

Abstract
A chronic infection with the parasite Toxoplasma gondii has previously been shown to protect mice against subsequent viral, bacterial, or protozoal infections. Here we have shown that a chronic T. gondii infection can prevent Plasmodium berghei ANKA-induced experimental cerebral malaria (ECM) in C57BL/6 mice. Treatment with soluble T. gondii antigens (STAg) reduced parasite sequestration and T cell infiltration in the brains of P. berghei-infected mice. Administration of STAg also preserved blood-brain barrier function, reduced ECM symptoms, and significantly decreased mortality. STAg treatment 24 h post-P. berghei infection led to a rapid increase in serum levels of interleukin 12 (IL-12) and gamma interferon (IFN-γ). By 5 days after P. berghei infection, STAg-treated mice had reduced IFN-γ levels compared to those of mock-treated mice, suggesting that reductions in IFN-γ at the time of ECM onset protected against lethality. Using IL-10- and IL-12βR-deficient mice, we found that STAg-induced protection from ECM is IL-10 independent but IL-12 dependent. Treatment of P. berghei-infected mice with recombinant IL-12 significantly decreased parasitemia and mortality. These data suggest that IL-12, either induced by STAg or injected as a recombinant protein, mediates protection from ECM-associated pathology potentially through early induction of IFN-γ and reduction in parasitemia. These results highlight the importance of early IL-12 induction in protection against ECM.
INTRODUCTION
Cerebral malaria (CM) is a fatal neurological complication that can arise during Plasmodium falciparum infection (1). Hallmarks of P. falciparum-induced CM that occur in the central nervous system include sequestration of parasitized erythrocytes, leukocytes, and platelets at the blood-brain barrier (BBB) (2–5). Even with current antimalaria drug treatment, progression to fatal CM remains high (2–7). Patients that survive CM infections can develop subsequent neurological complications (8, 9), highlighting the need for additional treatments. Because the exact contributions of immune and parasitic events to CM cannot be studied in humans (1, 10), relevant animal models are key for developing novel therapies.
The murine experimental cerebral malaria (ECM) model induced by Plasmodium berghei ANKA has many similarities to human CM, which include infected red blood cell (RBC) (iRBC) sequestration at the BBB, vascular leakage, and neurological symptoms (10–12). In addition, the ECM model has allowed the role of the immune response in ECM to be examined and dissected. Removal of immune cells, such as T or NK cells, or the cytokines gamma interferon (IFN-γ) and lymphotoxin alpha (LTα) prevents P. berghei ANKA-induced ECM (13–21). Understanding the immune response in ECM has allowed the investigation of potential treatments via regulation of innate immunity (22–27). Additionally, investigating the immune response during coinfection, either with other Plasmodium spp. (28, 29), helminths (30–33), or LP-BM5, the murine leukemia virus that induces murine AIDS (30, 34), has allowed the identification of immune components necessary for protection from ECM.
Toxoplasma gondii is an obligate intracellular apicomplexan parasite that infects any warm-blooded animal. The asexual life cycle consists of an acute systemic phase of disease with fast-replicating tachyzoites that transitions into a life-long chronic infection of bradyzoite cysts located primarily in striated muscle and brain tissue (35). Immune suppression of the host leads to bradyzoite cyst activation and reversion back to the tachyzoite stage of replication (36). T. gondii induces a robust Th1 immune response and stimulates innate Toll-like receptors, which leads to the production of interleukin 12 (IL-12), gamma interferon (IFN-γ), and a cytotoxic T cell response (37, 38). The induction of a Th1 immune response does not require live parasites (39–41). T. gondii infection also induces the proinflammatory cytokines IL-8 and IL-12, which play a major role in clearing the tachyzoite stage and maintaining the chronic stage (42–44). The anti-inflammatory cytokine IL-10 plays a critical role in limiting inflammation during T. gondii infection. Deletion of IL-10 results in enhanced mouse morbidity compared to that for control mice, and IL-10-dependent morbidity is reduced by IFN-γ and T cell depletion (45–47).
Animals with a chronic infection of the parasite Toxoplasma gondii can survive subsequent lethal challenges with bacteria, protozoa, or viruses (48–52). In addition, T. gondii chronic infection can reduce the number of Plasmodium yoelii blood-stage parasites (53) and prolong the survival of mice infected with P. berghei ANKA (54), suggesting a potential protective role during Plasmodium-induced disease. Furthermore, immunization with frozen and thawed T. gondii antigen or lysates induced an adaptive immune response that was protective against future P. berghei ANKA infections (55, 56). For this study, we examined how the immune response elicited by either chronic T. gondii infection or treatment with soluble T. gondii antigen (STAg) could inhibit P. berghei ANKA-induced ECM. We determined that IL-12 signaling plays a central role in the reduction of P. berghei parasitemia and the prevention of P. berghei-induced ECM.
MATERIALS AND METHODS
T. gondii cell culture and STAg preparation.
T. gondii parasites were serially passaged on human foreskin fibroblasts (HFF) in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum, 1% penicillin-streptomycin, and 2 mM l-glutamine. STAg was generated as previously described (57), using the PruΔHPT strain (58) with the following alterations. Parasites were egressed from HFF cells by replacing the DMEM with Hanks balanced salt solution (Thermo Scientific) containing 1 μM calcium ionophore (Sigma) for 6 min at 37°C. Freshly egressed parasites were collected by centrifugation at 550 × g for 10 min and washed twice with Dulbecco's phosphate-buffered saline (DPBS) (128 mM NaCl, 2.7 mM KCl, 8 mM NaH2PO4, and 1 mM KH2PO4). Parasites were suspended at a concentration of 4 × 108 parasites/ml in DPBS and sonicated. The protein mix was centrifuged at 100,000 × g for 45 min, and the soluble fraction was collected, aliquoted, and stored at −80°C.
ECM model and treatments.
P. berghei ANKA-induced ECM and the immune components involved in the induction of ECM are well documented. Thus, P. berghei ANKA was the preferred Plasmodium species/strain for our ECM studies. P. berghei ANKA was recovered from blood glycerol stocks in BALB/c mice, which do not develop ECM. C57BL/6J mice are highly susceptible to P. berghei ANKA-induced ECM, and numerous immune deletion mouse lines are available in this background. To examine ECM, experimental infections were initiated in C57BL/6J mice using blood from P. berghei ANKA-infected BALB/c mice. BALB/c and C57BL/6J mice were purchased from NCI (Frederick, MD). IL-12β2R−/− (B6.129S1-Il12b2tm1Jm/J) and IL-10−/− (B6.129P2-Il10tm1Cgn/J) mice were purchased from Jackson Laboratory (Bar Harbor, ME) and bred at the University of Wisconsin—Madison. All deletion strains were in a C57BL/6J background and backcrossed at least 10 generations. All animals use was approved by and in accordance with the policies of the Institutional Animal Care and Use Committee at the University of Wisconsin—Madison.
Percent parasitemia was determined by counting 500 to 1,000 RBC from Giemsa-stained thin blood smears and defined as the ratio of iRBC to total RBC. The universally lethal P. berghei ANKA clone 4 (59) was used in all experiments and was maintained as cryopreserved stabilates of mouse blood at ∼10% parasitemia. Infections were initiated by intraperitoneal (i.p.) injection with 50 μl of stabilate (∼106 iRBC) into BALB/c mice. At ∼10% parasitemia, blood was collected, and the ECM model was initiated by infecting age-matched C57BL/6J, IL-12β2R−/−, or IL-10−/− mice i.p. with 1 × 106 iRBC. Doses lower than 1 × 106 iRBC produced inconsistent ECM results. Experiments with the IL-12β2R−/− mice used 6-week-old mice, while all other experiments used ∼8-week-old mice. For STAg treatment, equivalent volumes of PBS or STAg were intravenously (i.v.) injected at the indicated times. Recombinant murine IL-12 (p70) derived from CHO cells had endotoxin levels of <1 endotoxin unit (EU)/μg or <0.1 ng/μg of protein (PeproTech, Rocky Hill, NJ) and was administered i.v. at the indicated amount 1 day after P. berghei ANKA infection. Mice were scored for ECM symptoms using a rapid behavioral scale from 1 to 18 and euthanized at scores of ≤5 (60). Lower scores were given for a loss of coordination (gait and balance) exploration (motor performance), strength and tone (body position and limb strength), reflexes/self-preservation (touch escape, pinna reflex, and toe pinch), and hygiene (grooming).
