invasins InlA (internalin) and InlB are known to stimulate the zipper mechanism of entry by activating the host cell receptors E-cadherin and c-Met (the hepatocyte growth factor receptor), respectively (15–17). In addition to InlA and InlB, LLO is sufficient to induce L. monocytogenes internalization via a pathway that requires the formation of the LLO pore complex, host cell tyrosine kinase signaling, F-actin polymerization, and dynamin (3). The detailed machinery underlying this internalization pathway remains to be elucidated. The ability to mediate bacterial internalization is one of several effects of LLO on host cells. Initially identified as a major virulence factor that is critical for intracellular survival (18, 19), LLO mediates the escape of L. monocytogenes from the endocytic vacuole to the cytosol, where the bacterium replicates (18, 20). While LLO displays optimal activity at an acidic pH, which facilitates the disruption of the endocytic vacuole, it is still active at neutral pH in the extracellular environment (3, 21). Indeed, LLO is released by L. monocytogenes in the extracellular environment (22, 23), eliciting various host cell responses. These responses include the activation of mitogen-activated protein (MAP) kinases (24, 25), the NLRP3 inflammasome (27, 28), caspase-1, and NF-κB (26). Extracellular LLO also decreases SUMOylation (29) and histone phosphorylation (28, 30) and causes mitochondrial fragmentation (31) as well as the arrest of protein synthesis (32). A fundamental question is, how can LLO exert so many activities? No protein receptor has been identified for LLO so far; therefore, it is likely that LLO acts nonspecifically by punching holes in host membranes. Perforation of the plasma membrane has multiple consequences, including but not limited to ion fluxes, membrane depolarization, and changes in redox potential and osmotic pressure. The LLO pore is very large (>30 nm in diameter), and therefore, even small proteins can be exchanged with the extracellular medium (33). These events likely elicit multiple signaling pathways, potentially explaining the diverse host cell responses to LLO. In particular, localized changes in the intracellular concentration of Ca2+ significantly affect host-pathogen interactions (34). LLO is known to stimulate influx of extracellular Ca2+ (35) and the release of Ca2+ from intracellular stores (36, 37). The influx of Ca2+ is crucial for repair pathways that restore plasma membrane integrity following perforation and for many other cellular processes, including gene transcription, intracellular trafficking, and cytoskeletal dynamics, all of which may affect the interaction of L. monocytogenes with host cells (34, 38, 39). The known Ca2+-dependent activities stimulated by LLO include mitochondrial fragmentation (31) and membrane repair (3). Variations in the intracellular concentration of K+ also affect host cell biology. Following host cell exposure to LLO, a decrease in the intracellular concentration of K+ leads to caspase-1 activation, histone H3 dephosphorylation, and the arrest of protein synthesis (28, 40). In the work described here, we evaluated the roles of Ca2+ and K+ fluxes triggered upon host cell perforation by LLO in L. monocytogenes internalization. Importantly, we also tested for plasma membrane damage caused by LLO in the absence of ion fluxes and adapted our methodology to mitigate such damage.
MATERIALS AND METHODS
Bacterial and mammalian cell cultures.
The wild-type (wt) L. monocytogenes strain 10403S and the isogenic Δhly deletion strain DP-L2161 were grown overnight at 37°C in brain heart infusion (BHI) (BD Biosciences). Overnight cultures were diluted 1/20 in BHI and grown at 37°C until the optical density at 600 nm (OD600) reached 0.7 to 0.8. Bacteria were washed three times in phosphate-buffered saline (PBS) or medium as indicated below. The human hepatocyte cell line HepG2 (ATCC HB-8065) was grown in minimum essential medium (MEM) plus Earle's salts and l-glutamine (Invitrogen) supplemented with 10% heat-inactivated fetal bovine serum (HI-FBS; Lonza), 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, 100 U/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). HepG2 cells (105 cells/well) were cultured in 24-well tissue culture plates on glass coverslips at 37°C in 5% CO2 atmosphere for 48 h before infection. To test the role of Ca2+ influx and K+ efflux, standard, Ca2+-free, and high-K+ media were prepared as follows. Standard medium (M1) contained 140 mM NaCl, 5 mM KCl, 10 mM HEPES, 1.5 mM CaCl2, 0.5 mM MgCl2, 0.36 mM K2HPO4, 0.44 mM KH2PO4, 5.5 mM d-glucose, and 4.2 mM NaHCO3 (pH 7.4). Ca2+-free medium (M2) was similar to M1 but lacked CaCl2. High-K+ medium (M3) was similar to M1 but contained 5 mM NaCl and 140 mM KCl (3, 41).
Toxin purification and polystyrene bead coating.
Recombinant six-His-tagged LLO and a monomer-locked variant of LLO that is unable to form pores (LLOml) were purified from E. coli BL21(DE3) as described previously (3, 41). Carboxylate microspheres (Alexa Fluor 350 labeled, 1-μm diameter; Molecular Probes) were covalently coated with bovine serum albumin (BSA) according to the manufacturer's instructions. LLOml was noncovalently bound to the surface of BSA-coated beads as previously described (3, 42).
Bacterial association with and internalization into host cells.
HepG2 cells were incubated with 106 bacteria per well (multiplicity of infection [MOI] of 2.5) in one of the media as indicated below. The cell culture plates were centrifuged for 5 min (230 × g) at room temperature and incubated for 30 min at 37°C. Cells were washed with PBS, fixed with 3% paraformaldehyde (PFA) in PBS for 15 min, washed with PBS, and blocked for 1 h in 0.1 M glycine, 10% HI-FBS in PBS, pH 7.4. Extracellular bacteria were labeled with anti-L. monocytogenes rabbit polyclonal antibodies (GeneTex) and then with anti-rabbit secondary antibodies conjugated to Alexa Fluor 488 (or Alexa Fluor 568) (Molecular Probes). Samples were then permeabilized with 0.2% Triton X-100 in PBS for 5 min, and total bacteria were labeled with anti-L. monocytogenes antibodies and secondary antibodies conjugated to Alexa Fluor 568 (or Alexa Fluor 647) (Molecular Probes). Slides were mounted in ProLong gold with DAPI (4′,6-diamidino-2-phenylindole; Molecular Probes) to stain host cell nuclei. To quantify the number of cells, 40 sets of images (phase-contrast, DAPI, Alexa Fluor 488 and Alexa Fluor 568, or Alexa Fluor 568 and Alexa Fluor 647) were automatically acquired for each condition using the 20× objective. MetaMorph imaging and analysis software was used to enumerate the total numbers of bacteria (Nt), extracellular bacteria (Ne), and mammalian cells (Nc) (43). The percentage of internalized bacteria was calculated as (Nt − Ne)/Nt × 100. Bacterial association with host cells was calculated as Nt/Nc. At least 100 bacteria were counted for each condition. In some instances, cells were labeled with an anti-TOMM20 mouse monoclonal antibody (Abcam) followed by anti-mouse Alexa Fluor 488-conjugated secondary antibodies. To quantify TOMM20 labeling, phase-contrast and TOMM20 fluorescence images were acquired with a 20× objective. Fluorescence images were background corrected, cell perimeters were traced based upon the phase-contrast images, and the pixel average fluorescence intensity (AFI) of TOMM20 labeling in at least 2,500 cells was quantified using Metamorph software.
