

Abstract
BALB/c mice are highly susceptible to experimental intraperitoneal Trypanosoma congolense infection. However, a recent report showed that these mice are relatively resistant to primary intradermal low-dose infection. Paradoxically, repeated low-dose intradermal infections predispose mice to enhanced susceptibility to an otherwise noninfectious dose challenge. Here, we explored the mechanisms responsible for this low-dose-induced susceptibility to subsequent low-dose challenge infection. We found that akin to intraperitoneal infection, low-dose intradermal infection led to production of interleukin-10 (IL-10), IL-6, IL-12, tumor necrosis factor alpha (TNF-α), transforming growth factor β (TGF-β), and gamma interferon (IFN-γ) by spleen and draining lymph node cells. Interestingly, despite the absence of parasitemia, low-dose intradermal infection led to expansion of CD4+ CD25+ Foxp3+ cells (T regulatory cells [Tregs]) in both the spleens and lymph nodes draining the infection site. Depletion of Tregs by anti-CD25 monoclonal antibody (MAb) treatment during primary infection or before challenge infection following repeated low-dose infection completely abolished the low-dose-induced enhanced susceptibility. In addition, Treg depletion was associated with dramatic reduction in serum levels of TGF-β and IL-10. Collectively, these findings show that low-dose intradermal infection leads to rapid expansion of Tregs, and these cells mediate enhanced susceptibility to subsequent infection.
INTRODUCTION
African trypanosomiasis (AT) is a disease that poses a serious threat to humans and livestock in sub-Saharan Africa. The disease is caused by several species of the extracellular hemoprotozoa belonging to the genus Trypanosoma. Although it is estimated that over 50 million people living in 25 countries are at risk of contracting the disease, the number of reported cases per year has dramatically decreased (∼10,000 new cases annually, although the actual number of cases might be up to 30,000 to 40,000 per year) due to increasing efforts to combat the disease (1–3). Trypanosoma congolense is one of the most important pathogens for cattle, and it is estimated that 3 million head of cattle die annually from the associated disease (4), leading to an annual loss of about U.S. $1.3 billion resulting directly from death, reduced meat and milk production, and control costs (5).
Efforts to control African trypanosomiasis have been hampered because of lack of understanding of the mechanisms that regulate disease pathogenesis and host protective immune response against the pathogen. In particular, the parasite's ability to undergo antigenic variation and our lack of understanding of the molecular mechanisms that regulate the process contribute to the failure to design an effective vaccine (6). During infection, trypanosomes constantly modify their variant surface glycoprotein (VSG) during host antibody response, resulting in the fluctuating waves of parasitemia that characterize African trypanosomiasis (7–9). Furthermore, infection with African trypanosomes is associated with profound immunosuppression, which increases the susceptibility of the host to the parasite and secondary infections (10–12). Understanding the mechanisms that regulate resistance and/or susceptibility to the disease could reveal novel interventions that might lead to effective disease control (13).
The murine model of experimental African trypanosomiasis has provided insights into the immunopathogenesis of the disease. In particular, C57BL/6 and BALB/c mice have mostly been used to study resistance and susceptibility to T. congolense infection. BALB/c mice are highly susceptible to intraperitoneal (i.p.) infection with T. congolense and die within 10 days after infection (14). In contrast, C57BL/6 mice are relatively resistant and are able to control several waves of parasitemia and survive for more than 4 months after infection (14, 15). Most studies in this model utilize the intraperitoneal route of infection and have led to some interesting discoveries (10–13). However, the fact that natural infection occurs naturally through the skin of the animal suggests that observations made with the intraperitoneal route of infection may not correctly reflect the real events that occur following skin infection. For example, following the bite of an infected tsetse fly and deposition of parasites in the host skin, the parasites first induce a local cutaneous inflammatory response (known as chancre) before migrating from the skin to the blood through the lymphatic system (16, 17). Thus, the intraperitoneal route of infection bypasses these early but important host responses that may ultimately dictate the outcome of infection. Indeed, a recent intradermal (i.d.) infection model shows that the outcome of i.d. infection is very different, with mice being relatively (about 1,000 times) more resistant to the intradermal than the intraperitoneal route (18). Paradoxically, primary low-dose intradermal infection predisposes to enhanced susceptibility following a challenge infection. However, the mechanisms of this low-dose intradermal infection-induced enhanced susceptibility are unknown.
CD4+ T cells that constitutively express CD25 and the transcription factor Foxp3 (called regulatory T cells [Tregs]) have been shown to play a major role in immune homeostasis by actively suppressing several pathological and physiological immune responses in the host (19–21). Although their primary role is to prevent autoimmunity and suppress inflammatory responses, Tregs have also been implicated in the pathogenesis of several infectious diseases, including those caused by parasites (22–24). In particular, increased numbers of CD4+ CD25+ Foxp3+ Tregs have been reported in experimental T. congolense infections (25, 26), and these cells have been implicated in enhanced susceptibility to the infection (24, 25), although the exact mechanisms remain unknown.
In this study, we investigated the mechanism through which low-dose intradermal T. congolense infection predisposes mice to enhanced susceptibility to subsequent reinfection. Our studies show that despite failure to establish parasitemia, repeated primary low-dose i.d. infection is associated with systemic increase in the percentages and absolute numbers of CD4+ CD25+ Foxp3+ regulatory T cells in infected mice. Depletion of these cells by treatment with anti-CD25 before and after repeated low-dose infection abolished the low-dose-induced susceptibility following rechallenge infection. In addition, Treg depletion was accompanied by a dramatic reduction in the serum levels of transforming growth factor β (TGF-β) and interleukin-10 (IL-10). We speculate that the induction of Tregs following low-dose intradermal infection could be an evasion strategy by the parasite to enhance its survival and transmission from one host to another.
MATERIALS AND METHODS
Mice.
The 6- to 8-week-old female BALB/c and outbred Swiss White (aka CD1) mice used in these experiments were purchased from Charles River, St. Constante, Quebec, Canada. Animals were housed at Central Animal Care Services (CACS), University of Manitoba, Winnipeg, Canada. Animal housing, handling, and feeding were done in accordance with the recommendations of the Canadian Council of Animal Care.
Parasites and mouse infection.
