

Abstract
Although Candida glabrata is an important pathogenic Candida species, relatively little is known about its innate immune recognition. Here, we explore the potential role of Dectin-2 for host defense against C. glabrata. Dectin-2-deficient (Dectin-2−/−) mice were found to be more susceptible to C. glabrata infections, showing a defective fungal clearance in kidneys but not in the liver. The increased susceptibility to infection was accompanied by lower production of T helper 1 (Th1) and Th17-derived cytokines by splenocytes of Dectin-2−/− mice, while macrophage-derived cytokines were less affected. These defects were associated with a moderate yet significant decrease in phagocytosis of the fungus by the Dectin-2−/− macrophages and neutrophils. Neutrophils of Dectin-2−/− mice also displayed lower production of reactive oxygen species (ROS) upon challenge with opsonized C. glabrata or C. albicans. This study suggests that Dectin-2 is important in host defense against C. glabrata and provides new insights into the host defense mechanisms against this important fungal pathogen.
INTRODUCTION
The opportunistic fungal pathogen Candida glabrata is the second most frequent cause of candidiasis after Candida albicans, accounting for approximately 15 to 25% of Candida infections (1, 2). Similar to other Candida species, C. glabrata resides as a commensal within the normal microbial flora of humans, but it may also cause serious infections in immunocompromised patients. In the last 3 decades, the number of infections due to non-albicans Candida species has increased significantly (3, 4). The current rise in the incidence of invasive C. glabrata infections is largely the result of the combinational increase in immunosuppressive infections and modern prolonged invasive medical interventions (5). Moreover, C. glabrata is of added concern due to the fact that a fraction of clinical isolates have been proven to be resistant to antifungal agents, such as azoles (6). The mortality rate associated with bloodstream infection with C. glabrata is approximately 50% in cancer patients and is even higher in bone marrow transplant patients (7, 8). However, compared to the literature on C. albicans, few studies have investigated the pathophysiology of C. glabrata infections, despite its high morbidity and mortality burden.
Pattern recognition of fungal components and activation of innate immunity are essential steps in the host defense against Candida species (9). The Candida cell wall is mainly composed of multiple layers of carbohydrates, such as mannan, β-glucan, and chitin, which are collectively recognized by C-type lectins, including macrophage mannose receptor (MR), SIGNR-1, Galectin-3, Mincle, Dectin-1, Dectin-2, and the Toll-like receptors. This recognition leads to activation of the host innate immune system. MR recognizes the N-linked mannans of fungal cell walls, SIGNR-1 recognizes branched α-mannans, and Galectin-3 recognizes β-(1,2)-mannans (9–11). It has recently been reported that Dectin-1 (Clec7a) and Dectin-2 (Clec4n) are specific receptors for β-glucans (12) and C. albicans-derived α-mannans (13), respectively.
Dectin-2 is a C-type lectin receptor primarily expressed by macrophages, dendritic cells and neutrophils, with specificity for structures rich in mannose (14, 15). Dectin-2 uses the FcRγ chain to signal via Syk- and caspase recruitment domain family member 9 (Card9)-dependent pathways (16–18). It represents an important receptor for the generation of the Th17-like adaptive immune response, coordinating the Th1-like responses together with Dectin-1 (17). Human Dectin-2 has a mannose recognition EPN motif (15, 19), strengthening the likelihood that Dectin-2 plays an important role in antifungal immunity. Additionally, Saijo et al. have reported that Dectin-2 is the functional receptor for α-mannans and that Dectin-2−/− mice are more susceptible to C. albicans (13). However, little is known regarding the specific role of Dectin-2 for the host defense against C. glabrata.
In this study, we have explored the contribution of Dectin-2 in the pathophysiology of murine C. glabrata systemic infection. We show that the burden of C. glabrata was significantly increased in kidneys of Dectin-2−/− mice, which was correlated with a decreased phagocytosis and killing of the fungus by the innate immune macrophages (as shown by live-cell video microscopy phagocytosis assays) and neutrophils (as shown by the ex vivo phagocytosis and killing assays). Moreover, the adaptive T helper-derived cytokine responses were also considerably affected in Dectin-2−/− mice. In conclusion, our data demonstrate an important role of Dectin-2 for innate immune responses and its significant contribution to the adaptive immune responses against C. glabrata.
MATERIALS AND METHODS
Generation of Dectin-2−/− mice.
Dectin-2−/− mice were constructed by Ozgene Pty Ltd., Australia, as described previously (20). Briefly, a targeting vector was designed to introduce a conditional mutation into the mouse Clec4n gene (Ensembl version no. ENSMUSG00000023349; http://www.ensembl.org/Mus_musculus/Gene/Summary?db=core;g=ENSMUSG00000023349;r=6:123229843-123247021). The strategy utilizes mutant loxP sites (lox66 and lox71) to enable an inversion of the flanked sequence in response to the expression of Cre recombinase. The recombination event inactivated the gene by switching the position and direction of the transcriptional termination (STOP) signal with Clec4n exon 2. The lox66 site was inserted upstream from Clec4n exon 2, and a neomycin selection cassette (PGK-Neo) was targeted into the intron between exon 2 and exon 3. The selection cassette was flanked with FLP recombination target (FRT) sites to enable removal by FlpE-mediated recombination. An inverted STOP element and a lox71 site were placed downstream from the selection marker. The 5′ and 3′ homology arms of the vector were approximately 6.1 kb and 5.9 kb, respectively. The targeting vector was linearized with PmeI and electroporated into the C57BL/6 embryonal stem (ES) cell line Bruce4. Neomycin-resistant clones were selected and screened by Southern blotting to identify homologous recombinants. Correctly targeted clones were microinjected into BALB/cJ blastocysts, which were transferred into pseudopregnant CBB6F1 foster females. Chimeric mice were obtained and outcrossed to C57BL/6J females to generate ES cell-derived targeted mutant progeny.
Eight- to 12-week-old female Dectin-2−/− (Clec4n−/−) mice on a C57BL/6J background were obtained from a breeding colony at the Central Animal Laboratory, Radboud University Nijmegen Medical Centre. Age-matched C57BL/6J female mice were obtained from Charles River Wiga (Sulzfeld, Germany). All mice weighed between 20 and 25 g. The animals were fed standard laboratory chow (Hope Farms, Woerden, The Netherlands) and water ad libitum. Most of the experiments were repeated at least twice with a minimum of five animals per time point. All experimentation conformed to the terms and conditions of the Ethics Committee on Animal Experiments of Radboud University, Nijmegen. Key experiments, such as for fungal outgrowth and phagocytosis and killing of C. glabrata, were repeated using Dectin-2−/− mice generated independently, kindly provided by Y. Iwakura (13). These validation studies were performed at the animal facility of the University of Aberdeen. Live-cell video microscopy was conducted in Aberdeen; C57BL/6 and Dectin-2−/− (provided by Y. Iwakura) mice were obtained from breeding colonies. All mice were used at 8 to 16 weeks of age. Animals were kept and handled in accordance with institutional guidelines.
Candida strains, culture media, and growth conditions.