Invasion assays using ionophores.
HepG2 cells were washed and incubated with BSA- or BSA-LLOml-coated beads (MOI of 20) in standard medium (M1) or with Δhly L. monocytogenes (MOI of 20) in standard medium (M1), Ca2+-free medium (M2), or high-K+ medium (M3). The cell culture plates were centrifuged (230 × g) for 5 min at 4°C (beads) or for 10 min at 22°C (bacteria) and then incubated at 37°C for 15 min before treatment with recombinant LLO, ionomycin (Sigma), and/or nigericin (Sigma) for 15 min. Cells were then washed, fixed with 3% PFA, and blocked. Extracellular beads were labeled with anti-BSA rabbit antiserum (Sigma B1520) followed by goat anti-rabbit secondary antibodies conjugated to Alexa Fluor 568. The percentage of internalized beads [(number of intracellular beads/total number of beads) × 100] was quantified by fluorescence microscopy based on their unique (Alexa Fluor 350 for intracellular beads) or dual (Alexa Fluor 350 plus Alexa Fluor 568 for extracellular beads) fluorescence. At least 100 beads were counted for each experimental condition. Extracellular and intracellular bacteria were differentially labeled and enumerated as described above in “Bacterial association with and internalization into host cells.”
Propidium iodide incorporation assays in fixed cells.
HepG2 cells were incubated with wt or Δhly L. monocytogenes at the MOIs indicated below for 30 min at 37°C in standard (M1) or Ca2+-free (M2) medium and were pulse labeled with 100 μM propidium iodide (PI) during the last 5 min of incubation. Cells were then washed and fixed with 3% PFA and labeled with DAPI. Thirty sets of phase-contrast and fluorescence images (DAPI and PI) were acquired with the 20× objective. Fluorescence images were background corrected, and the pixel AFI of the PI images was automatically quantified in the nuclear regions (defined by the DAPI staining) by using MetaMorph analysis software, with at least 2,000 cells represented for each experimental condition (43).
PI incorporation assays in living cells.
HepG2 cells were cultured in glass bottom culture dishes (35-mm petri dishes, 10-mm microwells; MatTek) for 48 h. Dishes were transferred to the stage of an inverted fluorescence microscope, and 20 μM PI was added. In live-cell-imaging experiments where PI is maintained in the cell culture medium throughout the course of the experiment, 20 μM PI is sufficient for efficient and reliable labeling. All movies were acquired on the microscope stage at 37°C using the 40× objective. Phase-contrast and fluorescence images were acquired every 20 s for 980 s. LLO (1 nM) was added 160 s after the initial acquisition. Cells were incubated in standard (M1), Ca2+-free (M2), or high-K+ (M3) medium. Fluorescence images were background corrected, cell perimeters were traced based upon the phase-contrast images, and the pixel AFI within the cell areas was quantified at each time point using the Metamorph software. At least 200 cells were analyzed for each experimental condition.
Microscope equipment.
Images were acquired on a motorized, inverted, wide-field fluorescence microscope (Axio Observer D1; Zeiss) equipped with 20× Plan Neofluar (numeric aperture [NA] of 0.5) and 40× Plan Neofluar (NA of 1.3) objectives, a high-speed Xenon fluorescence emission device (Lambda DG-4, 300 W; Sutter Instrument Company), a Lambda 10-3 optical emission filter wheel for the fluorescence imaging, a Smart shutter to control the illumination for phase-contrast imaging (Sutter Instrument Company), and a back-illuminated, frame-transfer electron-multiplying charge-coupled device (EMCCD) camera (Cascade II 512; Photometrics). The filter sets for fluorescence were purchased from Chroma Technology Corporation and were as follows: DAPI (49000), green fluorescent protein (GFP)/fluorescein isothiocyanate (FITC)/Alexa Fluor 488 (49002), Cy3/DsRed/Alexa Fluor 568 (49005), and Cy5 (49006). Images were acquired and analyzed using MetaMorph imaging software (Universal Imaging).
Statistics.
A minimum of three independent experiments were performed, each in duplicate, unless otherwise indicated. Data were expressed as means ± standard errors of the means (SEM). P values were calculated using a standard two-tailed Student's t test. Asterisks indicate a significant difference between the results for indicated experimental conditions (*, P < 0.05; **, P < 0.01).
RESULTS
Extracellular L. monocytogenes perforates host cells in an LLO-dependent manner.
To determine if the amount of LLO secreted by L. monocytogenes is sufficient to perforate the host cell plasma membrane, we incubated HepG2 cells for 30 min at 37°C with increasing concentrations of wild-type (wt) or LLO-deficient (Δhly) L. monocytogenes and measured plasma membrane perforation. The extent of plasma membrane perforation, which is proportional to the incorporation of the low-molecular-weight (668), cell-impermeable dye propidium iodide (PI), increased significantly with the bacterial concentration in an LLO-dependent manner (Fig. 1). HepG2 cells were significantly perforated even when incubated with wt L. monocytogenes at an MOI as low as 5 (Fig. 1). At all MOIs tested, HepG2 cells were morphologically unaffected and remained attached to the dish despite perforation of their plasma membrane, suggesting that an efficient membrane resealing process maintains cell integrity.
FIG 1.
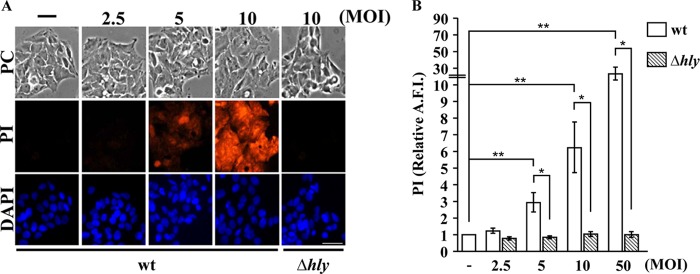
Extracellular L. monocytogenes bacteria perforate host cells in an LLO-dependent manner. Wild-type (wt) and LLO-deficient (Δhly) L. monocytogenes bacteria at various MOIs were incubated with HepG2 cells for 30 min at 37°C. The cell-impermeant dye propidium iodide (PI, 100 μM) was added to the cell culture medium during the last 5 min of incubation. After fixation and DAPI labeling, phase-contrast (PC) and fluorescence images were acquired with a 20× objective. (A) Representative images. Scale bar is 40 μm. (B) The average fluorescence intensity (A.F.I.) of PI was automatically measured in the nuclear region by quantitative fluorescence microscopy using Metamorph software. Results are expressed relative to the results for HepG2 cells incubated without bacteria (−). Results are the means ± SEM of at least three independent experiments performed in duplicate (*, P < 0.05; **, P < 0.01).
Host cells damaged by recombinant LLO reseal their plasma membrane via a Ca2+-dependent but K+-independent mechanism.
