

Abstract
Recent microbiome studies have implicated a role for Filifactor alocis in periodontal disease. In this study, we investigated the colonization and survival properties of F. alocis in a mouse subcutaneous chamber model of infection and characterized host innate immune responses. An infection of 109 F. alocis successfully colonized all chambers; however, the infection was cleared after 72 h. F. alocis elicited a local inflammatory response with neutrophils recruited into the chambers at 2 h postinfection along with an increase in levels of the proinflammatory cytokines interleukin 1β (IL-1β), IL-6, and tumor necrosis factor (TNF). F. alocis also induced apoptosis in chamber epithelial cells and neutrophils. Consistent with resolution of infection, neutrophil numbers and cytokine levels returned to baseline by 72 h. Fluorescent in situ hybridization (FISH) and quantitative PCR demonstrated that F. alocis exited the chambers and spread to the spleen, liver, lung, and kidney. Massive neutrophil infiltration was observed in the spleen and lungs, and the recruited neutrophils were in close proximity to the infecting bacteria. Significant epithelial injury was observed in the kidneys. Infection of all tissues was resolved after 7 days. This first in vivo study of the pathogenicity of F. alocis shows that in the chamber model the organism can establish a proinflammatory, proapoptotic local infection which is rapidly resolved by the host concordant with neutrophil influx. Moreover, F. alocis can spread to, and transiently infect, remote tissues where neutrophils can also be recruited.
INTRODUCTION
Periodontal diseases are a group of microbially driven, inflammatory-based diseases that afflict around half of the adult population in the United States alone (1). Heterotypic communities of organisms that exhibit polymicrobial synergy are responsible for the initiation and progression of disease (2). For a number of years, a small group of organisms, namely, Porphyromonas gingivalis, Tannerella forsythia, and Treponema denticola, were considered the primary pathogens in severe and chronic cases of adult periodontitis (3). However, data emerging from human microbiome projects have identified a wider range of uncultivable and fastidious bacteria associated with disease status (4–8).
Filifactor alocis, a Gram-positive, slow-growing, obligate anaerobic rod, is one of the newly appreciated potential periodontal pathogens. Several recent studies have found F. alocis at increased frequency and in higher numbers at periodontal disease sites than at healthy sites, leading to the proposal that F. alocis should be included as a diagnostic indicator of disease (4, 6, 7, 9–11). In addition, F. alocis is associated with aggressive periodontitis in children (12), endodontic lesions (13), and pericoronitis (14), indicating that the organism can display a range of pathogenic properties. F. alocis can form synergistic interactions with other common periodontal bacteria, such as Fusobacterium nucleatum, which may facilitate colonization by the organism and aid in the establishment of pathogenic periodontal communities (15). Studies of pathogenicity that have been performed to date show that F. alocis is relatively resistant to oxidative stress (16), can produce trypsin-like proteases (17), and can induce the secretion of proinflammatory cytokines from gingival epithelial cells (18). Furthermore, F. alocis and P. gingivalis interact synergistically in the invasion of epithelial cells in culture (16). However, the nature of the interaction between F. alocis and cells of the innate immune system has yet to be studied in vitro or in vivo.
A variety of animal models have been utilized to assess the pathogenic potential of periodontal bacteria, and different models recapitulate different stages of the disease process (19, 20). The murine subcutaneous chamber model is widely used to study bacterial survival and local inflammatory responses (19, 21). In addition, the interior of the coil becomes epithelialized, allowing in vivo responses of epithelial cells to be documented (19). While the model does not involve alveolar bone loss directly, previous studies have documented a correlation between the proinflammatory characteristics of P. gingivalis in the chamber model and alveolar bone loss in an oral infection model (22). In this study, we utilized the murine subcutaneous chamber model to investigate F. alocis survival and spreading, along with induction of proinflammatory and apoptotic responses in vivo.
MATERIALS AND METHODS
Bacterial strain and growth conditions.
F. alocis strain ATCC 38596 was cultivated in brain heart infusion (BHI) broth supplemented with l-cysteine (0.1%) and arginine (20%) for 7 days under anaerobic conditions. Numbers of bacteria were determined spectrophotometrically (optical density at 600 nm [OD600]).
Subcutaneous chamber model.
Eight- to 10-week-old female C57BL/6 mice were obtained from The Jackson Laboratory and housed under specific-pathogen-free conditions. All animal procedures were performed in accordance with the guidelines of Institutional Animal Care and Use Committee in compliance with established federal and state polices. Middorsal subcutaneous implantation of surgical-grade titanium coil chambers was performed under isoflurane anesthesia. Following a 7-day healing period, F. alocis (109 CFU in 100 μl of sterile pyrogen-free phosphate-buffered saline [PBS]) was injected into the chambers of each mouse by using a 25-gauge syringe. Sham-infected animals received PBS alone. Chamber exudates were harvested from mice at 2, 24, and 72 h postinfection by using a 25-gauge syringe (each chamber was sampled only once) and diluted 1:100 in PBS. Flow cytometry was used for phenotypic characterization of recruited cells. Aliquots of exudates were centrifuged, supernatants were used to analyze cytokine levels, and pellets were used to quantify F. alocis. After sampling and at 7 days postinfection, spleens, livers, lungs, and kidneys were surgically recovered.
Characterization of recruited cells in the chamber fluid.