C. glabrata CBS138 (ATCC 2001), a strain described elsewhere (21), was used in all experiments. C. glabrata was routinely grown and maintained on Sabouraud dextrose plates. For inoculum preparation, a single colony was grown in Sabouraud dextrose broth at 29°C for 24 h, with shaking. Cells were washed twice in sterile phosphate-buffered saline (PBS) and counted using a hemocytometer. Cell density was adjusted with PBS to the desired inoculum level. C. glabrata was heat killed for 30 min at 95°C. Candida albicans ATCC MYA-3573 (UC 820) was grown overnight to generate yeast cells in Sabouraud dextrose broth at 29°C, with shaking. Cells were harvested by centrifugation, washed twice with PBS, and resuspended in culture medium (RPMI 1640 Dutch modification). C. albicans yeast was heat killed for 30 min at 95°C. To generate hyphae, yeast cells were inoculated and grown overnight at 37°C in culture medium adjusted to pH 6.4 with hydrochloric acid. Hyphae were killed by exposure to 95°C for 30 min and resuspended in culture medium to a hyphal inoculum size that originated from 1 × 108 CFU/ml.
C. glabrata infection model and fungal burden.
A nonlethal experimental model of disseminated candidiasis was used, in which wild-type and Dectin-2−/− mice were injected intravenously, via the lateral tail vein, with C. glabrata (1 × 107 CFU/mouse) in a 100 μl volume of sterile pyrogen-free PBS. Mice were monitored daily. For survival studies, groups of 10 mice were followed-up for a period of 28 days. For immunological and histological studies, subgroups of 5 animals were killed on day 3, 7, or 14 postinfection. Tissues were collected and processed for fungal burden and cytokine analysis. To assess the tissue outgrowth of C. glabrata on these days, the liver and kidneys were removed aseptically, weighed, and homogenized in sterile PBS in a tissue grinder. The number of viable Candida cells in the tissues was determined by plating serial dilutions on Sabouraud dextrose agar plates, as described elsewhere (22). The CFU were counted after 24 h of incubation at 29°C and expressed as log CFU per gram of tissue.
Histopathology.
Kidney samples from infected mice were kept in formalin until processed. Sections were dehydrated with xylene, rehydrated through a graded series of ethanol solutions, and stained with hematoxylin and eosin (H&E) or periodic acid-Schiff (PAS) using conventional staining methods. All individual segments were evaluated for the presence and intensity of inflammation, as well as for the presence of fungi. Tissue sections were analyzed with a VisionTek digital microscope (Sakura), using VisionTek Live software.
In vitro cytokine production.
Peritoneal macrophages were isolated from mice by injecting 5 ml of ice-cold sterile PBS (pH 7.4) into the peritoneal cavity. After centrifugation and washing, cells were resuspended in RPMI 1640 culture medium containing 1 mM pyruvate, 2 mM l-glutamine, and 50 mg/liter gentamicin. Cells were counted using a Z1 Coulter particle counter (Beckman Coulter; Woerden, The Netherlands), adjusted to 1 × 106 cells/ml, and cultured in 96-well round-bottom microtiter plates (Costar, Corning, The Netherlands) at 1 × 105 cells/well in a final volume of 200 μl. After 24 h of incubation with different stimuli at 37°C and 5% CO2, the plates were centrifuged at 1,400 × g for 8 min, and the supernatants were collected and stored at −80°C until cytokine assays were performed.
Spleen cells were isolated by gently passing spleens through a sterile 200 μm filter chamber. After washing with sterile PBS and centrifugation at 4°C (1,200 rpm for 5 min), cells were resuspended in 2 ml RPMI 1640 in the presence of 20% fetal calf serum and counted, and concentrations were adjusted to 1 × 107 cells/ml. Cells were cultured in 24-well plates (Greiner, Alphen a/d Rijn, The Netherlands) at 5 × 106 cells/well, and different stimuli were added in a final volume of 1,000 μl. Supernatants were collected at two different time points (depending on the cytokine) as follows. After 48 h of incubation at 37°C and 5% CO2, 500 μl of supernatant per well was collected and stored at −80°C until cytokine assays were performed; thereafter, the plates were further incubated at 37°C and 5% CO2 for an additional 3 days. The plates were centrifuged at 1,400 × g for 8 min, and the remaining supernatants were collected and stored at −80°C until cytokine assays were performed. The concentrations of mouse tumor necrosis factor alpha (TNF-α), interleukin-1α (IL-1α), and IL-1β were determined by specific radioimmunoassay (RIA). Mouse IL-6, KC, IL-17, gamma interferon (IFN-γ), IL-22, and IL-10 concentrations were measured by commercial enzyme-linked immunosorbent assay (ELISA) kits (Biosource, Camarillo, CA) according to the instructions of the manufacturer.
Phagocytosis and killing of C. glabrata.
Peritoneal macrophages and neutrophils were recruited and phagocytosis and killing assays were performed according to a modification of a method described elsewhere (23). C. glabrata/macrophage or C. glabrata/neutrophil ratios of 10:1 were used in the phagocytosis and killing studies. Peritoneal macrophages or neutrophils from groups of 5 C57BL/6J (control) and 5 Dectin-2−/− mice were elicited by an intraperitoneal (i.p.) injection of heat-killed C. glabrata. Cells were collected in separate sterile tubes either 3 days (macrophages) or 4 h (neutrophils) after C. glabrata infection by washing the peritoneal cavity with 5 ml of ice-cold PBS. Phagocytes were centrifuged (for 10 min at 2,250 × g), counted in a Bürker counting chamber, and resuspended in RPMI 1640 Dutch modification (with 20 mM HEPES and without l-glutamine; ICN Biomedicals) supplemented with 5% heat-inactivated fetal calf serum, 1% gentamicin, 1% l-glutamine, and 1% pyruvate. The processes of phagocytosis and intracellular killing were studied in an adherent monolayer of phagocytes. To create a monolayer of phagocytes, 5 × 105 cells in 100 μl of RPMI 1640 were dispensed into the wells of a 96-well flat-bottom plate (Costar) and incubated at 37°C and 5% CO2. Macrophages were allowed to adhere for up to 2 h, but neutrophils for only 30 min before the monolayers were gently washed with culture medium to remove nonadherent cells. The percentage of adherence was calculated as follows: (1 − [number of nonadherent cells/5 × 105]) × 100. Subsequently, the cells were incubated with 1 × 104 CFU C. glabrata, which were opsonized for 30 min at 24°C in modified Eagle's medium (MEM; Gibco Life Technologies) that contained 2.5% fresh mouse serum (effector/target ratio, 40:1). After 15 min, supernatants were aspirated, and monolayers were gently washed with MEM to remove noningested C. glabrata. The supernatant and well washings that contained the noningested Candida were combined and plated in serial dilutions on Sabouraud agar plates. The percentage of phagocytosed microorganisms was defined as follows: [1 − (number of uningested CFU/CFU at the start of incubation)] × 100.