The influx of extracellular Ca2+ is critical for the activation of the plasma membrane repair response in cells injured by LLO and other pore-forming proteins or mechanical disruption (3, 39). We investigated whether K+ efflux is also required for resealing cells exposed to LLO. Membrane perforation was measured at 37°C by live-cell fluorescence imaging of HepG2 cells incubated with 1 nM recombinant LLO and 20 μM PI. In standard culture medium (M1), which contains physiological concentrations of Ca2+ and K+, we observed minimal incorporation of PI. In Ca2+-free medium (M2), we observed a rapid and massive influx of PI due to the absence of membrane resealing, as expected. In high-K+ culture medium (M3), we observed minimal incorporation of PI, demonstrating that blocking K+ efflux does not prevent membrane resealing (Fig. 2). Representative movies of each experimental condition are included in the supplemental material.
FIG 2.
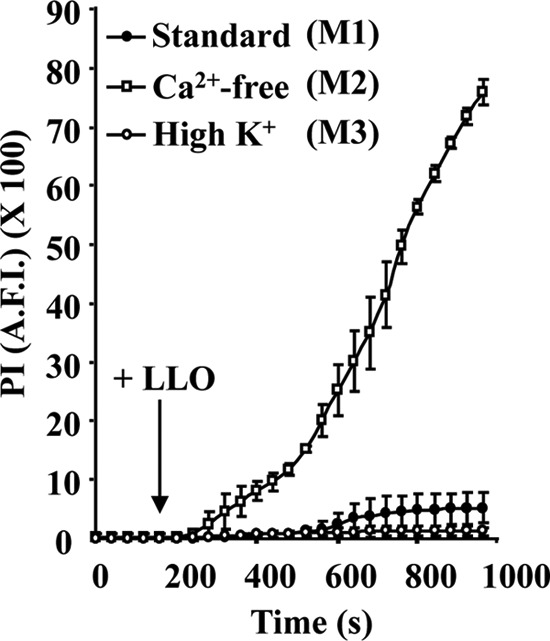
Host cells damaged by LLO undergo Ca2+-dependent but K+-independent membrane resealing. Perforation of HepG2 cells was measured by quantitative live-cell fluorescence microscopy. Cells were incubated on the microscope stage at 37°C for 980 s with 20 μM PI in standard medium (M1), Ca2+-free medium (M2), or high-K+ medium (M3). Phase-contrast and fluorescence images were recorded at regular time intervals using a 40× objective, and 1 nM LLO was added after 160 s of incubation. Results are expressed as the average fluorescence intensity (A.F.I.) of the cell area ± SEM. By 600 s, there was a statistically significant difference between the levels of PI incorporation in cells incubated in M1 and in M2. There was no significant difference between samples incubated in M1 and M3 at any time point.
Host cells damaged by L. monocytogenes undergo Ca2+-dependent but K+-independent membrane resealing.
It was unknown whether L. monocytogenes secretes enough LLO to activate the membrane-resealing pathway at the early stage of host cell invasion. We determined whether the Ca2+-dependent membrane-resealing pathway observed in cells exposed to recombinant LLO also occurs in cells exposed to L. monocytogenes. HepG2 cells were incubated with wt and Δhly L. monocytogenes for 30 min at 37°C in the presence (M1) or absence (M2) of extracellular Ca2+. Under these conditions, PI incorporation was significantly increased in cells incubated with wt L. monocytogenes in Ca2+-free buffer compared to the PI incorporation in cells incubated with wt bacteria in standard medium and with Δhly bacteria in Ca2+-free buffer (Fig. 3B), confirming that LLO secreted by a small number of L. monocytogenes organisms (MOI of 5) significantly perforates host cells and that host cells reseal their plasma membrane via a Ca2+-dependent process. At higher MOIs (MOI of ≥50), the differences between PI incorporation in cells incubated with wt L. monocytogenes with and without extracellular Ca2+ became less pronounced, likely due to more pronounced perforation that was less efficiently resealed and to loss of cells that detached because they were severely damaged in the absence of extracellular Ca2+. The PI incorporation was similar in cells incubated with L. monocytogenes in standard (M1) and high-K+ medium (M3), indicating that resealing of HepG2 cells damaged by L. monocytogenes does not require K+ efflux (Fig. 3C).
FIG 3.
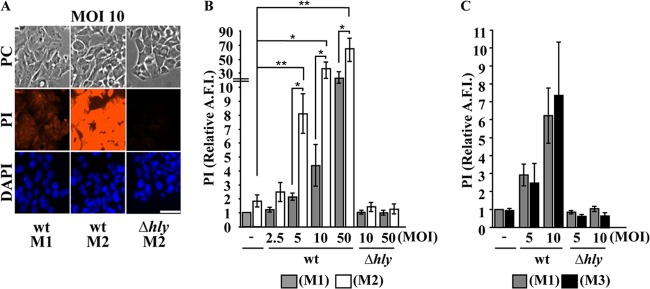
Host cells damaged by L. monocytogenes undergo Ca2+-dependent but K+-independent membrane resealing. Wild-type (wt) and LLO-deficient (Δhly) L. monocytogenes bacteria were incubated at various MOIs with HepG2 cells for 30 min at 37°C in standard medium (M1), Ca2+-free medium (M2), or high-K+ medium (M3). PI (100 μM) was added to the cell culture medium during the last 5 min of incubation. After fixation and DAPI labeling, phase-contrast (PC) and fluorescence images were acquired with a 20× objective. (A) Representative images. Images are displayed with the same scaling to show the relative intensities. Scale bar is 40 μm. (B and C) The average fluorescence intensity of PI was automatically measured in the nuclear region by quantitative fluorescence microscopy using Metamorph software. Results are expressed relative to the results for HepG2 cells incubated in M1 without bacteria. Results are the means ± SEM of at least three independent experiments performed in duplicate (*, P < 0.05; **, P < 0.01).
Ca2+ and K+ fluxes are necessary to activate LLO-dependent L. monocytogenes internalization.
We next investigated whether Ca2+ and/or K+ fluxes are critical for the activation of the LLO-dependent L. monocytogenes internalization pathway. A previous study, which used the gentamicin survival assay, proposed that Ca2+ influx is essential for L. monocytogenes entry into nonphagocytic cells (44). This conclusion was based upon the decreased recovery of viable intracellular bacteria incubated with host cells at an MOI of 50 in Ca2+-free buffer compared to the results with standard medium. A difficulty with this assay is that in Ca2+-free buffer, gentamicin (molecular weight [MW], 480) is expected to massively diffuse into the cytosol of damaged cells, similar to what we observed with PI (MW, 668) (Fig. 3A and B), possibly invalidating the gentamicin assay. To minimize the damage caused by LLO in the absence of extracellular Ca2+, we incubated HepG2 cells for 30 min at 37°C with L. monocytogenes bacteria at a low MOI (2.5) and used an immunofluorescence-based assay to quantify L. monocytogenes internalization (43). Indeed, this assay relies on fluorescent antibodies (molecular mass, 150 kDa) to distinguish extracellular from intracellular bacteria, the large size of which should limit their diffusion across perforated host cells. After cell fixation and blocking, we assessed whether or not plasma membrane perforation by LLO in Ca2+-free medium allowed the diffusion of antibodies. We labeled HepG2 cells with an antibody recognizing the cytosolic mitochondrial epitope TOMM20 and a secondary fluorescent antibody. We observed an increase, albeit statistically nonsignificant, in permeabilization to antibodies in cells incubated with wt L. monocytogenes in Ca2+-free medium in comparison to the results with standard medium (Fig. 4A). To ensure that this diffusion of antibodies did not affect the readout of bacterial internalization, we colabeled TOMM20 (Alexa Fluor 488), extracellular bacteria (Alexa Fluor 568), total bacteria (Alexa Fluor 647), and host cell nuclei. This approach allowed us to enumerate bacterial entry while determining whether cells were permeable or not to antibodies.