Assessment of BBB permeability.
Mice were administered 0.1 ml of 10 mg/ml Evans blue dye (MP Biomedicals) dissolved in DPBS 7 days after P. berghei ANKA infection or when the percent parasitemia was >8%. After 1 h, the mice were sacrificed and perfused with 0.9% NaCl saline, and tissues were removed and weighed. Evans blue was extracted by immersing the tissue in formamide and quantified by measuring absorbance of the formamide at 610 nm (61).
qPCR.
To measure the amount of P. berghei ANKA DNA associated with the brain, mice were sacrificed 7 days after P. berghei infection and perfused with 0.9% saline to remove P. berghei genomic DNA present in the circulatory system. Brain tissues were collected and minced, and genomic DNA was purified using the High Pure PCR template purification kit (Roche) from a portion of the minced brain. Quantitative PCR (qPCR) was performed using primer-probe sets specific to mouse albumin and P. berghei ANKA 18S rRNA and genomic DNA sequences as previously described (62). The previously published P. falciparum primers were modified to detect P. berghei 18S rRNA. The primer-probe sets used were as follows: P. berghei ANKA 18S primers, 5′-TCAACTACGAGCGTTTTAACTGCAAC-3′ and 5′-TTGGAATGATGGGAACTTAAAATCTTCCC-3′; probe, 5′-6-carboxyfluorescein (FAM) TGCCAGCAG ZEN CCGCGGTAATTC Zen Iowa BlackFQ (IBKFQ). Murine albumin primers were as follows: 5′-CAATCCTGAACCGTGTGTGTCT-3′ and 5′-TTCATCAACTGTCAGAGCAGAGAAG-3′; probe, 5′-FAM CCAAGTGCT ZEN GTAGTGGATCCCTGGTGG IBKFQ. Probes were labeled with the 6-FAM fluorophore and IBKFQ double quencher (Integrated DNA Technologies). Target genes were amplified using Absolute Blue QPCR mix (Thermo Scientific) using iCycler real-time PCR (Bio-Rad). Threshold cycle (CT) values were determined automatically by the Bio-Rad software. The relative levels of P. berghei genomic 18S rRNA (PbA18SCT) were normalized to mouse albumin levels (murine albuminCT) using the following equation: (Emurine albuminCT)/(EPbA18SCT), where E is efficiency, m is the slope of the dilution series/standard curve, and E = 10(−1/m) (63, 64). The efficiency of amplification was calculated to be 100% for both primer sets. The P. berghei ANKA 18S primers were not able to detect murine genomic DNA (gDNA). In addition, the amplification efficiency of the P. berghei ANKA 18S region was not altered when murine gDNA was included in efficiency curves. The limit of detection was approximately 20 pg of P. berghei purified gDNA in water or murine gDNA-spiked samples.
Cell purification and flow cytometry.
Brain mononuclear cells (BMNC) were isolated as previously described (47). Mice were perfused with 0.9% saline, and brains were collected, cut into small pieces, and further disrupted by passing through an 18-gauge needle. Brain tissues were digested with 25 μg/ml collagenase/dispase (Roche) and 750 μg/ml DNase I (Roche) for 45 min at 37°C. Cells were washed and passed through a 70-μm cell strainer (Falcon). BMNC were purified over a 30% to 60% discontinuous Percoll gradient centrifuged at 1,000 × g for 25 min at room temperature. Cells were collected from the interface, washed, and enumerated by trypan blue stain for viability. For flow cytometry, cells were washed with fluorescence-activated cell sorting (FACS) buffer (PBS [pH 7.4], 0.2% bovine serum albumin [BSA], and 1 mM EDTA) and incubated for 15 min with Fc block (0.1 μg/ml CD16/32; ebiosciences) prior to incubation with conjugated antibody. After antibody incubation, cells were fixed with 2% formaldehyde for 10 min. Cells were stained with CD3-eFluor 450, CD4-Alexa Fluor 700, and CD8 allophycocyanin (APC)-eFluor 780 (ebioscience) to monitor T cell accumulation at the brain. Events were collected on a BD LSR II flow cytometer (BD). Compensation and analyses were performed using the FlowJo software program (TreeStar).
Serum cytokine quantification.
Serum samples were obtained by tail bleed at the given time points. Cytokines were quantified from 12.5 μl of serum using a mouse inflammation cytokine bead array (CBA) kit (BD Biosciences), which measures IL-12p70, IFN-γ, tumor necrosis factor alpha (TNF-α), monocyte chemoattractant protein 1 (MCP-1), IL-6, and IL-10 in the same serum sample. Events were collected and gated using the BD LSR II flow cytometer and FACSDiva software program (BD Biosciences).
Statistical analysis.
A 2-way analysis of variance (ANOVA) analysis used to compare independent experimental repeat data sets to determine if significant variation between experiments existed and if the data could be merged. The dependent variables were parasitemia or time of death, and the independent variable were treatment, infection, and experiment. Kaplan-Meier survival curves were statistically compared using a Mantel-Cox log rank analysis. Log rank analysis P values was used to compare PBS-treated or untreated P. berghei ANKA-infected animals to coinfected animals or results with protein treatment. For single comparisons, significance was determined by the Mann-Whitney test. For multiple comparisons, data were compared by a Kruskal-Wallis test followed by Dunn's pairwise comparison. A P value of ≤0.05 was considered significant. All statistical analysis was performed in the Prism software program (GraphPad Software, Inc.).
RESULTS
Chronic T. gondii infection decreases P. berghei ANKA-induced morbidity and P. berghei parasitemia.
To determine if chronic T. gondii infection prevents P. berghei ANKA-induced disease, 7- to 8-week-old C57BL/6 mice were infected with 1,000 T. gondii parasites and allowed to established chronic infection for 28 days. T. gondii-infected or age-matched uninfected mice were then challenged with a lethal dose of P. berghei ANKA and monitored daily for ECM symptoms to determine if they needed to be euthanized. Ninety percent of the T. gondii-infected mice survived subsequent infection with P. berghei, whereas P. berghei challenge was uniformly lethal in mice not infected with T. gondii (Fig. 1A) (P < 0.01). Coinfection with T. gondii significantly reduced P. berghei parasitemia (Fig. 1B) (P < 0.05). Without T. gondii infection, P. berghei parasitemia reached ∼20% by the time the mice needed to be euthanized for ECM symptoms, whereas with chronic T. gondii infection, 95% of mice did not display ECM symptoms despite the fact that P. berghei parasitemia increased until it plateaued at ∼40%. These data suggest that T. gondii coinfection can reduce P. berghei ANKA parasitemia and prevent the onset of ECM.
FIG 1.
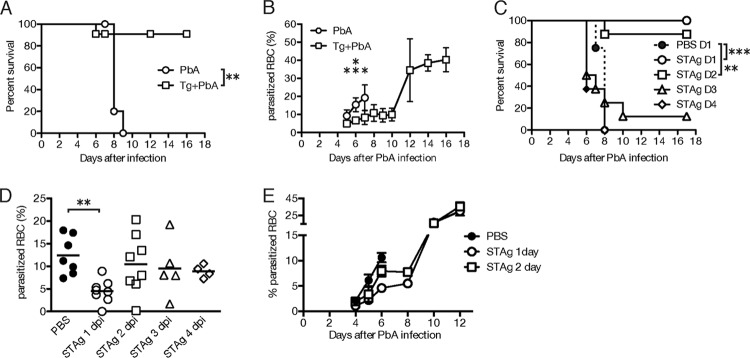
Chronic T. gondii infection or STAg prevents P. berghei ANKA (PbA)-induced ECM and lowers parasitemia. (A and B) Seven- or eight-week-old C57BL/6 mice inoculated with T. gondii (Tg+PbA) (n = 6) or medium (PbA) (n = 7) were challenged 28 days later with 1 × 106 P. berghei ANKA iRBC. Symptoms of ECM were monitored, and mice with scores ≤ 5 were euthanized (A). Parasitemia was determined from Giemsa-stained thin blood smears at various times postinfection (B). Data shown are from a representative experiment repeated twice. (C to E) Approximately 8-week-old C57BL/6 mice were inoculated with 1 × 106 iRBC and treated with PBS or STAg at days 1 (D1), 2 (D2), 3 (D3), or 4 (D4) post-P. berghei ANKA infection. The average data from two to five experiments are shown. (C) Symptoms of ECM were monitored, and mice with scores ≤ 5 were euthanized up to 17 days after P. berghei infection. (D and E) Thin blood smears were prepared at 6 days (D) or at the indicated days (E) after P. berghei infection. The average percentage of iRBC was determined after treatment with Giemsa stain. Survival significance was determined by Mantel-Cox log rank analysis comparing PBS or P. berghei ANKA alone to coinfection or STAg treatments. iRBC significance was determined with a Kruskal-Wallis test by comparing results for P. berghei ANKA alone or P. berghei ANKA plus PBS controls (PbA+PBS) to those for coinfection or STAg treatments (PbA/STAg). *, P < 0.05, **, P < 0.01; ***, P < 0.001.