The killing of C. glabrata by phagocytes was assessed in the same monolayers. After removal of the nonphagocytized Candida, 200 μl of culture medium, consisting of Sabouraud in MEM (50%, vol/vol), was added to the monolayers. After 3 h of incubation at 37°C and 5% CO2, the wells were gently detached with a cell scraper and washed with 200 μl distilled H2O to achieve lysis of phagocytes. This procedure was repeated 3 times, after which the pooled washes were adjusted to a final volume of 1 ml with distilled water. Microscopic examination of the culture plates showed that there was an almost complete removal of phagocytes. To quantify the viable intracellular Candida, 10-fold dilutions of each sample were spread on Sabouraud agar plates and incubated at 37°C for 24 h. The percentage of yeast killed by the phagocytes was determined as follows: [1 − (CFU after incubation/number of phagocytized CFU)] × 100. Phagocyte-free incubations of blastoconidia were included as a control for yeast viability.
Live-cell video microscopy phagocytosis assays.
Standard phagocytosis assays were performed as previously described (24). Briefly, 6 × 105 live C. glabrata yeasts were added to 2 × 105 macrophages in μ-Slide 8-well chambers (ibidi GmbH, Germany) immediately prior to imaging. Video microscopy experiments were performed using an Ultra View VoX live-cell imaging system (PerkinElmer) with the environmental control chamber set at 37°C. Images were captured at 1-min intervals for 3 h using an electron-multiplying charge-coupled device (EMCCD) camera. At least two independent experiments were conducted for each mouse strain, and two movies for each mouse were analyzed (n = 7 wild-type mice, and n = 8 Dectin-2−/− mice). Volocity 6.2.1 imaging analysis software was used to track macrophage migration at 1-min intervals for the first 60 min of the phagocytosis assay. The software enabled high-throughput analysis of macrophage migration, providing detailed information on the distances traveled and the directionality and velocity of hundreds of individual macrophages. The data were subsequently displayed in tracking diagrams and used to calculate the mean track velocity and track length of macrophages cultured with C. glabrata. The movies generated for migration analyses were also analyzed to determine phagocytic uptake at the 30-min, 60-min, and 180-min time points (25). For each mouse, 20 macrophages were analyzed for the percentage of uptake of live C. glabrata yeasts (calculated by determining the number of macrophages per 100 that had ingested at least one yeast). The phagocytic index was calculated by counting the total number of yeasts engulfed per hundred macrophages at each time point.
Reactive oxygen species (ROS) assay.
The spontaneous and stimulus-induced oxygen radical production levels of isolated neutrophils were evaluated using luminol-enhanced chemiluminescence and determined in an automated LB96V Microlumat plus luminometer (EG & G Berthold, Bald Wilberg, Germany) as previously described (26). In this study, neutrophils (2 × 105 per well) were seeded into 96-well plates and incubated in medium containing either RPMI, phorbol 12-myristate 13-acetate (PMA; 5 μg/ml), live opsonized C. albicans (107 CFU/ml) or live opsonized C. glabrata (107 CFU/ml). Luminol was added to each well in order to start the chemiluminescence reaction. Each measurement was carried out in at least duplicate repetitions. Chemiluminescence was determined every 145 s at 37°C for 1 h. Luminescence was expressed as relative light units (RLU) per second. Data were analyzed with Winglow software (EG & Berthold).
Ethics statement.
All experiments in this study were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health, the Dutch law on Animal Experiments, and FELASA regulations. The protocol was approved by the Ethics Committee on Animal Experiments of the Radboud University Nijmegen Medical Centre. All efforts were made to minimize suffering of the animals. All experiments performed in Aberdeen conformed to the terms and conditions of United Kingdom Home Office licenses for research on animals and the guidelines of the University of Aberdeen ethical review committee.
Statistical analysis.
Differences in phagocytosis, postphagocytic killing, and concentrations of cytokines were analyzed using the Mann-Whitney U test. Survival data were analyzed using the Kaplan-Meyer log rank test. Differences were considered significant at a P value of <0.05. The majority of the experiments was performed at least twice, and at least 5 mice/group/time point were used for the outgrowth, phagocytosis, killing, ROS, and cytokine synthesis experiments. A total of 10 mice/group were used for survival experiments. Data are presented as means ± standard errors of the means (SEM).
RESULTS
Increased susceptibility to C. glabrata systemic infection in Dectin-2−/− mice.
To determine the role of Dectin-2 in the host defense against C. glabrata, we first compared the susceptibility to infection of Dectin-2−/− and wild-type control mice. Following infection with C. glabrata, both groups of mice were able to effectively control and eradicate the fungus from the liver within 2 weeks (Fig. 1A). In contrast, while wild-type animals restrained the infection in their kidneys, significantly higher C. glabrata loads were observed in Dectin-2−/− mice (Fig. 1B). Similar increases in the fungal burdens were observed in the kidneys of the Dectin-2−/− mice provided by Y. Iwakura (Fig. 1C). Nonetheless, Dectin-2−/− mice were less susceptible to C. glabrata than has been reported for C. albicans, with no mortality recorded in any of the mouse strains during the 28 days following intravenous infection (data not shown). Compared with the results for noninfected mice (Fig. 2A), histopathological analysis identified zones with lymphocytic infiltration suggestive of chronic inflammation in both wild-type control (Fig. 2B) and Dectin-2−/− (Fig. 2C) mice. Sporadic neutrophilic granulocytes could be detected (Fig. 2D and E). At day 7 and day 14 postinfection, the kidneys of Dectin-2−/− mice displayed significantly increased numbers of inflammatory foci compared with the kidneys of wild-type mice. Interestingly, fungi were only partially associated with inflammation and were often present in the lumina of tubuli, not in the tissue itself (Fig. 2D and E). Therefore, the foci of chronic, lymphocytic inflammation were not associated with the presence of fungi. In conclusion, Dectin-2 appears to be an essential pattern recognition receptor for the adequate control of C. glabrata renal infection.
FIG 1.
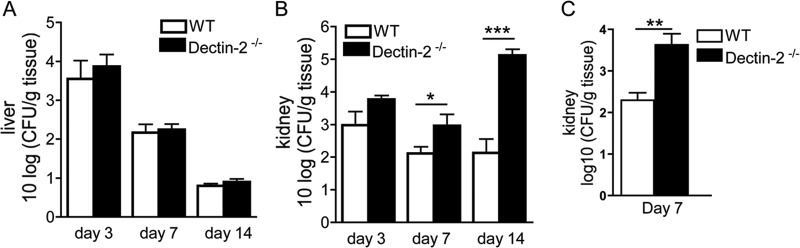
Greater C. glabrata loads in the kidneys of mice during systemic infection. (A and B) Quantification of Candida colonies from liver (A) and kidneys (B) of wild-type (WT) and Dectin-2−/− mice at days 3, 7, and 14 postinfection is shown. Wild-type and Dectin-2−/− mice received 107 CFU of C. glabrata per mouse intravenously. After 3, 7, and 14 days, organs were collected and serial dilutions were plated on Sabouraud solid medium. Results are means ± SEM (n = 10 to 14 mice per group from 3 independent experiments). Significance was determined with the Mann-Whitney U test. Statistically different groups are indicated as follows: *, P < 0.05; ***, P < 0.001. (C) Fungal burdens in the kidneys of wild-type and Dectin-2−/− mice provided by Y. Iwakura. Wild-type or Dectin-2−/− mice received 107 CFU of C. glabrata per mouse intravenously. After 7 days, kidneys were collected and serial dilutions were plated on Sabouraud agar medium. Results are means ± SEM (n = 12 mice per group from two independent experiments). Significance was determined with the Mann-Whitney U test. **, P < 0.01.