FIG 4.
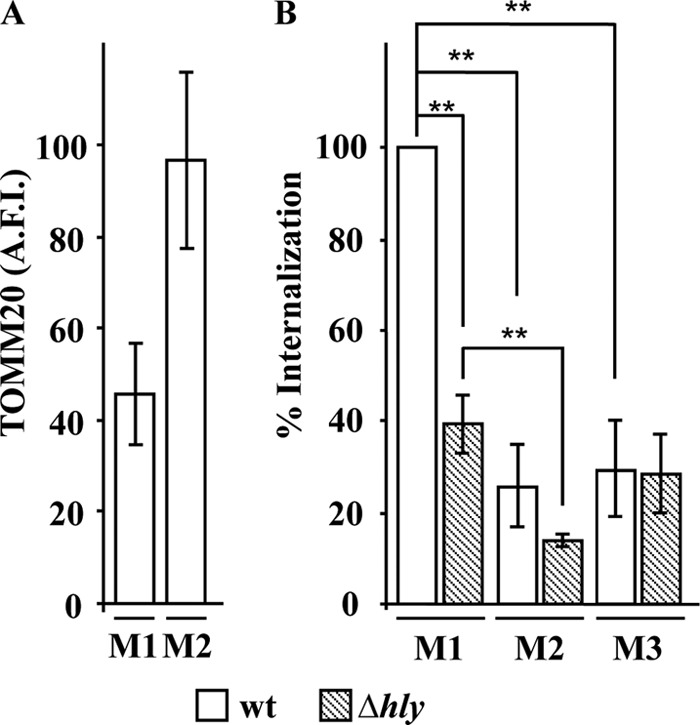
Ca2+ and K+ fluxes are necessary for LLO-dependent L. monocytogenes internalization. HepG2 cells were incubated with wild-type (wt) and LLO-deficient (Δhly) L. monocytogenes bacteria (MOI of 2.5) for 30 min at 37°C in standard medium (M1), Ca2+-free medium (M2), or high-K+ medium (M3). (A) After fixation and labeling, the entry of anti-TOMM20 antibody into HepG2 cells was measured by quantitative fluorescence microscopy in cells incubated with wt L. monocytogenes in M1 and M2. Results are the AFI of cells ± SEM from three independent experiments performed in duplicate. (B) Bacterial internalization was measured by quantitative fluorescence microscopy and expressed relative to the internalization of wt bacteria in M1. Results are the means ± SEM from three independent experiments performed in duplicate. (**, P < 0.01).
We found that in standard medium, LLO plays an important role in promoting efficient L. monocytogenes internalization, as previously reported (Fig. 4B) (3). In Ca2+-free medium, the internalization efficiency of wt L. monocytogenes was significantly decreased in comparison to that in cells incubated in standard medium. Indeed, under these conditions, the internalization level was as low as that of Δhly L. monocytogenes in standard medium (Fig. 4B). We also observed a decrease in the internalization of the LLO-deficient strain in Ca2+-free medium, which was not surprising given the importance of Ca2+ for interaction of the L. monocytogenes invasin InlA with its host cell receptor expressed on HepG2 cells, E-cadherin (16, 45, 46). Importantly, when comparing internalization in Ca2+-free medium, there was no longer a significant difference between the internalization of the wt and LLO-deficient strains, demonstrating that an influx of extracellular Ca2+ is required for the LLO-dependent internalization pathway (Fig. 4B). The results were unchanged when damaged cells, defined as TOMM20-positive cells, were excluded from the measurements (data not shown). Together, these results show unequivocally that L. monocytogenes internalization into HepG2 cells is Ca2+ dependent and that the level of perforation of the cells under these experimental conditions did not skew our ability to quantify L. monocytogenes internalization by fluorescence microscopy. To address the importance of K+ efflux, we compared the internalization efficiency of L. monocytogenes in standard (M1) and high-K+ (M3) media (Fig. 4B). The internalization efficiency of wt L. monocytogenes was significantly decreased in high-K+ medium, whereas the internalization efficiency of Δhly L. monocytogenes remained unchanged. We obtained similar results when we added 135 mM KCl to the medium without removing NaCl (data not shown). Modifying the ion concentrations in the cell culture medium did not affect L. monocytogenes association with host cells (data not shown). Together, these results support the hypothesis that simultaneous fluxes of Ca2+ and K+ are required for LLO-dependent internalization of L. monocytogenes.
Simultaneous Ca2+ and K+ fluxes are sufficient to activate bacterial internalization.
We investigated whether fluxes of Ca2+ and/or K+, in the absence of host cell perforation and any virulence factors, were sufficient to induce the internalization of large cargoes. We measured the internalization efficiency of 1-μm fluorescent polystyrene beads coated with bovine serum albumin (B-BSA) or with BSA and LLOml (B–BSA-LLOml) (LLOml is a monomer-locked variant of LLO that is unable to form pores). B-BSA and B–BSA-LLOml were previously shown to remain extracellular when incubated with HepG2 cells (3). Exogenously added LLO was sufficient to stimulate the internalization of the beads in a dose-dependent manner (Fig. 5A). This further demonstrated that the formation of LLO pores is required to induce the uptake of large particles and provided a convenient model to dissect how pore formation by LLO induces internalization. To establish whether LLO acted via generating ionic fluxes, we treated cells with Ca2+ and K+ ionophores in the absence of LLO. The Ca2+ ionophore ionomycin was sufficient to induce a low level of internalization of both BSA- and BSA-LLOml-coated beads, whereas the K+ ionophore nigericin did not induce internalization (Fig. 5B). We next treated cells with a mixture of ionomycin and nigericin to cause simultaneous fluxes of Ca2+ and K+. We found that bead internalization occurred to a much greater extent than with ionomycin alone. Importantly, in the presence of nigericin, the ionomycin concentration could be decreased to achieve an entry level similar to that induced by LLO (Fig. 5C). Therefore, simultaneous fluxes of Ca2+ and K+ are sufficient to induce the internalization of large cargoes.
FIG 5.