Trypanosoma congolense (Trans Mara strain) variant antigenic type TC13 was used in all experiments, and the origin of this strain has been previously reported (27). For expansion of frozen TC13 stabilates, CD1 mice were immunosuppressed by injection of cyclophosphamide (Cytoxan; 200 mg/kg) intraperitoneally, followed by infection with freshly thawed T. congolense stabilates from liquid nitrogen (27). Three days after infection, mice were anesthetized by isoflurane, and blood was collected by cardiac puncture. Parasites for infection were purified from the blood using DEAE-cellulose anion-exchange chromatography (28). Eluted parasites were washed in Tris-saline-glucose (TSG), counted with the hemocytometer, resuspended in TSG containing 10% heat-inactivated fetal bovine serum (FBS), and diluted to the desired concentrations. Mice were infected by intraperitoneal injection of 100 μl TSG-FBS containing 102 or 103 parasites. Intradermal (hind footpad) infections were made by injecting 50 μl TSG-FBS containing 102 or 103 parasites.
Estimation of parasitemia.
A drop of blood taken from the tail vain of every T. congolense-infected experimental mouse was placed on a microscope slide, and estimation was done by counting the number of parasites present in at least 10 fields at a ×400 magnification of light microscope. During the late stage of infections, when parasite loads are high, estimation was done using a technique described previously (29).
Cell culture.
At sacrifice, spleens and lymph nodes (popliteal lymph nodes) draining the intradermal infection site were processed into single-cell suspensions, treated with ACK lysis buffer, and washed two times with phosphate-buffered saline (PBS). The cells were counted, resuspended at final concentration of 4 × 106/ml in complete tissue culture medium (Dulbecco's modified Eagle's medium [DMEM] supplemented with 10% heat-inactivated FBS, 2 mmol l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin), and plated at 1 ml/well in 24-well tissue culture plates (Falcon; VWR Edmonton, AB, Canada) with or without whole trypanosome lysate (106/ml parasite equivalent). Cultured cell supernatant fluids were gently collected after 72 h and stored at −20°C until assayed for cytokines by enzyme-linked immunosorbent assay (ELISA).
Depletion of Tregs and macrophages.
In experiments requiring Treg depletion, each mouse received 100 μg of anti-CD25 monoclonal antibody (MAb) (clone PC61) intraperitoneally. Previous studies from our lab have shown that this dose of antibody causes sustained depletion of CD25+ Foxp3+ T cells (without affecting other cell populations) for up to 7 days in mice infected with T. congolense (25, 30). In vivo depletion of macrophages was done by intravenous injection of 200 μl of chlodronate-loaded liposomes (5 mg/ml; Encapsular NanoSciences LLC, Nashville, TN) 24 h before primary intradermal infection. The control group of mice was injected with liposomes. Macrophage depletion was confirmed by flow cytometric analysis of F4/80+ cell populations.
Direct ex vivo staining and flow cytometry.
Spleen and lymph node cells were directly stained ex vivo for CD4 and CD25 surface expression and intracellularly for Foxp3 using the Tregs staining kit (eBioscience) in accordance with the manufacturer's suggested protocols. Cell acquisition was made using the BD FACS (fluorescence-activated cell sorter) Canto II cytometer (BD Bioscience, San Diego, CA) and analyzed using FlowJo software (BD Bioscence).
ELISAs.
Cytokine (IL-6, IL-10, IL-12, gamma interferon [IFN-γ], tumor necrosis factor alpha [TNF-α], and TGF-β) levels in the serum and cell culture supernatant fluids were determined by sandwich ELISAs using antibody pairs purchased from BD Biosciences according to the manufacturer's suggested protocols. The sensitivities of the cytokine ELISAs range from 7.5 to 31 pg/ml.
Serum collection and measurement of trypanosome-specific antibodies.
Mice were anesthetized by intraperitoneal injection of xylazine (10 mg/kg) and ketamine (150 mg/kg). Blood was collected by cardiac puncture using a 1-ml syringe and 25-G needle. Blood samples were kept at 4°C for 4 h and spun at 2,400 rpm for 10 min, and serum was collected and stored at −20°C until used for antibody determination. Serum levels of Trypanosome-specific IgM and IgG antibodies in infected mice were determined by ELISA as previously described (12, 31).
DNA extraction and PCR.
The QIAamp DNA minikit (Qiagen, Mississauga, ON, Canada) was used to extract DNA from freshly collected whole-blood samples (200 μl each) of infected and uninfected mice according to the manufacturer's suggested protocol. The DNA concentration was estimated from 1 μl of each sample using the NanoVue Plus spectrophotometer (GE Health Care Life Sciences, Mississauga, ON, Canada). PCRs were performed in a 25-μl reaction mixture using Platinum PCR SuperMix (High Fidelity, Invitrogen Grand Island, NY) according to the manufacturer's suggested protocols. The amplification of T. congolense DNA was done using primers specific for a 314-bp satellite DNA of T. congolense (termed GOL) as previously described (32) (forward, 5′-GAGAACGGGCACTTTGCGATTTTC-3′; and reverse, 5′-GACAAACAAATCCCGCACAACCAT-3′). The amplified products were resolved by electrophoresis using a 2% agarose gel and visualized using an Alphaimager (Alpha Innotech, Fisher Scientific, Ottawa, ON, Canada).
Statistics.
Data are presented as means and standard errors of the means (SEM). A two-tailed Student's t test or analysis of variance (ANOVA) was used to compare means and SEM of cytokine production between groups. GraphPad Prism software was used. Differences were considered significant at a P value of <0.05.
RESULTS
Primary low-dose i.d. T. congolense infection and susceptibility to challenge infection due to repeated low-dose infection.
Although BALB/c mice are highly susceptible to intraperitoneal infection with Trypanosoma congolense (14), a recent report showed that these mice are relatively resistant to an intradermal infection route (18). Paradoxically, primary intradermal low-dose infection predisposes to enhanced susceptibility following rechallenge infection (18). However, the mechanisms that regulate low-dose intradermal infection-induced susceptibility are not known. In agreement with a previous report (18), we found that intraperitoneal infection of BALB/c mice with 102 to 103 T. congolense parasites leads to the development of fulminating parasitemia and death within 8 to 10 days (Fig. 1A and B). In contrast, mice infected intradermally with the same dose of parasites did not develop any parasitemia throughout the duration (21 days) of infection. Consistent with a previous report (18), a secondary low-dose (103 parasites) i.d. infection of these mice (which normally does not lead to parasitemia in naive mice) led to the development of fulminating parasitemia and death within 9 to 11 days (Fig. 1D and E). These results suggest that although BALB/c mice are resistance to primary low-dose intradermal infection, such resistance is associated with immunoregulatory mechanisms that predispose to enhanced susceptibility following reinfection.