FIG 2.

Dectin-2 deficiency affects kidney histopathology during systemic infection with C. glabrata. Histopathological assessment of kidneys in uninfected Dectin-2−/− (A), infected wild-type (B), and infected Dectin-2−/− (C) (hematoxylin-and-eosin [H&E]-stained sections) and infected Dectin-2−/− (D and E) (periodic acid-Schiff [PAS]-stained sections) mice. Representative localized lesions and C. glabrata colonies in the kidneys of Dectin-2−/− mice are shown (C, D, and E). All observations of infected kidneys were performed at day 14 after intravenous injection with C. glabrata.
Defective Th-associated cytokine production in Dectin-2−/− mice.
To further characterize the role of Dectin-2 for the recognition of C. glabrata, the ex vivo cytokine profile was assessed in peritoneal macrophages of naive wild-type and knockout mice. Interestingly, the innate response levels from uninfected Dectin-2−/− macrophages were slightly higher than the response levels of the macrophages isolated from the wild-type animals (TNF-α, IL-6, IL-1α, and IL-1β) (Fig. 3A). Only the IL-6 and KC production levels from Dectin-2−/− macrophages upon stimulation with C. albicans heat-killed hyphae were lower than the levels from wild-type macrophages, although the difference reached statistical significance only for KC (Fig. 3A). Regarding the T cell-derived cytokines (IL-10, IFN-γ, and IL-17) produced by the splenocytes of uninfected mice, no differences could be detected between the two groups of mice (Fig. 3B). The IL-17 production levels in the splenocytes from Dectin-2−/− mice were highly variable.
FIG 3.

Cytokine profile from naive mice. Cells from naive mice were stimulated for 24 h (peritoneal macrophages) (A) or for 48 h and 5 days (splenocytes) (B) with heat-killed (HK) or live C. glabrata (Cg), heat-killed C. albicans yeast (Cay) or hyphae (Cah), and live C. albicans (Ca). Levels of TNF-α, IL-6, KC, IL-1α, IL-1β, IL-10, IFN-γ, and IL-17 accumulation were measured by ELISA. Results are means ± SEM (n = 8 mice per group). Significance was determined with the Mann-Whitney U test. Statistically different groups are indicated as follows: *, P < 0.05; **, P < 0.01.
In a subsequent set of experiments, we studied the ex vivo cytokine responses of peritoneal macrophages and splenocytes isolated from mice infected with C. glabrata. No differences were detectable between the two groups of mice in most of the macrophage-derived cytokines assayed (intracellular/extracellular IL-1α and IL-1β and extracellular production of IL-6 and KC; data not shown), with the exception of TNF-α at day 14 postinfection (Fig. 4A). Similar to the results for peritoneal macrophages, ex vivo stimulation of splenocytes from C. glabrata-infected mice resulted in significantly lower levels of TNF-α production by Dectin-2−/−-deficient cells upon stimulation with either C. glabrata (Fig. 4B) or C. albicans (Fig. 4C), although dissimilarities were detectable earlier in the course of the infection. No significant differences could be detected in the amounts of the anti-inflammatory cytokine IL-10 produced by either strain. In addition to the responses of the innate cytokines, Th1 and Th17 responses at 7 days postinfection were also significantly reduced in Dectin-2−/− splenocytes, as mirrored by the low production of IFN-γ and IL-17 (Fig. 4B). The levels of IL-22 were moderately reduced at day 14 postinfection (Fig. 4B). A similar pattern with more pronounced differences was observed when splenocytes from infected mice were restimulated with C. albicans yeast (Fig. 4C). As such, Dectin-2 deficiency results in decreased production of protective T helper-derived cytokines.
FIG 4.

Dectin-2 is required during systemic C. glabrata infection for subsequent splenocyte recall responses. Wild-type or Dectin-2−/− mice received 107 CFU of C. glabrata per mouse intravenously. (A) After 3, 7, and 14 days, peritoneal macrophages were restimulated ex vivo with heat-killed C. glabrata (Cg), heat-killed C. albicans yeast (Cay), or hyphae (Cah) for 24 h. Levels of TNF-α accumulation in the supernatants were measured by ELISA. Results are means ± SEM (n = 9 or 10 mice per group) from 2 independent experiments. Significance was determined with the Mann-Whitney U test. Statistically different groups are indicated as follows: *, P < 0.05. (B and C) Wild-type or Dectin-2−/− mice received 107 CFU of C. glabrata per mouse intravenously. After 3, 7, and 14 days, splenocytes were restimulated ex vivo with either heat-killed C. glabrata (B) or heat-killed C. albicans yeast (C) for 48 h and 5 days. Levels of accumulation of TNF-α, IL-10, and IFN-γ (48 h) and of IL-17 and IL-22 (5 days) in the supernatants were measured by ELISA. Results are means ± SEM (n = 14 or 15 mice per group) from 3 independent experiments. Significance was determined with the Mann-Whitney U test. Statistically different groups are indicated as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Dectin-2−/− peritoneal macrophages are less efficient in the uptake and killing of C. glabrata ex vivo.
The phagocytic clearance of microorganisms, including yeasts, by professional phagocytes is mediated by receptor activation that drives the engulfment and internalization of the pathogen. We therefore assessed the ability of Dectin-2−/− peritoneal macrophages to recognize, phagocytose, and kill live opsonized C. glabrata. Using a classical ex vivo phagocytosis and killing assay (23), wild-type peritoneal macrophages were found to be slightly more efficient in the phagocytosis and subsequent killing of C. glabrata than Dectin-2−/− macrophages (Fig. 5A). We further investigated the migration of peritoneal macrophages toward C. glabrata by live-cell video microscopy using a phagocytosis assay described previously (25). Dynamic analysis of individual macrophages, achieved by tracking cells to determine their directionality, distance, and velocity for the first hour of the assay, demonstrated no major differences in migration dynamics between macrophages harvested from wild-type or Dectin-2−/− animals (Fig. 5B). However, in the same experiment, a complementary analysis revealed a significant difference between the two types of macrophages in terms of the uptake and phagocytosis of live C. glabrata. Following 30 min of coincubation with live C. glabrata, 51% of the wild-type macrophages had ingested at least one yeast, whereas Dectin-2−/− macrophages showed a significantly reduced uptake of 34% (Fig. 5C). This significantly reduced uptake capacity was noticeable early during the phagocytosis assay (30 min and 60 min) but not at a later time point (180 min) (Fig. 5C). Additionally, the total numbers of fungal cells phagocytosed by macrophages from Dectin-2−/− mice were also significantly lower than the numbers phagocytosed by wild-type macrophages at 30 min and 60 min of coincubation (Fig. 5D). Altogether, these results highlight a partial, yet significant role of Dectin-2 in mediating the uptake and engulfment of C. glabrata by macrophages. In addition, partial effects on fungal killing may also be envisioned, although more studies should be performed to establish that definitively.
FIG 5.