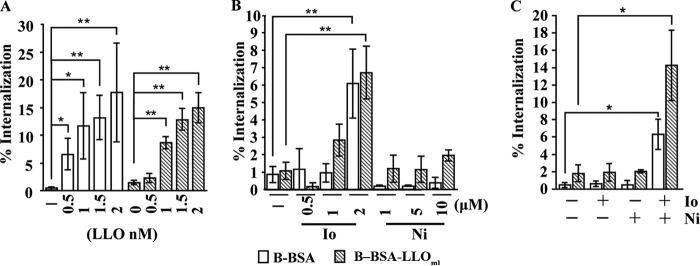
Ca2+ and K+ fluxes are sufficient to activate the internalization of large cargoes. Polystyrene beads coated with BSA (B-BSA) or BSA-LLOml (B–BSA-LLOml) (MOI of 20) were incubated with HepG2 cells for 15 min at 37°C, followed by treatment with LLO or ionophores for an additional 15 min. (A) Recombinant LLO was added to the cell culture medium at the indicated concentrations. (B) Ionomycin (Io) and/or nigericin (Ni) was added at the indicated concentrations. (C) Concentrations of 1 μM ionomycin and/or 10 μM nigericin were added to the cell culture medium as indicated. (A to C) Cells were washed and fixed, and extracellular beads were fluorescently labeled. Results are the means ± SEM of at least three independent experiments (*, P < 0.05; **, P < 0.01).
We next determined whether simultaneous Ca2+ and K+ fluxes were sufficient to enhance the entry of LLO-deficient L. monocytogenes. Neither the Ca2+ ionophore ionomycin nor the K+ ionophore nigericin alone was sufficient to increase L. monocytogenes internalization. However, bacterial internalization was significantly increased when the two ionophores were added together (Fig. 6). To verify that the ionophores affected cell signaling via generating Ca2+ and K+ fluxes, we repeated these experiments in Ca2+-free and K+-rich media. We found that when blocking Ca2+ influx or K+ efflux, the ionophores could no longer potentiate bacterial internalization, demonstrating that the effect of the ionophores on internalization was specific to their ability to induce an influx of extracellular Ca2+ and an efflux of intracellular K+ (Fig. 6).
FIG 6.
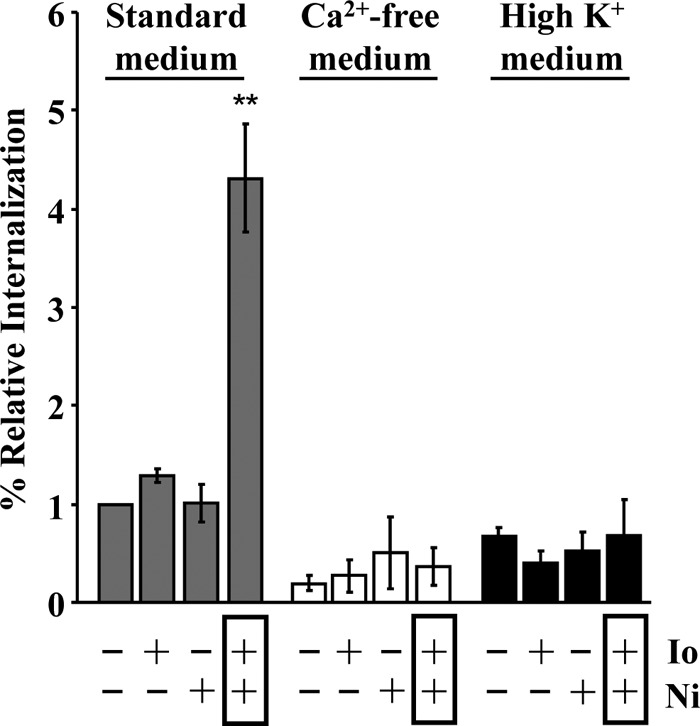
Ca2+ and K+ fluxes are sufficient to activate bacterial internalization. LLO-deficient (Δhly) L. monocytogenes bacteria (MOI of 20) were incubated with HepG2 cells with or without 1 μM ionomycin (Io) and/or 10 μM nigericin (Ni) in the indicated cell culture media. Bacterial internalization was measured by quantitative fluorescence microscopy and expressed relative to bacterial internalization in standard medium without ionophores. Results are expressed as the means ± SEM of at least three independent experiments (**, P < 0.01).
DISCUSSION
The present study demonstrates for the first time that host cells are significantly perforated by extracellular L. monocytogenes in an LLO-dependent manner and undergo Ca2+-dependent but K+-independent membrane resealing at early stages of infection. Importantly, perforation and repair are substantial even within 30 min and at a low multiplicity of infection (MOI of 5). These studies also show that Ca2+ influx alone, induced by ionomycin in the absence of plasma membrane perforation, is sufficient to induce plasma membrane rearrangements and internalization of polystyrene beads, with, however, a low efficiency. This suggested that in addition to Ca2+ influx, another signal is required for internalization. In support of this, K+ efflux is required for efficient internalization, and significant uptake of beads and bacteria was observed in cells treated with a combination of ionomycin and nigericin to induce simultaneous Ca2+ and K+ fluxes. Together, these results support the hypothesis that host cell plasma membrane perforation by pore-forming proteins induces nonspecific uptake of large cargoes, including bacteria, via inducing a combination of ion fluxes.
One important message of the present study is that LLO produced by L. monocytogenes perforates host cells during the early stage of invasion. This process was previously neglected, since perforation was unnoticed due to rapid Ca2+-dependent membrane resealing that maintains cell integrity and viability during L. monocytogenes infection. This finding further confirms that the activity of extracellular LLO during the intracellular life cycle of L. monocytogenes deserves further study. One such activity is the control of bacterial internalization. Previous studies demonstrated that host cell perforation by LLO activates the internalization of large cargoes, such as L. monocytogenes (3). This activation requires tyrosine kinase activity, F-actin polymerization, and dynamin, ultimately leading to the formation of an early endosome that can contain the bacterium or a 1-μm bead (3). The present work identified Ca2+ and K+ fluxes as initiators of the signaling pathway leading to bacterial internalization subsequent to host cell perforation.