To begin to elucidate the mechanism of T. gondii protection, we tested whether treatment with soluble T. gondii antigens, called STAg, could protect against P. berghei ANKA-induced ECM. We infected mice with P. berghei ANKA and administered STAg i.v. at various times post- P. berghei infection. Treatment of P. berghei-infected mice with STAg 1 day (P < 0.001 compared to results with PBS treatment) or 2 days (P < 0.01 compared to results with PBS treatment) after P. berghei infection resulted in ≥90% survival, but ECM was not prevented when mice were treated with STAg at 3 or 4 days post- P. berghei infection (Fig. 1C). STAg treatment 1 day after P. berghei infection significantly reduced parasitemia when it was measured 6 days after P. berghei infection (P < 0.01); however, parasitemia was not reduced when STAg treatment occurred 2 days after infection even though the mice were protected from ECM-induced disease (Fig. 1D). In addition, the STAg treatments on day 1 and to a lesser extent on day 2 reduced the level of parasitemia until 8 days after P. berghei infection (Fig. 1E), a time frame similar to that seen for P. berghei ANKA infection of mice with a chronic T. gondii infection (Fig. 1B). Treatment with a lysate of the host cells from which STAg was prepared did not prevent ECM or reduce parasitemia (data not shown). These data show that post- P. berghei infection, treatment with STAg can mimic T. gondii chronic infection and prevent P. berghei-induced ECM with or without reduction of P. berghei parasitemia.
STAg reduces P. berghei ANKA-induced vascular leakage and parasite sequestration in the brain.
Breakdown of the BBB and vascular leakage have been associated with CM in humans and ECM in mice (31, 65–67). To determine if STAg treatment could prevent this hallmark of P. berghei ANKA-induced ECM, we used Evans blue dye as an indicator of vascular leakage and BBB integrity (68). Mice were infected with P. berghei ANKA and treated with STAg 2 days postinfection to allow for similar parasitemia in infected mice. At 7 days post- P. berghei infection, PBS-treated mice showed vascular leakage of Evans blue into the brain tissue, whereas STAg-treated mice had reduced infiltration of dye into the brain, similar to results for naive mice (Fig. 2A). The reduced infiltration of dye was confirmed by quantifying Evans blue accumulation in brain tissue (Fig. 2B) (P < 0.05). As a control, we also measured vascular leakage into the mouse brain once their parasitemia was greater than 8% (Fig. 2C). Yet again, the accumulation of dye was significantly decreased by STAg treatment (P < 0.01 compared to results with PBS treatment). Histological examination of mouse brains detected vascular hemorrhage in both STAg- and PBS-treated mice, with a reduced hemorrhage size and frequency in brains of STAg-treated mice (Fig. 2D). The hemorrhage frequency and size were further reduced if mice were treated with STAg 1 day after infection (data not shown). These results suggest that STAg treatment of P. berghei ANKA-infected mice can preserve BBB integrity.
FIG 2.

STAg treatment of P. berghei ANKA (PbA)-infected mice maintains BBB integrity and reduces parasite sequestration. Approximately 8-week-old C57BL/6 mice were inoculated with 1 × 106 iRBC or PBS and then treated with PBS or STAg 2 days after infection. The percentage of iRBC was monitored after Giemsa stain treatment of thin blood smears. At 7 days postinfection (A, B, D, and E) or once parasitemia exceeded 8% (C), mice were injected with Evans blue dye (A to C and E). The mice were euthanized and perfused with saline, and the brains were collected, weighed, photographed, or processed for histology. (B and C) Evans blue was extracted from the tissue using formamide. The concentration of Evans blue per gram of brain tissue (μg of EB/g of brain) was determined by absorbance. Each symbol represents an individual mouse. (E) Brains from P. berghei ANKA-infected mice were collected, and genomic DNA was prepared. The numbers of P. berghei 18S and mouse albumin genomic DNA copies were determined by qPCR. The number of P. berghei copies was normalized to mouse albumin. Each symbol represents an individual mouse. Significance was determined by a Mann-Whitney test. *, P < 0.05. Shown are results from a representative experiment repeated two times.
Sequestration of iRBC and T cells in the brains of P. berghei ANKA-infected mice is required to induce ECM (15, 18, 20, 31, 65, 69). To determine if STAg treatment affected the ability of parasites to sequester in the brains of infected mice, the presence of P. berghei ANKA genomes in brain tissue was quantified by qPCR after intracardiac perfusion. Analysis of the brains from naive mice showed no P. berghei ANKA genomic DNA (data not shown). P. berghei genomic DNA was ∼4-fold less after STAg treatment than was seen for PBS-treated mice (Fig. 2E) (P < 0.01), even though the parasitemia was similar when the tissues were collected (data not shown). STAg treatment did not completely eliminate the sequestration of parasite genomes compared to that for naive animals, which suggests that STAg treatment reduces but does not eliminate the sequestration of iRBC in the brains of P. berghei ANKA-infected animals.
STAg reduces P. berghei ANKA-induced brain T cell localization and late levels of IFN-γ.
Sequestration of parasites in the brains of P. berghei ANKA-infected mice is enhanced by the presence of CD8+ and CD4+ T cells in the brain (31, 65, 70). Due to the reduction of parasites in the brains of STAg-treated mice, we hypothesized that STAg treatment would also reduce P. berghei-induced T cell accumulation in the brain. To this end, we monitored the numbers of CD4+ and CD8+ T cells in the brains by FACS analysis in naive mice, P. berghei-infected mice treated with STAg or PBS, or uninfected mice treated with STAg (Fig. 3A). Quantification of this FACS analysis showed that P. berghei ANKA infection increased the number of CD4+ and CD8+ T cells localized to the brain compared to results for naive animals or with STAg treatment alone, and STAg treatment of P. berghei-infected mice reduced brain-localized CD4+ and CD8+ T cells compared to results for PBS treatment (Fig. 3B and C). Although STAg treatment did not result in significant reductions in brain-localized T cells, the general reduction suggests that STAg treatment during P. berghei infection reduces T cell localization during P. berghei infection, which may in turn reduce damage to the brain vasculature.
FIG 3.

STAg reduces brain-localized T cells and late IFN-γ production. Approximately 8-week-old C57BL/6 mice were uninfected and treated with PBS (naive) or STAg (STAg) or inoculated with 1 × 106 P. berghei ANKA iRBC and treated with PBS (PbA/PBS) or STAg (PbA/STAg) 2 days post-P. berghei infection. At 6 days post-P. berghei infection, mice were euthanized and perfused, and brain tissue was collected. Brain mononuclear cells were isolated, and fluorescence-conjugated antibodies were used to detect CD3+ CD8+ or CD3+ CD4+ cells. (A) Representative CD4+ and CD8+ gates are shown from two experiments. The total number of brain CD8+ (B) or CD4+ (C) cells was determined. Each symbol represents an individual mouse. (D) Whole blood was collected 7 days after infection, and serum IFN-γ levels were quantitated by CBA (n = 6 to 7 for P. berghei-infected mice, and n = 4 for noninfected mice). Significance was determined by a Kruskal-Wallis test, but no comparisons were statistically significant.
The cytokine IFN-γ has been implicated in T cell-induced ECM pathology (19, 71, 72). Because mice deleted for the IFN-γ receptor chain (IFN-γ−/−) do not develop ECM (19, 71, 72), we measured serum cytokine levels of IFN-γ at 7 days post-P. berghei ANKA infection (5 days posttreatment), which is just prior to the onset of ECM symptoms. STAg-treated mice had lower levels of IFN-γ than PBS-treated animals (Fig. 3D). In combination with T cell reductions, these data suggest that STAg treatment may lower T cell localization by reducing late IFN-γ production, similar to results seen in IFN-γ−/− mice.
Analysis of early cytokine response.
