

Abstract
Neisseria meningitidis, the causative agent of meningitis and septicemia, attaches to and invades various cell types. Both steps induce and/or require tyrosine phosphorylation of host cell proteins. Here, we used a phospho array platform to identify active receptor tyrosine kinases (RTKs) and key signaling nodes in N. meningitidis-infected brain endothelial cells to decipher RTK-dependent signaling pathways necessary for bacterial uptake. We detected several activated RTKs, including the ErbB family receptors epidermal growth factor receptor (EGFR), ErbB2, and ErbB4. We found that pharmacological inhibition and genetic ablation of ErbB receptor tyrosine phosphorylation and expression resulted in decreased bacterial uptake and heterologous expression of EGFR, ErbB2, or ErbB4 in Chinese ovary hamster (CHO-K1) cells, which do not express of EGFR and ErbB4; the decrease caused a significant increase in meningococcal invasion. Activation of EGFR and ErbB4 was mediated by transactivation via the common ligand HB-EGF (heparin-binding EGF-like ligand), which was significantly elevated in infected cell culture supernatants. We furthermore determined that N. meningitidis induced phosphorylation of EGFR at Tyr845 independent of ligand binding, which required c-Src activation and was involved in mediating uptake of N. meningitidis into eukaryotic cells. Increased uptake was repressed by expression of EGFR Y845F, which harbored a point mutation in the kinase domain. In addition, activation of ErbB4 at its autophosphorylation site, Tyr1284, and phosphorylation of ErbB2 Thr686 were observed. Altogether, our results provide evidence that EGFR, ErbB2, and ErbB4 are activated in response to N. meningitidis infection and shed new light on the role of ErbB signaling in meningococcal infection biology.
INTRODUCTION
Neisseria meningitidis is a common colonizing bacterium of the human nasopharynx and can be found in 8 to 20% of healthy individuals (1). In rare cases, N. meningitidis overcomes the epithelial barrier and enters the bloodstream, followed by a severe septicemia or by an acute purulent meningitis (2). To cross cellular barriers, N. meningitidis has evolved with the ability to attach to and invade into a variety of cell types. N. meningitidis interacts with host cells by using several microbial structures, including type IV pili (TfP), the outer membrane adhesion proteins Opa and Opc, and the newly identified minor adhesion or adhesion-like proteins that mediate binding to different receptors (3–8). Subsequently, binding to receptors enables the pathogen to exploit the endocytotic capacity of the receptor to promote its internalization. In addition to the engagement of a specific receptor, microorganisms might indirectly activate signal transduction pathways and co-opt receptor signal transduction mechanisms to induce host cell signaling pathways that in turn lead to cytoskeleton rearrangements and bacterial uptake.
It has been established that N. meningitidis can signal through receptor tyrosine kinases (RTKs) and non-RTKs to promote their uptake into eukaryotic cells (9–11). RTKs catalyze the transfer of the γ-phosphate of ATP to the hydroxyl group of tyrosines on target proteins (12). A search of the human genome sequence has determined that 58 of the 90 tyrosine kinase sequences are RTKs and 32 are the nonreceptor type (13). Among all RTKs, the ErbB family (also called type I RTKs) is the prototypic member of the RTK superfamily. ErbB receptors play a crucial role in cell proliferation, differentiation, and motility and are expressed with different distributions and intensities in a variety of tissues. The ErbB receptor family includes four homologous members: EGFR (epidermal growth factor receptor, also termed ErbB1), ErbB2 (HER2/Neu), ErbB3 (HER3), and ErbB4 (HER4).
As is common for RTKs, ErbB receptors consist of a single membrane-spanning region, a cytoplasmatic region, and an extracellular ligand-binding domain (14, 15). Thirteen different EGF-related peptide growth ligand factors are currently known, including EGF, transforming growth factor-α, heparin-binding EGF-like ligand (HB-EGF), amphiregulin, betacellulin, epiregulin, epigen, and neuregulin (NRG) family members (16, 17). Ligand binding leads to homo- or heterodimer formation and activation of the intrinsic kinase domain, resulting in autophosphorylation of specific tyrosine residues within the cytoplasmatic domain. These phosphorylated residues then serve as docking sites for adapter molecules containing Src homology 2 (SH2) domains and phosphotyrosine-binding (PTB) domains, which include Shc, Grb2, and the p85 subunit of phosphatidylinositol kinase (18–20). This leads to activation of signaling pathways, such as the mitogen-activated protein kinase pathway and the S6 kinase cascade. EGFR and ErbB4 are fully functional RTKs, whereas ErbB2 has no endogenous ligand, suggesting that ErbB2 acts predominantly as a coreceptor (21). ErbB3 has a structurally impaired catalytic site and shows only residual kinase activity. Although the latter are “nonautonomous,” both ErbB2 and ErbB3 form heterodimers with other ErbB receptors that are capable of generating potent intracellular signals.
A total of 89 cytosolic tyrosine residues are found in the four ErbB members, with EGFR being the receptor of the ErbB family with the highest percentage of tyrosine residues and several binding sites for adapter proteins (22). In general, phosphorylation at the protein kinase domain is important for the regulation of its catalytic activity of the kinase. However, tyrosine phosphorylation of EGFR at residue 845 is not required for the regulation of the catalytic activity of EGFR (23), but it stabilizes the active conformation of the kinase domain. Interestingly, phosphorylation of EGFR at Tyr845 in the kinase domain is mediated by integrin engagement and known to involve c-Src activity (24).
The ErbB receptor family has been found to be important for interactions of viruses and bacteria with host cells. For example, EGFR mediates the uptake of cytomegalovirus by monocytes and of influenza A virus or Salmonella typhimurium by epithelial cells (25–27). Pseudomonas aeruginosa and Helicobacter pylori transactivate EGFR through HB-EGF ectodomain shedding to prevent host cell apoptosis (28–30), and Pasteurella multocida transactivates EGFR via G-protein-dependent mechanisms for inducing cell proliferation (31). In addition, Mycobacterium leprae activates ErbB2 to stimulate neuronal demyelination (32). A role of ErbB receptors in meningococcal pathogenesis has been recognized. It has been reported that EGFR clusters in A431, Chang, and HEC-1B cells underneath meningococcal microcolonies, a process that is dependent on type IV pilus expression (33). In addition, Hoffmann et al. reported ErbB2 clustering and activation in brain endothelial cells (11). The related species Neisseria gonorrhoeae has been shown to induce accumulation of both EGFR and ErbB2 at the site of bacterial attachment. Activation of EGFR is mediated through HB-EGF ectodomain shedding and is involved in uptake of N. gonorrhoeae by epithelial cells (34).
In this study we now show that infection of human brain-derived endothelial cells results in activation of EGFR, ErbB2, and ErbB4. Pharmacological inhibition and genetic ablation by RNA interference (RNAi) of these kinases result in decreased bacterial uptake. On the other hand, heterologous expression of EGFR, ErbB2, or ErbB4 in CHO-K1 cells causes a significant increase of meningococcal invasion. We demonstrate that phosphorylation of EGFR and ErbB4 is mediated by transactivation of their common ligand, HB-EGF. We furthermore demonstrate activation of three specific phosphorylation sites of EGFR, ErbB2, and ErbB4 and reveal that phosphorylation of EGFR at Tyr845 and phosphorylation of ErbB4 at Tyr 1284 are involved in mediating uptake of N. meningitidis into the eukaryotic cell.
MATERIALS AND METHODS
Bacteria strains and growth conditions.
N. meningitidis serogroup B strain MC58 (B:15: P1.7,16b) (Opc+ Opa+, class I pili) and unencapsulated mutant strain N. meningitidis MC58 siaD have been described previously (8). All strains were routinely cultured in proteose-peptone medium supplemented with 1% Polyvitex (bioMérieux, Lyon, France) to the mid-logarithmic phase and diluted to approximately 1 × 107 CFU in cell culture medium supplemented with 10% heat-inactivated (30 min at 56°C) pooled normal human serum for all cell culture experiments.
Cell lines.
Human brain microvascular endothelial cells (HBMEC) were kindly provided by K. S. Kim (Baltimore, MD) and were cultured as described recently (9). CHO-K1 cells (ATCC CRL-9618) were kindly provided by Thomas Rudel. CHO-K1 cells were cultured on gelatin-coated (0.1% in phosphate-buffered saline [PBS]; Biochrome AG, Berlin, Germany) cell culture dishes or flasks in Ham's F-12 nutrient mixture with Glutamax (Invitrogen, Carlsbad, CA) supplemented with 10% fetal calf serum (FCS). All cell lines were incubated at 37°C and 5% CO2.
Infection experiments and gentamicin protection assay.
For invasion assays, HBMEC and CHO-K1 cells were seeded onto 24-well tissue culture plates (Corning Costar) at a density of 5 × 104 cells and were grown to a concentration of ≈1 × 105 prior to infection. Cells were infected with bacteria at a multiplicity of infection (MOI) of 30 in the presence of the appropriate culture medium with 10% human serum (HS). After 4 h of infection, unbound bacteria were washed away and the number of total cell-associated bacteria was determined by lysis of cells with 1% saponin for 15 min and subsequent determination of the number of CFU by plating appropriate dilutions of the lysates on blood agar. The numbers of intracellular bacteria were determined after 2 h of incubation with cell culture medium supplemented with 200 μg/ml gentamicin (Biochrom, Berlin, Germany). The proportions of adhesive and invasive bacteria were calculated as the ratio of the number of intracellular bacteria versus the number of total associated bacteria. Results with control cells were set to 100%, and the results were calculated as the percentage of the control. All samples were tested in duplicate, and experiments were repeated at least three times. Bacterial viability was not affected by either saponin, lysis, or addition of inhibitors, as measured by colony plating. Effects of the inhibitors on cell viability were assessed by the trypan exclusion method (data not shown).
Inhibitors, EGF, and antibodies.