The influx of extracellular Ca2+ subsequent to plasma membrane injury caused by pore-forming toxins or mechanical disruption is essential for the activation of membrane repair (33, 47–50). If Ca2+ influx is prevented, the plasma membrane is not resealed and cells can rapidly lyse. The resealing machinery of cells exposed to diverse pore-forming toxins has recently been addressed, and it appears that plasma membrane trafficking is intimately linked to the resealing process via endocytosis or the shedding of microvesicles that contain the toxin pores (51–55). It was proposed that cells injured by the CDC streptolysin O (SLO), produced by Streptococcus pyogenes, are repaired via Ca2+-dependent endocytosis of SLO pores (39). In this model, an increase in cytosolic Ca2+ activates rapid exocytosis of lysosomes, which release acid sphingomyelinase (ASM). ASM catalyzes the synthesis of ceramides on the outer leaflet of the plasma membrane, leading to endocytosis of the SLO pores via a clathrin-, dynamin-, and F-actin-independent route (56). We expect that cells injured by LLO and SLO, which form a similar pore, use a similar repair mechanism (11). In support of this, we reported that resealing of cells injured by LLO, similar to resealing after damage by SLO, is Ca2+ dependent but clathrin, dynamin, and F-actin independent (3). We now show that resealing in response to LLO is K+ independent. It was proposed that T. cruzi injures host cells to exploit the Ca2+- and ASM-dependent but clathrin-, dynamin-, and F-actin-independent resealing process to gain entry into host cells (4). Based upon the findings reported in the present manuscript and past studies (3), this model is not sufficient to account for LLO-dependent L. monocytogenes internalization into host cells. While LLO-mediated internalization and plasma membrane resealing have a shared requirement for Ca2+ influx and are both independent of clathrin, they differ in important ways. Unlike the membrane-resealing process, LLO-induced L. monocytogenes internalization is F-actin, dynamin, and K+ dependent (3). Therefore, our results confirm that ion fluxes are critical regulators of endocytosis following host cell perforation but reveal important differences between the sensu stricto plasma membrane-resealing process and LLO-mediated L. monocytogenes internalization. It will be necessary to further dissect the events occurring during the membrane repair process in order to establish whether additional events beyond sensu stricto resealing involve F-actin, dynamin, and K+ efflux. Alternatively, LLO-induced bacterial uptake may use a pathway that occurs downstream from or is distinct from repair of the plasma membrane.
K+ efflux subsequent to cell perforation by LLO and other pore-forming toxins is known to be responsible for the activation of caspase-1 (30, 57), histone modification (28), autophagy (32), and arrest in protein synthesis (32). The finding reported here that K+ efflux also regulates the uptake of the pathogen is novel. How host cells detect and respond to variations in intracellular K+ is also unsettled. Whereas Ca2+ is a central regulator involved in a large variety of processes, including endocytosis, few studies have addressed the role of K+ in endocytosis. It was previously shown that lowering intracellular K+ concentrations decreases clathrin-coated pit formation and receptor endocytosis (58); however, other cargoes can still be internalized (59). The LLO-dependent uptake of L. monocytogenes is clathrin independent. Therefore, it is likely that low K+ levels facilitate an alternative or compensatory, clathrin-independent pathway. Also, K+ efflux subsequent to cell attack by LLO was shown to activate multiple protein kinases (31).
It appears that signaling downstream of Ca2+ and K+ fluxes is complex and involves several pathways that may or may not cross-talk with one another. For example, histone dephosphorylation induced by LLO is Ca2+ independent and K+ dependent, whereas bacterial internalization is both Ca2+ and K+ dependent. When cells were treated with ionophores in an attempt to reproduce the effect of LLO without creating membrane pores, the Ca2+ ionophore ionomycin was sufficient to induce the internalization of polystyrene beads. However, the ionomycin concentration required to induce this event approached amounts that were detrimental to the cells, as cells started to round up during the course of the experiment. The Ca2+ influx stimulated by this concentration of ionomycin was capable of influencing membrane dynamics and internalization but with low efficiency. In contrast, treating the cells with the K+ ionophore nigericin had no such effect, even at high concentrations. In an attempt to initiate multiple fluxes, as occurs following perforation of the plasma membrane by LLO, we treated cells with a combination of nigericin and ionomycin. The addition of both ionophores at concentrations that were insufficient to induce internalization when added alone led to enhanced internalization of bacteria and beads, approaching the levels induced by LLO. This result provides evidence that host cells likely integrate responses generated from multiple signals that are mobilized by ion fluxes. The way in which host cells sense changes in K+ levels and the nature and extent of cross-talk between the response pathways induced by ion fluxes following membrane perforation are exciting avenues for future work. Finally, it will be important to determine whether other pathogens also cause local plasma membrane lesions to facilitate their internalization into host cells.
Supplementary Material
ACKNOWLEDGMENT
We are grateful to Daniel Portnoy for providing the bacterial strains used in this study.
Footnotes
Published ahead of print 23 December 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01067-13.
REFERENCES
- 1.Cossart P, Sansonetti PJ. 2004. Bacterial invasion: the paradigms of enteroinvasive pathogens. Science 304:242–248. 10.1126/science.1090124 [DOI] [PubMed] [Google Scholar]
- 2.Galan JE, Wolf-Watz H. 2006. Protein delivery into eukaryotic cells by type III secretion machines. Nature 444:567–573. 10.1038/nature05272 [DOI] [PubMed] [Google Scholar]
- 3.Vadia S, Arnett E, Haghighat AC, Wilson-Kubalek EM, Tweten RK, Seveau S. 2011. The pore-forming toxin listeriolysin O mediates a novel entry pathway of L. monocytogenes into human hepatocytes. PLoS Pathog. 7:e1002356. 10.1371/journal.ppat.1002356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fernandes MC, Cortez M, Flannery AR, Tam C, Mortara RA, Andrews NW. 2011. Trypanosoma cruzi subverts the sphingomyelinase-mediated plasma membrane repair pathway for cell invasion. J. Exp. Med. 208:909–921. 10.1084/jem.20102518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sukeno A, Nagamune H, Whiley RA, Jafar SI, Aduse-Opoku J, Ohkura K, Maeda T, Hirota K, Miyake Y, Kourai H. 2005. Intermedilysin is essential for the invasion of hepatoma HepG2 cells by Streptococcus intermedius. Microbiol. Immunol. 49:681–694. 10.1111/j.1348-0421.2005.tb03647.x [DOI] [PubMed] [Google Scholar]