FIG 1.

Low-dose i.d. T. congolense infection and susceptibility to challenge infection. Groups of BALB/c mice were infected intraperitoneally or intradermally (4 mice per group) with 102 and 103 Trypanosoma congolense (clone TC13) parasites. At indicated time points, parasitemia (A) was monitored by microscopy by counting the number of parasites in the blood taken from the tail vein of infected mice. The survival of infected mice was also determined (B). Note that all mice infected i.d. with 102 and 103 TC13 parasites controlled the infection, while all mice infected i.p. with 102 and 103 parasites developed infection and died within 8 to 10 days postinfection. In another experiment, groups of mice were infected i.d. (C) with 102 T. congolense parasites once a week for 2 weeks (which does not lead to parasitemia). One week after the last parasite injection, mice were reinfected (challenged) intradermally with 103 T. congolense parasites. Four naive age-matched mice were also infected with 103 T. congolense parasites i.d. or i.p. as controls. Parasitemia (D) and survival period (E) were determined. The results presented are representative of 4 different experiments with similar results. Error bars show means ± SEM.
Induction of proinflammatory cytokines after low-dose i.d. infection with T. congolense.
The enhanced susceptibility to experimental intraperitoneal T. congolense infection in mice has been associated with overproduction of proinflammatory cytokines, which leads to cytokine storm and the systemic inflammatory response syndrome (33). Therefore, we compared the levels of proinflammatory cytokines after low-dose i.d. and i.p. T. congolense infections in order to assess whether differences in the production of these cytokines could account for the apparent enhanced resistance to primary i.d. infection. As shown in Fig. 2A to L, intradermal low-dose infection (akin to intraperitoneal infection) was associated with the production of IL-6, IL-10, IL-12, IFN-γ, TNF-α, and TGF-β by spleen (Fig. 2A to F) and draining lymph node (Fig. 2G to L) cells. Interestingly, spleen cells from mice infected intradermally produced comparable levels of these cytokines (including TGF-β), with the exception of IFN-γ and TNF-α, which were significantly higher in the culture supernatant fluids of cells from intraperitoneally infected mice (Fig. 2D and E). In contrast, the draining lymph node cells from intradermally infected mice produced more IL-10 than those from mice infected intraperitoneally (Fig. 2H).
FIG 2.
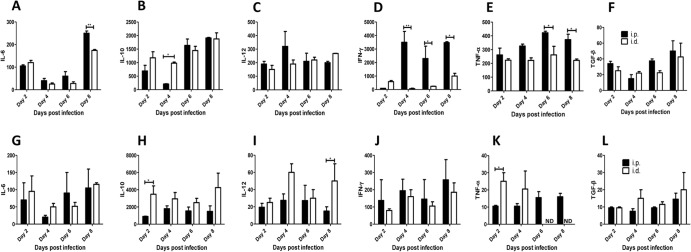
Cytokine profile in mice infected i.d. and i.p. with 103 T. congolense parasites. Groups of mice (4 to 5 per group) were infected either i.d. or i.p. with 103 T. congolense parasites, and at indicated times, the mice were sacrificed and the spleen and draining lymph node cells were cultured in complete medium in the presence of freeze-thawed T. congolense. After 72 h, the levels of IL-6, IL-10, IL-12, IFN-γ, TNF-α, and TGF-β in the cell culture supernatant fluids were determined by ELISA and expressed as change (pg/ml) over levels in cells from uninfected (naive) animals. Panels A to F represent cytokine levels in the spleen, and panels G to L represent cytokine levels in the lymph node. The data presented are representative of 3 different experiments with similar results. Error bars show means ± SEM. *, P < 0.05; **, P < 0.01. ND, not detected (i.e., below the ELISA sensitivity).
A previous report showed that B cells and antibodies do not contribute to the enhanced resistance following intradermal T. congolense infection (18). Consistent with this, we found that primary intradermal infection did not induce significant levels of parasite-specific antibodies (see Fig. S1A and B in the supplemental material). Interestingly, despite the absence of parasitemia, repeated low-dose intradermal infection induced significant levels of parasite-specific antibodies that were comparable to those induced following primary intraperitoneal infection (see Fig. S1A and B). Taken together, these results indicate that despite the absence of parasitemia, low-dose intradermal T. congolense infection results in some level of immune response (induction of antibodies and the production of proinflammatory cytokines by the spleen and lymph node cells). This is in agreement with a previous study that reported priming of the adaptive immune response very early after intradermal infection (18) even in the absence of parasitemia.
Primary low-dose intradermal infection does not cause latent parasitemia but induces durable susceptibility.
Unlike intraperitoneal infection, primary low-dose (102 to 103 parasites) intradermal infection does not lead to parasitemia (Fig. 1A). However, intradermal infection leads to a robust cytokine response (Fig. 2A to L), comparable to intraperitoneal infection, suggesting that it might cause low-level (latent) infection undetectable by the conventional method of assessing parasitemia. To investigate this, we treated low-dose-infected mice with cyclophosphamide (3 injections given at 3-day intervals) after 30 days of primary intradermal infection and monitored them for parasitemia for an additional 4 weeks. Low-dose i.d.-infected mice treated with cyclophosphamide did not develop parasitemia (Fig. 3A). Additionally, we assessed the blood of mice infected by the intradermal route for parasite DNA by PCR. As shown in Fig. 3B, while we could readily amplify parasite DNA in the blood of mice infected i.p., we were unable to amplify parasite DNA in mice infected by the intradermal route at different times after infection. Collectively, these results indicate that primary low-dose i.d. infection is completely cleared by the immune system.
FIG 3.

Primary low-dose intradermal infection-induced susceptibility is durable. BALB/c mice (n = 6) were infected (inf.) intradermally with 102 T. congolense parasites weekly (for 2 consecutive weeks [Fig. 1C]). After 30 days of no detectable parasitemia, some mice (n = 3) were treated three times with cyclophosphamide (at 3-day intervals), while others were challenged (i.d.) with 103 T. congolense parasites and monitored for parasitemia (A). In addition, blood samples from uninfected (naive) or infected mice (i.d. or i.p.) were assessed for the presence of T. congolense DNA by PCR. Parasites purified from the blood of i.p.-infected mice by diethylaminoethyl (DEAE)-cellulose anion-exchange chromatography were used as the positive control (B). In another experiment, BALB/c mice (n = 8) were infected (i.d.) twice with 102 T. congolense parasites (once a week for 2 weeks [Fig. 1C]). After 40 days of no detectable parasitemia, some mice (n = 4) and some age-matched controls (n = 3) were reinfected with 103 T. congolense parasites, and parasitemia (C) and survival (D) were monitored. chall. inf., challenge infection. The results presented are representative of 2 different experiments with similar results. Error bars show means ± SEM.