EGFR inhibitor II (BIBX1382), ErbB2 inhibitor II, the tyrosine kinase inhibitor PP2, phorbol-12-myristate-13-acetate (PMA), and the diphtheria toxin CRM197 mutant were purchased from Calbiochem (La Jolla, CA). The EGFR/ErbB2/ErbB4 inhibitor Pelitinib (EKB-569) was obtained from Selleck Chemicals (Houston, TX). All inhibitors were reconstituted and stored as recommended. Inhibitors were added to the cells 1 h prior to infection and were present during the experimental period. EGF was purchased from Sigma Chemie GmbH (Steinheim, Germany), reconstituted in 1× PBS–10% FCS, and stored at −20°C. Recombinant EGF was as also purchased from Sigma Chemie GmbH.
The following antibodies were used for Western blotting: monoclonal antibody (MAb) phosphotyrosine antibody p-Tyr-100 (9411), MAb EGF receptor (D38B1 XP), MAb ErbB2 (29D8), and polyclonal antibody (pAB) β-actin (all from Cell Signaling Technology, Danvers, MA), pAB p-EGFR (Tyr845) and MAb ErbB4 (C-7) (all from Santa Cruz Biotechnology, Santa Cruz, CA), anti-phospho-ErbB2 (Thr686) from Millipore (Billerica, MA), and MAb ErbB4 Phospho (pY1284) from Epitomics (Burlingame, CA). The MAb anti-EGFR (Ab-1) from Calbiochem (La Jolla, CA), MAb anti-human ErbB4 from US Biological (Swampscott, MA), and Neu (C-3) from Santa Cruz Biotechnology (Santa Cruz, CA) were used for flow cytometry, immunfluorescence, and neutralization studies. For neutralization studies, cells were incubated with antibody 1 h prior to infection, and neutralizing antibody remained during infection at a final concentration of 5 μg/ml.
DNA expression plasmids, small interfering RNAs (siRNAs), and transfection of cells.
Expression constructs encoding EGFR (Addgene plasmid 11011) (35), ErbB2 (Addgene plasmid 16257) (36), and ErbB4 (Addgene plasmid 29527) (37) were kindly provided by M. Meyerson, M.-C. Hung, and Y. Samuels. Expression constructs for wild-type Src and Src K297M were kindly provided by David D. Schlaepfer (University of California at San Diego, La Jolla, CA). pcDNA3.1 was purchased from Invitrogen (Carlsbad, CA) and was used as the negative control in infection experiments.
Expression constructs encoding EGFR and ErbB4 (Addgene plasmids 11011 and 29527) were used as the wild type or modified by site-directed mutagenesis to introduce the EGFR Y845F and ErbB4 Y1284F mutations. Mutations were verified by sequence analysis.
For transfection experiments, CHO-K1 cells were seeded onto 24-well tissue culture plates, grown to semiconfluence, and transfected with 1 μl transfection reagent and 0.5 μl booster reagent from the NovaCHOice transfection kit (EMD Millipore, Billerica, MA). Control nonsilencing siRNA and siRNA against EGFR, ErbB2, and ErbB4 were synthesized by Santa Cruz Biotechnology (Santa Cruz, CA). siRNA transfections were carried out as described recently (38). Protein knockdown efficiencies were verified by immunoblotting 72 h after transfection.
PathScan RTK antibody array kit.
To analyze the receptor tyrosine kinases involved in meningococcal infection, the PathScan RTK signaling antibody array kit from Cell Signaling Technology (Danvers, MA) was used. The PathScan RTK signaling array kit is based on a chemiluminescent sandwich enzyme-linked immunosobent assay (ELISA) format and contains 39 fixed antibodies against phosphorylated forms of kinases and key signaling nodes, and it was used according to the manufacturer's recommendation. HBMEC were infected with N. meningitidis at an MOI of 30 and were washed at 3 h postinfection (p.i.). HBMEC treated with 100 ng EGF for 30 min served as the positive control, and cells without bacteria were the negative control. Cells were lysed in nondenaturing lysis buffer (purchased with the PathScan RTK signaling antibody array kit and containing 20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM Na2 EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM beta-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin) and a protease inhibitor cocktail tablet (Roche Diagnostics, Mannheim, Germany). To quantify cell (HBMEC) protein load, a probe of each sample was combined with 2× SDS sample buffer, boiled, and used for a β-actin alignment using Western blot analysis. Cell lysates were adjusted to 1 μg/ml. Further staining, washing, and development were carried out according to the manufacturer's instructions. Images were analyzed with ImageJ software (http://rsbweb.nih.gov/ij/) by loading the image as a gray-scale picture. Each kinase array dot was manually selected, and an average intensity for each kinase was calculated. Normalization was done by subtracting the intensity of the negative control dot from each value. For comparisons of different infection experiments, sets were normalized so that the positive controls had equal intensities.
Human EGFR phosphorylation array C1.
For screening of additional phosphorylation sites of EGFR and search of ErbB4 activation, the human EGFR phosphorylation antibody array C1 from RayBio (Norcross, GA) was employed according to the manufacturer's instructions. Images were analyzed with ImageJ software as outlined for the PathScan RTK antibody array kit.
RT-PCR.
HBMEC were infected with bacteria, and total RNA was extracted with the RNeasy minikit (Invitrogen, Carlsbad, CA) at the time points indicated below. RNA was converted to cDNA with SuperScript II (Invitrogen) according to the manufacturer‘s protocol. The RNA was digested with RNase, and cDNA was purified with the PCR purification kit (Invitrogen). Five nanograms of cDNA was amplified with 1× Power SYBR green master mix (Applied Systems, Darmstadt, Germany) and 25 μM each primer in the StepOnePlus cycler (Applied Systems) under the following conditions: 50°C for 2 min, 95°C for 2 min, followed by 40 cycles with 95°C for 15 s, 56°C for 15 s, and 72°C for 1 min. The sequences of the HB-EGF primers used were 5′-GAGGAGCACGGGAAAAGAA-3′ and 5′-CCGGAGCTCCTTCACATATT-3′. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was amplified as the internal control for normalization with primers 5′-GCACCGTCAAGGCTGAGAAC-3′ and 5′-ATGGTGGTGAAGACGCCAGT-3′.
HB-EGF ELISA.
To analyze release of secretory HB-EGF during infection, cells were infected with N. meningitidis as described above. After the indicated time points, cell supernatants were discarded and cells were washed with 1.5 M NaCl–1× PBS–1% bovine serum albumin (BSA) to dissolve heparin-bound HB-EGF. Secretory HB-EGF levels were then quantified from the wash buffer using human HB-EGF ELISA (Sigma Chemie GmbH, Steinheim, Germany).
Immunoblotting and immunoprecipitation (IP).
Expression levels of ErbB family members were examined by using cell extracts prepared from transfected cells as described recently (38). Membranes were probed with primary antibody overnight at 4°C. The intensities of bands on the Western blot were quanitified by using ImageJ software. The area under the curve (AUC) of the specific signal was corrected for the AUC of the β-actin loading control.
For immunoprecipitation, the Pierce cross-link magnetic IP/co-IP kit from Santa Cruz Biotechnology was used according to the manufacturer‘s protocol. In brief, cells were lysed in lysis buffer containing a protein inhibitor cocktail tablet, and supernatants were incubated with anti-EGFR, anti-ErbB2, and anti-ErbB4 antibody-coated magnetic beads. After washing, bound proteins were dissolved from the beads and protein concentrations were quantified by using the Lowry method with BSA as the standard. Equal protein concentrations were applied in Western blot analyses, and membranes were incubated with the appropriate ErbB antibodies and secondary antibodies as described above. To analyze ErbB phosphorylation, membranes were stripped and counterstained with the MAb phosphotyrosine antibody p-Tyr-100.
Flow cytometry analyses.
For flow cytometry analyses, CHO-K1 cells were released from confluent monolayer cultures with trypsin, washed, and resuspended in fluorescence-activated cell sorting (FACS) buffer (5% FCS–0.1% sodium azide in PBS). A total of 1 × 106 cells were added to each vial and incubated with the indicated ErbB antibody (each at 1:200) for 90 min on ice. Cells were washed twice and were resuspended in FACS buffer. Subsequently, cells were incubated with a tetramethyl rhodamine isothiocyanate (TRITC)-conjugated secondary anti-mouse antibody (1:500) and washed twice after 30 min. The analysis was performed using CellQuest Pro software (version 5.2) with a FACSCalibur apparatus (Becton, Dickinson, Heidelberg, Germany). Positive fluorescence was determined using a 4-decade log scale, and TRITC fluorescence (FL4) was expressed as the mean channel number for 10,000 cells.
Confocal microscopy.
For colocalization analysis, HBMEC were seeded on glass coverslips and were grown to semiconfluence. Cells were infected with N. meningitidis at an MOI of 30. After infection, cells were washed once in PBS and fixed with 3.7% paraformaldehyde–PBS for 20 min. Fixed cells were washed three times with PBS. Samples were blocked in blocking buffer (2% FCS–PBS) for 1 h and then incubated with ErbB antibodies (anti-EGFR, anti-ErbB2, and ErbB4, all at a 1:50 dilution) and antimeningococcal serum (1:50) for another 1 h. Cells were washed three times for 5 min with PBS and incubated with Cy3- or Cy5-conjugated secondary antibodies (diluted 1:200 in blocking buffer) for 1 h at room temperature. After three washes in PBS, coverslips were mounted in quick-hardening mounting medium (Fluka, Steinheim, Germany). Colocalization was monitored with a TCS SP5 confocal laser scanning microscope (Leica, Wetzlar, Germany) with a 63×, 1.4-numerical aperture Plapo oil immersion objective lens. Fluorescence signals of labeled specimens (ErbB receptor and bacteria) were serially recorded with appropriate excitation wavelengths and emission bands for Cy3 or Cy5, respectively, to avoid bleedthrough. Images were processed with LAS AF (Leica) and ImageJ software and documented using Adobe Photoshop CS.
Statistical analysis.
A two-tailed Student's t test was used to calculate statistical significance. P values of ≤0.05 were considered significant; P values of ≤0.01 were considered highly significant.