STAg is known to induce a strong Th1 response that includes initial induction of interleukin 12 (IL-12), which in turn stimulates IFN-γ production (73, 74). To confirm that STAg treatment increased Th1 cytokine production and to understand how STAg treatment was affecting the early stages of P. berghei ANKA infection, we measured serum levels of IFN-γ, IL-12p70, IL-10, monocyte chemoattractant protein 1 (MCP-1), and IL-6 by CBA. Mice were infected with P. berghei ANKA and treated with STAg or PBS 1 day post-P. berghei infection, and serum cytokines were measured at 2 h, 14 h, and 4 days after treatment. Two hours after STAg treatment, P. berghei-infected mice had increases in IL-12p70, MCP-1, and IL-6 compared to levels for P. berghei-infected mice treated with PBS (Fig. 4). By 14 h after STAg treatment, IFN-γ had also increased. In contrast, STAg increased serum levels of IL-10 and TNF-α by 14 h after infection, but this elevation was not seen in STAg-treated P. berghei-infected animals. By 4 days after treatment, STAg treatment reduced the level of IFN-γ compared that for to PBS-treated animals, similar to the results day 7 after infection. However, the levels of IL-12, IL-10, IL-6, MCP-1, or TNF-α were not different. These results suggest that STAg treatment of P. berghei-infected mice stimulates a rapid Th1 response that can reduce parasitemia and subsequent ECM.
FIG 4.

STAg treatment increases serum cytokine levels. Approximately 8-week-old C57BL/6 mice were uninfected or inoculated with 1 × 106 P. berghei ANKA iRBC and treated with PBS or STAg 1 day post-P. berghei infection. Blood was collected at 2 or 14 h after treatment, and serum cytokine levels of IL-12p70, IFN-γ, MCP-1, IL-6, TNF-α, and IL-10 were quantitated by CBA (n = 3 per time point). A representative of two experiments is shown. Significance was determined by a Kruskal-Wallis test, but no comparisons were statistically significant.
STAg-induced protection requires IL-12 but not IL-10.
IL-10 suppresses a Th1 response and has been shown in multiple studies to play a major role in preventing ECM (28, 33, 34, 75). To investigate whether STAg-induced protection against ECM requires IL-10, we infected IL-10−/− mice with P. berghei ANKA and treated mice with STAg after 24 h. STAg treatment was just as effective in wild-type (wt) and IL-10−/− mice, as evidenced by similar percent survivals (Fig. 5A) (P < 0.001 when results for PBS were compared with those for STAg treatment within each genotype) and parasitemias (Fig. 5B) (P < 0.01), suggesting that IL-10 is not required for STAg-induced protection.
FIG 5.

IL-12 but not IL-10 contributes to STAg-induced protection. Approximately 8-week-old C57BL/6 wt or IL-10−/− (A and B) or 6-week-old wt or IL-12βR−/− (C and D) mice were inoculated with 1 × 106 iRBCs and treated with PBS or STAg 24 h later. (E and F) wt C57BL/6 mice were inoculated with 1 × 106 iRBCs and treated with increasing amounts of recombinant purified IL-12p70 at 24 h postinfection. Symptoms of ECM were monitored, and mice with scores ≤ 5 were euthanized (A, C, and E). Thin blood smears were prepared 6 days after P. berghei ANKA infection, and the percentage of iRBC was determined after treatment with Giemsa stain (B, D, and F). Survival significance was determined by Mantel-Cox log rank analysis comparing PBS or P. berghei alone to coinfection or STAg treatments. iRBC significance was determined by a Kruskal-Wallis test. *, P < 0.05; **, P < 0.01; ***, P < 0.001. Two independent experiments are shown in panels A to C, and a representative experiment is shown for panels D to F.
Analysis of the STAg-induced cytokine response showed a substantial induction of IL-12 (Fig. 4). To understand the contribution of IL-12 to STAg-induced protection from ECM, we infected IL-12βR−/− mice with P. berghei ANKA and monitored mice for ECM symptoms after STAg or PBS treatment. In contrast to results for the IL-10−/− mice, STAg treatment did not prevent P. berghei-induced ECM (Fig. 5C) or reduce and delay parasitemia (Fig. 5D; see also Fig. S1A in the supplemental material) in IL-12β2R−/− mice. Thus, STAg-induced protection from ECM requires IL-12. To test whether IL-12 alone was sufficient to prevent P. berghei ANKA-induced ECM, we treated with increasing amounts of recombinant murine IL-12p70 24 h after P. berghei infection and monitored for ECM symptoms. Murine IL-12p70 is a 75-kDa heterodimeric glycoprotein consisting of disulfide-linked 35-kDa and 40-kDa subunits. While mice that received less than 0.1 μg of recombinant IL-12p70 succumbed to infection at rates similar to those for PBS-treated animals, 50% of mice that received 0.1 μg and 100% of mice that received 1 μg (P < 0.01 compared to results for PBS treatment) of recombinant IL-12p70 survived P. berghei infection (Fig. 5E). Treatments with 1 μg and 0.1 μg of IL-12 showed significant reductions in parasitemia at 6 days post-P. berghei ANKA infection compared to that for PBS-treated mice (Fig. 5F). In addition, these two doses of IL-12 delayed the accumulation of parasitemia, similar to results seen during stag treatment (see Fig. S1B in the supplemental material). Contaminating endotoxins, including lipopolysaccharide (LPS), were below the limit of detection of <0.1 ng/μg of protein, or <0.01 ng endotoxin per treatment as reported by the manufacturer. These results demonstrate that exogenous IL-12p70 is sufficient to prevent P. berghei ANKA-induced ECM.
To further investigate the signaling cascade induced by IL-12 treatment of a P. berghei ANKA infection and how it compares to results with STAg treatment, we treated P. berghei ANKA-infected mice 1 day after infection with STAg or 1 μg IL-12 and compared the levels of serum cytokines at 2 h, 14 h, and 5 days after treatment (Fig. 6). Importantly, STAg induced concentrations of IL-12 in serum similar to those with IL-12 treatment alone, which suggests that the 1-μg dose was biologically relevant. STAg and IL-12 treatments both induced IFN-γ and MCP-1, which suggests that the induction of IL-12 by STAg induces the downstream production of IFN-γ and MCP-1 and that these cytokines can contribute to STAg-induced protection.
FIG 6.

STAg treatment increases serum cytokine levels. Approximately 8-week-old C57BL/6 mice were uninfected or inoculated with 1 × 106 P. berghei ANKA iRBC and treated with PBS or STAg 1 day post-P. berghei infection. Blood was collected at 2 h, 14 h, or 5 days after treatment, and serum cytokine levels of IL-12p70, IFN-γ, MCP-1, IL-6, TNF-α, and IL-10 were quantitated by CBA (n = 2 to 4 per time point). Significance was determined by a Kruskal-Wallis test followed by a Dunn's pairwise comparison. *, P < 0.05; **, P < 0.01.
DISCUSSION
These studies have shown that T. gondii chronic infection and the subset of soluble T. gondii antigens called STAg can lessen the disease induced by Plasmodium species in rodents. Specifically, a single STAg treatment protected mice against P. berghei ANKA-induced ECM symptoms by reducing parasite sequestration and T cell accumulation in the brains. Early induction of Th1 cytokines likely lead to decreases in overall parasite levels in the blood, as well as reductions in IFN-γ levels at the time ECM symptoms are normally induced. Previous studies have shown that STAg is a potent inducer of IL-12 responses in vivo (74). In this study, we showed that STAg treatment is not effective at preventing ECM symptoms in IL-12β2R−/− mice and that treatment with recombinant IL-12 alone was sufficient to reduce parasitemia and prevent ECM. These results stress the role of IL-12 in the development of ECM.
To determine how STAg prevented P. berghei ANKA-induced ECM, we evaluated the role of STAg in BBB permeability and discovered that the permeability induced by P. berghei infection was reduced by STAg treatment. Consistent with this result, we found that STAg treatment reduced the level of P. berghei genomes in the brain after perfusion, suggesting that STAg treatment reduced parasite sequestration. This reduction in parasite sequestration is especially relevant because mice treated with STAg on day 2 postinfection do not show a reduction in parasitemia compared to controls (Fig. 1D). It is possible that STAg directly influences parasite cytoadherence. Binding of P. berghei iRBCs to brain vascular endothelial cells is dependent on vascular cell adhesion protein 1 (VCAM-1) (76). Our future studies will examine the effect of STAg on VCAM-1 levels in brain vascular endothelial cells and if STAg blocks cytoadherence of P. berghei ANKA.