Impact of Dectin-2 deficiency on phagocytosis and killing of C. glabrata by peritoneal macrophages. (A) Phagocytosis and killing of C. glabrata cells by murine peritoneal macrophages. The results are represented as percentage of phagocytosis (the percentage of fungal cells engulfed by macrophages present in the well) and percentage of killing (the percentage of killed fungal cells among the phagocytosed yeasts). The C. glabrata/macrophage ratio was 10:1. Significance was determined with the Mann-Whitney U test. Values represent means ± SEM (n = 5 mice per group). (B) Migration of peritoneal macrophages was determined using live-cell video microscopy. Tracking diagrams illustrate the distances traveled and the directionality and velocity of macrophages exposed to C. glabrata at a fungal particle/macrophage ratio of 3:1. Due to the large number of macrophages tracked per video, the data were filtered to show only macrophages whose tracks lasted until 30 min to 60 min, as previous work has shown that migration activity is greater early on (25). Tracks represent the movement of individual macrophages relative to their starting position, symbols indicate the location of macrophages at 1-min intervals, and arrows represent directionality. (n = 7 control mice and n = 8 Dectin-2−/− mice for 4 independent experiments). (C and D) Percentage of uptake (C) and phagocytic index (D) determined using live-cell video microscopy. The percentage of macrophages that have ingested at least one C. glabrata and the number of C. glabrata per 100 macrophages are depicted. Significance was determined with the Mann-Whitney U test. Values represent means ± SEM. *, P < 0.05.
Dectin-2−/− neutrophils are less efficient in phagocytosing and killing C. glabrata ex vivo.
We next assessed the recognition and killing of C. glabrata by neutrophils, pivotal players in the clearance of invading fungal cells from tissues. Neutrophils were harvested from the peritoneal cavity of both wild-type and Dectin-2−/− mice and further subjected to an ex vivo phagocytosis and killing assay (23). The phagocytosis of C. glabrata by the Dectin-2−/− neutrophils was considerably less effective than the phagocytosis of C. glabrata by neutrophils of wild-type mice (Fig. 6A). The killing of C. glabrata by the Dectin-2−/− neutrophils was slightly diminished compared with the killing by control neutrophils (Fig. 6A). Importantly, neutrophils from Dectin-2−/− mice showed a defect in secretion of ROS upon interaction with either live opsonized C. glabrata or C. albicans compared with the ROS secretion of wild-type neutrophils (Fig. 6B). Interestingly, the defect was specific for fungi, as no differences in ROS production were observed when cells were stimulated with PMA (Fig. 6B). Thus, Dectin-2 is important in both the uptake of C. glabrata by neutrophils and the subsequent ROS production and killing of the yeast.
FIG 6.
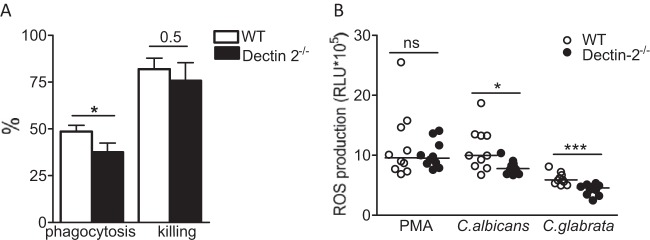
Impact of Dectin-2 deficiency on phagocytosis and killing of C. glabrata by neutrophils. (A) Phagocytosis and killing of C. glabrata cells by murine neutrophils. The results are represented as percentage of phagocytosis (the percentage of fungal cells engulfed by neutrophils present in the well) and percentage of killing (the percentage of killed fungal cells among the phagocytosed yeast). The C. glabrata/neutrophil ratio was 10:1. Values represent means ± SEM (n = 10 mice per group) for 2 independent experiments. P values are for phagocytosis or killing induced by wild-type and Dectin-2−/− macrophages. *, P < 0.05. (B) Dectin-2−/− neutrophils cannot induce the respiratory burst to the same extent as the wild type. Dectin-2−/− or wild-type neutrophils were stimulated with RPMI, phorbol 12-myristate 13-acetate (PMA), live opsonized C. glabrata, or live opsonized C. albicans, and overall oxygen radical production (area under the curve after 60 min) was measured with luminol-enhanced chemiluminescence in isolated neutrophils. Data are represented as means ± SEM (n = 10 mice per group) for 2 independent experiments. Significance was determined with the Mann-Whitney U test. Statistically different groups are indicated as follows: *, P < 0.05; ***, P < 0.001.
DISCUSSION
Dectin-2 is one of the main pattern recognition receptors for fungal mannans, with recent studies demonstrating an important role for this receptor in host defense against C. albicans (13). In the present study, we show that Dectin-2 is also an important component of the host defense against C. glabrata. The increased susceptibility of Dectin-2−/− mice to a systemic infection with C. glabrata is associated with a combination of decreased production of protective T helper-derived cytokines and defective phagocytosis and killing of the yeast by both neutrophils and macrophages.
Little is known of the role of the C-type lectin receptor, Dectin-2. A recent study reported that Dectin-2−/− mice are highly susceptible to a systemic infection with C. albicans (13). Moreover, it has been shown that Dectin-2 plays an important role in the induction of protective Th17 responses (17) and that, together with Dectin-3, it forms a heterodimeric pattern recognition receptor for host defense against C. albicans (27). The intracellular signaling induced by Dectin-2 involves activation of CARD9 (28), phospholipase C-γ (29), and protein kinase C-σ (30). Our study demonstrates that Dectin-2 has a nonredundant role for host defense against C. glabrata. This is an important observation not only for our improved understanding of C. glabrata infection but also for Dectin-2 biology: while Dectin-2 has been suggested to recognize mainly C. albicans hyphae (13), its importance for recognition of a Candida species that does not usually develop hyphae is a broadening of the biological importance of Dectin-2. The increased susceptibility to C. glabrata was demonstrated in two independently generated groups of Dectin-2−/− mice, reinforcing this conclusion. However, these mice were less susceptible to C. glabrata than to C. albicans, as reported earlier (13). The increased susceptibility of Dectin-2−/− mice to C. glabrata was apparent through their incapacity to eliminate the fungus from the kidneys, the target organ of disseminated candidiasis (31). Previously reported studies already showed that systemic candidiasis increased the fungal burden in kidneys of Dectin-1−/− mice compared with the fungal burden in kidneys of the wild-type mice (12). The fact that the animals were able to eradicate C. glabrata from the liver, in comparison with the results for the kidneys, confirms recent findings concerning organ-specific Candida infection and immune responses mediated primarily by neutrophils and monocytes, as well as by various molecular factors (32–34).