RESULTS
Survey of active RTKs in human brain-derived endothelial cells infected with N. meningitidis.
We have recently shown that N. meningitidis invades human nonprofessional phagocytes, such as endothelial cells, by endocytosis, a process that involves activation of non-RTKs (10, 38). In our study, we aimed to analyze the capacity of N. meningitidis to alter the main RTK pathways to support internalization into HBMEC. Therefore, an activity assay using the commercially available PathScan RTK signaling antibody array kit was conducted; the kit includes 39 different RTKs, including EGFR, ErbB2, and ErbB3, non-receptor tyrosine kinases (e.g., Src, Lck), and key signaling nodes (e.g., Erk1/2, Akt, and S6). HBMEC were infected with N. meningitidis MC58 and an unencapsulated, more invasive isogenic mutant, MC58 siaD, for 3 h. In strain MC58, the serogroup B capsule has an inhibitory determinant that affects bacterial invasion of endothelial cells. The unencapsulated mutant strain was therefore included in this screen to search for differences of the phosphorylation pattern of RTKs in HBMEC due to the higher invasive capacity of strain N. meningitidis MC58 siaD. A total protein concentration of 1 μg/ml per sample was applied. Untreated cells and cells treated with 100 ng EGF served as controls. Figure 1 shows the kinases for which we obtained an antibody signal after normalization to the negative control signal. The highest RTK phosphorylation signals were obtained relative to background for tyrosine-related kinase B receptor (TrkB), FMS-like tyrosine kinase receptor 3 (FLT-3), ephrin receptors (EphA3 and EphB1), tyrosine-protein kinase UFO (Axl), and insulin receptor (InsR). Moreover, a significant increase of phosphorylation of key signaling proteins was observed with Akt (pT308 and pS473), extracellular signal-regulated kinases (Erk1/2 [pT202/pY204]), ribosomal S6 kinase (pS235/236), Abelson murine leukemia viral oncogene homolog 1 (c-Abl), and insulin receptor substrate 1 (IRS-1). Interestingly, detection of c-Src activation with the PathScan RTK signaling antibody array confirmed the data of our previous study, in which we demonstrated the important role of c-Src in meningococcal uptake into HBMEC (10). Since c-Src mediates EGFR phosphorylation followed by EGFR activation and with regard to our previous study, we decided to investigate the role of ErbB receptors and the synergism between c-Src and EGFR in meningococcal internalization in this study (39, 40).
FIG 1.
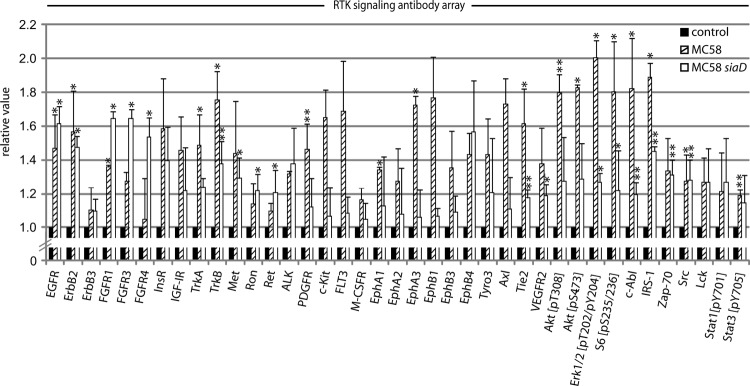
Evidence for phosphorylation of RTKs and key signal proteins in N. meningitidis-infected HBMEC. The PathScan RTK signaling antibody array kit was utilized to analyze the phosphorylated signal levels of kinases and key signaling nodes (see Materials and Methods). HBMEC were infected with MC58 or MC58 siaD at an MOI of 30 or were left uninfected (control), and cell lysates were collected after 3 h. Dot densities were analyzed with ImageJ software. The values for control cells were set as 1, and the values of MC58 and MC58 siaD are shown relative to the control. Results represent mean values ± standard deviations of three independent experiments. *, P < 0.05; **, P < 0.01 relative to untreated cells.
N. meningitidis activates the receptor tyrosine kinases EGFR, ErbB2, and ErbB4.
Interestingly, we observed significant phosphorylation signals for EGFR and ErbB2, but only a slight phosphorylation signal for ErbB3, in HBMEC cell lysates infected with N. meningitidis (Fig. 1 and 2A). Cells infected with bacteria revealed phosphorylation levels comparable to those for cells treated with EGF as a control (Fig. 2A). As ErbB4 was not included in this array, a second ELISA, the commercially available human EGFR phosphorylation antibody array C1′, was carried out (Fig. 2B). HBMEC were infected as described above with both N. meningitidis strains, and a 1 μg/ml total protein concentration was applied. As shown in Fig. 2B, an increased phosphorylation signal for ErbB4 was detected in infected cells. Cells treated with EGF did not show significant activation of ErbB4. Since no significant phosphorylation of ErbB3 could be detected in N. meningitidis-infected HBMEC, further analysis focused on the role of EGFR, ErbB2, and ErbB4 in the infection process.
FIG 2.
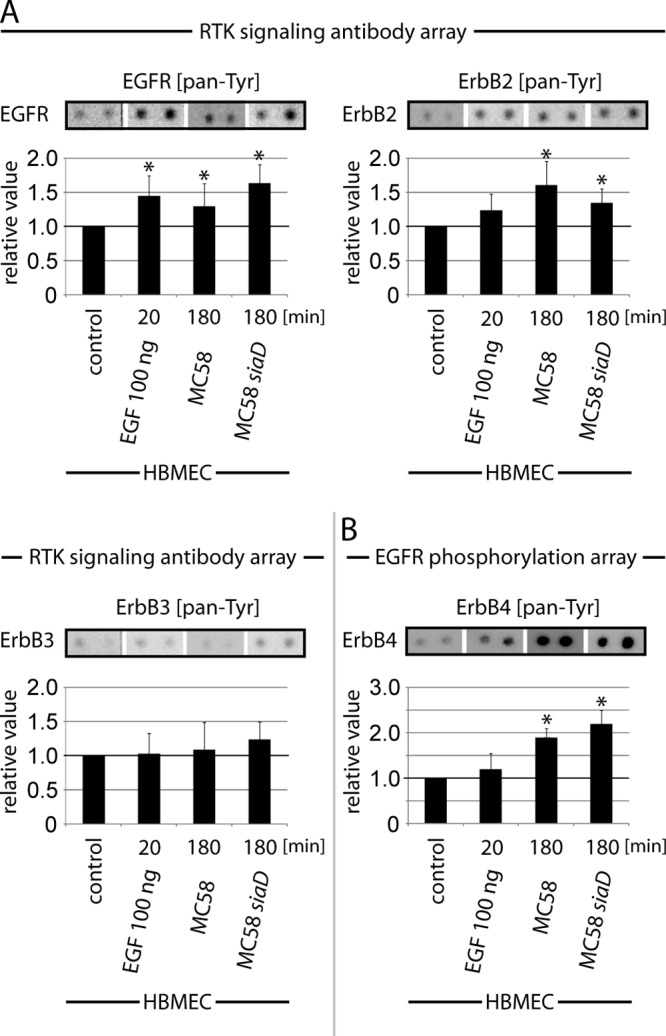
N. meningitidis infection causes activation of EGFR, ErbB2, and ErbB4. HBMEC either were left uninfected (control) or were incubated for 20 min with 100 ng/ml EGF (positive control) or infected for 180 min with N. meningitidis strain MC58 or MC58 siaD, and then cell lysates were analyzed using the PathScan RTK signaling antibody array (A) or the human EGFR phosphorylation antibody array (B) as described in Materials and Methods. A chemiluminescent film image (upper panel) and the quantification of that image (lower panel) are shown. The chemiluminescent array images were captured following 60-s film exposures. Dots were quantified by densitometric analysis using ImageJ. Graphs represent data from three independent experiments. *, P < 0.05.
EGFR, ErbB2, and ERbB4 accumulate at the site of bacterial attachment.
EGFR has been shown to cluster at the site of bacterial attachment in A431, Chang, and HEC-1B cells (33), and an accumulation of ErbB2 has been observed in infected brain endothelial cells underneath the meningococcal microcolonies (11). To determine the cellular localization of ErbB receptors during meningococcal infection in our cell culture model, confocal microscopy analyses were performed. As shown in Fig. 3, EGFR, ErbB2, and ErbB4 accumulated in the vicinity of attached bacteria on the cell surfaces. In uninfected control cells, EGFR, ErbB2, and ErbB4 were evenly distributed on the cell surface.
FIG 3.