Total cell counts (red blood cells excluded) were determined manually using a hemocytometer. Phenotypic analysis of cells was performed by reacting cells with fluorescein isothiocyanate-conjugated anti-mouse F4/80 monoclonal antibody (MAb), CD3 MAb, or phycoerythrin-conjugated anti-mouse Ly6G MAb (LifeSpan Biosciences) at 4°C for 30 min. The antibody-labeled cells were washed twice in flow cytometry staining buffer (eBioscience), fixed in 1% paraformaldehyde, and analyzed with the appropriate isotype controls by flow cytometry.
Quantification of F. alocis.
Chamber sample pellets were lysed and DNA was extracted using a Wizard genomic DNA purification kit (Promega). Quantitative PCR (qPCR) (23) was performed by using primers specific for F. alocis 16S rRNA (24): forward, 5′-CAGGTGGTTTAACAAGTTAGTGG-3′; reverse, 5′-CTAAGTTGTCCTTAGCTGTCTCG-3′. For quantitation, genomic DNA from laboratory cultures of F. alocis was isolated, quantity and purity were determined spectrophotometrically, and a series of dilutions were prepared. Each dilution was amplified with the 16S primers, and the number of gene copies in the chamber sample was determined by comparison with this standard curve.
Fluorescent in situ hybridization (FISH).
Tissues were fixed in 4% paraformaldehyde for 24 h, dehydrated in a graded ethanol series, and clarified in xylene. Paraffin wax-embedded sections (5 μm) were mounted on slides and deparaffinized in xylene, followed by a graded ethanol series. Samples were treated with lysozyme (Sigma; 70,000 U ml−1) in Tris-HCl (pH 7.5) for 10 min at 3°C. An F. alocis-specific probe, 5′-TCTTTGTCCACTATCGTTTTGA-3′ (16), was 5′ labeled with Oregon Green 488. A P. gingivalis-specific probe, 5′-CAATACTCGTATCGCCCGTTATTC-3′, was 5′ labeled with Cy3 as a negative control. Hybridization was performed in 10 μl hybridization buffer (0.9 M sodium chloride, 20 mM Tris-HCl, 0.01% sodium dodecyl sulfate, pH 7.2) containing 20 ng of species-specific probes. After incubation overnight in a dark humidified chamber at 37°C, slides were rinsed with 50% formamide and 2× saline-sodium citrate buffer (0.3 M NaCl, 0.03 M trisodium citrate dehydrate Na3C6H5O7 · 2H2O, pH 7.0) and mounted with Vectashield (Vector). The slides were visualized using a confocal microscope (Olympus; FV100).
Tissue histology and assessment of neutrophil infiltration.
For histological assessment, spleens, livers, lungs, and kidneys were paraffin embedded. Sections of embedded tissues were stained with hematoxylin and eosin (H&E) and examined by light microscopy for tissue morphology. To determine neutrophil infiltration, staining for the leukocyte-specific esterase, naphthol AS-D chloroacetate esterase (NACE; Sigma), was performed on tissue sections. Slides were incubated in a solution of sodium nitrate, fast red violet BL base solution, TRIZMAL 6.3 buffer, and naphthol AS-D chloroacetate solution in deionized water for 15 min at 37°C. After being rinsed, slides were counterstained with Gills hematoxylin solution and coverslipped. Stained tissue sections were examined microscopically for morphology and positively stained cells. Visualization was with an Olympus BX51 microscope and Image Pro 6.2 software. To establish the percentage of area present in the tissue sample that was esterase positive (neutrophils), tissue sections were randomly screened (4 fields/slide, at ×20 magnification, approximately 0.55 mm2 each), and Image J-Fiji software was used to calculate the area percentage.
Immunohistochemistry.
Tissues were fixed in 4% paraformaldehyde for 24 h, washed with PBS (pH 7.4), and frozen in OCT compound at −80°C. Serial sections (7 to 8 μm) were cut using a cryostat. For double immunofluorescence staining, sections were blocked in 10% bovine serum albumin (BSA) for 2 h and simultaneously incubated with rabbit primary antibodies to active caspase-3 (Abcam) and either mouse pan-cytokeratin (Invitrogen) or Ly6G. After being washed in PBS, sections were stained with goat anti-rabbit IgG Alexa Fluor 594 (Invitrogen) or rabbit anti-mouse IgG Alexa Fluor 488 (Invitrogen) for 30 min at room temperature. Images were captured by confocal microscopy.
Cytokine ELISA.
Local (chamber exudate) cytokine levels of tumor necrosis factor (TNF), interleukin 1β (IL-1β), and IL-6 were determined by enzyme-linked immunosorbent assay (ELISA) using mouse OptEIA sets (BD Biosciences, San Jose, CA) according to the manufacturer's directions.
RESULTS
Colonization and spreading of F. alocis.
