

Abstract
The simultaneous interaction of poly(A)-binding protein (PABP) with eukaryotic translation initiation factor 4G (eIF4G) and the mRNA 3′ poly(A) tail promotes translation initiation. We previously showed that the interaction of PABP-interacting protein 1 (Paip1) with PABP and eukaryotic translation initiation factor 3 (eIF3; via the eIF3g subunit) further stimulates translation. Here, we demonstrate that the interaction of eIF3 with Paip1 is regulated by amino acids through the mTORC1 signaling pathway. The Paip1-eIF3 interaction is impaired by the mTORC1 inhibitors, rapamycin and PP242. We show that ribosomal protein S6 kinases 1 and 2 (S6K1/2) promote the interaction of eIF3 with Paip1. The enhancement of Paip1-eIF3 interaction by amino acids is abrogated by an S6K inhibitor or shRNA against S6K1/2. S6K1 interacts with eIF3f and, in vitro, phosphorylates eIF3. Finally, we show that S6K inhibition leads to a reduction in translation by Paip1. We propose that S6K1/2 phosphorylate eIF3 to stimulate Paip1-eIF3 interaction and consequent translation initiation. Taken together, these data demonstrate that eIF3 is a new translation target of the mTOR/S6K pathway.
INTRODUCTION
Translational control provides a rapid response to growth and proliferation stimuli, stress, and nutrient availability. Consequently, it plays a critical role in cell growth and proliferation (for reviews, see references 1 and 2). mTOR (mechanistic/mammalian target of rapamycin complex), a phosphatidylinositol 3-kinase (PI3K)-like serine/threonine protein kinase also controls cell growth and proliferation (3). It forms two different complexes, mTORC1, whose activity is inhibited by the drug, rapamycin, and mTORC2, which is not. Hypoxia, energy status, growth factors, and amino acids signals converge on mTORC1 (for a review, see reference 4). Amino acids play an important permissive role in insulin-mediated stimulation of translation initiation (5). Branched-chain amino acids, especially leucine, activate mTORC1 (for a review, see reference 6). Amino acid signaling to mTOR is independent of the PI3K/Akt/TSC1-TSC2/Rheb pathway. Amino acids mediate mTORC1 activation mainly via the Rag GTPases (for a review, see reference 7).
mTORC1 controls translation initiation through phosphorylation of two main targets, p70-S6Ks and the inhibitory 4E-binding proteins (4E-BPs) (2). mTORC1 phosphorylation of 4E-BPs causes their dissociation from the cap binding protein, eukaryotic translation initiation factor 4E (eIF4E), allowing the association of eIF4G, an adaptor protein, together with eIF4A, an RNA helicase, to form the eIF4F complex (eIF4E/eIF4A/eIF4G). Poly(A)-binding protein (PABP) binds to the poly(A) tail at the mRNA 3′ end. The interaction between eIF4G and PABP brings about the circularization of the mRNA and activates translation. eIF4G also interacts with eIF3, which promotes the recruitment of the 43S preinitiation complex (PIC) to the mRNA. The 48S ribosomal complex then scans the mRNA 5′ untranslated region for the AUG initiation codon (2).
Another important target of mTORC1 are the S6Ks: S6K1 and S6K2. S6Ks control ribosome biogenesis and the activity of eIF4B, which stimulates eIF4A (8, 9). In addition, S6K1 and 2 phosphorylate ribosomal protein S6, elongation factor 2 kinase (eEF2K), and programmed cell death protein 4 (PDCD4), a tumor suppressor) (10–12). S6K1 and mTOR associate with the eIF3 complex, in a growth factor-dependent and rapamycin-sensitive manner (13–15).
Because of the large size of the eIF3 complex (∼800 kDa), which is composed of 13 subunits, it has been difficult to study and to fully understand the roles of the individual subunits. As a versatile scaffold for the translation initiation complex, eIF3 stimulates most of the reactions in the initiation pathway, including binding of ternary complex and other components of the 43S PIC to the 40S subunit, mRNA recruitment, and ribosome scanning on the mRNA and selection of the translation start codon (16). Importantly, eIF3 is phosphorylated in mammalian cells on seven subunits (a, b, c, f, g, h, and j) on 29 phosphorylation sites (17) with no associated functions, except for the oncogenic properties of eIF3h (18).