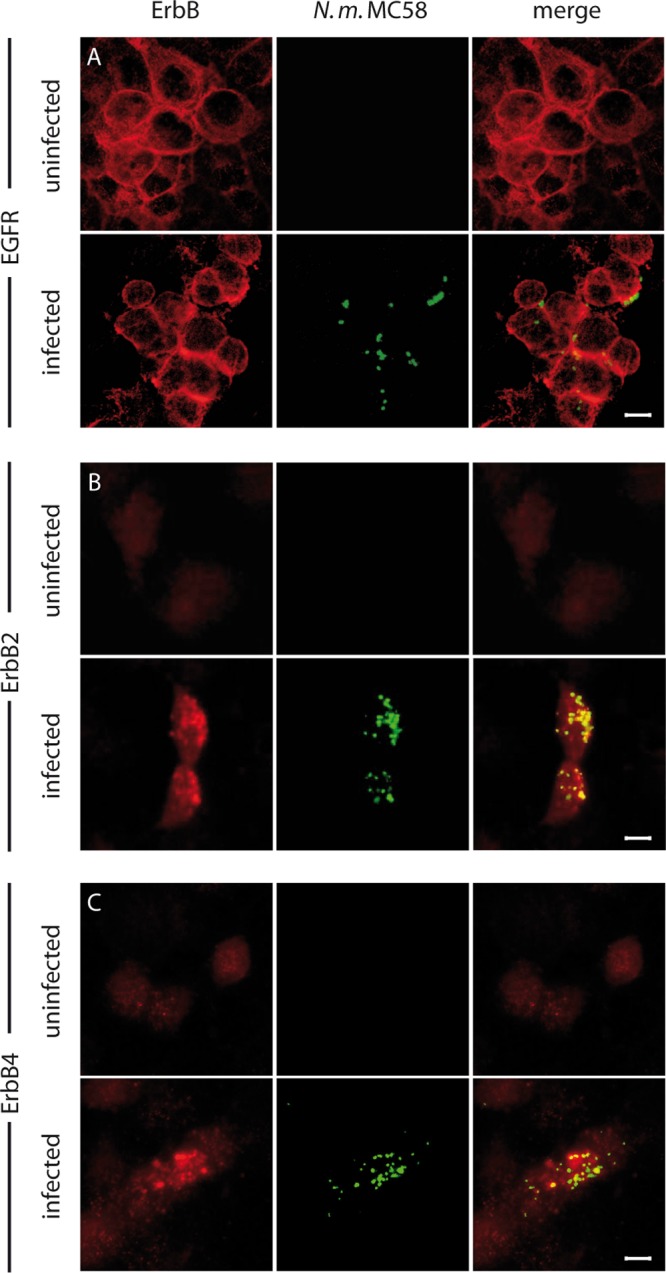
EGFR, ErbB2, and ErbB4 accumulated at the site of meningococcal adhesion. HBMEC were infected with N. meningitidis at an MOI of 30 for 1 h, fixed, and examined by confocal microscopy. Bacteria were stained with a rabbit antimeningococcal serum and counterstained with an anti-rabbit Cy3 antibody (green). ErbB receptors were stained with either anti-EGFR (A), anti-ErbB2 (B), or anti-ErbB4 (C) followed by counterstaining with anti-mouse Cy5 antibody (red). Uninfected HBMEC served as controls. Optical sections were acquired using a confocal microscope (Leica SP5). The figure shows representative images of single optical sections from three independent experiments. Bar, 5 μm.
Activation of the ErbB receptor family promotes meningococcal invasion into HBMEC.
EGFR, ErbB2, and ErbB4 are recruited to the site of adhering meningococci and are activated during infection, but the role for the signaling process remained to be understood. Recent data demonstrated that inhibition of protein tyrosine kinases by genistein significantly interfered with meningococcal uptake (9). We therefore addressed the question of whether activation of EGFR, ErbB2, or ErbB4 is involved in meningococcal uptake. We first determined the distribution patterns of the ErbB receptors on the HBMEC outer cell membranes via flow cytometry analyses. EGFR was found in high concentrations on the cell surface, whereas ErbB2 and ErbB4 showed significantly lower concentrations there (Fig. 4A). To analyze the impact of EGFR/ErbB2/ErbB4 activation on N. meningitidis invasion into the host cell, gentamicin protection assays were performed in the presence of pharmacological inhibitors of either ErB1/2/4 (Pelitinib; EKB-569), EGFR (EGFR inhibitor II; BIBX1382), or ErbB2 (ErbB2 inhibitor II). All inhibitors are cell membrane permeable and prevent EGFR, ErbB2, or EGFR/ErbB2/ErbB4 phosphorylation, respectively, at the cytoplasmic domain. Inhibitors were added 1 h prior to infection at the indicated concentrations, and cells were infected with N. meningitidis MC58 and MC58 siaD for 4 h. Pelitinib caused a significant decrease of bacterial uptake in a dose-dependent manner for the unencapsulated strain MC58 siaD (∼30% inhibition with 5 μM Pelitinib and ∼80% inhibition with 10 μM Pelitinib at 4 h p.i. (Fig. 4B), whereas uptake of strain MC58 decreased only slightly (data not shown). At the same time, bacterial adherence slightly increased. Next, treatment of HBMEC with a specific EGFR inhibitor (EGFR inhibitor II; BIBX1382) was assessed and also showed decreased invasion of N. meningitidis MC58 siaD into HBMEC in a dose-dependent manner (Fig. 4B). We could not directly test the role of ErbB2 on meningococcal invasion, because the ErbB2 inhibitor (ErbB2 inhibitor II) did not block phosphorylation of ErbB2 after the cells were exposed to strains MC58 or MC58 siaD (data not shown).
FIG 4.

N. meningitidis internalization by HBMEC requires ErbB receptor family activity. (A) Flow cytometry analyses were performed to determine EGFR, ErbB2, and ErbB4 distribution patterns on the cell surface of HBMEC. Cells were stained with extracellular-binding anti-EGFR, anti-ErbB2, and anti-ErbB4 antibodies or an isotype control antibody and counterstained with a TRITC-labeled secondary antibody. The data show mean values ± standard deviations (SD) of three independent experiments performed in duplicate. (B) HBMEC were preincubated with the indicated concentrations of either ErB1/2/4 inhibitor (Pelitinib; EKB-569) or EGFR inhibitor II (BIBX1382), and cells were then infected for 4 h with the unencapsulated strain, MC58 siaD. The numbers of adherent (black bars) and invasive (gray bars) bacteria were determined in a gentamicin protection assay. The graphs show the percentages of adhesion and invasion of inhibitor-treated cells, relative to control cells (means ± SD of three independent experiments performed in duplicate). *, P < 0.05. (C) HBMEC were transfected with 100 nM siRNA specific for EGFR, ErbB2, or ErbB4 to limit ErbB expression or were transfected with a scrambled control siRNA. siRNA-transfected cells were infected with MC58 (left panel) or the isogenic unencapsulated strain MC58 siaD (right panel). Percentages of adhesive (black bars) and invasive (gray bars) bacteria were measured in a gentamicin protection assay at 4 h p.i. The graphs show means ± SD of three independent experiments performed in duplicate. *, P < 0.05, relative to cells transfected with scrambled control siRNA. In parallel, whole-cell lysate (WCL) extracts were prepared and analyzed by Western blotting. Staining with anti-EGFR, anti-ErbB2, and anti-ErbB4 antibodies demonstrated genetic knockdown expression of the corresponding ErbB receptor in cells transfected with EGFR, ErbB2, or ErbB4 siRNA, compared to levels in control siRNA-transfected cells.
To further corroborate these inhibitor data, we next took several independent genetic ErbB receptor ablation approaches to verify the importance of these receptors in the N. meningitidis invasion process. RNAi-mediated ablation of EGFR, ErbB2, and ErbB4 reduced meningococcal uptake by HBMEC by about 18%, 19%, and 19% for strain MC58 and by about 40%, 30%, and 20% for the unencapsulated mutant strain MC58 siaD, respectively, while this uptake was not affected by scrambled control siRNA (Fig. 4C). Western blot analysis demonstrated genetic knockdown expression of the respective ErbB receptor in EGFR, ErbB2, or ErB4 siRNA-transfected cells compared to control siRNA-transfected cells (Fig. 4C, lower panel).
Meningococcal uptake is increased in CHO-K1 cells expressing EGFR, ErB2, or ErbB4.
ErbB receptors form homo- and heterodimers during activation. The assembly of the dimers differentiates cell types and is dependent on ErbB distribution and expression intensity (14, 16, 41). To analyze the contribution of EGFR, ErbB2, and ErbB4 homodimers to the meningococcal infection process separately, without combinatorial properties of induced receptor dimerization, we employed wild-type CHO-K1 cells, which do not express EGFR and ErbB4, whereas they show low expression of ErbB2 (see Fig. S1A in the supplemental material) (42). Although CHO cells do not support adherence via pili, N. meningitidis can adhere and invade CHO-K1 cells via its Opa or Opc proteins, as demonstrated recently by Bradley et al. (43). We repeated these findings and infected CHO-K1 cells with the encapsulated wild-type strain MC58, the isogenic unencapsulated strain MC58 siaD (Opa+ Opc+), and two isogenic mutant strains lacking Opc expression (MC58 opc and MC58 siaD opc) to rule out a possible role of Opc in adhesion and invasion. Opa-mediated binding could be excluded, based on the lack of CEACAM1 receptor expression. Opc− derivatives showed significantly low levels of adhesion and invasion (see Fig. S2 in the supplemental material), demonstrating Opc-mediated adhesion and invasion to CHO cells. CHO-K1 cells were transfected with mammalian expression vectors that encoded either wild-type EGFR, ErbB2, or ErbB4. This system permitted us to determine the specific role of each ErbB member, its activation, and its downstream stimulation. Transfection efficiencies were controlled in parallel with a green fluorescent protein (GFP)-actin-expressing plasmid, and fluorescence was subsequently analyzed by flow cytometry. The transfection efficiencies varied between 75 and 83%. Western blot analysis of cells transfected with EGFR, ErbB2, or ErbB4 demonstrated overexpression of all three ErbB family receptors 48 h after transfection. Moreover, ErbB family members expressed in CHO-K1 cells were present on the cell surface, as demonstrated by flow cytometry, establishing their ability to participate in ligand binding and signaling events (see Fig. S1A). Moreover, we proved the functionality of heterologous EGFR expression in CHO-K1 cells by treatment with EGF. HBMEC and EGFR-transfected CHO-K1 cells were preincubated with 100 ng EGF for 30 min, and cell lysates were stained with an anti-EGFR antibody. Subsequently, membranes were stripped and counterstained with an antiphosphotyrosine antibody, demonstrating activation of EGFR after EGF treatment in transfected cells (see Fig. S1B). We then determined meningococcal adhesion and invasion to CHO-EGFR, CHO-ErbB2, and CHO-ErbB4 in gentamicin protection assays. At 48 h after transfection, cells were infected with N. meningitidis MC58 at an MOI of 30, and bacterial uptake was determined after a further incubation of 4 h. As shown in Fig. 5A, heterologous expression of EGFR led to a significant increase of meningococcal invasion to about 260% above control cells. Moreover, ErbB2 as well as ErbB4 overexpression increased bacterial invasion to 260% and 220%, respectively, compared to pcDNA-transfected control cells (Fig. 5A). To confirm the phosphorylation signal data was from the PathScan RTK signaling antibody array, we determined phosphorylation of the respective receptors in response to N. meningitidis infection in CHO-K1 transfected cells. CHO-K1 cells were transfected with the constructs as described above, and ErbB receptors were precipitated from cell lysates and stained with anti-EGFR, anti-ErbB2, and anti-ErbB4 antibodies, followed by stripping and counterstaining with the antiphosphotyrosine antibody p-Tyr-100. Indeed, infection with N. meningitidis resulted in increased phosphorylation of EGFR, ErbB2, and ErbB4, which could be abrogated in the presence of the respective inhibitor (Fig. 5B). In contrast to HBMEC, ErbB2 inhibitor II efficiently blocked phosphorylation in the CHO transfection model.