The mouse subcutaneous chamber model was used to examine the ability of F. alocis to colonize in vivo and to characterize the acute inflammatory response to F. alocis infection. As the culture requirements of the organism are not fully defined and growth on solid medium is inefficient, numbers of F. alocis were determined by quantitative PCR. F. alocis inocula of 1 × 109 viable bacteria successfully colonized all of the chambers. Bacterial levels in 50 μl of chamber fluid were in the 105 to 106 range after 2 h (Fig. 1); however, by 72 h, only low levels of F. alocis could be detected. The gradual reduction in numbers of F. alocis recovered from the chambers could be the result of killing by the host or F. alocis exiting the chambers and spreading systemically. To begin to distinguish between these possibilities, we investigated spreading of F. alocis to the spleen, liver, lung, and kidney. FISH analysis revealed detectable levels of F. alocis in the liver, lung, and kidney at 2 h postinoculation (Fig. 2A). At 24 h following chamber inoculation, all the tissues sampled had detectable F. alocis colonization, indicating that F. alocis can exit the subcutaneous chambers and spread to remote tissues. After 72 h, amounts of F. alocis were reduced in the spleen, liver, and lung. Quantitative PCR (Fig. 2B) corroborated the presence of F. alocis DNA in the tissues at 2 h and 24 h and showed reduced levels in the liver and lung by 72 h. To reveal the ultimate fate of F. alocis in these tissues, we repeated the experiment over a 7-day time period. By 7 days after infection, F. alocis DNA was undetectable by FISH and at very low levels by PCR in all tissues (Fig. 2A and B).
FIG 1.
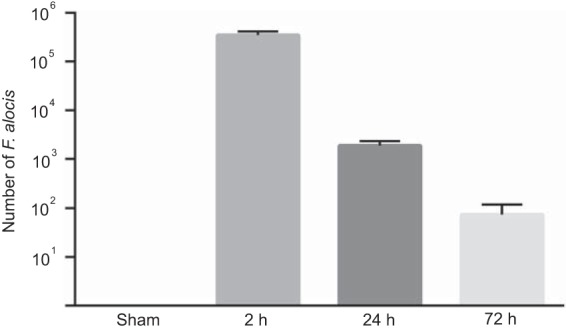
F. alocis successfully colonizes subcutaneous chambers after inoculation of 109 cells. Bars represent the mean numbers determined by qPCR in 50 μl from each of five chambers ± standard deviations at each time point. One representative experiment of three is shown.
FIG 2.

F. alocis spreads to remote tissues. (A) FISH analysis of spleen, liver, lung, and kidney tissues at the indicated times postinfection. Images are representative of organs at each time point and were collected by confocal microscopy. Bar represents 50 μm. (B) qPCR of F. alocis DNA in tissues. Data are expressed as mean numbers from tissues ± standard deviations at each time point. One representative experiment of three is shown.
These results show that F. alocis can establish a local infection in subcutaneous chambers which is rapidly resolved. However, the organism can spread from the site of local infection and colonize organs such as the spleen, liver, lung, and kidney. The host is also able to resolve the infection at remote tissues, and the organism is eliminated within 7 days.
Cellular and cytokine inflammatory response to F. alocis.
To investigate the nature of the innate immune response to local infection with F. alocis, the cellular infiltrate and cytokine levels in the chamber fluid were characterized. As shown in Fig. 3, a peak of neutrophil (Ly6G+) infiltration occurred at 2 h postinfection, which gradually declined to basal levels by 72 h. Up to 72 h, no CD3+ T cells or F4/80+ macrophages were recruited to the chamber (data not shown). Minimal neutrophils and other leukocytes were detected in the sham animals. We hypothesized that this granulocytic infiltration would instigate a proinflammatory cytokine profile, and indeed challenge with F. alocis incited robust levels of IL-1β, TNF, and IL-6, within 2 h postinfection (Fig. 4). The IL-1β response was the most robust and, although reduced at 72 h, remained statistically elevated. TNF and IL-6 returned to baseline levels by 24 h postinfection, contemporaneous with the reduction in bacterial levels and neutrophil recruitment in the chambers and consistent with the resolution of local infection.
FIG 3.
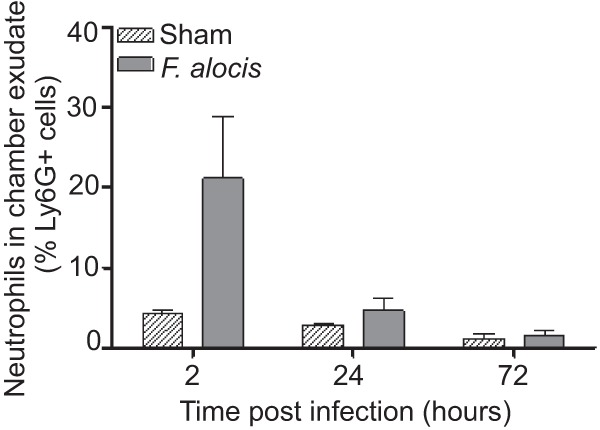
Time course of neutrophil recruitment in chamber exudates in response to challenge with F. alocis. The percentage of Ly6G+-recruited cells in chamber fluid was determined by flow cytometry. Data are expressed as means ± standard errors of the means (SEM) of the percentage of Ly6G-positive cells from 4 mice per time point. One representative experiment of three is shown.
FIG 4.

Cytokine levels in chamber exudates in response to challenge with F. alocis. IL-1β (A), TNF (B), and IL-6 (C) expression levels were determined by ELISA. Chamber fluids were measured at 2, 24, and 72 h postinfection with F. alocis and in sham-infected animals. Data are expressed as means ± SEM (n = 5 mice per group). *, P < 0.0001, significant enhancement of cytokine induction compared with sham infection using analysis of variance (ANOVA) followed by Tukey posttest. One representative experiment of three is shown.
Neutrophil infiltration of tissues.