The interaction of Paip1 (PABP-interacting protein 1) and eIF3 is important for the translation promoting activity of Paip1, by stabilization of the circular mRNA conformation via the PABP-eIF4G interaction (19). We previously showed that Paip1-eIF3 interaction is mediated by eIF3g (19). Paip1 is poorly phosphorylated (global mass spectrometry data from www.phosphosite.org and unpublished data from our lab), while several eIF3 subunits, including eIF3g, are phosphorylated on multiple sites (17, 20). Phosphorylation of eIF3 subunits could stabilize the eIF3 complex and subsequently enhance interactions with eIF3-binding partners (20). Numerous proteins implicated in the translation initiation process are regulated by mTOR-dependent phosphorylation signals, notably 4E-BP, S6Ks which act on eIF4B, but surprisingly not eIF3. Due to the simultaneous interactions of eIF3 with mTORC1 and S6K1 (13–15), we wanted to determine whether mTOR/S6K signaling regulates eIF3 phosphorylation.
To address this question, we used amino acid-mediated stimulation of mTOR, which is entirely independent of the PI3K/Akt/TSC1-TSC2/Rheb signaling and does not activate the Ras/Raf/Mek/ERK/RSK pathway (6). Here, we show that amino acids enhance Paip1-eIF3 interaction through S6Ks. The interaction of S6K1 with eIF3f/p47, together with eIF3 phosphorylation by S6K1 in vitro, indicates that eIF3 phosphorylation controls the Paip1-eIF3 interaction.
MATERIALS AND METHODS
Reagents.
The antibodies used were obtained as follows. Anti-eIF3g was obtained from T. K. Tang (Academia Sinica, Taipei, Taiwan). Anti-Paip1 was generated by immunizing rabbits with recombinant glutathione S-transferase (GST)–Paip1 p65 (21). Anti-phospho-Akt (Ser473), -phospho-S6K1 (Thr389), -S6K1, -phospho-S6 (Ser240/244), -phospho-4E-BP1 (Ser65), -4E-BP1, and -PABP were from Cell Signaling Technology. Anti-S6, GAPDH, and eIF3b were from Santa Cruz. Antihemagglutinin (anti-HA) was from Covance Technology, anti-β-actin was from Sigma-Aldrich, and anti-S6K2 was from Bethyl Laboratories. Rapamycin was from LC Laboratories, PP242 and doxycycline were from Sigma-Aldrich, and U0126 was from Cell Signaling Technology. His6-tagged p70-S6 kinase 1 (amino acids 1 to 421) and S6K inhibitor, DG2, were from Merck-Millipore. Purified rabbit reticulocyte eIF3 was a gift from W. C. Merrick (Case Western Reserve University, Cleveland, OH).
Cell culture.
HeLa S3 and HEK 293 cells were obtained from the American Tissue Culture Collection and maintained in Dulbecco minimum essential medium (DMEM; Wisent) supplemented with 10% fetal bovine serum (Wisent) and 5 U of penicillin-streptomycin (Wisent)/ml in 5% CO2. Wild-type (WT) and S6K1/2 double-knockout mouse embryonic fibroblasts (MEFs) and ectopic reexpression of S6K1 and S6K2 were previously described (22). S6K1/2 double-knockdown HeLa S3 cells were obtained by lentiviral shRNA silencing strategy as described previously (23). The Sigma MISSION shRNA lentiviral vector accession numbers were: human S6K1 (TRCN0000003162), human S6K2 (TRCN0000000729), and nontarget shRNA control (SHC002). Cells were starved amino acid starved in Hanks balanced salt solution (HBSS; Wisent) containing calcium and magnesium supplemented with MEM vitamins (Invitrogen), 4.5 g of d-glucose/liter, and 5 U of penicillin-streptomycin/ml. For amino acid stimulation, HBSS media were replaced by DMEM or supplemented with amino acids solution. MEM essential and nonessential amino acid solutions (Invitrogen) were mixed with glutamine and adjusted at pH 7.4 to obtain a 25×-concentrated amino acid solution. Starved cells were pretreated with 20 nM rapamycin, 2.5 μM PP242, or 5 μM DG2 for 1 h before stimulation or left untreated. For experiments conducted using MEFs, full dephosphorylation of S6 was achieved after 8 h of treatment.