FIG 5.

Heterologous expression of EGFR, ErbB2, and ErbB4 results in increased bacterial uptake. (A) CHO-K1 cells were transfected with control plasmid or 1 μg mammalian expression vector encoding EGFR, ErbB2, or ErbB4, and cells were infected with strain MC58. Percentages of adherent (black bars) and invasive (gray bars) bacteria were determined. Percentages of adhesion and invasion for ErbB-transfected cells were compared to results with pcDNA-transfected cells. The data are means ± SD of three independent experiments performed in duplicate. (B) In parallel, ErB receptors were immunoprecipitated from infected cell lysates (IP), and samples were analyzed by Western blotting with anti-EGFR, anti-ErbB2, and anti-ErbB4 antibodies (upper panel). Following stripping, the blots were reprobed with p-Tyr-100 antibody (lower panel). In addition, cells were preincubated with the ErbB inhibitors EGFR inhibitor II (BIBX1382), ErbB2 inhibitor II, or ErB1/2/4 inhibitor (Pelitinib; EKB-569).
ErbB activation based on HB-EGF release during infection.
Overexpression of the three ErbB members in CHO-K1 cells demonstrated a critical role of activated EGFR, ErbB2, and ErbB4 in the N. meningitidis uptake process. To rule out whether direct binding of N. meningitidis to EGFR or ErbB4 activates these receptors, we performed additionally gentamicin protection assays in the presence of neutralizing antibodies that bound to the extracellular ligand-binding domain of EGFR and ErbB4. Cells were left untreated or preincubated with the neutralizing antibodies for 1 h and infected with N. meningitidis, and bacterial adhesion and uptake were determined. Pretreatment of cells with anti-EGFR or anti-ErbB4 had no influence on bacterial adherence to HBMEC, whereas bacterial uptake was inhibited by about 40% in the presence of 5 μg/ml anti-EGFR and about 20% when cells were preincubated with anti-ErbB4 (Fig. 6A).
FIG 6.

ErbB activation follows HB-EGF release during infection. (A) HBMEC were incubated with neutralization antibodies against EGFR or ErbB4 (each at 5 μg/ml), respectively, for 1 h and then infected with MC58. Adhesive and invasive bacteria were determined in a gentamicin protection assay as described above. Percentages of adhesion and invasion of untreated cells were compared to results with antibody-treated cells. *, P < 0.05. (B) HB-EGF mRNA levels in HBMEC infected with N. meningitidis. HBMEC were infected with N. meningitidis MC58 or left uninfected (control). mRNAs were extracted at the indicated time points of infection, and quantitative real-time reverse transcription-PCR was performed. Target gene, HB-EGF; reference gene, GAPDH. Means and SD were calculated from the results of three independent experiments. *, P < 0.05. (C) Estimation of released HB-EGF after infection of HBMEC with MC58. At the indicated time points, membrane-bound HB-EGF was dissolved by washing with 1.5 M NaCl–PBS–1% BSA, and the HB-EGF concentration was quantified in a commercial HB-EGF ELISA. (D) HBMEC were pretreated with the HB-EGF inhibitor CRM197 and infected with N. meningitidis MC58, and percentages of intracellular bacteria were determined after a further 4-h incubation. Means and SD results are from three independent experiments performed in duplicate. *, P < 0.05, relative to untreated cells.
Since the neutralizing antibodies interfered with the binding sites for growth factors, we hypothesized that N. meningitidis might transactivate EGFR and ErbB4 via activation of endogenous ligands, as described for H. pylori and the related species N. gonorrhoeae (34, 44), supporting bacterial uptake. Numerous ligands are known to cause transactivation of the EGFR family, and previous studies have shown that EGFR transactivation could be linked to cleavage of the membrane-bound pro-heparin-binding EGF-like growth factor (pro-HB-EGF) that activates EGFR in a number of systems (45). Moreover, HB-EGF serves as a common ligand for EGFR and ErbB4. We first addressed the question of whether N. meningitidis increases expression and release of HB-EGF. Total RNA samples of uninfected and N. meningitidis-infected cells were recovered, and the mRNA level of HB-EGF was analyzed using real-time PCR. The transcription levels were normalized against the mRNA level of GAPDH. The mRNA level for HB-EGF significantly increased over time after infection with meningococci. HB-EGF transcripts increased about 2.5-fold compared to uninfected control cells (Fig. 6B). In parallel, the release of cleaved, secretory HB-EGF in the supernatant during infection was estimated. HBMEC were infected with N. meningitidis, and cell supernatants were collected and analyzed for secretory HB-EGF by ELISA. By introducing an additional wash step with 1.5 M sodium chloride, we were able to release HSPG-bound HB-EGF in the supernatant (46): release of HB-EGF increased by about 22% at 4 h p.i. and by about 41% at 8 h p.i. (Fig. 6C). To determine whether HB-EGF-mediated activation of EGFR and ErbB4 contributes to bacterial uptake, we assessed whether pretreatment with CRM197 affected meningococcal invasion. CRM197 is a diphtheria toxin mutant with weak toxicity that blocks HB-EGF activity by preventing pro-HB-EGF shedding and binding (47–49). HBMEC were preincubated with 10 and 20 μg/ml CRM197 and infected with N. meningitidis MC58 at an MOI of 30, and bacterial uptake was determined at 4 h p.i. As shown in Fig. 6D, CRM197 significantly blocked meningococcal uptake in a dose-dependent manner, to about ∼40% at 4 h p.i., demonstrating that an increase of HB-EGF by about 22% in the supernatant was able to support meningococcal internalization. PMA was included in these experiments to serve as a positive control, because PMA can induce cleavage of HB-EGF in HBMEC. Pretreatment of PMA-stimulated HBMEC with 10 μg/ml CRM197 verified that the concentration was sufficient to inhibit pro-HB-EGF shedding in HBMEC (see Fig. S3 in the supplemental material).
ErbB2 activity is regulated by phosphorylation at Thr686.
Screening of specific phosphorylation sites by using the EGFR phosphorylation antibody array revealed activation of ErbB2 at Thr686 (Fig. 7A), a docking site for protein kinase A (PKA)/PKC (50). To verify ErbB2 activation at this specific site, CHO-K1 cells were transfected with ErbB2 and were infected with N. meningitidis for a 1-h period, and cell lysates were collected at 15 min, 30 min, and 60 min p.i. and analyzed by Western blotting using anti-Thr686 phospho-specific antibody. As shown in Fig. 7A, ErbB2-transfected CHO-K1 cells demonstrated a significant increase of Thr686 activation in response to N. meningitidis infection.
FIG 7.

N. meningitidis induces ErbB phosphorylation at specific sites. (A) HBMEC were infected with MC58 for 3 h, and cell lysates were analyzed using the human EGFR phosphorylation antibody array C1′ from RayBio, demonstrating phosphorylation at Thr686. For Western blot analysis, ErbB2-transfected CHO-K1 cells were infected, and cell lysates were probed with the anti-pErbB2 (T686) antibody. (B) Src-dependent activation of EGFR at Tyr845. HBMEC were infected, and cell lysates were collected at the indicated time points. EGFR phosphorylation was examined with an anti-pEGFR (Y845) antibody. EGFR-transfected CHO-K1 cells were incubated with the Src inhibitor PP2 and the pan-EGFR inhibitor. After infection, WCL extracts were used for Western blot analysis with the anti-pEGFR (Y845) antibody. CHO-K1 cells were cotransfected with EGFR and c-Src or rather the catalytically inactive c-Src K297M point mutant. After 48 h, cells were infected, and cell lysates were probed with the pEGFR (Y845) antibody. To determine the role of Tyr845 EGFR phosphorylation during meningococcal infection, CHO-K1 cells were transfected with EGFR wild-type (wt) and the EGFR Y845F mutant. After 48 h, cells were infected with N. meningitidis MC58, and the percentages of adhesive (black bars) and intracellular (gray bars) bacteria were determined. *, P < 0.05, relative to cells transfected with EGFR wt. In parallel, WCL extracts of transfected cells showed comparable expression levels of EGFR wt and the EGFR Y845F mutant.
Phosphorylation of EGFR on Tyr845 is involved in N. meningitidis uptake.
Since we recently observed activation of the non RTK c-Src as a response of brain endothelial cells to N. meningitidis infection and found that c-Src plays an essential role during bacterial uptake, we analyzed cells for activation of EGFR at Tyr854, which is a c-Src-binding site of EGFR (24). As described above, either HBMEC or CHO-K1 cells transfected with EGFR were infected with N. meningitidis for a 1-h period, and cell lysates were collected at 15 min, 30 min, and 60 min p.i. and analyzed by Western blotting using anti-Tyr845 phospho-specific antibody. Both HBMEC- and EGFR-transfected CHO-K1 cells revealed an increase of Tyr845 activation after infection with meningococci (Fig. 7B; data shown for HBMEC). To analyze a possible transactivation of EGFR via Src family PTK, cells were pretreated with PP2, a cell membrane-permeable selective Src family PTK inhibitor, 1 h prior to infection with N. meningitidis. Cell lysates were collected and analyzed with an anti-Tyr845 antibody, and this showed significantly less phosphorylation of EGRF at Tyr845. To determine the specific role of c-Src, CHO-K1 cells were transfected with c-Src wild type and with the catalytic inactive point mutant c-Src K297M (10). Phosphorylation of EGFR at Tyr845 was only observed in cells transfected with the c-Src wild type, demonstrating that c-Src acts upstream of EGFR (Fig. 7B, right panel). To evaluate the role of Tyr845 phosphorylation downstream of c-Src in the infection process, this site was mutated to phenylalanine to produce the EGFR Y845F mutant. CHO-K1 cells were infected with wild-type EGFR and the EGFR Y845F mutant, and invasion was assayed in gentamicin protection experiments. Indeed, overexpression of EGFR Y845F in CHO-K1 cells resulted in a significant decrease of bacterial uptake by about 30% compared to wild-type EGFR-transfected cells (Fig. 7B, lower panels).