Next, we investigated whether low-dose intradermal infection-induced susceptibility is durable. We infected mice with 102 T. congolense parasites once a week for 2 weeks, waited for an additional 40 days, and rechallenged them with 103 T congolense parasites. The results presented in Fig. 3 show that low-dose intradermally infected mice were still susceptible to rechallenge infection after 40 days of primary infection, as evidenced by development of fulminating parasitemia (Fig. 3C) and death within 12 to 14 days postchallenge (Fig. 3D). Taken together, these results show that low-dose intradermal infection does not lead to latent infection, and the susceptibility it induces is long lasting.
Induction of regulatory T cells following low-dose injection of mice with T. congolense.
Because we and others have previously shown that regulatory T cells (Tregs) prevent the control of infection with T. congolense (24, 25), we wondered whether low-dose intradermal infection was associated with expansion of these cells. Therefore, we infected mice intradermally with 103 T. congolense parasites and on the indicated days assessed the percentages and absolute numbers of CD4+ CD25+ Foxp3+ regulatory T cells in the spleens and draining lymph nodes by flow cytometry. For comparison, we also included mice infected intraperitoneally in this study. Our results show that despite the absence of parasitemia, the percentages and absolute numbers of Tregs in the spleens (Fig. 4A to D) and lymph nodes (Fig. 4E to H) significantly increased after low-dose intradermal infection akin to intraperitoneal infection. This increased Tregs expansion was sustained such that after 3 weeks of primary intradermal infection, the number of Tregs was similar to that observed after 4 days of primary intradermal infection (Fig. 5A and B). In addition, Tregs were further expanded following secondary low-dose intradermal reinfection (Fig. 5A and B). Collectively, these results show a strong association between intradermal low-dose T. congolense infection and increased numbers of Tregs in the spleen and draining lymph node. These data suggest that the expansion of Tregs in mice after low-dose intradermal injection could play a role in the enhanced susceptibility following challenge infection.
FIG 4.

Low-dose intradermal T. congolense infection leads to systemic expansion of CD4+ CD25+ Foxp3+ cells (Tregs). Groups of mice (4 or 5 mice per group) were infected either i.d. or i.p. with 103 T. congolense parasites. At the indicated days, infected mice were sacrificed, and single-cell suspensions of the spleen and draining lymph nodes were stained with fluorochrome-conjugated antibodies against CD4, CD25, and Foxp3, and the percentages (A, B, C, E, F, and G) and absolute numbers (D and H) of Tregs were assessed by flow cytometry. Panels A, B, E, and F are representative dot plots (percentages and standard deviations [SD]), while panels C and G are line graphs of the means ± SEM of 4 or 5 mice per group. The data presented are representative of 3 different experiments with similar results. Bars show means ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
FIG 5.

Expansion of Tregs following primary intradermal infection is sustained. Eight mice were infected intradermally with 102 T. congolense parasites weekly (for 2 consecutive weeks). One week after the last infection, some mice (n = 4) were sacrificed to determine the baseline levels of Tregs. The remaining mice (n = 4) and some age-matched naive controls were challenged (chall. inf.) intradermally with 103 T. congolense parasites and sacrificed 4 days postchallenge to determine the levels of Tregs. Shown are the percentages of CD4+ CD25+ Foxp3+ cells (Tregs) in the spleens (A) and the lymph nodes (B) draining the infection site, presented as percent fold increase over the level in naive (uninfected) mice. pri. inf., primary infection. The data presented are representative of 2 different experiments with similar results. Error bars show means ± SEM. *, P < 0.05; **, P < 0.01. ns, not significant.
Depletion of Tregs abolished the susceptibility observed following intradermal challenge infection of mice.
We found that low-dose intradermal T. congolense infection of mice led to expansion of Tregs in the spleen and draining lymph nodes of mice infected intradermally (Fig. 4B, C, F, and G; Fig. 5A and B). Because previous studies showed that Tregs negatively influence the outcome of T. congolense infection in mice (24, 25), we hypothesized that depletion of Tregs during challenge infection will abrogate the enhanced susceptibility mediated by repeated low-dose intradermal infection. We therefore injected mice repeatedly with 102 T. congolense parasites (2 times with a 1-week interval), and on the third week injected them with anti-CD25 monoclonal antibody (PC61) to deplete Tregs as we have done previously (25, 30). Twenty-four hours later, mice were challenged intradermally with 103 T. congolense parasites. As shown in Fig. 6A and B, and consistent with our findings (Fig. 1D and E), repeated primary low-dose intradermal infection led to enhanced susceptibility to challenge infection with 103 parasites, with all mice developing parasitemia and dying by day 12 postchallenge infection. In contrast, depletion of CD25+ cells (by anti-CD25 monoclonal antibody treatment) prior to challenge infection completely abolished this susceptibility, such that mice treated with anti-CD25 MAb (but not isotype control) did not develop any parasitemia and survived for over 21 days after challenge infection when the experiment was terminated (Fig. 6A and B). Furthermore, sera from mice treated with anti-CD25 produced significantly smaller amounts of TGF-β (Fig. 6C) and IL-10 (Fig. 6D) compared with the isotype control-treated group. However, the production of TNF-α and IL-6 by these cells did not change (data not shown). Collectively, these results indicate that Tregs expand after low-dose intradermal infection and mediate susceptibility to intradermal challenge infection of mice.
FIG 6.

Depletion of Tregs abolishes intradermal low-dose-enhanced susceptibility to T. congolense infection. Groups of mice (n = 4 or 5) were injected weekly with 102 T. congolense parasites or PBS (for 2 consecutive weeks). On the 3rd week, mice were treated with anti-CD25 MAb PC61 (100 μg/mouse) or the isotype-matched control MAb and rechallenged (chall. inf.) with 103 T. congolense parasites the next day. Daily parasitemia (A) and survival period (B) were monitored as described in Materials and Methods. At the humane endpoint, mice were sacrificed, and sera were assessed for TGF-β (C) and IL-10 (D) by ELISA. In another experiment, groups of mice were injected with anti-CD25 or isotype control MAb (n = 3 or 4 mice/group) at each point of the primary (pri. inf.) low-dose (102 parasites) T. congolense infection (once weekly for 2 weeks). On the 3rd week, the mice were challenged with 103 T. congolense parasites, and parasitemia (E) and survival period (F) were determined. The data presented are representative of 3 (A to D) and 2 (E and F) different experiments with similar results. Bars show means ± SEM. *, P < 0.05; **, P < 0.01. ns, not significant; ND, not detected (i.e., below the ELISA sensitivity).