Plasmid construction.
Human eIF3g and eIF3f cDNAs were amplified from pGEX-eIF3g and pGEX-eIF3f plasmids (19) and subcloned into pcDNA-3HA using EcoRI sites. pcDNA-3HA-eIF3g was used as the template for PCR-based site-directed mutagenesis to produce the nonphosphorylatable mutant form of eIF3g (T41A and S42A).
Protein purification and GST pulldown.
Purification of recombinant GST-Paip1 and GST pulldowns were performed as previously described (19).
Transfection and immunoprecipitation.
pcDNA3-HA-eIF3g (WT or mutants), pcDNA-HA-eIF3f, or pcDNA3-HA-eIF4E (24) transfection was performed using Lipofectamine Plus reagent (Invitrogen) according to the manufacturer's protocol. At 48 h after transfection, cells were lysed in buffer C (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1% NP-40) containing protease and phosphatase inhibitors. First, cell extracts were transferred onto protein G-Sepharose beads (GE Healthcare) with 2 μl of HA antibody and incubated for 2 h at 4°C. Beads were washed four times in buffer C containing protease and phosphatase inhibitors. Bound proteins were processed for Western blotting. The same protocol was applied for S6K1-eIF3 interaction using buffer composed of 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 10% glycerol, 0.2% NP-40, 0.5 mM sodium orthovanadate, 5 mM NaF, 80 μM β-glycerophosphate, 10 mM sodium pyrophosphate, 1 mM dithiothreitol (DTT), and 1 mM EGTA as described previously (13).
Two-dimensional electrophoresis.
HeLa cells were lysed on ice in buffer B containing protease inhibitors. Half of the cell extract was treated with lambda protein phosphatase (New England BioLabs) at room temperature for 30 min. The other half was supplemented with phosphatase inhibitors (2 mM Na3VO4, 2 mM NaF, 25 mM β-glycerophosphate). Then, 200 μg of each extract was resolved on Immobiline dry strips (pH 4-7 [linear], 13 cm; GE Healthcare) according to the IPGphor isoelectric focusing (IEF) protocol. Then strips were equilibrated in sodium dodecyl sulfate (SDS) equilibration buffer prior second dimension SDS-PAGE and Western blotting.
In vitro kinase assay.
In vitro kinase assays were performed in a 25-μl reaction volume containing 60 mM HEPES-KOH (pH 7.6), 3 mM MgCl2, 3 mM MnCl2, and 1.2 mM DTT containing 50 μM ATP and 0.03 μCi of [γ-32P]ATP/μl. Briefly, 2 μg of RRL-purified eIF3 were incubated for 15 min at room temperature to inhibit kinase activity copurified with eIF3 complex in the presence of 500 ng of His6-S6K1 and/or 100 nM DG2. Reaction mixtures were transferred to ice, supplemented with ATP and [γ32P]ATP, further incubated at 30°C for 20 min, and stopped by the addition of 25 μl of Laemmli buffer. Proteins were resolved by SDS-PAGE and transferred onto a nitrocellulose membrane. Membranes were processed for 32P detection, followed by Western blotting with the indicated antibodies.
Translation assays.