N. meningitidis induces phosphorylation of ErbB4 on Tyr1284.
ErbB4 activation in response to N. meningitidis infection has not been recognized before. Interestingly, the human EGFR phosphorylation antibody array demonstrated phosphorylation of ErbB4 at its autophosphorylation site, Tyr1284, which is known to be essential for coupling to downstream signaling pathways. To confirm the data for the EGFR antibody array, HBMEC and CHO-K1 cells transfected with ErbB4 were infected with N. meningitidis for a 1-h period, and cell lysates were collected at 15 min, 30 min, and 60 min p.i. and analysis by Western blotting using anti-Tyr1284 antibody. Indeed, HBMEC revealed a significant increase of Tyr1284 activation after infection, with a maximal peak at 60 min p.i. (Fig. 8, upper left panel). To analyze the impact of this specific site in meningococcal uptake, tyrosine was mutated to phenylalanine. The overexpression of ErbB4 Y1284F in CHO-K1 cells showed a significant decrease in bacterial internalization compared to cells transfected with ErbB4 wild type (Fig. 8).
FIG 8.
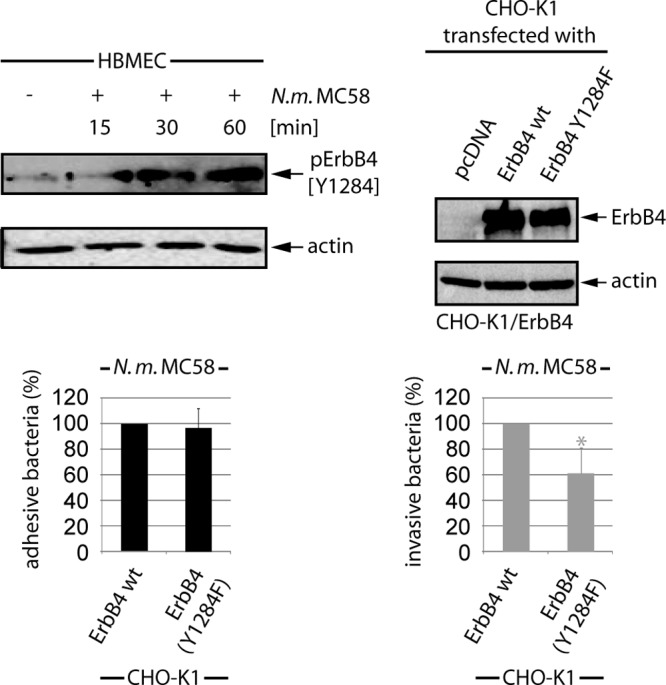
N. meningitidis induces ErbB4 phosphorylation at site Tyr1284. WCL extracts of MC58-infected HBMEC were analyzed using the pErbB4 (Y1284) antibody. To specify the role of ErbB4 Tyr1284, CHO-K1 cells were transfected with ErbB4 wild type (wt) and the ErbB4 Y1284F point mutant. At 24 h posttransfection, cells were infected with N. meningitidis, and percentages of adhesive (black bars) and invasive (gray bars) bacteria were determined. *, P < 0.05, relative to cells transfected with ErbB4 wt. Western blot analysis demonstrated comparable expression levels of ErbB4 and ErbB4 Y1284F.
DISCUSSION
We and others have previously reported that N. meningitidis induce tyrosine phosphorylation of host cell proteins in infected cells and that activation of non-RTKs plays a key role in mediating bacterial uptake (10, 11, 38). We have now shown that N. meningitidis can induce phosphorylation of EGFR, ErbB2, and ErbB4 in eukaryotic cells and that ErbB activation promotes meningococcal invasion. Activation of ErbB receptors occurred at specific residues at their cytoplasmatic tail. We determined three specific sites on EGFR, ErbB2, and ErbB4 that are phosphorylated in response to meningococcal infection, including Tyr845 of EGFR, Thr686 of ErbB2, and Tyr1284 of ErbB4. Moreover, we determined that the mechanism underlying the activation of EGFR and ErbB4 receptor phosphorylation involves activation of the common receptor ligand HB-EGF, as recently demonstrated for the related species N. gonorrhoeae (34). Furthermore, EGFR phosphorylation at Tyr845 occurrs downstream of c-Src.
RTKs form a large family of receptor molecules on the plasma membranes of eukaryotic cells. They play an essential role in signal transduction pathways that mediate cell-to-cell communication, and several studies have shown that RTKs are involved in the interactions of bacteria with the host cell (25–31). To obtain a comprehensive picture of functionally activated RTKs in N. meningitidis-infected cells, we surveyed receptor activation by using a commercially available phosphoarray that captured 39 RTKs and key signaling nodes from cell lysates. We found that members of the EGFRs (EGFR, ErbB2), the tyrosine-related kinase B receptor, and the ephrin receptor EphA3, as well as Akt, extracellular signal-regulated kinases, ribosomal S6, Abelson murine leukemia viral oncogene homolog 1, insulin receptor substrate 1, and c-Src were significantly activated in infected cells (Fig. 1). Moreover, high phosphorylation signals were obtained for FMS-like tyrosine kinase receptor 3, tyrosine-protein kinase UFO (Axl), and insulin receptor (InsR), but these signals were not statistically significantly different. Activation of c-Src confirmed the data of our previous study, in which we demonstrated the important role of c-Src in meningococcal uptake into HBMEC (10).
The ErbB family has been recognized before to play a major role in N. meningitidis host cell interactions. Receptor accumulation and/or activation have been described. EGFR accumulation and clustering underneath meningococcal microcolonies occurs in epithelial cells, which is dependent on type IV pilus expression (33). In addition, ErbB2 accumulation has been observed in endothelial cells. Moreover, ErbB2 phosphorylation has been demonstrated to be involved in meningococcal uptake by endothelial cells (11). Through induction of ErbB2 receptor phosphorylation, N. meningitidis is capable of activating c-Src and cortactin, which links the host cell cytoskeleton to host cell membrane receptors (10, 11, 34, 36).
We now report that N. meningitidis can induce phosphorylation not only of the ErbB2 receptor but also of EGFR and ErbB4 in endothelial cells. From our study, we furthermore conclude that all three members mediate uptake by endothelial cells. Initial inhibitor studies using tyrosine kinase inhibitor specific for EGFR and a pan-ErbB tyrosine kinase inhibitor suggested that EGFR and ErbB4, respectively, participate in the bacterial uptake process. More importantly, inhibitor data were confirmed by genetic ablation of EGFR, ErbB2, and ErbB4 expression by using specific siRNAs. We then established CHO-K1 cells transfected with each of the three ErbB receptors as a model system that allowed us to analyze receptor phosphorylation separately without the influence of the combinatorial properties of induced receptor dimerization. We found that heterologous expression of all three ErbB receptors in CHO-K1 cells caused significant tyrosine phosphorylation after infection with N. meningitidis, confirming our phospho array data. Moreover, heterologous expression of ErbB receptors in CHO-K1 cells increased bacterial uptake, supporting the role of EGFR, ErbB2, and ErbB4 in the meningococcal invasion process. However, since ErbB family members form hetero- as well as homodimers and the nature of the dimers greatly influences downstream signaling cascades, data observed for the CHO transfected cells might not necessarily reflect the observations found in endothelial cells.
By using the EGFR phosphorylation antibody array, we examined the levels of phosphorylation of specific sites for human ErbB receptors in N. meningitidis-infected host cells. We now report the novel finding that ErbB4 is phosphorylated in response to infection with N. meningitidis. To our knowledge this is the first description of ErbB4 phosphorylation initiated by a bacterial pathogen. Tyrosine residues 1022, 1056, 1150, 1162, 1188, 1202, 1242, 1258, and 1284 were identified as autophosphorylation sites (51, 52). Here, we have demonstrated that N. meningitidis infection induces significant phosphorylation at the autophosphorylation site Tyr1284 and that this site is also involved in bacterial uptake. This tyrosine residue, when phosphorylated, serves as a binding site for the adapter protein Shc (22), after which Ras is recruited to the receptor and activation of the intracellular MAP kinase pathway takes place. The mechanism for how ErbB4 Tyr1284 phosphorylation leads to bacterial uptake is unknown. Tyr1284 phosphorylation most likely is crucial to couple to required signaling events that are necessary for the cytoskeletal rearrangement resulting in meningococcal uptake. Interestingly, we noted threonine phosphorylation of ErbB2 at position 686. Recent data have demonstrated that the activity of ErbB2 is attenuated by phosphorylation of the Thr686 residue, which causes tyrosine dephosphorylation and receptor endocytosis (50, 53, 54). The precise role of ErbB2 Thr686 phosphorylation in N. meningitidis interaction with endothelial cells, however, remains to be elucidated.