While STAg treatment did not significantly reduce T cell localization in the brain, there is a trend toward reduction in brain-localized T cells. Perforin and granzyme B produced by CD8+ T cells are major contributors to the disruption of BBB integrity during P. berghei ANKA infection (18, 77). The reduced permeability observed upon STAg treatment may result from the limited presence of these enzymes due to the STAg-dependent decrease in the numbers of CD8+ T cells in the brain. The reduction of localized T cells also correlated with reduced serum IFN-γ levels. IFN-γ contributes to the localization of T cells to the brain during P. berghei ANKA infection (71, 78, 79). Thus, reductions in IFN-γ levels following STAg treatment may contribute to the prevention of ECM symptoms by reducing the number of T cells localized to the brain during P. berghei infection.
Knowing that STAg is a strong inducer of IL-12 (74), we investigated the role of IL-12 in ECM. STAg treatment of P. berghei ANKA-infected IL-12βR−/− mice did not prevent P. berghei ANKA-induced ECM. While the IL-12 receptor has been reported to be required for the induction of ECM (13), we found that IL-12β2R−/− mice are susceptible to P. berghei ANKA-induced ECM at 6 weeks of age. When infected with P. berghei after 7 weeks of age, IL-12βR−/− mice were resistant to ECM induction. Why the IL-12βR−/− mice become resistant after 7 weeks of age is unclear, because other IL-12 components and receptors can be deleted with an ECM response still being produced (13). Using 6-week-old IL-12βR−/− mice, we showed that STAg-induced protection from ECM is dependent on IL-12 signaling. Treatment of P. berghei ANKA-infected mice with recombinant IL-12 prevented ECM symptoms, lethality, and reduced parasitemia. STAg and IL-12 treatment alone induced similar concentrations of IL-12 in serum, suggesting that the 1-μg dose determined to be 100% effective (Fig. 5E) was appropriate. Along with IFN-γ, both STAg and IL-12 treatments induced early MCP-1 production, suggesting that these cytokines likely contribute to STAg-induced protection. Other cytokines, such as TNF-α, are highly induced only in response to IL-12 treatments and thus are unlikely to be essential for STAg-induced prevention of ECM symptoms and reduction of parasitemia.
While the importance of IL-12 for the prevention of P. berghei ANKA-induced ECM is clear, the precise mechanism of action is not fully understood. IL-12 induces downstream cytokines that mediate the immune response during Plasmodium infection. IL-12 induces IL-10 production (80), and IL-10 plays a major role in preventing ECM (28, 33, 34, 76). Due to the immunosuppressive nature of IL-10, the induction of IL-10 could have prevented the localization of T cells to the brain. However, STAg treatment did not change serum levels of IL-10 at any of the time points we examined, and STAg treatment was still protective in IL-10−/− mice. Thus, IL-10 was not a major contributor to STAg-induced protection.
Early induction of IL-12 stimulates IFN-γ production (73, 74), and IFN-γ was observed in response to STAg treatment (Fig. 4 and 6). IL-12-induced IFN-γ plays a major role in protecting against blood-stage parasite replication during Plasmodium chabaudi infection as well as that with human malaria parasites (81–83). We tried to prevent ECM with a wide dose range of exogenous IFN-γ treatments (0.001 to 1 μg/mouse) without success (data not shown). IFN-γ levels were higher in STAg- than in PBS-treated mice 14 h after treatment; however, this trend reverses by 4 days posttreatment, when IFN-γ levels were higher in PBS- than in STAg-treated mice (Fig. 3D and 4). Furthermore, we see that the Th1 cytokine response remains low in P. berghei ANKA-infected PBS-treated mice for at least the first 38 h postinfection (Fig. 4). The correct source and/or tissue concentration of IFN-γ may be critical in order to reduce parasitemia and prevent ECM, and this may explain why i.v. inoculation of IFN-γ was not successful. In contrast, bloodstream inoculation of IL-12 is able to recapitulate the systemic response induced by STAg. IL-12 may induce an additional factor, such as the chemokine MCP-1, which may be necessary either alone or in combination with IFN-γ to protect against ECM by decreasing parasitemia or directing immune cell localization. This factor may not be induced by exogenous IFN-γ and thus may account for the lack of protection with IFN-γ treatment.
STAg has been shown to induce paralysis of IL-12 production from dendritic cells (DCs), which protects animals from lethal parasite-induced immunopathology (84). In isolated spleen cells, IL-12 production in response to STAg is transient, with peak production 6 to 12 h poststimulation and a return to baseline levels by 24 h (74). We see similar results for IL-12 stimulation in mice after STAg treatment with or without P. berghei ANKA infection (Fig. 4 and 6). STAg also induces systemic paralysis of IL-12 production from DCs in vivo, since stimulation with a second dose of STAg 24 h after the first does not induce IL-12 production from DCs (84). DC paralysis upon STAg reinjection has been connected to the functional downregulation of the chemokine receptor CCR5 by endogenously produced lipoxin A4 (85, 86). Because mice deficient in the enzyme responsible for lipoxin A4 synthesis (5-LO−/−) are unable to induce DC paralysis, future studies of the IL-12 signaling cascades during P. berghei ANKA infection will examine STAg protection in 5-LO−/− mice. Likewise, because STAg contains cyclophilin 18, a chemokine mimic that signals through CCR5 (87), future studies will examine the ability of purified cyclophilin 18 to protect against P. berghei-induced ECM. Together, these studies will further elucidate the role of IL-12 in STAg-mediated protection from ECM.
Supplementary Material
ACKNOWLEDGMENTS
We sincerely thank Bill Weidanz for the P. berghei ANKA strain used in these studies and Chris Hunter and Kasturi Haldar for training in brain-specific flow cytometry and the ECM rapid behavioral screen, respectively.
This research was supported by the American Heart Association, 0840059N (to L.J.K.) and 5 T32 HL007899 (to E.W.S.), and a Burroughs Wellcome travel grant (to E.W.S.).
Footnotes
Published ahead of print 6 January 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01259-13.