Our data showed that F. alocis has the ability to spread systemically. Hence, to assess the inflammatory response at these sites, tissue sections from spleen, liver, lung, and kidney were stained for the neutrophil-specific esterase to determine the degree of neutrophil infiltration. Although F. alocis was detected by FISH in all four tissues, a massive neutrophil infiltration was observed only in the lung and spleen (Fig. 5). Lung tissue sections from F. alocis-infected animals showed signs of inflammation, such as alveolar thickening, as early as 2 h postinfection, and these were still visible by 72 h (Fig. 5A; see also Fig. S1 in the supplemental material). Moreover, in lung tissue sections, there was a significant (P < 0.05, t test) increase in neutrophil-specific esterase-positive cell infiltration from 0.4% ± 0.03 of the observed area positive for neutrophils in sham control animals to 1% ± 0.1 in the 2-h-postinfection animals. By 24 h, there was only a minimal further increase of neutrophil infiltration (1.2% ± 0.1), and the neutrophil numbers gradually declined by 72 h (0.2% ± 0.03). Lung tissue sections from 7 days postinfection showed no neutrophil infiltration and normal lung architecture, similar to the sham control group (data not shown).
FIG 5.

F. alocis dissemination into distal organs induces neutrophil influx. Lung tissue sections (A) or spleen tissue sections (B) from 2, 24, or 72 h after infection with F. alocis or from after sham infection were stained with naphthol AS-D chloroacetate esterase (NACE) for neutrophil-specific esterase (strongly purple-stained cells). The top panel represents lower magnification (×20 main image), and the lower panel shows an enlargement (magnification, ×40 of box). White ovals in panel A show areas of alveolar thickening. (C) Positive FISH and NACE of lung, spleen, kidney, and liver tissue sections; original magnification, ×20. Arrows indicate examples of positive neutrophil esterase cells showing darker stain than that of red blood cells. Bar represents 50 μm. One representative experiment of three is shown.
In the spleen, there was an increase in neutrophil-specific esterase-positive cell infiltration from 4.5% ± 0.4 in sham control animals to 8.2% ± 0.6 (P < 0.05, t test) in the perimarginal zone and marginal zone (MZ) in the 2-h-postinfection animals (Fig. 5B). The peak of neutrophil infiltration in the MZ was observed by 24 h (11.5% ± 2), and some follicular neutrophil infiltration was also observed. By 72 h postinfection, there was a marked reduction (9% ± 0.6) in the number of splenic neutrophils (Fig. 5B), and spleen tissue sections from 7 days postinfection showed a neutrophil distribution similar to the sham controls (data not shown).
Since high F. alocis numbers as well as neutrophil infiltration in the lung and spleen occurred 24 h postinfection, we examined the physical association between bacteria and neutrophils. Figure 5C depicts F. alocis-positive FISH in the bronchiole epithelial areas as well as in the alveoli of infected lungs. Neutrophil-specific esterase-positive cells were not observed in the bronchiole area but in close proximity and dispersed in the alveolar space. In the spleen, although it is more difficult to distinguish the precise architecture, F. alocis colonized the MZ and red pulp area, and the massive neutrophil infiltration was observed in the MZ area. F. alocis was also detected in the kidney, primarily in the tubular epithelial cells, and in the liver sinusoids. However, there was minimal neutrophils infiltration in these areas (Fig. 5C). These results indicate that in the lung and spleen tissue areas where F. alocis locates, neutrophils are present in the same area or in very close proximity and probably contribute to the ultimate elimination of the bacteria.
The histological architecture of the liver from the F. alocis-infected animals did not show significant tissue damage compared to that of the sham-infected control group, and only minimal neutrophil recruitment was observed (Fig. 5C; see also Fig. S1 in the supplemental material).
The kidneys of the F. alocis-infected animals showed more tissue injury than the lung, spleen, and liver. H&E staining revealed a marked tubular epithelial injury in the infected animals at 2 h (Fig. 6). The epithelial injury spreads from parietal epithelium to distal epithelium, and the degree of injury increased by 24 h postinfection, with more debris observed in the tubular cells. The progression of tubular damage increased by 72 h postinfection, when many of the nuclei appeared pyknotic, with a marked amount of debris in the epithelial lumens (Fig. 6). By 7 days postinfection, there were signs of recovery of the kidney tubular cells (data not shown).
FIG 6.
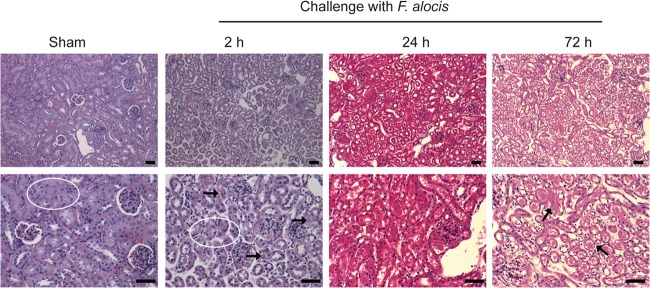
F. alocis induced significant tubular epithelial injury in the kidney. Hematoxylin and eosin (H&E) staining of kidney tissue sections from sham or 2, 24, or 72 h after infection with F. alocis; original magnification, ×20 (top) and ×40 (bottom). White oval in sham controls depicts normal tubular epithelial cell architecture; at 2 h after F. alocis infection, white oval and black arrows depict an area of tubular epithelial damage; at 72 h after F. alocis infection, black arrows depict pyknotic nuclei and cellular debris in tubular cell lumens. Bar represents 50 μm. One representative experiment of three is shown.