Fungal pathogens have developed a number of strategies to improve their chance of survival in the host environment; e.g., C. albicans and Cryptococcus neoformans either destroy macrophages or escape via nonlytic exocytosis (35, 36). Interestingly, C. glabrata engulfed by macrophages does not undergo a morphological transition but survives and multiplies within mammalian macrophages (37, 38), which are ultimately damaged by ingested C. glabrata (38). In our study, we assessed the role of Dectin-2 for the phagocytosis and killing of C. glabrata and observed that neutrophils and macrophages lacking this receptor were less capable of ingesting and killing the yeast. Using live-cell video microscopy, we found that significantly fewer Dectin-2−/− macrophages took up C. glabrata, in accordance with a lower phagocytic index where phagocytosis had occurred. These defects in the uptake and phagocytosis of C. glabrata could not be attributed to a defect in mobility. Finally, signaling through C-type lectin receptors induces cytokines and ROS that both activate the spleen tyrosine kinase (Syk)–CARD9–nuclear factor-κB (NF-κB) pathway (39) and, most likely, induce the intraphagosomal damage of fungal cells. In line with a role of the CARD9 pathway, Drewniak et al. showed that human CARD9 deficiency resulted in a selective defect in the host defense against invasive fungal infection, caused by impaired phagocyte killing in unopsonized samples compared with the phagocyte killing in opsonized samples (40). In the present study, Dectin-2−/− neutrophils produced less ROS than wild-type neutrophils after stimulation of cells with either opsonized C. glabrata or C. albicans, suggesting a role of Dectin-2 in the killing of Candida species despite the presence of opsonins.
The induction of proinflammatory cytokines is an important component of anti-Candida host defense. Macrophage-derived proinflammatory cytokines like IL-1, KC, or IL-6 activate the recruitment of phagocytes and the phagocytosis and killing of fungi by phagocytes, and mice deficient in these cytokines have an increased susceptibility to systemic candidiasis (41, 42). No major defects in the production of these cytokines were observed in cells isolated from Candida-challenged Dectin-2−/− mice. In contrast, the secretion of cytokines produced by Th1 and Th17 lymphocytes was significantly decreased in Dectin-2−/− mice. IFN-γ is the main product of Th1 lymphocytes, and it has been proven to have a crucial role in antifungal host defense in mice (43). The defective production of IFN-γ by splenocytes of Dectin-2−/− mice may represent one of the mechanisms determining the increased susceptibility to C. glabrata in these mice. Similarly, the Th17 cytokine, IL-17, represents an important component of antifungal host defense through its activation of neutrophils (44), while patients with defective Th17 responses develop chronic mucocutaneous candidiasis (45). The level of IL-17 produced by Dectin-2−/− splenocytes was significantly decreased at day 7 postinfection, suggesting a role for Dectin-2 in regulating splenocyte recall responses to fungal infection. The defective Th17 responses in Dectin-2−/− mice could therefore contribute to their susceptibility to C. glabrata. These results underline the conclusions of Saijo et al. (13) concerning Dectin-2-mediated immunity to C. albicans. The IL-17 defect in response to C. glabrata observed in our study was, however, less pronounced than that previously reported for C. albicans (13). Moreover, we should observe that the cytokine differences were most pronounced earlier during infection. At later time points during the infection, the picture starts to be biased by the higher fungal burden in the KO mice, which may counterbalance the defective cytokine production capacity.
The known fungal ligands recognized by Dectin-2 are α-mannans (13). Whereas significant research has been conducted on the structure of the C. albicans cell wall, much less is known regarding the cell wall components of C. glabrata. While recognition of mannans of C. glabrata by Dectin-2 is most likely, future studies should define the precise ligand-receptor interaction profile, especially as C. glabrata is less likely to form hyphae than C. albicans. Moreover, both endogenous (T cell) and exogenous (microbial carbohydrate) ligands for Dectin-2 may be important for the generation and maintenance of tolerance or the recognition of carbohydrate structures. Of particular interest is that previous reports have highlighted the ability of Dectin-2 to specifically signal after recognition of the hyphal form of C. albicans (28). The interaction between C. glabrata and Dectin-2 is different, as C. glabrata does not form traditional compartmentalized hyphae. Still, previous reports have described tubes and pseudohypha formation in several clinical isolates of C. glabrata (46), and these might be recognized by Dectin-2.
In conclusion, we demonstrate that the immune responses triggered by Dectin-2 during infection with C. glabrata might link innate immune recognition, phagocytosis, and killing of the fungus with the adaptive T helper-dependent responses. A combination of these mechanisms controls C. glabrata during systemic infection. Deciphering the precise mechanisms responsible for host defense against C. glabrata represents an important step in understanding of the pathophysiology of the disease and for the possibility to design future novel immunotherapeutic strategies.
ACKNOWLEDGMENTS
We thank A. Whittington for the useful discussion and H. M. J. Roelofs and E. D. Olthof for the assistance with ROS production. We thank the University of Aberdeen Animal Facility and Microscopy Facility and also D. MacCallum and R. Drummond for technical assistance.
This work was supported by European Union ALLFUN (FP7/2007 2013, HEALTH-2010-260338) (Fungi in the setting of inflammation, allergy and autoimmune diseases: Translating basic science into clinical practices ALLFUN) to D.C.I., M.G.N., and N.A.R.G. M.G.N and J.Q. were also supported by a Vici grant of the Netherlands Organization of Scientific Research (to M.G.N.). N.A.R.G. was also supported by the Wellcome Trust (080088, 086827, 075470, and 097377). L.P.E. is a CSO Senior Clinical Fellow and supported by WT project, programme, and equipment grants (089930 and 094847). G.D.B. was funded by the Wellcome Trust (086558 and 097377).
There is no conflict of interest.