REFERENCES
- 1.Anonymous. 2000. Severe falciparum malaria. World Health Organization, Communicable Diseases Cluster. Trans. R. Soc. Trop. Med. Hyg. 94(Suppl 1):S1–S90 [PubMed] [Google Scholar]
- 2.Medana IM, Turner GDH. 2006. Human cerebral malaria and the blood-brain barrier. Int. J. Parasitol. 36:555–568. 10.1016/j.ijpara.2006.02.004 [DOI] [PubMed] [Google Scholar]
- 3.Silamut K, Phu NH, Whitty C, Turner GD, Louwrier K, Mai NT, Simpson JA, Hien TT, White NJ. 1999. A quantitative analysis of the microvascular sequestration of malaria parasites in the human brain. Am. J. Pathol. 155:395–410. 10.1016/S0002-9440(10)65136-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clark IA, Awburn MM, Whitten RO, Harper CG, Liomba NG, Molyneux ME, Taylor TE. 2003. Tissue distribution of migration inhibitory factor and inducible nitric oxide synthase in falciparum malaria and sepsis in African children. Malar. J. 2:6. 10.1186/1475-2875-2-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grau GE, Mackenzie CD, Carr RA, Redard M, Pizzolato G, Allasia C, Cataldo C, Taylor TE, Molyneux ME. 2003. Platelet accumulation in brain microvessels in fatal pediatric cerebral malaria. J. Infect. Dis. 187:461–466. 10.1086/367960 [DOI] [PubMed] [Google Scholar]
- 6.Krishna S. 2012. Adjunctive management of malaria. Curr. Opin. Infect. Dis. 25:484–488. 10.1097/QCO.0b013e3283567b20 [DOI] [PubMed] [Google Scholar]
- 7.John CC, Kutamba E, Mugarura K, Opoka RO. 2010. Adjunctive therapy for cerebral malaria and other severe forms of Plasmodium falciparum malaria. Expert Rev. Anti Infect. Ther. 8:997–1008. 10.1586/eri.10.90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kihara M, Carter JA, Newton CRJC. 2006. The effect of Plasmodium falciparum on cognition: a systematic review. Trop. Med. Int. Health 11:386–397. 10.1111/j.1365-3156.2006.01579.x [DOI] [PubMed] [Google Scholar]
- 9.Idro R, Ndiritu M, Ogutu B, Mithwani S, Maitland K, Berkley J, Crawley J, Fegan G, Bauni E, Peshu N, Marsh K, Neville B, Newton C. 2007. Burden, features, and outcome of neurological involvement in acute falciparum malaria in Kenyan children. JAMA 297:2232–2240. 10.1001/jama.297.20.2232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rénia L, Potter SM, Mauduit M, Rosa DS, Kayibanda M, Deschemin J-C, Snounou G, Grüner AC. 2006. Pathogenic T cells in cerebral malaria. Int. J. Parasitol. 36:547–554. 10.1016/j.ijpara.2006.02.007 [DOI] [PubMed] [Google Scholar]
- 11.de Souza JB, Hafalla JCR, Riley EM, Couper KN. 2010. Cerebral malaria: why experimental murine models are required to understand the pathogenesis of disease. Parasitology 137:755–772. 10.1017/S0031182009991715 [DOI] [PubMed] [Google Scholar]
- 12.Hunt NH, Grau GE, Engwerda C, Barnum SR, van der Heyde H, Hansen DS, Schofield L, Golenser J. 2010. Murine cerebral malaria: the whole story. Trends Parasitol. 26:272–274. 10.1016/j.pt.2010.03.006 [DOI] [PubMed] [Google Scholar]
- 13.Fauconnier M, Palomo J, Bourigault M-L, Meme S, Szeremeta F, Beloeil JC, Danneels A, Charron S, Rihet P, Ryffel B, Quesniaux VFJ. 2012. IL-12Rβ2 is essential for the development of experimental cerebral malaria. J. Immunol. 188:1905–1914. 10.4049/jimmunol.1101978 [DOI] [PubMed] [Google Scholar]
- 14.Amani V, Vigário AM, Belnoue E, Marussig M, Fonseca L, Mazier D, Rénia L. 2000. Involvement of IFN-gamma receptor-medicated signaling in pathology and anti-malarial immunity induced by Plasmodium berghei infection. Eur. J. Immunol. 30:1646–1655. [DOI] [PubMed] [Google Scholar]
- 15.Hermsen C, van de Wiel T, Mommers E, Sauerwein R, Eling W. 1997. Depletion of CD4+ or CD8+ T-cells prevents Plasmodium berghei induced cerebral malaria in end-stage disease. Parasitology 114(Part 1):7–12. 10.1017/S0031182096008293 [DOI] [PubMed] [Google Scholar]
- 16.Sun G, Chang W-L, Li J, Berney SM, Kimpel D, van der Heyde HC. 2003. Inhibition of platelet adherence to brain microvasculature protects against severe Plasmodium berghei malaria. Infect. Immun. 71:6553–6561. 10.1128/IAI.71.11.6553-6561.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grau GE, Heremans H, Piguet PF, Pointaire P, Lambert PH, Billiau A, Vassalli P. 1989. Monoclonal antibody against interferon gamma can prevent experimental cerebral malaria and its associated overproduction of tumor necrosis factor. Proc. Natl. Acad. Sci. U. S. A. 86:5572–5574. 10.1073/pnas.86.14.5572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nitcheu J, Bonduelle O, Combadiere C, Tefit M, Seilhean D, Mazier D, Combadiere B. 2003. Perforin-dependent brain-infiltrating cytotoxic CD8+ T lymphocytes mediate experimental cerebral malaria pathogenesis. J. Immunol. 170:2221–2228 [DOI] [PubMed] [Google Scholar]
- 19.Hansen DS, Bernard NJ, Nie CQ, Schofield L. 2007. NK cells stimulate recruitment of CXCR3+ T cells to the brain during Plasmodium berghei-mediated cerebral malaria. J. Immunol. 178:5779–5788 [DOI] [PubMed] [Google Scholar]
- 20.Belnoue E, Kayibanda M, Vigário AM, Deschemin J-C, van Rooijen N, Viguier M, Snounou G, Rénia L. 2002. On the pathogenic role of brain-sequestered alphabeta CD8+ T cells in experimental cerebral malaria. J. Immunol. 169:6369–6375 [DOI] [PubMed] [Google Scholar]
- 21.Engwerda CR, Mynott TL, Sawhney S, de Souza JB, Bickle QD, Kaye PM. 2002. Locally up-regulated lymphotoxin alpha, not systemic tumor necrosis factor alpha, is the principle mediator of murine cerebral malaria. J. Exp. Med. 195:1371–1377. 10.1084/jem.20020128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martins YC, Zanini GM, Frangos JA, Carvalho LJM. 2012. Efficacy of different nitric oxide-based strategies in preventing experimental cerebral malaria by Plasmodium berghei ANKA. PLoS One 7:e32048. 10.1371/journal.pone.0032048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Penet M-F, Abou-Hamdan M, Coltel N, Cornille E, Grau GE, de Reggi M, Gharib B. 2008. Protection against cerebral malaria by the low-molecular-weight thiol pantethine. Proc. Natl. Acad. Sci. U. S. A. 105:1321–1326. 10.1073/pnas.0706867105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reis PA, Comim CM, Hermani F, Silva B, Barichello T, Portella AC, Gomes FCA, Sab IM, Frutuoso VS, Oliveira MF, Bozza PT, Bozza FA, Dal-Pizzol F, Zimmerman GA, Quevedo J, Castro-Faria-Neto HC. 2010. Cognitive dysfunction is sustained after rescue therapy in experimental cerebral malaria, and is reduced by additive antioxidant therapy. PLoS Pathog. 6:e1000963. 10.1371/journal.ppat.1000963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Serghides L, Kim H, Lu Z, Kain DC, Miller C, Francis RC, Liles WC, Zapol WM, Kain KC. 2011. Inhaled nitric oxide reduces endothelial activation and parasite accumulation in the brain, and enhances survival in experimental cerebral malaria. PLoS One 6:e27714. 10.1371/journal.pone.0027714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morrell CN, Srivastava K, Swaim A, Lee MT, Chen J, Nagineni C, Hooks JJ, Detrick B. 2011. Beta interferon suppresses the development of experimental cerebral malaria. Infect. Immun. 79:1750–1758. 10.1128/IAI.00810-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Franklin BS, Ishizaka ST, Lamphier M, Gusovsky F, Hansen H, Rose J, Zheng W, Ataíde MA, de Oliveira RB, Golenbock DT, Gazzinelli RT. 2011. Therapeutical targeting of nucleic acid-sensing Toll-like receptors prevents experimental cerebral malaria. Proc. Natl. Acad. Sci. U. S. A. 108:3689–3694. 10.1073/pnas.1015406108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Niikura M, Kamiya S, Nakane A, Kita K, Kobayashi F. 2010. IL-10 plays a crucial role for the protection of experimental cerebral malaria by co-infection with non-lethal malaria parasites. Int. J. Parasitol. 40:101–108. 10.1016/j.ijpara.2009.08.009 [DOI] [PubMed] [Google Scholar]