Induction of apoptosis by F. alocis.
F. alocis has been found to be proapoptotic toward epithelial cells (18), and to determine the effect of F. alocis infection on apoptosis of neutrophils and keratinocytes in the chamber model, chambers were excised and probed with caspase-3 antibodies. Neutrophils and epithelial cells were distinguished by labeling with Ly6G or pan-cytokeratin antibodies, respectively. Sham-infected chambers served as controls and demonstrated little caspase-3 activity. There was a consistent increase in the number of keratinocytes and neutrophils expressing active caspase-3 over the 72-h-postinfection period (Fig. 7). Keratinocytes were more resistant to apoptosis compared to neutrophils in this model. Only a small fraction of keratinocytes expressed active caspase-3 at 24 h, which increased to less than half by 72 h. In contrast, at 2 h, only a small fraction of neutrophils expressed caspase-3, but this increased to approximately half by 24 h, and the majority of neutrophils were apoptotic at 72 h.
FIG 7.
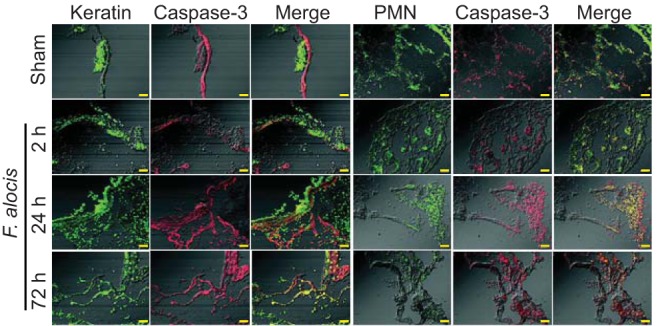
F. alocis induces apoptosis in keratinocytes and neutrophils within chambers. Sections of excised subcutaneous chambers were stained with antibodies to active caspase-3 and pan-cytokeratin (keratin) (A) or Ly6G (PMN) (B). Following reaction with fluorescent secondary antibodies, images were collected by confocal microscopy. Bar represents 50 μm. Data are representative of five chambers at each time point. One representative experiment of three is shown.
DISCUSSION
A strong association has emerged between F. alocis and oral diseases, including periodontitis (4, 11); however, little is known regarding the pathogenic mechanisms of the organism. In this study, we examined the behavior of F. alocis in mouse subcutaneous chambers which allow modeling of bacterial colonization, survival, and spreading, along with inflammatory responses. An inoculum of 109 bacteria was sufficient to reproducibly establish colonization of the chambers. This is consistent with the properties of other recognized periodontal pathogens, such as P. gingivalis and T. forsythia, which can also colonize subcutaneous chambers with inocula of 109 viable bacteria (25, 26). The local infection with F. alocis was rapidly resolved, and bacterial levels in the chamber decreased after 2 h and were almost undetectable by 72 h. Over this time period, F. alocis spread systemically and colonized remote tissues, including the spleen, liver, lung, and kidney. Interestingly, the organism was cleared from these tissues by 7 days, indicating that in this model system F. alocis can cause an acute rapidly spreading infection that is controlled by the host. Given the epidemiological association between periodontal diseases and serious systemic conditions such as cardiovascular disease and preterm delivery of low-birth-weight infants (27), the ability of F. alocis to spread systemically may allow the organisms to access remote sites in humans and contribute to disease either alone or in combination with other oral bacteria.
The healthy gingival crevice is colonized by a variety of microorganisms that assemble into heterotypic communities (28). These communities are, in general, proinflammatory over time, which facilitates host control of the microbial challenge by various mechanisms, including the recruitment of neutrophils into the crevice (29). Overt or keystone pathogens disrupt this balanced immuno-inflammatory state by either induction of destructive inflammatory responses or by targeted immune suppression and subversion (30). In the chamber model, F. alocis elicited the recruitment of neutrophils, coincident with a decrease in bacterial numbers, although the neutrophils were unable to control the spread of the organism. The sensitivity or resistance of F. alocis to neutrophil killing is a topic for further investigation. F. alocis induced the secretion of the proinflammatory cytokines IL-1β, IL-6, and TNF in the chamber fluid, cytokines that are derived predominantly from innate immune cells such as neutrophils, and also Th1 cells, although the latter cell type was not detected in the chamber exudates. TNF activates the transcription factor NF-κB, which controls expression of the neutrophil chemokine IL-8 (31) and could thus contribute to further neutrophil influx. Proinflammatory cytokines IL-1β, IL-6, and TNF also have potential tissue destructive capability. In the gingival crevice, these cytokines can stimulate pathways that activate osteoclasts and elevate alveolar bone resorption (32). IL-1β, IL-6, and TNF also activate matrix metalloproteases and other immune effectors, such as PGE2, both of which can contribute to tissue breakdown and failure to repair (19, 33, 34). During the disease process, IL-1β, IL-6, and TNF levels in the periodontal pocket are elevated (35, 36), and inhibition of IL-1 and TNF can reduce the severity of experimental periodontitis (34). F. alocis can also induce the expression of IL-1β, TNF, and IL-6 from gingival epithelial cells maintained in culture (18), consistent with an overall proinflammatory nature of the organism.