Footnotes
Published ahead of print 16 December 2013
REFERENCES
- 1.Pfaller MA, Messer SA, Moet GJ, Jones RN, Castanheira M. 2011. Candida bloodstream infections: comparison of species distribution and resistance to echinocandin and azole antifungal agents in intensive care unit (ICU) and non-ICU settings in the SENTRY Antimicrobial Surveillance Program (2008-2009). Int. J. Antimicrob. Agents 38:65–69. 10.1016/j.ijantimicag.2011.02.016 [DOI] [PubMed] [Google Scholar]
- 2.Abi-Said D, Anaissie E, Uzun O, Raad I, Pinzcowski H, Vartivarian S. 1997. The epidemiology of hematogenous candidiasis caused by different Candida species. Clin. Infect. Dis. 24:1122–1128. 10.1086/513663 [DOI] [PubMed] [Google Scholar]
- 3.Kauffman CA, Vazquez JA, Sobel JD, Gallis HA, McKinsey DS, Karchmer AW, Sugar AM, Sharkey PK, Wise GJ, Mangi R, Mosher A, Lee JY, Dismukes WE. 2000. Prospective multicenter surveillance study of funguria in hospitalized patients. The National Institute for Allergy and Infectious Diseases (NIAID) Mycoses Study Group. Clin. Infect. Dis. 30:14–18. 10.1086/313583 [DOI] [PubMed] [Google Scholar]
- 4.Ruan SY, Chu CC, Hsueh PR. 2008. In vitro susceptibilities of invasive isolates of Candida species: rapid increase in rates of fluconazole susceptible-dose dependent Candida glabrata isolates. Antimicrob. Agents Chemother. 52:2919–2922. 10.1128/AAC.00323-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hajjeh RA, Sofair AN, Harrison LH, Lyon GM, Arthington-Skaggs BA, Mirza SA, Phelan M, Morgan J, Lee-Yang W, Ciblak MA, Benjamin LE, Sanza LT, Huie S, Yeo SF, Brandt ME, Warnock DW. 2004. Incidence of bloodstream infections due to Candida species and in vitro susceptibilities of isolates collected from 1998 to 2000 in a population-based active surveillance program. J. Clin. Microbiol. 42:1519–1527. 10.1128/JCM.42.4.1519-1527.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fidel PL, Jr, Vazquez JA, Sobel JD. 1999. Candida glabrata: review of epidemiology, pathogenesis, and clinical disease with comparison to C. albicans. Clin. Microbiol. Rev. 12:80–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anaissie EJ, Vartivarian SE, Abi-Said D, Uzun O, Pinczowski H, Kontoyiannis DP, Khoury P, Papadakis K, Gardner A, Raad II, Gilbreath J, Bodey GP. 1996. Fluconazole versus amphotericin B in the treatment of hematogenous candidiasis: a matched cohort study. Am. J. Med. 101:170–176. 10.1016/S0002-9343(96)80072-6 [DOI] [PubMed] [Google Scholar]
- 8.Goodman JL, Winston DJ, Greenfield RA, Chandrasekar PH, Fox B, Kaizer H, Shadduck RK, Shea TC, Stiff P, Friedman DJ, Powderly WG, Silber JL, Horowitz H, Lichtin A, Wolff SN, Mangan KF, Silver SM, Weisdorf D, Ho WG, Gilbert G, Buell D. 1992. A controlled trial of fluconazole to prevent fungal infections in patients undergoing bone marrow transplantation. N. Engl. J. Med. 326:845–851. 10.1056/NEJM199203263261301 [DOI] [PubMed] [Google Scholar]
- 9.Netea MG, Brown GD, Kullberg BJ, Gow NA. 2008. An integrated model of the recognition of Candida albicans by the innate immune system. Nat. Rev. Microbiol. 6:67–78. 10.1038/nrmicro1815 [DOI] [PubMed] [Google Scholar]
- 10.Poulain D, Jouault T. 2004. Candida albicans cell wall glycans, host receptors and responses: elements for a decisive crosstalk. Curr. Opin. Microbiol. 7:342–349. 10.1016/j.mib.2004.06.011 [DOI] [PubMed] [Google Scholar]
- 11.Cambi A, Netea MG, Mora-Montes HM, Gow NA, Hato SV, Lowman DW, Kullberg BJ, Torensma R, Williams DL, Figdor CG. 2008. Dendritic cell interaction with Candida albicans critically depends on N-linked mannan. J. Biol. Chem. 283:20590–20599. 10.1074/jbc.M709334200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taylor PR, Tsoni SV, Willment JA, Dennehy KM, Rosas M, Findon H, Haynes K, Steele C, Botto M, Gordon S, Brown GD. 2007. Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nat. Immunol. 8:31–38. 10.1038/ni1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saijo S, Ikeda S, Yamabe K, Kakuta S, Ishigame H, Akitsu A, Fujikado N, Kusaka T, Kubo S, Chung SH, Komatsu R, Miura N, Adachi Y, Ohno N, Shibuya K, Yamamoto N, Kawakami K, Yamasaki S, Saito T, Akira S, Iwakura Y. 2010. Dectin-2 recognition of alpha-mannans and induction of Th17 cell differentiation is essential for host defense against Candida albicans. Immunity 32:681–691. 10.1016/j.immuni.2010.05.001 [DOI] [PubMed] [Google Scholar]
- 14.Ariizumi K, Shen GL, Shikano S, Ritter R, III, Zukas P, Edelbaum D, Morita A, Takashima A. 2000. Cloning of a second dendritic cell-associated C-type lectin (Dectin-2) and its alternatively spliced isoforms. J. Biol. Chem. 275:11957–11963. 10.1074/jbc.275.16.11957 [DOI] [PubMed] [Google Scholar]
- 15.McGreal EP, Rosas M, Brown GD, Zamze S, Wong SY, Gordon S, Martinez-Pomares L, Taylor PR. 2006. The carbohydrate-recognition domain of Dectin-2 is a C-type lectin with specificity for high mannose. Glycobiology 16:422–430. 10.1093/glycob/cwj077 [DOI] [PubMed] [Google Scholar]
- 16.Barrett NA, Maekawa A, Rahman OM, Austen KF, Kanaoka Y. 2009. Dectin-2 recognition of house dust mite triggers cysteinyl leukotriene generation by dendritic cells. J. Immunol. 182:1119–1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Robinson MJ, Osorio F, Rosas M, Freitas RP, Schweighoffer E, Gross O, Verbeek JS, Ruland J, Tybulewicz V, Brown GD, Moita LF, Taylor PR, Reis e Sousa C. 2009. Dectin-2 is a Syk-coupled pattern recognition receptor crucial for Th17 responses to fungal infection. J. Exp. Med. 206:2037–2051. 10.1084/jem.20082818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sato K, Yang XL, Yudate T, Chung JS, Wu J, Luby-Phelps K, Kimberly RP, Underhill D, Cruz PD, Jr, Ariizumi K. 2006. Dectin-2 is a pattern recognition receptor for fungi that couples with the Fc receptor gamma chain to induce innate immune responses. J. Biol. Chem. 281:38854–38866. 10.1074/jbc.M606542200 [DOI] [PubMed] [Google Scholar]
- 19.Gavino AC, Chung JS, Sato K, Ariizumi K, Cruz PD., Jr 2005. Identification and expression profiling of a human C-type lectin, structurally homologous to mouse Dectin-2. Exp. Dermatol. 14:281–288. 10.1111/j.0906-6705.2005.00312.x [DOI] [PubMed] [Google Scholar]
- 20.Zhang Z, Lutz B. 2002. Cre recombinase-mediated inversion using lox66 and lox71: method to introduce conditional point mutations into the CREB-binding protein. Nucleic Acids Res. 30:e90. 10.1093/nar/gnf089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koszul R, Malpertuy A, Frangeul L, Bouchier C, Wincker P, Thierry A, Duthoy S, Ferris S, Hennequin C, Dujon B. 2003. The complete mitochondrial genome sequence of the pathogenic yeast Candida (Torulopsis) glabrata. FEBS Lett. 534:39–48. 10.1016/S0014-5793(02)03749-3 [DOI] [PubMed] [Google Scholar]