The procedure for the translation assay for Paip1 was previously described (19). Briefly, HeLa cells were seeded in 12-well tissue culture dishes 1 day prior to transfection. Cells were cotransfected with 125 ng of pTet-HA-Paip1 or the control vector, 125 ng of pUHD-15-1, which expresses the Tet-controlled transactivator (tTA) and 25 ng of pRL-CMV (Promega) per well as a Renilla luciferase reporter construct. pBI-L vector (Stratagene) expressing firefly luciferase was used as a control vector. For assays measuring inhibitor effect, after transfection, the cells were incubated 24 h with 300 ng of doxycycline (Dox)/ml, 20 nM rapamycin, and/or 20 μM DG2 or left untreated. For assays measuring nutrient deprivation, after transfection, cells were placed in serum-free medium containing 300 ng of Dox/ml or left untreated for 24 h to obtain a homogenous expression of HA-Paip1. Cell media were then supplemented with 10% serum, replaced by HBSS, or left unchanged, in the presence or absence of Dox for an additional 8 h before harvesting. Cells were lysed in 1× passive lysis buffer (Promega) and Renilla and firefly luciferase activities were quantified with a dual-luciferase reporter assay system (Promega). The Renilla luciferase activity was corrected based on protein concentration, as measured with Bio-Rad protein assay reagent. The relative induction of translation by Paip1 was determined by calculating the ratio of Renilla luciferase activity between the induced condition (no Dox) and the repressed condition (300 ng of Dox/ml).
RESULTS
Paip1-eIF3 interaction is regulated by amino acid availability.
To study how Paip1-eIF3 interaction is regulated by amino acid availability, we first starved HeLa cells of amino acids, and the effect on mTOR signaling was determined by Western blotting with phospho-specific antibodies against S6K1 (Thr389) and S6 (Ser240/Ser244) (referred to here as phospho-S6) as a readout of mTOR activity. After 18 h of amino acid deprivation, both phospho-S6K and phospho-S6 decreased to minimal levels, indicating mTORC1 was fully inactivated (Fig. 1A, Time zero). Amino acids were added back for 2 to 24 h and changes in Paip1-eIF3 interaction were monitored. As a control, cells were maintained for 24 h in complete medium (DMEM supplemented with 10% serum). In agreement with earlier reports (6), amino acid addition induced S6K1 and S6 phosphorylation, which reached a maximum level after 2 h. While S6K1 phosphorylation returned to control levels after 24 h, S6 phosphorylation remained elevated compared to the control level (Fig. 1A, top panel). Previously, Paip1 was shown to interact with the eIF3 complex along with PABP, and all eIF3 subunits were present in stoichiometric amounts (17, 19). To determine whether the Paip1-eIF3 interaction is regulated by amino acids, we performed GST pulldowns with recombinant GST-Paip1 (p65 isoform) (19). Paip1 interaction with eIF3 was gradually enhanced from 2 to 24 h and reached a maximal level, similar to that observed in cells maintained in control conditions after 24 h, as determined by GST pulldown assays (Fig. 1A, bottom panel). For most of the experiments described below, we used the 4-h time point, since Paip1-eIF3 interaction was markedly enhanced at this time compared to amino acid-starved cells. Taken together, these data demonstrate that Paip1-eIF3 interaction is stimulated by amino acids.
FIG 1.

Paip1-eIF3 interaction is stimulated by amino acids and inhibited by mTOR inhibitors. (A) HeLa cells were grown in DMEM with 10% serum (Ctrl, control condition) or amino acid starved overnight. Starved HeLa cells were stimulated with amino acids (AA) for the indicated times or left untreated. GST pulldown experiments were conducted with GST-Paip1 (p65 isoform) or GST alone using whole-cell lysates (WCL). WCL (top panel) and GST pulldown eluates (bottom panel) were processed for Western blotting with the indicated antibodies: phospho-Ser240/444 S6 (p-S6) or phospho-Thr389 S6K (p-S6K). (B) Experiments were performed as in panel A. Cells were treated with different mTOR inhibitors (1 h): 20 nM rapamycin (Rapa.) or 2.5 μM PP242, followed by 4 h of amino acid stimulation.
mTOR inhibition blocks the Paip1-eIF3 interaction.