- 29.Voza T, Vigário AM, Belnoue E, Grüner AC, Deschemin J-C, Kayibanda M, Delmas F, Janse CJ, Franke-Fayard B, Waters AP, Landau I, Snounou G, Rénia L. 2005. Species-specific inhibition of cerebral malaria in mice coinfected with Plasmodium spp. Infect. Immun. 73:4777–4786. 10.1128/IAI.73.8.4777-4786.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hochman S, Kim K. 2012. The impact of HIV coinfection on cerebral malaria pathogenesis. J. Neuroparasitol. 3:235547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Amante FH, Haque A, Stanley AC, Rivera FDL, Randall LM, Wilson YA, Yeo G, Pieper C, Crabb BS, de Koning-Ward TF, Lundie RJ, Good MF, Pinzon-Charry A, Pearson MS, Duke MG, McManus DP, Loukas A, Hill GR, Engwerda CR. 2010. Immune-mediated mechanisms of parasite tissue sequestration during experimental cerebral malaria. J. Immunol. 185:3632–3642. 10.4049/jimmunol.1000944 [DOI] [PubMed] [Google Scholar]
- 32.Bucher K, Dietz K, Lackner P, Pasche B, Fendel R, Mordmüller B, Ben-Smith A, Hoffmann WH. 2011. Schistosoma co-infection protects against brain pathology but does not prevent severe disease and death in a murine model of cerebral malaria. Int. J. Parasitol. 41:21–31. 10.1016/j.ijpara.2010.06.008 [DOI] [PubMed] [Google Scholar]
- 33.Specht S, Ruiz DF, Dubben B, Deininger S, Hoerauf A. 2010. Filaria-induced IL-10 suppresses murine cerebral malaria. Microbes Infect. 12:635–642. 10.1016/j.micinf.2010.04.006 [DOI] [PubMed] [Google Scholar]
- 34.Eckwalanga M, Marussig M, Tavares MD, Bouanga JC, Hulier E, Pavlovitch JH, Minoprio P, Portnoï D, Rénia L, Mazier D. 1994. Murine AIDS protects mice against experimental cerebral malaria: down-regulation by interleukin 10 of a T-helper type 1 CD4+ cell-mediated pathology. Proc. Natl. Acad. Sci. U. S. A. 91:8097–8101. 10.1073/pnas.91.17.8097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dubey JP. 1998. Advances in the life cycle of Toxoplasma gondii. Int. J. Parasitol. 28:1019–1024. 10.1016/S0020-7519(98)00023-X [DOI] [PubMed] [Google Scholar]
- 36.Dubey JP, Jones JL. 2008. Toxoplasma gondii infection in humans and animals in the United States. Int. J. Parasitol. 38:1257–1278. 10.1016/j.ijpara.2008.03.007 [DOI] [PubMed] [Google Scholar]
- 37.Pollard AM, Knoll LJ, Mordue DG. 2009. The role of specific Toxoplasma gondii molecules in manipulation of innate immunity. Trends Parasitol. 25:491–494. 10.1016/j.pt.2009.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yarovinsky F, Sher A. 2006. Toll-like receptor recognition of Toxoplasma gondii. Int. J. Parasitol. 36:255–259. 10.1016/j.ijpara.2005.12.003 [DOI] [PubMed] [Google Scholar]
- 39.Mason NJ, Liou H-C, Hunter CA. 2004. T cell-intrinsic expression of c-Rel regulates Th1 cell responses essential for resistance to Toxoplasma gondii. J. Immunol. 172:3704–3711 [DOI] [PubMed] [Google Scholar]
- 40.Yarovinsky F, Zhang D, Andersen JF, Bannenberg GL, Serhan CN, Hayden MS, Hieny S, Sutterwala FS, Flavell RA, Ghosh S, Sher A. 2005. TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science 308:1626–1629. 10.1126/science.1109893 [DOI] [PubMed] [Google Scholar]
- 41.Aliberti J, Valenzuela JG, Carruthers VB, Hieny S, Andersen J, Charest H, Reis e Sousa C, Fairlamb A, Ribeiro JM, Sher A. 2003. Molecular mimicry of a CCR5 binding-domain in the microbial activation of dendritic cells. Nat. Immunol. 4:485–490. 10.1038/ni915 [DOI] [PubMed] [Google Scholar]
- 42.Alexander J, Hunter CA. 1998. Immunoregulation during toxoplasmosis. Chem. Immunol. 70:81–102. 10.1159/000058701 [DOI] [PubMed] [Google Scholar]
- 43.Denkers EY, Gazzinelli RT. 1998. Regulation and function of T-cell-mediated immunity during Toxoplasma gondii infection. Clin. Microbiol. Rev. 11:569–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yap GS, Sher A. 1999. Cell-mediated immunity to Toxoplasma gondii: initiation, regulation and effector function. Immunobiology 201:240–247. 10.1016/S0171-2985(99)80064-3 [DOI] [PubMed] [Google Scholar]
- 45.Neyer LE, Grunig G, Fort M, Remington JS, Rennick D, Hunter CA. 1997. Role of interleukin-10 in regulation of T-cell-dependent and T-cell-independent mechanisms of resistance to Toxoplasma gondii. Infect. Immun. 65:1675–1682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suzuki Y, Sher A, Yap G, Park D, Neyer LE, Liesenfeld O, Fort M, Kang H, Gufwoli E. 2000. IL-10 is required for prevention of necrosis in the small intestine and mortality in both genetically resistant BALB/c and susceptible C57BL/6 mice following peroral infection with Toxoplasma gondii. J. Immunol. 164:5375–5382 [DOI] [PubMed] [Google Scholar]
- 47.Wilson EH, Wille-Reece U, Dzierszinski F, Hunter CA. 2005. A critical role for IL-10 in limiting inflammation during toxoplasmic encephalitis. J. Neuroimmunol. 165:63–74. 10.1016/j.jneuroim.2005.04.018 [DOI] [PubMed] [Google Scholar]
- 48.O'Brien KB, Schultz-Cherry S, Knoll LJ. 2011. Parasite-mediated upregulation of NK cell-derived gamma interferon protects against severe highly pathogenic H5N1 influenza virus infection. J. Virol. 85:8680–8688. 10.1128/JVI.05142-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ruskin J, Remington JS. 1968. Immunity and intracellular infection: resistance to bacteria in mice infected with a protozoan. Science 160:72–74. 10.1126/science.160.3823.72 [DOI] [PubMed] [Google Scholar]
- 50.Mahmoud AA, Warren KS, Strickland GT. 1976. Acquired resistance to infection with Schistosoma mansoni induced by Toxoplasma gondii. Nature 263:56–57. 10.1038/263056a0 [DOI] [PubMed] [Google Scholar]
- 51.Gentry LO, Remington JS. 1971. Resistance against Cryptococcus conferred by intracellular bacteria and protozoa. J. Infect. Dis. 123:22–31. 10.1093/infdis/123.1.22 [DOI] [PubMed] [Google Scholar]
- 52.Remington JS, Merigan TC. 1969. Resistance to virus challenge in mice infected with protozoa or bacteria. Proc. Soc. Exp. Biol. Med. 131:1184–1188. 10.3181/00379727-131-34066 [DOI] [PubMed] [Google Scholar]
- 53.Charest H, Sedegah M, Yap GS, Gazzinelli RT, Caspar P, Hoffman SL, Sher A. 2000. Recombinant attenuated Toxoplasma gondii expressing the Plasmodium yoelii circumsporozoite protein provides highly effective priming for CD8+ T cell-dependent protective immunity against malaria. J. Immunol. 165:2084–2092 [DOI] [PubMed] [Google Scholar]
- 54.Mengs U, Pelster B. 1982. The course of Plasmodium berghei infection in mice latently infected with Toxoplasma gondii. Cell. Mol. Life Sci. 38:570–571. 10.1007/BF02327054 [DOI] [PubMed] [Google Scholar]
- 55.Omata Y, Yagami K, Takei Y, Suzuki N, Nakabayashi T. 1981. Protective reaction against malaria infection in mice sensitized with frozen-thawed toxoplasma tachyzoites. Zentralbl. Bakteriol. Mikrobiol. Hyg. A 250:223–235 [PubMed] [Google Scholar]
- 56.Suzuki N, Kikushima K, Miyagami T, Igarashi I, Sakurai H, Saito A, Osaki H. 1987. Modulator effect of Toxoplasma lysate antigen in mice experimentally infected with Plasmodium berghei. Zentralbl. Bakteriol. Mikrobiol. Hyg. A 264:422–434 [DOI] [PubMed] [Google Scholar]
- 57.Denkers EY, Gazzinelli RT, Hieny S, Caspar P, Sher A. 1993. Bone marrow macrophages process exogenous Toxoplasma gondii polypeptides for recognition by parasite-specific cytolytic T lymphocytes. J. Immunol. 150:517–526 [PubMed] [Google Scholar]