- 6.Sanchez-Martinez S, Madan V, Carrasco L, Nieva JL. 2012. Membrane-active peptides derived from picornavirus 2B viroporin. Curr. Protein Pept. Sci. 13:632–643. 10.2174/138920312804142165 [DOI] [PubMed] [Google Scholar]
- 7.Ishino T, Chinzei Y, Yuda M. 2005. A Plasmodium sporozoite protein with a membrane attack complex domain is required for breaching the liver sinusoidal cell layer prior to hepatocyte infection. Cell. Microbiol. 7:199–208. 10.1111/j.1462-5822.2004.00447.x [DOI] [PubMed] [Google Scholar]
- 8.Fernandez-Puentes C, Carrasco L. 1980. Viral infection permeabilizes mammalian cells to protein toxins. Cell 20:769–775. 10.1016/0092-8674(80)90323-2 [DOI] [PubMed] [Google Scholar]
- 9.Hayes CS, Aoki SK, Low DA. 2010. Bacterial contact-dependent delivery systems. Annu. Rev. Genet. 44:71–90. 10.1146/annurev.genet.42.110807.091449 [DOI] [PubMed] [Google Scholar]
- 10.Kafsack BF, Carruthers VB. 2010. Apicomplexan perforin-like proteins. Commun. Integr. Biol. 3:18–23. 10.4161/cib.3.1.9794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dunstone MA, Tweten RK. 2012. Packing a punch: the mechanism of pore formation by cholesterol dependent cytolysins and membrane attack complex/perforin-like proteins. Curr. Opin. Struct. Biol. 22:342–349. 10.1016/j.sbi.2012.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vazquez-Boland JA, Dominguez-Bernal G, Gonzalez-Zorn B, Kreft J, Goebel W. 2001. Pathogenicity islands and virulence evolution in Listeria. Microbes Infect. 3:571–584. 10.1016/S1286-4579(01)01413-7 [DOI] [PubMed] [Google Scholar]
- 13.Seveau S, Pizarro-Cerda J, Cossart P. 2007. Molecular mechanisms exploited by Listeria monocytogenes during host cell invasion. Microbes Infect. 9:1167–1175. 10.1016/j.micinf.2007.05.004 [DOI] [PubMed] [Google Scholar]
- 14.Pizarro-Cerda J, Kuhbacher A, Cossart P. 2012. Entry of Listeria monocytogenes in mammalian epithelial cells: an updated view. Cold Spring Harb. Perspect. Med. 2:a010009. 10.1101/cshperspect.a010009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gaillard JL, Berche P, Frehel C, Gouin E, Cossart P. 1991. Entry of L. monocytogenes into cells is mediated by internalin, a repeat protein reminiscent of surface antigens from gram-positive cocci. Cell 65:1127–1141 [DOI] [PubMed] [Google Scholar]
- 16.Mengaud J, Ohayon H, Gounon P, Mege RM, Cossart P. 1996. E-cadherin is the receptor for internalin, a surface protein required for entry of L. monocytogenes into epithelial cells. Cell 84:923–932 [DOI] [PubMed] [Google Scholar]
- 17.Shen Y, Naujokas M, Park M, Ireton K. 2000. InIB-dependent internalization of Listeria is mediated by the Met receptor tyrosine kinase. Cell 103:501–510. 10.1016/S0092-8674(00)00141-0 [DOI] [PubMed] [Google Scholar]
- 18.Gaillard JL, Berche P, Mounier J, Richard S, Sansonetti P. 1987. In vitro model of penetration and intracellular growth of Listeria monocytogenes in the human enterocyte-like cell line Caco-2. Infect. Immun. 55:2822–2829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Portnoy DA, Jacks PS, Hinrichs DJ. 1988. Role of hemolysin for the intracellular growth of Listeria monocytogenes. J. Exp. Med. 167:1459–1471. 10.1084/jem.167.4.1459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schnupf P, Portnoy DA. 2007. Listeriolysin O: a phagosome-specific lysin. Microbes Infect. 9:1176–1187. 10.1016/j.micinf.2007.05.005 [DOI] [PubMed] [Google Scholar]
- 21.Bavdek A, Gekara NO, Priselac D, Gutierrez Aguirre I, Darji A, Chakraborty T, Macek P, Lakey JH, Weiss S, Anderluh G. 2007. Sterol and pH interdependence in the binding, oligomerization, and pore formation of Listeriolysin O. Biochemistry 46:4425–4437. 10.1021/bi602497g [DOI] [PubMed] [Google Scholar]
- 22.Bubert A, Sokolovic Z, Chun SK, Papatheodorou L, Simm A, Goebel W. 1999. Differential expression of Listeria monocytogenes virulence genes in mammalian host cells. Mol. Gen. Genet. 261:323–336. 10.1007/PL00008633 [DOI] [PubMed] [Google Scholar]
- 23.Moors MA, Levitt B, Youngman P, Portnoy DA. 1999. Expression of listeriolysin O and ActA by intracellular and extracellular Listeria monocytogenes. Infect. Immun. 67:131–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tang P, Rosenshine I, Cossart P, Finlay BB. 1996. Listeriolysin O activates mitogen-activated protein kinase in eucaryotic cells. Infect. Immun. 64:2359–2361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang P, Sutherland CL, Gold MR, Finlay BB. 1998. Listeria monocytogenes invasion of epithelial cells requires the MEK-1/ERK-2 mitogen-activated protein kinase pathway. Infect. Immun. 66:1106–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kayal S, Lilienbaum A, Poyart C, Memet S, Israel A, Berche P. 1999. Listeriolysin O-dependent activation of endothelial cells during infection with Listeria monocytogenes: activation of NF-kappa B and upregulation of adhesion molecules and chemokines. Mol. Microbiol. 31:1709–1722. 10.1046/j.1365-2958.1999.01305.x [DOI] [PubMed] [Google Scholar]
- 27.Meixenberger K, Pache F, Eitel J, Schmeck B, Hippenstiel S, Slevogt H, N′Guessan P, Witzenrath M, Netea MG, Chakraborty T, Suttorp N, Opitz B. 2010. Listeria monocytogenes-infected human peripheral blood mononuclear cells produce IL-1beta, depending on listeriolysin O and NLRP3. J. Immunol. 184:922–930. 10.4049/jimmunol.0901346 [DOI] [PubMed] [Google Scholar]
- 28.Hamon MA, Cossart P. 2011. K+ efflux is required for histone H3 dephosphorylation by Listeria monocytogenes listeriolysin O and other pore-forming toxins. Infect. Immun. 79:2839–2846. 10.1128/IAI.01243-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ribet D, Hamon M, Gouin E, Nahori MA, Impens F, Neyret-Kahn H, Gevaert K, Vandekerckhove J, Dejean A, Cossart P. 2010. Listeria monocytogenes impairs SUMOylation for efficient infection. Nature 464:1192–1195. 10.1038/nature08963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hamon MA, Batsche E, Regnault B, Tham TN, Seveau S, Muchardt C, Cossart P. 2007. Histone modifications induced by a family of bacterial toxins. Proc. Natl. Acad. Sci. U. S. A. 104:13467–13472. 10.1073/pnas.0702729104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stavru F, Bouillaud F, Sartori A, Ricquier D, Cossart P. 2011. Listeria monocytogenes transiently alters mitochondrial dynamics during infection. Proc. Natl. Acad. Sci. U. S. A. 108:3612–3617. 10.1073/pnas.1100126108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gonzalez MR, Bischofberger M, Freche B, Ho S, Parton RG, van der Goot FG. 2011. Pore-forming toxins induce multiple cellular responses promoting survival. Cell. Microbiol. 13:1026–1043. 10.1111/j.1462-5822.2011.01600.x [DOI] [PubMed] [Google Scholar]