Cytokines such as IL-1β, IL-6, and TNF are also proapoptotic, and F. alocis induced both keratinocyte and neutrophil apoptosis in infected chambers. Previous reports have established that F. alocis can also induce apoptosis in gingival epithelial cells maintained in culture (18); thus, F. alocis can cause epithelial apoptosis both in vitro and in vivo. Furthermore, apoptosis can be demonstrated in periodontal lesions (37, 38), and apoptosis may be the direct result of bacterial action or the indirect result of proinflammatory cytokine secretion. Following 72 h of infection with F. alocis, the majority of, but not all, neutrophils were apoptotic. Surviving neutrophils could be involved in transport of F. alocis to remote sites, as has been demonstrated for other pathogenic microorganisms, such as Mycobacterium tuberculosis, Burkholderia pseudomallei, and Leishmania major (39–42). It has been reported that during inflammation, a portion of the neutrophils that have already left the circulation and transmigrated to the tissues can migrate back to the circulation (43). This process of reverse migration has been linked to dissemination of inflammation into other tissues. While a link between neutrophils and dissemination of F. alocis to distant organs remains to be established, by 24 h postinfection, we observed a peak of bacterial number in all four tissues collected as well as an increase neutrophil infiltration in spleen and lung. It has been shown recently that splenic neutrophils (NBH) have a distinct phenotype and can release activating signals, such as the cytokine BAFF (BLyS) and the proliferation-inducing ligand APRIL, which will promote survival and differentiation of B cells both in a T cell-dependent and T cell-independent manner (44). The NBH cells are located in the MZ of the spleen and infiltrate into the follicular areas upon lipopolysaccharide (LPS) challenge and infection. Furthermore, the cross talk between splenic neutrophils and MZ B cells is an important mechanism that allows the initiation of a rapid antibody response (45). In addition, upon LPS challenge in mice, there is a marked increase of the keratinocyte-derived chemokine (KC) in the MZ and red pulp of the spleen, accompanied by a significant influx of neutrophils, which are involved in clearance of the infection (46). Our data also showed a marked increase in neutrophil influx into the MZ of the spleen by 2 h postinfection, which further increased by 24 h postinfection. This observation indicates that following F. alocis dissemination and infection of the spleen, the neutrophil influx could lead to clearance of the infection.
In summary, this study shows that F. alocis is able to cause a local and systemic infection which results primarily in a rapid neutrophil infiltration to the site of infection, accompanied by a significant increase of proinflammatory cytokines. By 72 h, the local chamber infection is resolved and F. alocis disseminates into distal organs, resulting in neutrophil recruitment in the lung and spleen and a marked tubular epithelial injury in the kidneys. The tubular kidney cells were able to regenerate, and by 7 days postinfection, the kidneys and the rest of the organs had no detectable bacteria or tissue injury. The implications of this work, along with that of other recent studies (16, 18, 47), begin to establish the pathogenic credentials of F. alocis. As antimicrobial therapeutic strategies in periodontal disease are based to a large extent on traditional pathogens such as P. gingivalis, the emergence of virulent organisms such as F. alocis may lead to a reevaluation of treatment and management of the disease.
Supplementary Material
ACKNOWLEDGMENTS
The support of the NIH through DE022867, DE017921 (R.J.L.), DE023207 (J.P.), and HL087924 (S.M.U.) and the European Union through grant FP7-HEALTH-F3-2012-306029 “TRIGGER” (J.P.) is gratefully acknowledged.
Footnotes
Published ahead of print 30 December 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01434-13.
REFERENCES
- 1.Eke PI, Dye BA, Wei L, Thornton-Evans GO, Genco RJ. 2012. Prevalence of periodontitis in adults in the United States: 2009 and 2010. J. Dent. Res. 91:914–920. 10.1177/0022034512457373 [DOI] [PubMed] [Google Scholar]
- 2.Hajishengallis G, Lamont RJ. 2012. Beyond the red complex and into more complexity: the polymicrobial synergy and dysbiosis (PSD) model of periodontal disease etiology. Mol. Oral Microbiol. 27:409–419. 10.1111/j.2041-1014.2012.00663.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL., Jr 1998. Microbial complexes in subgingival plaque. J. Clin. Periodontol. 25:134–144. 10.1111/j.1600-051X.1998.tb02419.x [DOI] [PubMed] [Google Scholar]