- 58.Mordue DG, Scott-Weathers CF, Tobin CM, Knoll LJ. 2007. A patatin-like protein protects Toxoplasma gondii from degradation in activated macrophages. Mol. Microbiol. 63:482–496. 10.1111/j.1365-2958.2006.05538.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Amani V, Boubou MI, Pied S, Marussig M, Walliker D, Mazier D, Renia L. 1998. Cloned lines of Plasmodium berghei ANKA differ in their abilities to induce experimental cerebral malaria. Infect. Immun. 66:4093–4099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Carroll RW, Wainwright MS, Kim K-Y, Kidambi T, Gómez ND, Taylor T, Haldar K. 2010. A rapid murine coma and behavior scale for quantitative assessment of murine cerebral malaria. PLoS One 5:e13124. 10.1371/journal.pone.0013124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hed J, Dahlgren C, Rundquist I. 1983. A simple fluorescence technique to stain the plasma membrane of human neutrophils. Histochemistry 79:105–110. 10.1007/BF00494347 [DOI] [PubMed] [Google Scholar]
- 62.Hermsen CC, Telgt DS, Linders EH, van de Locht LA, Eling WM, Mensink EJ, Sauerwein RW. 2001. Detection of Plasmodium falciparum malaria parasites in vivo by real-time quantitative PCR. Mol. Biochem. Parasitol. 118:247–251. 10.1016/S0166-6851(01)00379-6 [DOI] [PubMed] [Google Scholar]
- 63.Rooney PJ, Ayong L, Tobin CM, Moreno SNJ, Knoll LJ. 2011. TgVTC2 is involved in polyphosphate accumulation in Toxoplasma gondii. Mol. Biochem. Parasitol. 176:121–126. 10.1016/j.molbiopara.2010.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Payne TM, Payne AJ, Knoll LJ. 2011. A Toxoplasma gondii mutant highlights the importance of translational regulation in the apicoplast during animal infection. Mol. Microbiol. 82:1204–1216. 10.1111/j.1365-2958.2011.07879.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Baptista FG, Pamplona A, Pena AC, Mota MM, Pied S, Vigário AM. 2010. Accumulation of Plasmodium berghei-infected red blood cells in the brain is crucial for the development of cerebral malaria in mice. Infect. Immun. 78:4033–4039. 10.1128/IAI.00079-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Thumwood CM, Hunt NH, Clark IA, Cowden WB. 1988. Breakdown of the blood-brain barrier in murine cerebral malaria. Parasitology 96:579–589. 10.1017/S0031182000080203 [DOI] [PubMed] [Google Scholar]
- 67.Medana IM, Chaudhri G, Chan-Ling T, Hunt NH. 2001. Central nervous system in cerebral malaria: “innocent bystander” or active participant in the induction of immunopathology? Immunol. Cell Biol. 79:101–120. 10.1046/j.1440-1711.2001.00995.x [DOI] [PubMed] [Google Scholar]
- 68.Hawkins BT, Egleton RD. 2006. Fluorescence imaging of blood-brain barrier disruption. J. Neurosci. Methods 151:262–267. 10.1016/j.jneumeth.2005.08.006 [DOI] [PubMed] [Google Scholar]
- 69.Grau GE, Piguet PF, Engers HD, Louis JA, Vassalli P, Lambert PH. 1986. L3T4+ T lymphocytes play a major role in the pathogenesis of murine cerebral malaria. J. Immunol. 137:2348–2354 [PubMed] [Google Scholar]
- 70.Claser C, Malleret B, Gun SY, Wong AYW, Chang ZW, Teo P, See PCE, Howland SW, Ginhoux F, Rénia L. 2011. CD8+ T Cells and IFN-γ mediate the time-dependent accumulation of infected red blood cells in deep organs during experimental cerebral malaria. PLoS One 6:e18720. 10.1371/journal.pone.0018720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Belnoue E, Potter SM, Rosa DS, Mauduit M, Grüner AC, Kayibanda M, Mitchell AJ, Hunt NH, Rénia L. 2008. Control of pathogenic CD8+ T cell migration to the brain by IFN-gamma during experimental cerebral malaria. Parasite Immunol. 30:544–553. 10.1111/j.1365-3024.2008.01053.x [DOI] [PubMed] [Google Scholar]
- 72.Grau GE, Fajardo LF, Piguet PF, Allet B, Lambert PH, Vassalli P. 1987. Tumor necrosis factor (cachectin) as an essential mediator in murine cerebral malaria. Science 237:1210–1212. 10.1126/science.3306918 [DOI] [PubMed] [Google Scholar]
- 73.Gazzinelli RT, Wysocka M, Hayashi S, Denkers EY, Hieny S, Caspar P, Trinchieri G, Sher A. 1994. Parasite-induced IL-12 stimulates early IFN-gamma synthesis and resistance during acute infection with Toxoplasma gondii. J. Immunol. 153:2533–2543 [PubMed] [Google Scholar]
- 74.Reis e Sousa C, Hieny S, Scharton-Kersten T, Jankovic D, Charest H, Germain RN, Sher A. 1997. In vivo microbial stimulation induces rapid CD40 ligand-independent production of interleukin 12 by dendritic cells and their redistribution to T cell areas. J. Exp. Med. 186:1819–1829. 10.1084/jem.186.11.1819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.El-Assaad F, Wheway J, Mitchell AJ, Lou J, Hunt NH, Combes V, Grau GE. 2013. Cytoadherence of Plasmodium berghei-infected red blood cells to murine brain and lung microvascular endothelial cells in vitro. Infect. Immun. 81:3984–3991. 10.1128/IAI.00428-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kossodo S, Monso C, Juillard P, Velu T, Goldman M, Grau GE. 1997. Interleukin-10 modulates susceptibility in experimental cerebral malaria. Immunology 91:536–540. 10.1046/j.1365-2567.1997.00290.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Haque A, Best SE, Unosson K, Amante FH, de Labastida F, Anstey NM, Karupiah G, Smyth MJ, Heath WR, Engwerda CR. 2011. Granzyme B expression by CD8+ T cells is required for the development of experimental cerebral malaria. J. Immunol. 186:6148–6156. 10.4049/jimmunol.1003955 [DOI] [PubMed] [Google Scholar]
- 78.Nie CQ, Bernard NJ, Norman MU, Amante FH, Lundie RJ, Crabb BS, Heath WR, Engwerda CR, Hickey MJ, Schofield L, Hansen DS. 2009. IP-10-mediated T cell homing promotes cerebral inflammation over splenic immunity to malaria infection. PLoS Pathog. 5:e1000369. 10.1371/journal.ppat.1000369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Van den Steen PE, Deroost K, van Aelst I, Geurts N, Martens E, Struyf S, Nie CQ, Hansen DS, Matthys P, van Damme J, Opdenakker G. 2008. CXCR3 determines strain susceptibility to murine cerebral malaria by mediating T lymphocyte migration toward IFN-gamma-induced chemokines. Eur. J. Immunol. 38:1082–1095. 10.1002/eji.200737906 [DOI] [PubMed] [Google Scholar]
- 80.Perona-Wright G, Mohrs K, Szaba FM, Kummer LW, Madan R, Karp CL, Johnson LL, Smiley ST, Mohrs M. 2009. Systemic but not local infections elicit immunosuppressive IL-10 production by natural killer cells. Cell Host Microbe 6:503–512. 10.1016/j.chom.2009.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Normaznah Y, Halim AA, Outhayphone M, Zamri MR. 1999. Protective immunity against Plasmodium berghei malaria after administration of interleukin-12. Malays. J. Pathol. 21:123–125 [PubMed] [Google Scholar]
- 82.Sedegah M, Finkelman F, Hoffman SL. 1994. Interleukin 12 induction of interferon gamma-dependent protection against malaria. Proc. Natl. Acad. Sci. U. S. A. 91:10700–10702. 10.1073/pnas.91.22.10700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stevenson MM, Tam MF, Wolf SF, Sher A. 1995. IL-12-induced protection against blood-stage Plasmodium chabaudi AS requires IFN-gamma and TNF-alpha and occurs via a nitric oxide-dependent mechanism. J. Immunol. 155:2545–2556 [PubMed] [Google Scholar]
- 84.Reis e Sousa C, Yap G, Schulz O, Rogers N, Schito M, Aliberti J, Hieny S, Sher A. 1999. Paralysis of dendritic cell IL-12 production by microbial products prevents infection-induced immunopathology. Immunity 11:637–647. 10.1016/S1074-7613(00)80138-7 [DOI] [PubMed] [Google Scholar]
- 85.Aliberti J, Reis e Sousa C, Schito M, Hieny S, Wells T, Huffnagle GB, Sher A. 2000. CCR5 provides a signal for microbial induced production of IL-12 by CD8 alpha+ dendritic cells. Nat. Immunol. 1:83–87. 10.1038/76957 [DOI] [PubMed] [Google Scholar]
- 86.Aliberti J, Hieny S, Reis e Sousa C, Serhan CN, Sher A. 2002. Lipoxin-mediated inhibition of IL-12 production by DCs: a mechanism for regulation of microbial immunity. Nat. Immunol. 3:76–82. 10.1038/ni745 [DOI] [PubMed] [Google Scholar]
- 87.Aliberti J, Valenzuela JG, Carruthers VB, Hieny S, Andersen J, Charest H, Reis e Sousa C, Fairlamb A, Ribeiro JM, Sher A. 2003. Molecular mimicry of a CCR5 binding-domain in the microbial activation of dendritic cells. Nat. Immunol. 4:485–490. 10.1038/ni915 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.