- 33.Walev I, Bhakdi SC, Hofmann F, Djonder N, Valeva A, Aktories K, Bhakdi S. 2001. Delivery of proteins into living cells by reversible membrane permeabilization with streptolysin-O. Proc. Natl. Acad. Sci. U. S. A. 98:3185–3190. 10.1073/pnas.051429498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.TranVan Nhieu G, Clair C, Grompone G, Sansonetti P. 2004. Calcium signalling during cell interactions with bacterial pathogens. Biol. Cell 96:93–101. 10.1016/j.biolcel.2003.10.006 [DOI] [PubMed] [Google Scholar]
- 35.Repp H, Pamukci Z, Koschinski A, Domann E, Darji A, Birringer J, Brockmeier D, Chakraborty T, Dreyer F. 2002. Listeriolysin of Listeria monocytogenes forms Ca2+-permeable pores leading to intracellular Ca2+ oscillations. Cell. Microbiol. 4:483–491. 10.1046/j.1462-5822.2002.00207.x [DOI] [PubMed] [Google Scholar]
- 36.Gekara NO, Westphal K, Ma B, Rohde M, Groebe L, Weiss S. 2007. The multiple mechanisms of Ca2+ signalling by listeriolysin O, the cholesterol-dependent cytolysin of Listeria monocytogenes. Cell. Microbiol. 9:2008–2021. 10.1111/j.1462-5822.2007.00932.x [DOI] [PubMed] [Google Scholar]
- 37.Gekara NO, Groebe L, Viegas N, Weiss S. 2008. Listeria monocytogenes desensitizes immune cells to subsequent Ca2+ signaling via listeriolysin O-induced depletion of intracellular Ca2+ stores. Infect. Immun. 76:857–862. 10.1128/IAI.00622-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berridge MJ, Lipp P, Bootman MD. 2000. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 1:11–21. 10.1038/35036035 [DOI] [PubMed] [Google Scholar]
- 39.Idone V, Tam C, Goss JW, Toomre D, Pypaert M, Andrews NW. 2008. Repair of injured plasma membrane by rapid Ca2+-dependent endocytosis. J. Cell Biol. 180:905–914. 10.1083/jcb.200708010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bischofberger M, Iacovache I, van der Goot FG. 2012. Pathogenic pore-forming proteins: function and host response. Cell Host Microbe 12:266–275. 10.1016/j.chom.2012.08.005 [DOI] [PubMed] [Google Scholar]
- 41.Glomski IJ, Gedde MM, Tsang AW, Swanson JA, Portnoy DA. 2002. The Listeria monocytogenes hemolysin has an acidic pH optimum to compartmentalize activity and prevent damage to infected host cells. J. Cell Biol. 156:1029–1038. 10.1083/jcb.200201081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gedde MM, Higgins DE, Tilney LG, Portnoy DA. 2000. Role of listeriolysin O in cell-to-cell spread of Listeria monocytogenes. Infect. Immun. 68:999–1003. 10.1128/IAI.68.2.999-1003.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haghighat AC, Seveau S. 2010. Quantification of host-microbe interactions by automated fluorescence microscopy. J. Immunol. Methods 352:186–191. 10.1016/j.jim.2009.11.013 [DOI] [PubMed] [Google Scholar]
- 44.Dramsi S, Cossart P. 2003. Listeriolysin O-mediated calcium influx potentiates entry of Listeria monocytogenes into the human Hep-2 epithelial cell line. Infect. Immun. 71:3614–3618. 10.1128/IAI.71.6.3614-3618.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.da Silva Tatley F, Aldwell FE, Dunbier AK, Guilford PJ. 2003. N-terminal E-cadherin peptides act as decoy receptors for Listeria monocytogenes. Infect. Immun. 71:1580–1583. 10.1128/IAI.71.3.1580-1583.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Theard D, Raspe MA, Kalicharan D, Hoekstra D, van IJzendoorn SC. 2008. Formation of E-cadherin/beta-catenin-based adherens junctions in hepatocytes requires serine-10 in p27(Kip1). Mol. Biol. Cell 19:1605–1613. 10.1091/mbc.E07-07-0661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bi GQ, Alderton JM, Steinhardt RA. 1995. Calcium-regulated exocytosis is required for cell membrane resealing. J. Cell Biol. 131:1747–1758. 10.1083/jcb.131.6.1747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Terasaki M, Miyake K, McNeil PL. 1997. Large plasma membrane disruptions are rapidly resealed by Ca2+-dependent vesicle-vesicle fusion events. J. Cell Biol. 139:63–74. 10.1083/jcb.139.1.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abreu-Blanco MT, Verboon JM, Parkhurst SM. 2011. Single cell wound repair: dealing with life's little traumas. Bioarchitecture 1:114–121. 10.4161/bioa.1.3.17091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McNeil PL, Steinhardt RA. 2003. Plasma membrane disruption: repair, prevention, adaptation. Annu. Rev. Cell Dev. Biol. 19:697–731. 10.1146/annurev.cellbio.19.111301.140101 [DOI] [PubMed] [Google Scholar]
- 51.Husmann M, Beckmann E, Boller K, Kloft N, Tenzer S, Bobkiewicz W, Neukirch C, Bayley H, Bhakdi S. 2009. Elimination of a bacterial pore-forming toxin by sequential endocytosis and exocytosis. FEBS Lett. 583:337–344. 10.1016/j.febslet.2008.12.028 [DOI] [PubMed] [Google Scholar]
- 52.Husmann M, Dersch K, Bobkiewicz W, Beckmann E, Veerachato G, Bhakdi S. 2006. Differential role of p38 mitogen activated protein kinase for cellular recovery from attack by pore-forming S. aureus alpha-toxin or streptolysin O. Biochem. Biophys. Res. Commun. 344:1128–1134. 10.1016/j.bbrc.2006.03.241 [DOI] [PubMed] [Google Scholar]
- 53.Thiery J, Keefe D, Saffarian S, Martinvalet D, Walch M, Boucrot E, Kirchhausen T, Lieberman J. 2010. Perforin activates clathrin- and dynamin-dependent endocytosis, which is required for plasma membrane repair and delivery of granzyme B for granzyme-mediated apoptosis. Blood 115:1582–1593. 10.1182/blood-2009-10-246116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Los FC, Kao CY, Smitham J, McDonald KL, Ha C, Peixoto CA, Aroian RV. 2011. RAB-5- and RAB-11-dependent vesicle-trafficking pathways are required for plasma membrane repair after attack by bacterial pore-forming toxin. Cell Host Microbe 9:147–157. 10.1016/j.chom.2011.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Babiychuk EB, Monastyrskaya K, Potez S, Draeger A. 2011. Blebbing confers resistance against cell lysis. Cell Death Differ. 18:80–89. 10.1038/cdd.2010.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tam C, Idone V, Devlin C, Fernandes MC, Flannery A, He X, Schuchman E, Tabas I, Andrews NW. 2010. Exocytosis of acid sphingomyelinase by wounded cells promotes endocytosis and plasma membrane repair. J. Cell Biol. 189:1027–1038. 10.1083/jcb.201003053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gurcel L, Abrami L, Girardin S, Tschopp J, van der Goot FG. 2006. Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell 126:1135–1145. 10.1016/j.cell.2006.07.033 [DOI] [PubMed] [Google Scholar]
- 58.Larkin JM, Brown MS, Goldstein JL, Anderson RG. 1983. Depletion of intracellular potassium arrests coated pit formation and receptor-mediated endocytosis in fibroblasts. Cell 33:273–285. 10.1016/0092-8674(83)90356-2 [DOI] [PubMed] [Google Scholar]
- 59.Moya M, Dautry-Varsat A, Goud B, Louvard D, Boquet P. 1985. Inhibition of coated pit formation in Hep2 cells blocks the cytotoxicity of diphtheria toxin but not that of ricin toxin. J. Cell Biol. 101:548–559. 10.1083/jcb.101.2.548 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.