Interestingly, neither ErbB overexpression nor blocking of the extracellular domain of EGFR by using a neutralizing antibody showed any effect on bacterial attachment. We therefore hypothesized that ErbB activation is mediated by transactivation via activation of endogenous ligands, as described for H. pylori and the related species N. gonorrhoeae (34, 44). Phosphorylation of EGFR mediated through release of HB-EGF has also been observed after incubation of epithelial cells with Gram-positive lipoteichoic acid (44, 55). All EGFR ligands are synthesized as membrane-spanning precursor molecules that have to be proteolytically processed by metalloproteinases of the ADAM (a disintegrin and metalloprotease) family to become fully active (56, 57). As shown for N. gonorrhoeae, N. meningitidis is able to upregulate HB-EGF mRNA levels. Swanson et al. (34), moreover, reported that by incubating HEC-1-B cells with N. gonorrhoeae they were unable to detect soluble HB-EGF in the supernatant in an ELISA. By introducing an additional wash step with 1.5 M sodium chloride, we were able to recover HSPG-bound HB-EGF in the supernatant and detected significant levels of HB-EGF in the supernatant of infected HBMEC and CHO-K1 cells. We recently showed that MMP-8 (ADAM-8) is secreted in the supernatant of infected HBMEC participating in the cleavage of the tight junction protein occludin (58). Whether MMPs are involved in shedding of the membrane-bound precursors of HB-EGFR as demonstrated for N. gonorrhoeae and whether MMP-8 in particular acts as a shedding agent on the release of HB-EGF are interesting subjects for further analysis.
In addition to the release of growth factor receptor ligands, transactivation may occur by activation of catalytic sites at the cytoplasmatic domains of ErbB receptors via kinases. Interestingly, we noted phosphorylation of EGFR at Tyr845, the Src-binding site. Since we recently observed activation of the non-RTK c-Src as a response of endothelial cells to N. meningitidis infection (10), we analyzed the role of activation of EGFR at Tyr845 in the meningococcal uptake process. We found that c-Src acts upstream of EGFR. Furthermore, our data showed that overexpression of a mammalian expression vector encoding an EGFR Y845F mutant decreased meningococcal uptake, demonstrating that EGFR Tyr845 phosphorylation participates in the uptake process. Tyr845 of EGFR is located in the activation segment of the kinase domain. In general, activation segments of protein kinases contain a phosphorylation site(s) that is important for regulation of catalytic activity. However, in the case of EGFR, it has been reported that Tyr845 is not required for the regulation of catalytic activity of EGFR but is required for stabilization of the active conformation of the kinase domain. In summary, our data provide evidence that, besides ErbB2, EGFR and ErbB4 are activated in response to N. meningitidis endothelial cell infection. These findings shed new light on the role of specific ErbB phosphorylation sites in meningococcal infection biology.
Supplementary Material
ACKNOWLEDGMENTS
We thank Michael Schmutzler for technical assistance and Hilde Merkert for expert assistance with the confocal microscope. We are indebted to Frank Günther for helpful discussions.
This work was supported by grant SCHU 2394/1-2 from the Deutsche Forschungsgemeinschaft.
We have no conflicts of interest that are directly relevant to the content of this article.
Footnotes
Published ahead of print 30 December 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01346-13.
REFERENCES
- 1.Caugant DA, Maiden MCJ. 2009. Meningococcal carriage and disease: population biology and evolution. Vaccine 27:B64–B70. 10.1016/j.vaccine.2009.04.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nassif X, Bourdoulous S, Eugene E, Couraud PO. 2002. How do extracellular pathogens cross the blood-brain barrier? Trends Microbiol. 10:227–232. 10.1016/S0966-845X(02)02649-1 [DOI] [PubMed] [Google Scholar]
- 3.Schmitt C, Turner D, Boesl M, Abele M, Frosch M, Kurzai O. 2007. A functional two-partner secretion system contributes to adhesion of Neisseria meningitidis to epithelial cells. J. Bacteriol. 189:7968–7976. 10.1128/JB.00851-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Turner DP, Marietou AG, Johnston L, Ho KK, Rogers AJ, Wooldridge KG, Ala'Aldeen DA. 2006. Characterization of MspA, an immunogenic autotransporter protein that mediates adhesion to epithelial and endothelial cells in Neisseria meningitidis. Infect. Immun. 74:2957–2964. 10.1128/IAI.74.5.2957-2964.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scarselli M, Serruto D, Montanari P, Capecchi B, Adu-Bobie J, Veggi D, Rappuoli R, Pizza M, Arico B. 2006. Neisseria meningitidis NhhA is a multifunctional trimeric autotransporter adhesin. Mol. Microbiol. 61:631–644. 10.1111/j.1365-2958.2006.05261.x [DOI] [PubMed] [Google Scholar]
- 6.Virji M. 2009. Pathogenic neisseriae: surface modulation, pathogenesis and infection control. Nat. Rev. Microbiol. 7:274–286. 10.1038/nrmicro2097 [DOI] [PubMed] [Google Scholar]
- 7.Sa E Cunha C, Griffiths NJ, Virji M. 2010. Neisseria meningitidis Opc invasin binds to the sulphated tyrosines of activated vitronectin to attach to and invade human brain endothelial cells. PLoS Pathog. 6(5):e1000911. 10.1371/journal.ppat.1000911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Unkmeir A, Latsch K, Dietrich G, Wintermeyer E, Schinke B, Schwender S, Kim KS, Eigenthaler M, Frosch M. 2002. Fibronectin mediates Opc-dependent internalization of Neisseria meningitidis in human brain microvascular endothelial cells. Mol. Microbiol. 46:933–946. 10.1046/j.1365-2958.2002.03222.x [DOI] [PubMed] [Google Scholar]
- 9.Sokolova O, Heppel N, Jagerhuber R, Kim KS, Frosch M, Eigenthaler M, Schubert-Unkmeir A. 2004. Interaction of Neisseria meningitidis with human brain microvascular endothelial cells: role of MAP- and tyrosine kinases in invasion and inflammatory cytokine release. Cell. Microbiol. 6:1153–1166. 10.1111/j.1462-5822.2004.00422.x [DOI] [PubMed] [Google Scholar]
- 10.Slanina H, Konig A, Hebling S, Hauck CR, Frosch M, Schubert-Unkmeir A. 2010. Entry of Neisseria meningitidis into mammalian cells requires the Src family protein tyrosine kinases. Infect. Immun. 78:1905–1914. 10.1128/IAI.01267-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoffmann I, Eugene E, Nassif X, Couraud PO, Bourdoulous S. 2001. Activation of ErbB2 receptor tyrosine kinase supports invasion of endothelial cells by Neisseria meningitidis. J. Cell Biol. 155:133–143. 10.1083/jcb.200106148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hunter T. 1998. The Croonian Lecture 1997. The phosphorylation of proteins on tyrosine: its role in cell growth and disease. Philos. Trans. R. Soc. London B Biol. Sci. 353:583–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robinson DR, Wu YM, Lin SF. 2000. The protein tyrosine kinase family of the human genome. Oncogene 19:5548–5557. 10.1038/sj.onc.1203957 [DOI] [PubMed] [Google Scholar]
- 14.Yarden Y, Sliwkowski MX. 2001. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2:127–137. 10.1038/35052073 [DOI] [PubMed] [Google Scholar]
- 15.Hynes NE, Lane HA. 2005. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat. Rev. Cancer 5:341–354. 10.1038/nrc1609 [DOI] [PubMed] [Google Scholar]
- 16.Citri A, Yarden Y. 2006. EGF-ERBB signalling: towards the systems level. Nat. Rev. Mol. Cell Biol. 7:505–516. 10.1038/nrm1962 [DOI] [PubMed] [Google Scholar]
- 17.Linggi B, Carpenter G. 2006. ErbB receptors: new insights on mechanisms and biology. Trends Cell Biol. 16:649–656. 10.1016/j.tcb.200610.008 [DOI] [PubMed] [Google Scholar]
- 18.Soltoff SP, Cantley LC. 1996. p120cbl is a cytosolic adapter protein that associates with phosphoinositide 3-kinase in response to epidermal growth factor in PC12 and other cells. J. Biol. Chem. 271:563–567 [DOI] [PubMed] [Google Scholar]
- 19.Yarden Y, Schlessinger J. 1987. Epidermal growth factor induces rapid, reversible aggregation of the purified epidermal growth factor receptor. Biochemistry 26:1443–1451 [DOI] [PubMed] [Google Scholar]
- 20.Yarden Y, Schlessinger J. 1987. Self-phosphorylation of epidermal growth factor receptor: evidence for a model of intermolecular allosteric activation. Biochemistry 26:1434–1442 [DOI] [PubMed] [Google Scholar]
- 21.Graus-Porta D, Beerli RR, Daly JM, Hynes NE. 1997. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 16:1647–1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schulze WX, Deng L, Mann M. 2005. Phosphotyrosine interactome of the ErbB-receptor kinase family. Mol. Syst. Biol. 1:2005.0008. 10.1038/msb4100012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ware MF, Tice DA, Parsons SJ, Lauffenburger DA. 1997. Overexpression of cellular Src in fibroblasts enhances endocytic internalization of epidermal growth factor receptor. J. Biol. Chem. 272:30185–30190 [DOI] [PubMed] [Google Scholar]
- 24.Biscardi JS, Maa MC, Tice DA, Cox ME, Leu TH, Parsons SJ. 1999. c-Src-mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor function. J. Biol. Chem. 274:8335–8343 [DOI] [PubMed] [Google Scholar]