Depletion of Tregs at each point of repeated low-dose infection before challenge also abolished susceptibility to challenge infection.
We found that repeated low-dose intradermal infection leads to expansion of Tregs, and enhanced susceptibility to challenge infection and depletion of these cells at the time of challenge infection abolished this susceptibility (Fig. 4 and 6). Next, we wondered whether depletion of Tregs at the time of primary low-dose infections would also abolish low-dose-induced susceptibility to challenge infection. We therefore treated mice with anti-CD25 MAb 24 h before each 102-parasite low-dose intradermal infection (once weekly for 2 weeks). On the 3rd week, the mice were challenged with 103 T. congolense parasites to determine the outcome of infection following anti-CD25 MAb treatment. In contrast to isotype-treated controls, anti-CD25 MAb-treated mice remained disease free following challenge infection (Fig. 6E and F). These results strongly suggest that low-dose intradermal infection leads to induction of Tregs that mediate susceptibility to T. congolense challenge infection.
Depletion of macrophages does not alter the course of primary intradermal infection.
Because it was shown that inducible nitric oxide synthase (iNOS)-deficient mice were susceptible to primary intradermal infection with T. congolense, macrophages were proposed to be an important cell type in the innate control of primary intradermal infection (18). Therefore, we investigated the role of macrophages during primary and challenge intradermal infection with T. congolense. Results presented in Fig. 7A show that depletion of macrophages (using chlodronate liposomes) did not alter the resistance observed in primary intradermal infection, as mice depleted of macrophages were still resistant to primary low-dose i.d. infection and survived until the end of the experiment (Fig. 7B). Furthermore, when these mice were challenged after 3 weeks with 103 T. congolense parasites, they developed parasitemia and succumbed to the infection within 13 days, akin to mice that were not depleted of macrophages. Collectively, these results suggest that macrophages are not primarily responsible for the relative resistance to low-dose intradermal T. congolense infection.
FIG 7.
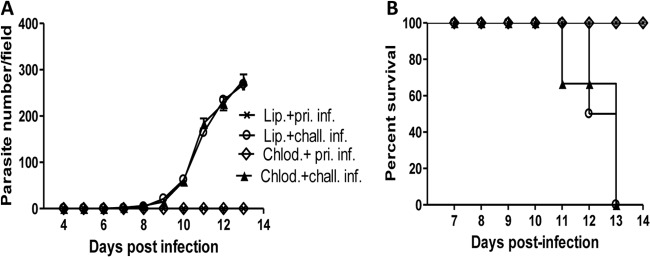
Depletion of macrophages does not abolish primary low-dose i.d. resistance and susceptibility to rechallenge infection. Groups of BALB/c mice (n = 4) were treated with liposomal chlodronate (Chlod.) to deplete macrophages or with control liposome (Lip). After 24 h, all mice were infected i.d. with 102 T. congolense parasites and monitored for parasitemia (A). Three weeks after primary infection (pri. inf.) (without parasitemia), all mice were rechallenged (chall. inf.) with 103 T. congolense parasites, and parasitemia (A) and survival (B) were monitored. The results presented are representative of 2 different experiments with similar results. Error bars show means ± SEM.
DISCUSSION
The primary aim of this study was to investigate the mechanism through which repeated intradermal low-dose infection enhances susceptibility to T. congolense infection of mice. Wei et al. reported that infection of mice with a low number of T. congolense enhanced susceptibility to reinfection (18). However, the mechanism(s) behind this important observation was not addressed. Here, we focused on determining the role of regulatory T cells in this process. We found that despite the absence of parasitemia and disease, primary infection of mice with low-dose T. congolense led to expansion of regulatory T cells in the spleens and lymph nodes draining the infection sites. Depletion of these cells by anti-CD25 MAb treatment during the time of repeated low-dose infection or at the time of secondary reinfection abolished the low-dose-induced susceptibility to T. congolense infection. This was associated with dramatic reductions in serum levels of IL-10 and TGF-β, key cytokines that mediate Treg-induced dampening of immune responses. Collectively, these findings show that intradermal low-dose T. congolense infection leads to expansion of Tregs that mediate the enhanced susceptibility to subsequent low-dose reinfection.
Immunosuppression is a hallmark of African trypanosomiasis. Trypanosome-infected animals (including humans, cattle, and mice) show marked suppression of the immune response to vaccines as well as to T-cell mitogens, including concanavalin A (ConA) and phytohemagglutinin (PHA) (34–43). In addition, infected mice also show a reduced antibody response to sheep red blood cells (SRBC) following immunization (44–48). Several mechanisms have been proposed as the cause of trypanosome-induced immunosuppression, including macrophages (49), suppressor T cells (50), nitric oxide (NO) (51), and Tregs (24, 25). Recent reports (including those from our laboratory) have shown that conventional and naturally occurring Tregs (characterized as CD4+ CD25+ Foxp3+) contribute to enhanced susceptibility to experimental T. congolense infection in mice (24, 25). Depletion of these cells prior to infection significantly enhances the resistance of the highly susceptible BALB/c mice. We found here that intradermal low-dose infection leads to systemic expansion of CD4+ CD25+ Foxp3+ cells, and this was associated with enhanced susceptibility following reinfection. Depletion of these cells completely abolished this susceptibility, directly confirming the role of the expanded population of Tregs in intradermal low-dose infection-induced susceptibility.
How does low-dose intradermal infection lead to induction of Tregs? A previous report shows that low-dose intradermal infection is controlled primarily by NO and TNF-α produced presumably by activated macrophages (innate immunity) (18). However, our results clearly rule out macrophages as the primary mediator of either increased resistance to primary i.d. T. congolense infection or enhanced susceptibility to rechallenge infection following repeated low-dose i.d. infection. There is evidence that natural killer T cells (NKT cells) play a major role in suppressing immunity following intradermal infection (18). It has been proposed that the presentation of trypanosomal glycosylphosphatidylinositol (GPI)-derived antigens by CD1d molecules to a subpopulation of NKT cells leads to alternative activation of macrophages and a concomitant production of large amounts of TGF-β (52). This microenvironment would favor preferential development of Tregs from naive T cells, which in turn, further promotes the alternative activation of macrophages leading to more IL-10 and TGF-β production. It is also conceivable that the expansion of Tregs after low dose intradermal infection is associated with the production of TGF-β and IL-10, which contribute to enhanced susceptibility during challenge infection. In line with this, we found that depletion of Tregs by anti-CD25 MAb treatment was associated with reductions in TGF-β and IL-10 production in infected mice (Fig. 6C and D).