Amino acids stimulate mTORC1 activity, and consequently the phosphorylation of the downstream targets 4E-BP1 and S6K1 (25). To determine whether mTOR controls the Paip1-eIF3 interaction, we used two mTOR inhibitors, rapamycin (an allosteric mTORC1-specific inhibitor) and PP242 (an active site mTOR inhibitor, which inhibits mTORC1 and mTORC2) (26). Amino acid-starved HeLa cells were pretreated with each inhibitor for 1 h, followed by the addition of amino acids for 4 h. Rapamycin and PP242 strongly diminished amino acid-induced phosphorylation of S6K1 and S6 after amino acid stimulation (Fig. 1B, top panel) and the Paip1-eIF3 interaction (bottom panel). In contrast, the Paip1-PABP interaction remained unchanged. The amino acid-enhanced Paip1-eIF3 interaction was not sensitive to a MEK inhibitor (U0126; data not shown), indicating that the Ras/mitogen-activated protein kinase (MAPK) signaling pathway does not play a role in controlling the Paip1-eIF3 interaction. Thus, mTOR inhibitors impair the interaction of Paip1 with eIF3, by interdicting the amino acid-induced mTORC1 pathway.
eIF3g phosphorylation on Thr41 and Ser42 is not stimulated by amino acids, nor does it affect Paip1-eIF3 interaction.
We hypothesized that the Paip1-eIF3 interaction was regulated by Paip1 and/or eIF3 phosphorylation. On the one hand, mass spectrometry failed to identify kinases interacting with recombinant GST-Paip1 (19). Furthermore, the Paip1-PABP interaction was not controlled by amino acids (Fig. 1B). The PABP- and eIF3-binding regions on Paip1 are partially overlapping, near the PAM2 motif, suggesting that Paip1-eIF3 interaction is regulated not through Paip1 but rather through eIF3 (19). On the other hand, eIF3 interaction with mTORC1 and S6K1 (13–15) was previously reported, and several eIF3 subunits are phosphorylated in response to cell growth and proliferation-promoting signals (17). The Paip1-binding subunit, eIF3g, contains two phosphorylation sites: Thr41 and Ser42 (17). Consequently, we investigated whether amino acids induce eIF3g phosphorylation to enhance Paip1-eIF3 interaction. To this end, we performed two-dimensional (2D) electrophoresis, mass spectrometry, and phosphorylation site mutagenesis analysis. To map the phosphorylation site(s) on eIF3g, we used whole lysates from HeLa cells grown overnight in complete or amino acid-free medium. A λ protein phosphatase-treated extract was used to visualize unphosphorylated proteins. The λ protein phosphatase dephosphorylated S6K1 and S6 in the control lysate (Fig. 2A). In response to amino acid deprivation, both S6K1 and S6 became completely dephosphorylated (Fig. 2A). The same samples were analyzed by 2D IEF–SDS-PAGE. Under standard growth conditions or after amino acid deprivation, the phosphoprotein map of eIF3g contained high levels of nonphosphorylated protein (Fig. 2B, black arrow) and two major phosphorylated species (gray arrow). Indeed, after phosphatase treatment, all of the eIF3g isoforms shifted to the nonphosphorylated state (Fig. 2B). We failed to detect doubly phosphorylated eIF3g at the predicted isoelectric point under any condition (Fig. 2B, dashed arrow). The two phosphorylated proteins most probably correspond to phosphorylation on either Thr41 or Ser42. Thus, the 2D electrophoresis analysis suggests that eIF3g is phosphorylated only on one amino acid, and this phosphorylation is independent of amino acid stimulation.
FIG 2.