- 22.Kullberg BJ, van't Wout JW, van Furth R. 1990. Role of granulocytes in increased host resistance to Candida albicans induced by recombinant interleukin-1. Infect. Immun. 58:3319–3324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kullberg BJ, van't Wout JW, Hoogstraten C, van Furth R. 1993. Recombinant interferon-gamma enhances resistance to acute disseminated Candida albicans infection in mice. J. Infect. Dis. 168:436–443. 10.1093/infdis/168.2.436 [DOI] [PubMed] [Google Scholar]
- 24.McKenzie CG, Koser U, Lewis LE, Bain JM, Mora-Montes HM, Barker RN, Gow NA, Erwig LP. 2010. Contribution of Candida albicans cell wall components to recognition by and escape from murine macrophages. Infect. Immun. 78:1650–1658. 10.1128/IAI.00001-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lewis LE, Bain JM, Okai B, Gow NA, Erwig LP. 2013. Live-cell video microscopy of fungal pathogen phagocytosis. J. Vis. Exp. 2013(71):50196. 10.3791/50196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Versleijen MW, Oyen WJ, Roelofs HM, van Emst-de Vries SE, Willems PH, Jansen JB, Wanten GJ. 2008. Immune function and leukocyte sequestration under the influence of parenteral lipid emulsions in healthy humans: a placebo-controlled crossover study. Am. J. Clin. Nutr. 87:539–547 [DOI] [PubMed] [Google Scholar]
- 27.Zhu LL, Zhao XQ, Jiang C, You Y, Chen XP, Jiang YY, Jia XM, Lin X. 2013. C-type lectin receptors Dectin-3 and Dectin-2 form a heterodimeric pattern-recognition receptor for host defense against fungal infection. Immunity 39:324–334. 10.1016/j.immuni.2013.05.017 [DOI] [PubMed] [Google Scholar]
- 28.Bi L, Gojestani S, Wu W, Hsu YM, Zhu J, Ariizumi K, Lin X. 2010. CARD9 mediates Dectin-2-induced IkappaBalpha kinase ubiquitination leading to activation of NF-kappaB in response to stimulation by the hyphal form of Candida albicans. J. Biol. Chem. 285:25969–25977. 10.1074/jbc.M110.131300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gorjestani S, Yu M, Tang B, Zhang D, Wang D, Lin X. 2011. Phospholipase Cgamma2 (PLCgamma2) is key component in Dectin-2 signaling pathway, mediating anti-fungal innate immune responses. J. Biol. Chem. 286:43651–43659. 10.1074/jbc.M111.307389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Strasser D, Neumann K, Bergmann H, Marakalala MJ, Guler R, Rojowska A, Hopfner KP, Brombacher F, Urlaub H, Baier G, Brown GD, Leitges M, Ruland J. 2012. Syk kinase-coupled C-type lectin receptors engage protein kinase C-sigma to elicit Card9 adaptor-mediated innate immunity. Immunity 36:32–42. 10.1016/j.immuni.2011.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spellberg B, Ibrahim AS, Edwards JE, Jr, Filler SG. 2005. Mice with disseminated candidiasis die of progressive sepsis. J. Infect. Dis. 192:336–343. 10.1086/430952 [DOI] [PubMed] [Google Scholar]
- 32.Lionakis MS, Lim JK, Lee CC, Murphy PM. 2011. Organ-specific innate immune responses in a mouse model of invasive candidiasis. J. Innate Immun. 3:180–199. 10.1159/000321157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lionakis MS, Fischer BG, Lim JK, Swamydas M, Wan W, Richard Lee CC, Cohen JI, Scheinberg P, Gao JL, Murphy PM. 2012. Chemokine receptor Ccr1 drives neutrophil-mediated kidney immunopathology and mortality in invasive candidiasis. PLoS Pathog. 8:e1002865. 10.1371/journal.ppat.1002865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ngo LY, Kasahara S, Kumasaka DK, Knoblaugh SE, Jhingran A, Hohl TM. 2014. Inflammatory monocytes mediate early and organ-specific innate defense during systemic candidiasis. J. Infect. Dis. 209:109–119. 10.1093/infdis/jit413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alvarez M, Casadevall A. 2006. Phagosome extrusion and host-cell survival after Cryptococcus neoformans phagocytosis by macrophages. Curr. Biol. 16:2161–2165. 10.1016/j.cub.2006.09.061 [DOI] [PubMed] [Google Scholar]
- 36.Bain JM, Lewis LE, Okai B, Quinn J, Gow NA, Erwig LP. 2012. Non-lytic expulsion/exocytosis of Candida albicans from macrophages. Fungal Genet. Biol. 49:677–678. 10.1016/j.fgb.2012.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kaur R, Ma B, Cormack BP. 2007. A family of glycosylphosphatidylinositol-linked aspartyl proteases is required for virulence of Candida glabrata. Proc. Natl. Acad. Sci. U. S. A. 104:7628–7633. 10.1073/pnas.0611195104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seider K, Brunke S, Schild L, Jablonowski N, Wilson D, Majer O, Barz D, Haas A, Kuchler K, Schaller M, Hube B. 2011. The facultative intracellular pathogen Candida glabrata subverts macrophage cytokine production and phagolysosome maturation. J. Immunol. 187:3072–3086. 10.4049/jimmunol.1003730 [DOI] [PubMed] [Google Scholar]
- 39.Drummond RA, Saijo S, Iwakura Y, Brown GD. 2011. The role of Syk/CARD9 coupled C-type lectins in antifungal immunity. Eur. J. Immunol. 41:276–281. 10.1002/eji.201041252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Drewniak A, Gazendam RP, Tool AT, van Houdt M, Jansen MH, van Hamme JL, van Leeuwen EM, Roos D, Scalais E, de Beaufort C, Janssen H, van den Berg TK, Kuijpers TW. 2013. Invasive fungal infection and impaired neutrophil killing in human CARD9 deficiency. Blood 121:2385–2392. 10.1182/blood-2012-08-450551 [DOI] [PubMed] [Google Scholar]
- 41.Basu S, Quilici C, Zhang HH, Grail D, Dunn AR. 2008. Mice lacking both G-CSF and IL-6 are more susceptible to Candida albicans infection: critical role of neutrophils in defense against Candida albicans. Growth Factors 26:23–34. 10.1080/08977190801987513 [DOI] [PubMed] [Google Scholar]
- 42.Gorjestani S, Darnay BG, Lin X. 2012. Tumor necrosis factor receptor-associated factor 6 (TRAF6) and TGFbeta-activated kinase 1 (TAK1) play essential roles in the C-type lectin receptor signaling in response to Candida albicans infection. J. Biol. Chem. 287:44143–44150. 10.1074/jbc.M112.414276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou P, Sieve MC, Bennett J, Kwon-Chung KJ, Tewari RP, Gazzinelli RT, Sher A, Seder RA. 1995. IL-12 prevents mortality in mice infected with Histoplasma capsulatum through induction of IFN-gamma. J. Immunol. 155:785–795 [PubMed] [Google Scholar]
- 44.Dejima T, Shibata K, Yamada H, Hara H, Iwakura Y, Naito S, Yoshikai Y. 2011. Protective role of naturally occurring interleukin-17A-producing gammadelta T cells in the lung at the early stage of systemic candidiasis in mice. Infect. Immun. 79:4503–4510. 10.1128/IAI.05799-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van de Veerdonk FL, Plantinga TS, Hoischen A, Smeekens SP, Joosten LA, Gilissen C, Arts P, Rosentul DC, Carmichael AJ, Smits-van der Graaf CA, Kullberg BJ, van der Meer JW, Lilic D, Veltman JA, Netea MG. 2011. STAT1 mutations in autosomal dominant chronic mucocutaneous candidiasis. N. Engl. J. Med. 365:54–61. 10.1056/NEJMoa1100102 [DOI] [PubMed] [Google Scholar]
- 46.Lachke SA, Joly S, Daniels K, Soll DR. 2002. Phenotypic switching and filamentation in Candida glabrata. Microbiology 148:2661–2674 [DOI] [PubMed] [Google Scholar]