Previous studies (25, 30) showed that the anti-CD25 MAb is effective at depleting mostly CD25+ Foxp3+ Treg cells. Another study found that while this antibody effectively depletes over 80% of Foxp3+ cells, only about 45 to 50% of CD25+ (mostly CD25high) cells were depleted, suggesting that the antibody is highly selective and does not globally deplete CD25+ T cells. This is very significant because CD25 is the high-affinity IL-2 receptor α chain, which is universally expressed and critically important for T-cell responses. Following anti-CD25 MAb treatment, Treg depletion lasts for 7 days in both the spleens and draining lymph nodes, after which repopulation of CD25+ cells is initiated. This suggests that the dose of antibody used in this study is incapable of causing permanent depletion of CD25+ cells, which might inhibit subsequent T-cell activation and contribute to suppressed immunity.
B cells (via the production of antibodies) are important in immunity to experimental African trypanosome infection. B-cell-deficient mice are highly susceptible to various strains of African trypanosomes, and this susceptibility is reversed by passive transfer of VSG-specific antibody or primed B cells (53). Both IgM and IgG antibody subclasses are important in mediating antitrypanosome clearance (54), although IgG antibodies may be qualitatively more important (12, 31). Hence, we investigated whether low-dose intradermal infection elicits a strong antibody response that could account for the enhanced resistance following primary infection. In contrast to a previous report (18), repeated primary intradermal infection elicited significant antibody (IgM and IgG) responses. Despite this antibody response, these mice became susceptible following low-dose rechallenge, suggesting that the antibodies were ineffective at mediating resistance to intradermal T. congolense infection.
In conclusion, we have shown that low-dose intradermal T. congolense infection leads to systemic and sustained expansion of Tregs, which is responsible for mediating enhanced susceptibility to repeated low-dose T. congolense reinfection. Depletion of Tregs by anti-CD25 MAb treatment completely abolished the low-dose intradermal infection-induced susceptibility to secondary reinfection. We speculate that low-dose intradermal induction of Tregs and subsequent enhanced susceptibility may be an evasion strategy by the parasite to enhance its survival and transmission in the host. Thus, it will be interesting to determine whether low-dose intradermal infection-induced expansion of regulatory T cells occurs in other parasitic diseases, like malaria, leishmaniasis, and babesiosis, which are usually transmitted by low-dose injection of the infective stage into the dermis of mammalian hosts by their respective vectors.
Supplementary Material
ACKNOWLEDGMENTS
We thank the members of the Parasite Vaccines Development Laboratory for some technical assistance. We are grateful to Henry Tabel for critically reading the manuscript and providing insightful comments.
Funding for this study was provided by The National Sciences and Engineering Research Council (NSERC) Discovery Grant and The Manitoba Health Research Council Research Chair Award (J.U.).
Footnotes
Published ahead of print 16 December 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01028-13.
REFERENCES
- 1.Simarro PP, Cecchi G, Paone M, Franco JR, Diarra A, Ruiz JA, Fevre EM, Courtin F, Mattioli RC, Jannin JG. 2010. The atlas of human African trypanosomiasis: a contribution to global mapping of neglected tropical diseases. Int. J. Health Geogr. 9:57. 10.1186/1476-072X-9-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stuart K, Brun R, Croft S, Fairlamb A, Gurtler RE, McKerrow J, Reed S, Tarleton R. 2008. Kinetoplastids: related protozoan pathogens, different diseases. J. Clin. Invest. 118:1301–1310. 10.1172/JCI33945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brun R, Don R, Jacobs RT, Wang MZ, Barrett MP. 2011. Development of novel drugs for human African trypanosomiasis. Future Microbiol. 6:677–691. 10.2217/fmb.11.44 [DOI] [PubMed] [Google Scholar]
- 4.Hursey BS. 2001. The programme against African trypanosomiasis: aims, objectives and achievements. Trends Parasitol. 17:2–3. 10.1016/S1471-4922(00)01851-1 [DOI] [PubMed] [Google Scholar]
- 5.Kristjanson PM, Swallow BM, Rowlands GJ, Kruska RL, de Leeuw PN. 1999. Measuring the costs of African animal trypanosomosis: the potential benefits of control and returns to research. Agric. Syst. 59:79–98. 10.1016/S0308-521X(98)00086-9 [DOI] [Google Scholar]
- 6.Magez S, Truyens C, Merimi M, Radwanska M, Stijlemans B, Brouckaert P, Brombacher F, Pays E, De Baetselier P. 2004. P75 tumor necrosis factor-receptor shedding occurs as a protective host response during African trypanosomiasis. J. Infect. Dis. 189:527–539. 10.1086/381151 [DOI] [PubMed] [Google Scholar]
- 7.Nakamura Y, Naessens J, Takata M, Taniguchi T, Sekikawa K, Gibson J, Iraqi F. 2003. Susceptibility of heat shock protein 70.1-deficient C57BL/6 J, wild-type C57BL/6J and A/J mice to Trypanosoma congolense infection. Parasitol. Res. 90:171–174. 10.1007/s00436-003-0844-3 [DOI] [PubMed] [Google Scholar]
- 8.Shi M, Wei G, Pan W, Tabel H. 2006. Experimental African trypanosomiasis: a subset of pathogenic, IFN-gamma-producing, MHC class II-restricted CD4+ T cells mediates early mortality in highly susceptible mice. J. Immunol. 176:1724–1732 [DOI] [PubMed] [Google Scholar]