- 4.Griffen AL, Beall CJ, Campbell JH, Firestone ND, Kumar PS, Yang ZK, Podar M, Leys EJ. 2012. Distinct and complex bacterial profiles in human periodontitis and health revealed by 16S pyrosequencing. ISME J. 6:1176–1185. 10.1038/ismej.2011.191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dewhirst FE, Chen T, Izard J, Paster BJ, Tanner AC, Yu WH, Lakshmanan A, Wade WG. 2010. The human oral microbiome. J. Bacteriol. 192:5002–5017. 10.1128/JB.00542-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kumar PS, Griffen AL, Moeschberger ML, Leys EJ. 2005. Identification of candidate periodontal pathogens and beneficial species by quantitative 16S clonal analysis. J. Clin. Microbiol. 43:3944–3955. 10.1128/JCM.43.8.3944-3955.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar PS, Leys EJ, Bryk JM, Martinez FJ, Moeschberger ML, Griffen AL. 2006. Changes in periodontal health status are associated with bacterial community shifts as assessed by quantitative 16S cloning and sequencing. J. Clin. Microbiol. 44:3665–3673. 10.1128/JCM.00317-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paster BJ, Boches SK, Galvin JL, Ericson RE, Lau CN, Levanos VA, Sahasrabudhe A, Dewhirst FE. 2001. Bacterial diversity in human subgingival plaque. J. Bacteriol. 183:3770–3783. 10.1128/JB.183.12.3770-3783.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dahlen G, Leonhardt A. 2006. A new checkerboard panel for testing bacterial markers in periodontal disease. Oral Microbiol. Immunol. 21:6–11. 10.1111/j.1399-302X.2005.00243.x [DOI] [PubMed] [Google Scholar]
- 10.Colombo AP, Boches SK, Cotton SL, Goodson JM, Kent R, Haffajee AD, Socransky SS, Hasturk H, Van Dyke TE, Dewhirst F, Paster BJ. 2009. Comparisons of subgingival microbial profiles of refractory periodontitis, severe periodontitis, and periodontal health using the human oral microbe identification microarray. J. Periodontol. 80:1421–1432. 10.1902/jop.2009.090185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abusleme L, Dupuy AK, Dutzan N, Silva N, Burleson JA, Strausbaugh LD, Gamonal J, Diaz PI. 2013. The subgingival microbiome in health and periodontitis and its relationship with community biomass and inflammation. ISME J. 7:1016–1025. 10.1038/ismej.2012.174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shaddox LM, Huang H, Lin T, Hou W, Harrison PL, Aukhil I, Walker CB, Klepac-Ceraj V, Paster BJ. 2012. Microbiological characterization in children with aggressive periodontitis. J. Dent. Res. 91:927–933. 10.1177/0022034512456039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Montagner F, Jacinto RC, Signoretti FG, Sanches PF, Gomes BP. 2012. Clustering behavior in microbial communities from acute endodontic infections. J. Endod. 38:158–162. 10.1016/j.joen.2011.09.029 [DOI] [PubMed] [Google Scholar]
- 14.Mansfield JM, Campbell JH, Bhandari AR, Jesionowski AM, Vickerman MM. 2012. Molecular analysis of 16S rRNA genes identifies potentially periodontal pathogenic bacteria and archaea in the plaque of partially erupted third molars. J. Oral Maxillofac. Surg. 70:1507–1514.e1-6. 10.1016/j.joms.2011.09.049 [DOI] [PubMed] [Google Scholar]
- 15.Wang Q, Wright CJ, Dingming H, Uriarte SM, Lamont RJ. 2013. Oral community interactions of Filifactor alocis in vitro. PLoS One 8:e76271. 10.1371/journal.pone.0076271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aruni AW, Roy F, Fletcher HM. 2011. Filifactor alocis has virulence attributes that can enhance its persistence under oxidative stress conditions and mediate invasion of epithelial cells by porphyromonas gingivalis. Infect. Immun. 79:3872–3886. 10.1128/IAI.05631-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maiden MF, Tanner A, Macuch PJ. 1996. Rapid characterization of periodontal bacterial isolates by using fluorogenic substrate tests. J. Clin. Microbiol. 34:376–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moffatt CE, Whitmore SE, Griffen AL, Leys EJ, Lamont RJ. 2011. Filifactor alocis interactions with gingival epithelial cells. Mol. Oral Microbiol. 26:365–373. 10.1111/j.2041-1014.2011.00624.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Graves DT, Fine D, Teng YT, Van Dyke TE, Hajishengallis G. 2008. The use of rodent models to investigate host-bacteria interactions related to periodontal diseases. J. Clin. Periodontol. 35:89–105. 10.1111/j.1600-051X.2007.01172.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Graves DT, Kang J, Andriankaja O, Wada K, Rossa C., Jr 2012. Animal models to study host-bacteria interactions involved in periodontitis. Front. Oral Biol. 15:117–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Genco CA, Cutler CW, Kapczynski D, Maloney K, Arnold RR. 1991. A novel mouse model to study the virulence of and host response to Porphyromonas (Bacteroides) gingivalis. Infect. Immun. 59:1255–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilensky A, Polak D, Awawdi S, Halabi A, Shapira L, Houri-Haddad Y. 2009. Strain-dependent activation of the mouse immune response is correlated with Porphyromonas gingivalis-induced experimental periodontitis. J. Clin. Periodontol. 36:915–921. 10.1111/j.1600-051X.2009.01464.x [DOI] [PubMed] [Google Scholar]
- 23.Ammann TW, Bostanci N, Belibasakis GN, Thurnheer T. 2013. Validation of a quantitative real-time PCR assay and comparison with fluorescence microscopy and selective agar plate counting for species-specific quantification of an in vitro subgingival biofilm model. J. Periodont. Res. 48:517–526. 10.1111/jre.12034 [DOI] [PubMed] [Google Scholar]