- 25.Chan G, Nogalski MT, Yurochko AD. 2009. Activation of EGFR on monocytes is required for human cytomegalovirus entry and mediates cellular motility. Proc. Natl. Acad. Sci. U. S. A. 106:22369–22374. 10.1073/pnas.0908787106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eierhoff T, Hrincius ER, Rescher U, Ludwig S, Ehrhardt C. 2010. The epidermal growth factor receptor (EGFR) promotes uptake of influenza A viruses (IAV) into host cells. PLoS Pathog. 6(9):e1001099. 10.1371/journal.ppat.1001099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Galan JE, Pace J, Hayman MJ. 1992. Involvement of the epidermal growth factor receptor in the invasion of cultured mammalian cells by Salmonella typhimurium. Nature 357:588–589 [DOI] [PubMed] [Google Scholar]
- 28.Zhang J, Li H, Wang J, Dong Z, Mian S, Yu FS. 2004. Role of EGFR transactivation in preventing apoptosis in Pseudomonas aeruginosa-infected human corneal epithelial cells. Invest. Ophthalmol. Vis. Sci. 45:2569–2576. 10.1167/iovs.03-1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yan F, Cao H, Chaturvedi R, Krishna U, Hobbs SS, Dempsey PJ, Peek RM, Jr, Cover TL, Washington MK, Wilson KT, Polk DB. 2009. Epidermal growth factor receptor activation protects gastric epithelial cells from Helicobacter pylori-induced apoptosis. Gastroenterology 136:1297–1307. 10.1053/j.gastro.2008.12.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tabassam FH, Graham DY, Yamaoka Y. 2009. Helicobacter pylori activate epidermal growth factor receptor- and phosphatidylinositol 3-OH kinase-dependent Akt and glycogen synthase kinase 3β phosphorylation. Cell. Microbiol. 11:70–82. 10.1111/j.1462-5822.2008.01237.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seo B, Choy EW, Maudsley S, Miller WE, Wilson BA, Luttrell LM. 2000. Pasteurella multocida toxin stimulates mitogen-activated protein kinase via G(q/11)-dependent transactivation of the epidermal growth factor receptor. J. Biol. Chem. 275:2239–2245. 10.1074/jbc.275.3.2239 [DOI] [PubMed] [Google Scholar]
- 32.Tapinos N, Ohnishi M, Rambukkana A. 2006. ErbB2 receptor tyrosine kinase signaling mediates early demyelination induced by leprosy bacilli. Nat. Med. 12:961–966. 10.1038/nm1433 [DOI] [PubMed] [Google Scholar]
- 33.Merz AJ, Enns CA, So M. 1999. Type IV pili of pathogenic Neisseriae elicit cortical plaque formation in epithelial cells. Mol. Microbiol. 32:1316–1332 [DOI] [PubMed] [Google Scholar]
- 34.Swanson KV, Griffiss JM, Edwards VL, Stein DC, Song W. 2011. Neisseria gonorrhoeae-induced transactivation of EGFR enhances gonococcal invasion. Cell. Microbiol. 13:1078–1090. 10.1111/ju.1462-5822.2011.01603.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Greulich H, Chen TH, Feng W, Janne PA, Alvarez JV, Zappaterra M, Bulmer SE, Frank DA, Hahn WC, Sellers WR, Meyerson M. 2005. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2(11):e313. 10.1371/journal.pmed.0020313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li YM, Pan Y, Wei Y, Cheng X, Zhou BP, Tan M, Zhou X, Xia W, Hortobagyi GN, Yu D, Hung MC. 2004. Upregulation of CXCR4 is essential for HER2-mediated tumor metastasis. Cancer Cell 6:459–469. 10.1016/j.ccr.2004.09.027 [DOI] [PubMed] [Google Scholar]
- 37.Prickett TD, Agrawal NS, Wei X, Yates KE, Lin JC, Wunderlich JR, Cronin JC, Cruz P, Rosenberg SA, Samuels Y. 2009. Analysis of the tyrosine kinome in melanoma reveals recurrent mutations in ERBB4. Nat. Genet. 41:1127–1132. 10.1038/ng.438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Slanina H, Hebling S, Hauck CR, Schubert-Unkmeir A. 2012. Cell invasion by Neisseria meningitidis requires a functional interplay between the focal adhesion kinase, Src and cortactin. PLoS One 7(6):e39613. 10.1371/journal.pone.0039613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moro L, Dolce L, Cabodi S, Bergatto E, Boeri Erba E, Smeriglio M, Turco E, Retta SF, Giuffrida MG, Venturino M, Godovac-Zimmermann J, Conti A, Schaefer E, Beguinot L, Tacchetti C, Gaggini P, Silengo L, Tarone G, Defilippi P. 2002. Integrin-induced epidermal growth factor (EGF) receptor activation requires c-Src and p130Cas and leads to phosphorylation of specific EGF receptor tyrosines. J. Biol. Chem. 277:9405–9414. 10.1074/jbc.M109101200 [DOI] [PubMed] [Google Scholar]
- 40.Kloth MT, Laughlin KK, Biscardi JS, Boerner JL, Parsons SJ, Silva CM. 2003. STAT5b, a mediator of synergism between c-Src and the epidermal growth factor receptor. J. Biol. Chem. 278:1671–1679. 10.1074/jbc.M207289200 [DOI] [PubMed] [Google Scholar]
- 41.Tao RH, Maruyama IN. 2008. All EGF(ErbB) receptors have preformed homo- and heterodimeric structures in living cells. J. Cell Sci. 121:3207–3217. 10.1242/jcs.033399 [DOI] [PubMed] [Google Scholar]
- 42.Shi W, Fan H, Shum L, Derynck R. 2000. The tetraspanin CD9 associates with transmembrane TGF-alpha and regulates TGF-alpha-induced EGF receptor activation and cell proliferation. J. Cell Biol. 148:591–602. 10.1083/jcb.148.3.591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bradley CJ, Griffiths NJ, Rowe HA, Heyderman RS, Virji M. 2005. Critical determinants of the interactions of capsule-expressing Neisseria meningitidis with host cells: the role of receptor density in increased cellular targeting via the outer membrane Opa proteins. Cell. Microbiol. 7:1490–1503. 10.1111/j.1462-5822.2005.00572.x [DOI] [PubMed] [Google Scholar]
- 44.Keates S, Sougioultzis S, Keates AC, Zhao D, Peek RM, Jr, Shaw LM, Kelly CP. 2001. cag+ Helicobacter pylori induce transactivation of the epidermal growth factor receptor in AGS gastric epithelial cells. J. Biol. Chem. 276:48127–48134. 10.1074/jbc.M107630200 [DOI] [PubMed] [Google Scholar]
- 45.Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, Ullrich A. 1999. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature 402:884–888 [DOI] [PubMed] [Google Scholar]
- 46.Xu KP, Ding Y, Ling J, Dong Z, Yu FS. 2004. Wound-induced HB-EGF ectodomain shedding and EGFR activation in corneal epithelial cells. Invest. Ophthalmol. Vis. Sci. 45:813–820. 10.1167/iovs.03-0851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kageyama T, Ohishi M, Miyamoto S, Mizushima H, Iwamoto R, Mekada E. 2007. Diphtheria toxin mutant CRM197 possesses weak EF2-ADP-ribosyl activity that potentiates its anti-tumorigenic activity. J. Biochem. 142:95–104. 10.1093/jb/mvm116 [DOI] [PubMed] [Google Scholar]
- 48.Mitamura T, Higashiyama S, Taniguchi N, Klagsbrun M, Mekada E. 1995. Diphtheria toxin binds to the epidermal growth factor (EGF)-like domain of human heparin-binding EGF-like growth factor/diphtheria toxin receptor and inhibits specifically its mitogenic activity. J. Biol. Chem. 270:1015–1019 [DOI] [PubMed] [Google Scholar]
- 49.Hamaoka M, Chinen I, Murata T, Takashima S, Iwamoto R, Mekada E. 2010. Anti-human HB-EGF monoclonal antibodies inhibiting ectodomain shedding of HB-EGF and diphtheria toxin binding. J. Biochem. 148:55–69. 10.1093/jb.mvq033 [DOI] [PubMed] [Google Scholar]
- 50.Ouyang X, Gulliford T, Epstein RJ. 1998. The duration of phorbol-inducible ErbB2 tyrosine dephosphorylation parallels that of receptor endocytosis rather than threonine-686 phosphorylation: implications for the physiological role of protein kinase C in growth factor receptor signalling. Carcinogenesis 19:2013–2019 [DOI] [PubMed] [Google Scholar]
- 51.Kainulainen V, Sundvall M, Maatta JA, Santiestevan E, Klagsbrun M, Elenius K. 2000. A natural ErbB4 isoform that does not activate phosphoinositide 3-kinase mediates proliferation but not survival or chemotaxis. J. Biol. Chem. 275:8641–8649. 10.1074/jbc.275.12.8641 [DOI] [PubMed] [Google Scholar]
- 52.Carraway KL, III, Cantley LC. 1994. A neu acquaintance for erbB3 and erbB4: a role for receptor heterodimerization in growth signaling. Cell 78:5–8 [DOI] [PubMed] [Google Scholar]
- 53.Tan M, Li P, Sun M, Yin G, Yu D. 2006. Upregulation and activation of PKC alpha by ErbB2 through Src promotes breast cancer cell invasion that can be blocked by combined treatment with PKC alpha and Src inhibitors. Oncogene 25:3286–3295. 10.1038/sj.onc.1203961 [DOI] [PubMed] [Google Scholar]
- 54.Ouyang X, Gulliford T, Zhang H, Huang GC, Epstein R. 1996. Human cancer cells exhibit protein kinase C-dependent c-erbB-2 transmodulation that correlates with phosphatase sensitivity and kinase activity. J. Biol. Chem. 271:21786–21792 [DOI] [PubMed] [Google Scholar]
- 55.Basbaum C, Li D, Gensch E, Gallup M, Lemjabbar H. 2002. Mechanisms by which gram-positive bacteria and tobacco smoke stimulate mucin induction through the epidermal growth factor receptor (EGFR). Novartis Found. Symp. 248:171–176 [PubMed] [Google Scholar]
- 56.Fischer OM, Hart S, Gschwind A, Ullrich A. 2003. EGFR signal transactivation in cancer cells. Biochem. Soc. Trans. 31:1203–1208 [DOI] [PubMed] [Google Scholar]
- 57.Sanderson MP, Dempsey PJ, Dunbar AJ. 2006. Control of ErbB signaling through metalloprotease mediated ectodomain shedding of EGF-like factors. Growth Factors 24:121–136. 10.1080/08977190600634373 [DOI] [PubMed] [Google Scholar]
- 58.Schubert-Unkmeir A, Konrad C, Slanina H, Czapek F, Hebling S, Frosch M. 2010. Neisseria meningitidis induces brain microvascular endothelial cell detachment from the matrix and cleavage of occludin: a role for MMP-8. PLoS Pathog. 6(4):e1000874. 10.1371/journal.ppat.1000874 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.