- 9.Uzonna JE, Kaushik RS, Gordon JR, Tabel H. 1998. Immunoregulation in experimental murine Trypanosoma congolense infection: anti-IL-10 antibodies reverse trypanosome-mediated suppression of lymphocyte proliferation in vitro and moderately prolong the lifespan of genetically susceptible BALB/c mice. Parasite Immunol. 20:293–302 [DOI] [PubMed] [Google Scholar]
- 10.Askonas BA. 1985. Macrophages as mediators of immunosuppression in murine African trypanosomiasis. Curr. Top. Microbiol. Immunol. 117:119–127 [DOI] [PubMed] [Google Scholar]
- 11.Roelants GE, Pinder M. 1984. Immunobiology of African trypanosomiasis. Contemp. Top. Immunobiol. 12:225–274 [DOI] [PubMed] [Google Scholar]
- 12.Uzonna JE, Kaushik RS, Gordon JR, Tabel H. 1999. Cytokines and antibody responses during Trypanosoma congolense infections in two inbred mouse strains that differ in resistance. Parasite Immunol. 21:57–71. 10.1046/j.1365-3024.1999.00202.x [DOI] [PubMed] [Google Scholar]
- 13.Tabel H, Kaushik RS, Uzonna JE. 2000. Susceptibility and resistance to Trypanosoma congolense infections. Microbes Infect. 2:1619–1629. 10.1016/S1286-4579(00)01318-6 [DOI] [PubMed] [Google Scholar]
- 14.Jennings FW, Whitelaw DD, Holmes PH, Urquhart GM. 1978. The susceptibility of strains of mice to infection with Trypanosoma congolense. Res. Vet. Sci. 25:399–400 [PubMed] [Google Scholar]
- 15.Morrison WI, Murray M. 1979. Trypanosoma congolense: inheritance of susceptibility to infection in inbred strains of mice. Exp. Parasitol. 48:364–374. 10.1016/0014-4894(79)90121-8 [DOI] [PubMed] [Google Scholar]
- 16.Barry JD, Emergy DL. 1984. Parasite development and host responses during the establishment of Trypanosoma brucei infection transmitted by tsetse fly. Parasitology 88:67–84. 10.1017/S0031182000054354 [DOI] [PubMed] [Google Scholar]
- 17.Gray AR, Luckins AG. 1980. The initial stage of infection with cyclically-transmitted Trypanosoma congolense in rabbits, calves and sheep. J. Comp. Pathol. 90:499–512. 10.1016/0021-9975(80)90099-7 [DOI] [PubMed] [Google Scholar]
- 18.Wei G, Bull Zhou HX, Tabel H. 2011. Intradermal infections of mice by low numbers of African trypanosomes are controlled by innate resistance but enhance susceptibility to reinfection. J. Infect. Dis. 203:418–429. 10.1093/infdis/jiq051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sakaguchi S. 2004. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu. Rev. Immunol. 22:531–562. 10.1146/annurev.immunol.21.120601.141122 [DOI] [PubMed] [Google Scholar]
- 20.Sakaguchi S, Setoguchi R, Yagi H, Nomura T. 2006. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in self-tolerance and autoimmune disease. Curr. Top. Microbiol. Immunol. 305:51–66. 10.1007/3-540-29714-6_3 [DOI] [PubMed] [Google Scholar]
- 21.Sakaguchi S. 2000. Regulatory T cells: key controllers of immunologic self-tolerance. Cell 101:455–458. 10.1016/S0092-8674(00)80856-9 [DOI] [PubMed] [Google Scholar]
- 22.Sarfo BY, Wilson NO, Bond VC, Stiles JK. 2011. Plasmodium berghei ANKA infection increases Foxp3, IL-10 and IL-2 in CXCL-10 deficient C57BL/6 mice. Malar. J. 10:69. 10.1186/1475-2875-10-69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Okwor I, Liu D, Beverley SM, Uzonna JE. 2009. Inoculation of killed Leishmania major into immune mice rapidly disrupts immunity to a secondary challenge via IL-10-mediated process. Proc. Natl. Acad. Sci. U. S. A. 106:13951–13956. 10.1073/pnas.0905184106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wei G, Tabel H. 2008. Regulatory T cells prevent control of experimental African trypanosomiasis. J. Immunol. 180:2514–2521 [DOI] [PubMed] [Google Scholar]
- 25.Okwor I, Onyilagha C, Kuriakose S, Mou Z, Jia P, Uzonna JE. 2012. Regulatory T cells enhance susceptibility to experimental Trypanosoma congolense infection independent of mouse genetic background. PLoS Negl. Trop. Dis. 6:e1761. 10.1371/journal.pntd.0001761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuriakose S, Muleme HM, Onyilagha C, Singh R, Jia P, Uzonna JE. 2012. Diminazene aceturate (Berenil) modulates the host cellular and inflammatory responses to Trypanosoma congolense infection. PLoS One 7:e48696. 10.1371/journal.pone.0048696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tabel H. 1982. Activation of the alternative pathway of bovine complement by Trypanosoma congolense. Parasite Immunol. 4:329–335. 10.1111/j.1365-3024.1982.tb00444.x [DOI] [PubMed] [Google Scholar]
- 28.Lanham SM, Godfrey DG. 1970. Isolation of salivarian trypanosomes from man and other mammals using DEAE-cellulose. Exp. Parasitol. 28:521–534. 10.1016/0014-4894(70)90120-7 [DOI] [PubMed] [Google Scholar]
- 29.Herbert WJ, Lumsden WH. 1976. Trypanosoma brucei: a rapid “matching” method for estimating the host's parasitemia. Exp. Parasitol. 40:427–431. 10.1016/0014-4894(76)90110-7 [DOI] [PubMed] [Google Scholar]
- 30.Okwor I, Muleme H, Jia P, Uzonna JE. 2009. Altered proinflammatory cytokine production and enhanced resistance to Trypanosoma congolense infection in lymphotoxin beta-deficient mice. J. Infect. Dis. 200:361–369. 10.1086/599792 [DOI] [PubMed] [Google Scholar]
- 31.Morrison WI, Roelants GE, Mayor-Withey KS, Murray M. 1978. Susceptibility of inbred strains of mice to Trypanosoma congolense: correlation with changes in spleen lymphocyte populations. Clin. Exp. Immunol. 32:25–40 [PMC free article] [PubMed] [Google Scholar]
- 32.Pereira de Almeida PJ, Ndao M, Goossens B, Osaer S. 1998. PCR primer evaluation for the detection of trypanosome DNA in naturally infected goats. Vet. Parasitol. 80:111–116. 10.1016/S0304-4017(98)00205-2 [DOI] [PubMed] [Google Scholar]