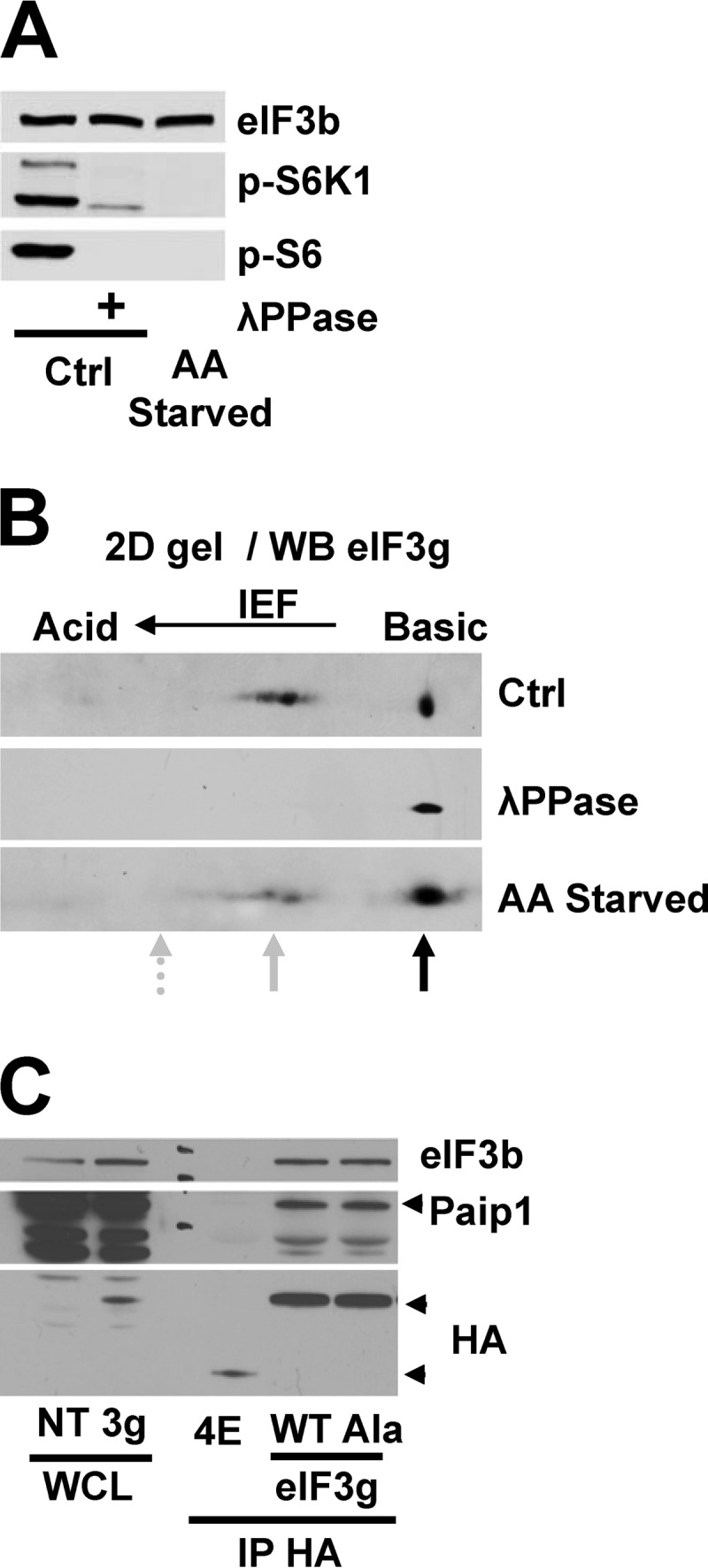
eIF3g phosphorylation is not affected by amino acids deprivation and does not alter Paip1-eIF3 interaction. (A) Whole-cell lysates from HeLa cells grown in control conditions (Ctrl) or amino acid starved overnight were treated with lambda phosphatase (λPPase) and processed for Western blotting. (B) Whole-cell extracts from panel A were subjected to 2D IEF–SDS-PAGE, followed by immunoblotting with anti-eIF3g antibody. The black arrow indicates nonphosphorylated proteins, and the solid gray arrow indicates monophosphorylated proteins. The dashed arrow indicates the predicted position of doubly phosphorylated proteins based on the calculated isoelectric point. (C) Exogenous HA-eIF4E or HA-eIF3g wild-type (WT) or nonphosphorylatable mutant T41A-S42A (Ala) were expressed in HeLa cells. WCL were subjected to immunoprecipitation with anti-HA antibody (IP-HA) and processed for Western blotting.