- 24.Siqueira JF, Jr, Rocas IN. 2004. Simultaneous detection of Dialister pneumosintes and Filifactor alocis in endodontic infections by 16S rDNA-directed multiplex PCR. J. Endod. 30:851–854. 10.1097/01.DON.0000132300.13023.5D [DOI] [PubMed] [Google Scholar]
- 25.Metzger Z, Lin YY, Dimeo F, Ambrose WW, Trope M, Arnold RR. 2009. Synergistic pathogenicity of Porphyromonas gingivalis and Fusobacterium nucleatum in the mouse subcutaneous chamber model. J. Endod. 35:86–94. 10.1016/j.joen.2008.10.015 [DOI] [PubMed] [Google Scholar]
- 26.Gosling PT, Gemmell E, Carter CL, Bird PS, Seymour GJ. 2005. Immunohistological analysis of Tannerella forsythia-induced lesions in a murine model. Oral Microbiol. Immunol. 20:25–30. 10.1111/j.1399-302X.2004.00188.x [DOI] [PubMed] [Google Scholar]
- 27.Oppermann RV, Weidlich P, Musskopf ML. 2012. Periodontal disease and systemic complications. Braz. Oral Res. 26(Suppl 1):39–47 [DOI] [PubMed] [Google Scholar]
- 28.Kuboniwa M, Lamont RJ. 2010. Subgingival biofilm formation. Periodontol. 2000 52:38–52. 10.1111/j.1600-0757.2009.00311.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Curtis MA, Zenobia C, Darveau RP. 2011. The relationship of the oral microbiotia to periodontal health and disease. Cell Host Microbe 10:302–306. 10.1016/j.chom.2011.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, McIntosh ML, Alsam A, Kirkwood KL, Lambris JD, Darveau RP, Curtis MA. 2011. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe 10:497–506. 10.1016/j.chom.2011.10.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vallabhapurapu S, Karin M. 2009. Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 27:693–733. 10.1146/annurev.immunol.021908.132641 [DOI] [PubMed] [Google Scholar]
- 32.Preshaw PM, Taylor JJ. 2011. How has research into cytokine interactions and their role in driving immune responses impacted our understanding of periodontitis? J. Clin. Periodontol. 38(Suppl 11):60–84. 10.1111/j.1600-051X.2010.01671.x [DOI] [PubMed] [Google Scholar]
- 33.Birkedal-Hansen H. 1993. Role of cytokines and inflammatory mediators in tissue destruction. J. Periodontal Res. 28:500–510. 10.1111/j.1600-0765.1993.tb02113.x [DOI] [PubMed] [Google Scholar]
- 34.Graves DT, Cochran D. 2003. The contribution of interleukin-1 and tumor necrosis factor to periodontal tissue destruction. J. Periodontol. 74:391–401. 10.1902/jop.2003.74.3.391 [DOI] [PubMed] [Google Scholar]
- 35.Howells GL. 1995. Cytokine networks in destructive periodontal disease. Oral Dis. 1:266–270 [DOI] [PubMed] [Google Scholar]
- 36.Okada H, Murakami S. 1998. Cytokine expression in periodontal health and disease. Crit. Rev. Oral Biol. Med. 9:248–266. 10.1177/10454411980090030101 [DOI] [PubMed] [Google Scholar]
- 37.Tonetti MS, Cortellini D, Lang NP. 1998. In situ detection of apoptosis at sites of chronic bacterially induced inflammation in human gingiva. Infect. Immun. 66:5190–5195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vitkov L, Krautgartner WD, Hannig M. 2005. Bacterial internalization in periodontitis. Oral Microbiol. Immunol. 20:317–321. 10.1111/j.1399-302X.2005.00233.x [DOI] [PubMed] [Google Scholar]
- 39.John B, Hunter CA. 2008. Immunology. Neutrophil soldiers or Trojan horses? Science 321:917–918 [DOI] [PubMed] [Google Scholar]
- 40.Liu PJ, Chen YS, Lin HH, Ni WF, Hsieh TH, Chen HT, Chen YL. 2013. Induction of mouse melioidosis with meningitis by CD11b(+) phagocytic cells harboring intracellular B. pseudomallei as a Trojan horse. PLoS Negl. Trop. Dis. 7:e2363. 10.1371/journal.pntd.0002363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lowe DM, Redford PS, Wilkinson RJ, O'Garra A, Martineau AR. 2012. Neutrophils in tuberculosis: friend or foe? Trends Immunol. 33:14–25. 10.1016/j.it.2011.10.003 [DOI] [PubMed] [Google Scholar]
- 42.Thwaites GE, Gant V. 2011. Are bloodstream leukocytes Trojan horses for the metastasis of Staphylococcus aureus? Nat. Rev. Microbiol. 9:215–222. 10.1038/nrmicro2508 [DOI] [PubMed] [Google Scholar]
- 43.Kolaczkowska E, Kubes P. 2013. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 13:159–175. 10.1038/nri3399 [DOI] [PubMed] [Google Scholar]
- 44.Cerutti A, Cols M, Puga I. 2013. Marginal zone B cells: virtues of innate-like antibody-producing lymphocytes. Nat. Rev. Immunol. 13:118–132. 10.1038/nri3383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cerutti A, Puga I, Magri G. 2013. The B cell helper side of neutrophils. J. Leukoc. Biol. 94:1–6. 10.1189/jlb.0113024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pan H, Ma Y, Wang D, Wang J, Jiang H, Pan S, Zhao B, Wu Y, Xu D, Sun X, Liu L, Xu Z. 2013. Effect of IFN-alpha on KC and LIX expression: role of STAT1 and its effect on neutrophil recruitment to the spleen after lipopolysaccharide stimulation. Mol. Immunol. 56:12–22. 10.1016/j.molimm.2013.04.001 [DOI] [PubMed] [Google Scholar]
- 47.Aruni AW, Roy F, Sandberg L, Fletcher HM. 2012. Proteome variation among Filifactor alocis strains. Proteomics 12:3343–3364. 10.1002/pmic.201200211 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.