- 33.Kaushik RS, Uzonna JE, Zhang Y, Gordon JR, Tabel H. 2000. Innate resistance to experimental African trypanosomiasis: differences in cytokine (TNF-alpha, IL-6, IL-10 and IL-12) production by bone marrow-derived macrophages from resistant and susceptible mice. Cytokine 12:1024–1034. 10.1006/cyto.2000.0685 [DOI] [PubMed] [Google Scholar]
- 34.Uzonna JE, Kaushik RS, Gordon JR, Tabel H. 1998. Experimental murine Trypanosoma congolense infections. I. Administration of anti-IFN-gamma antibodies alters trypanosome-susceptible mice to a resistant-like phenotype. J. Immunol. 161:5507–5515 [PubMed] [Google Scholar]
- 35.Uzonna JE, Kaushik RS, Zhang Y, Gordon JR, Tabel H. 1998. Experimental murine Trypanosoma congolense infections. II. Role of splenic adherent CD3+Thy1.2+ TCR-alpha beta- gamma delta- CD4+8- and CD3+Thy1.2+ TCR-alpha beta- gamma delta- CD4-8- cells in the production of IL-4, IL-10, and IFN-gamma and in trypanosome-elicited immunosuppression. J. Immunol. 161:6189–6197 [PubMed] [Google Scholar]
- 36.Sileghem M, Darji A, Hamers R, Van de Winkel M, De Baetselier P. 1989. Dual role of macrophages in the suppression of interleukin 2 production and interleukin 2 receptor expression in trypanosome-infected mice. Eur. J. Immunol. 19:829–835. 10.1002/eji.1830190508 [DOI] [PubMed] [Google Scholar]
- 37.Sileghem M, Darji A, Remels L, Hamers R, De Baetselier P. 1989. Different mechanisms account for the suppression of interleukin 2 production and the suppression of interleukin 2 receptor expression in Trypanosoma brucei-infected mice. Eur. J. Immunol. 19:119–124. 10.1002/eji.1830190119 [DOI] [PubMed] [Google Scholar]
- 38.Sileghem M, Flynn JN. 1992. Suppression of interleukin 2 secretion and interleukin 2 receptor expression during tsetse-transmitted trypanosomiasis in cattle. Eur. J. Immunol. 22:767–773. 10.1002/eji.1830220321 [DOI] [PubMed] [Google Scholar]
- 39.Sternberg J, McGuigan F. 1992. Nitric oxide mediates suppression of T cell responses in murine Trypanosoma brucei infection. Eur. J. Immunol. 22:2741–2744. 10.1002/eji.1830221041 [DOI] [PubMed] [Google Scholar]
- 40.Pearson TW, Roelants GE, Pinder M, Lundin LB, Mayor-Withey KS. 1979. Immune depression in trypanosome-infected mice. III. Suppressor cells. Eur. J. Immunol. 9:200–204. 10.1002/eji.1830090306 [DOI] [PubMed] [Google Scholar]
- 41.Kierszenbaum F, Muthukkumar S, Beltz LA, Sztein MB. 1991. Suppression by Trypanosoma brucei rhodesiense of the capacities of human T lymphocytes to express interleukin-2 receptors and proliferate after mitogenic stimulation. Infect. Immun. 59:3518–3522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Borowy NK, Sternberg JM, Schreiber D, Nonnengasser C, Overath P. 1990. Suppressive macrophages occurring in murine Trypanosoma brucei infection inhibit T-cell responses in vivo and in vitro. Parasite Immunol. 12:233–246. 10.1111/j.1365-3024.1990.tb00951.x [DOI] [PubMed] [Google Scholar]
- 43.Murray PK, Jennings FW, Murray M, Urquhart GM. 1974. The nature of immunosuppression in Trypanosoma brucei infections in mice. II. The role of the T and B lymphocytes. Immunology 27:825–840 [PMC free article] [PubMed] [Google Scholar]
- 44.Askonas BA, Corsini AC, Clayton CE, Ogilvie BM. 1979. Functional depletion of T- and B-memory cells and other lymphoid cell subpopulations during trypanosomiasis. Immunology 36:313–321 [PMC free article] [PubMed] [Google Scholar]
- 45.Goodwin LG, Green DG, Guy MW, Voller A. 1972. Immunosuppression during trypanosomiasis. Br. J. Exp. Pathol. 53:40–43 [PMC free article] [PubMed] [Google Scholar]
- 46.Hudson KM, Byner C, Freeman J, Terry RJ. 1976. Immunodepression, high IgM levels and evasion of the immune response in murine trypanosomiasis. Nature 264:256–258. 10.1038/264256a0 [DOI] [PubMed] [Google Scholar]
- 47.Sacks DL, Selkirk M, Ogilvie BM, Askonas BA. 1980. Intrinsic immunosuppressive activity of different trypanosome strains varies with parasite virulence. Nature 283:476–478. 10.1038/283476a0 [DOI] [PubMed] [Google Scholar]
- 48.Wellhausen SR, Mansfield JM. 1979. Lymphocyte function in experimental African trypanosomiasis. II. Splenic suppressor cell activity. J. Immunol. 122:818–824 [PubMed] [Google Scholar]
- 49.Grosskinsky CM, Askonas BA. 1981. Macrophages as primary target cells and mediators of immune dysfunction in African trypanosomiasis. Infect. Immun. 33:149–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jayawardena AN, Waksman BH. 1977. Suppressor cells in experimentally trypanosomiasis. Nature 265:539–541. 10.1038/265539a0 [DOI] [PubMed] [Google Scholar]
- 51.Schleifer KW, Mansfield JM. 1993. Suppressor macrophages in African trypanosomiasis inhibit T cell proliferative responses by nitric oxide and prostaglandins. J. Immunol. 151:5492–5503 [PubMed] [Google Scholar]
- 52.Tabel HWG, Bull H. 2012. Immunosuppression, induced upon intradermal infections, causes failures of vaccines against African trypanosomiases. Western College of Veterinary Medicine, University of Saskatchewan, Saskatoon, Saskatchewan, Canada: http://www.usask.ca/wcvm/departments/microbiology/African%20trypanosomiasis%20vaccine%20II%2015-9-2012.pdf [Google Scholar]
- 53.Campbell GH, Phillips SM. 1976. Adoptive transfer of variant-specific resistance to Trypanosoma rhodesiense with B lymphocytes and serum. Infect. Immun. 14:1144–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dempsey WL, Mansfield JM. 1983. Lymphocyte function in experimental African trypanosomiasis. V. Role of antibody and the mononuclear phagocyte system in variant-specific immunity. J. Immunol. 130:405–411 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.