Damoc et al. (17) showed that either Thr41 and Ser42 or both can be phosphorylated on eIF3g in the presence of serum. We therefore searched for phosphopeptides by mass spectrometry in immunoprecipitated HA-tagged eIF3g of HeLa cells grown in complete or amino acid-free medium. In both conditions, eIF3g was phosphorylated on either Thr41 or Ser42, but peptides carrying both of the phosphorylated amino acids were not detected (data not shown). To further corroborate our mass spectrometry results, we generated a phosphomutant eIF3g, by mutating both Thr41 and Ser42 to alanines. HA immunoprecipitations were performed in HeLa cells transfected with plasmids expressing HA-tagged wild-type (WT) eIF3g, nonphosphorylatable mutant (Ala) eIF3g, or HA-tagged eIF4E as a negative control for the Paip1 interaction. Equal amounts of Paip1 were coimmunoprecipitated with both WT and the Ala mutant HA-eIF3g (Fig. 2C), demonstrating a similar interaction with Paip1. Similar data were obtained using recombinant GST-tagged wild-type and nonphosphorylatable (Ala) mutant eIF3g (data not shown). From these experiments, we conclude that phosphorylation of eIF3g at either Thr41 or Ser42 is neither regulated by amino acids nor controlling its interaction with Paip1.
S6K activity promotes Paip1-eIF3 interaction.
It is conceivable that the phosphorylation of one or more of the other 12 subunits of eIF3 affects its interaction with Paip1. Of special interest, eIF3b and eIF3c directly interact with eIF3g and contain multiple phosphorylation sites (17, 20). eIF3 interacts with both mTORC1 and S6K1 (13–15). We therefore hypothesized that mTORC1 and/or S6Ks are the kinases that phosphorylate eIF3 and induce the interaction of Paip1 with eIF3. To test this hypothesis, we used an S6K-specific inhibitor, DG2 (27). Upon amino acid stimulation, DG2 completely blocked S6 phosphorylation and caused increased phosphorylation of S6K1 (Fig. 3A, top panel). The DG2-mediated hyperphosphorylation of S6K1 might be due to the elimination of the S6K negative feedback on an upstream kinase and thus maintaining mTORC1 active. In the presence of DG2, Paip1-eIF3 interaction was no longer enhanced by amino acid (Fig. 3A, bottom panel), indicating that S6K could be a potential kinase of eIF3. Because the above-described experiments were performed using cell extracts incubated with purified recombinant GST-Paip1 fusion protein, we wanted to determine whether the results could be recapitulated in cells. To this end, Flag-tagged Paip1 was expressed in HeLa cells, and Paip1-eIF3 interaction was analyzed by Western blotting following anti-Flag immunoprecipitation. As observed with recombinant GST-Paip1, Flag-Paip1 interaction with endogenous eIF3 was sensitive to DG2 (data not shown), bolstering the idea that S6K could be a potential kinase of eIF3.
FIG 3.

Paip1-eIF3 interaction requires S6K activity. (A) HeLa cells were grown in DMEM with 10% serum (Ctrl, control condition) or amino acid starved overnight. Amino acid-starved HeLa cells were pretreated with DG2 at 5 μM (1 h) or left untreated, followed by amino acid stimulation (4 h). Whole-cell lysates (WCL) were subjected to GST-Paip1 pulldown. WCL (top panel) and GST-Paip1 pulldown eluates (bottom panel) were processed for Western blotting with the indicated antibodies: phospho-Ser240/444 S6 (p-S6) or phospho-Thr389 S6K (p-S6K). (B) HeLa cells expressing either S6K1 and S6K2 shRNAs (S6K1/2 shRNA) or Scramble shRNA were treated as in panel A using 20 nM rapamycin (Rapa.). WCL (top panel) and GST-Paip1 pulldown eluates (bottom panel) were processed for Western blotting using phospho-Ser65 4E-BP1 (p-4E-BP1). Paip1-eIF3 interaction is expressed as a percentage (±the standard deviation) of Paip1-eIF3 interaction in HeLa cells in a control condition. Quantifications were performed using ImageJ software on replicate experiments. (C) WT MEFs or S6K1/2 double-knockout MEFs were amino acid starved for 8 h and left untreated. WCL (top panel) and GST-Paip1 pulldown eluates (bottom panel) were processed for Western blotting. (D) S6K1/2 double-knockout MEFs were transduced with vector expressing S6K1, S6K2, S6K1, and S6K2 or empty vector (Mock). Cells were treated as in panel C, and WCL (top panel) and GST-Paip1 pulldown eluates (bottom panel) were processed for Western blotting. The asterisk indicates a nonspecific band.
To further corroborate this finding, we used HeLa cells depleted of S6K1(>90%) and S6K2 (>80%) (Fig. 3B, top panel). Knockdown of S6K1/2 had no effect on the quantity of S6 protein but greatly reduced phosphorylation, as expected (Fig. 3B, top panel). However, amino acids increased 4E-BP1 phosphorylation to the same extent in control and S6K1/2 knockdown cells, indicating that the mTORC1 kinase activity directed toward these targets remained unchanged. As such, rapamycin-mediated inhibition of mTOR reduced the amino acid-induced phosphorylation of 4E-BP1 and S6 (Fig. 3B, top panel). In control cells, amino acid-stimulated Paip1-eIF3 interaction was sensitive to rapamycin. However, in the S6K1/2 knockdown cells, Paip1-eIF3 interaction was markedly reduced and was neither affected by amino acid deprivation nor by rapamycin treatment (Fig. 3B, bottom panel). These data strongly support the conclusion that S6Ks play a central role in amino acid regulation of the Paip1-eIF3 interaction.
We further investigated the role of S6Ks in Paip1-eIF3 interaction using S6K1/2 double-knockout (DKO) and WT MEFs. As expected, S6 was phosphorylated on Ser240-244 only in WT MEFs in complete media but not in extracts of cells lacking S6Ks or amino acid-starved for 8 h (Fig. 3C). The Paip1-eIF3 interaction was much weaker in the S6K1/2 DKO MEFs than in the WT MEFs, and amino acid deprivation had no additional effect on Paip1-eIF3 dissociation in S6K1/2 DKO MEFs (Fig. 3C, bottom panel). Similar experiments were performed using DKO MEFs that had S6K1, S6K2, or S6K1/2 reexpressed (Fig. 3D). Although reexpression of S6K1 or S6K2 alone increased S6 phosphorylation and Paip1-eIF3 interaction in DKO MEFs, reexpression of both S6K1 and S6K2 fully restored S6 phosphorylation, Paip1-eIF3 interaction, and sensitivity to amino acid (Fig. 3D, top panel). Thus, S6K activity promotes the interaction between Paip1 with eIF3.
S6K interacts with eIF3f, phosphorylates eIF3 in vitro, and regulates Paip1 activity in vivo.
We next sought to determine whether S6K1 and eIF3 directly interact. Immunoprecipitation experiments were conducted on extracts from cells expressing HA-tagged eIF3g or green fluorescent protein (GFP). We also expressed HA-tagged eIF3f as a direct interaction between eIF3f with S6K1 was previously reported (13). Both transfected eIF3f and eIF3g were integrated into the eIF3 complex as they interacted with subunits 3a, 3b, and 3c (Fig. 4A). We detected an S6K1 interaction mostly with eIF3f and to a much lesser extent with eIF3g, suggesting that eIF3f is the direct interactor of S6K1 and that some eIF3 subcomplexes could be more prone to phosphorylation by S6Ks.
FIG 4.

S6K interacts with and phosphorylates eIF3 in vitro. (A) HEK-293T cells were transfected with constructs expressing HA-tagged eIF3f (3f), eIF3g (3g), GFP, or an empty vector. Whole-cell lysates (WCL) were subjected to immunoprecipitation with an anti-HA antibody. WCL and HA immunoprecipitates were processed for Western blotting with the indicated antibodies. IgB, light-chain immunoglobulin. (B) Kinase assays were performed with recombinant His-tagged S6K1 (HIS-S6K) and RRL-purified eIF3 (eIF3). Proteins were incubated for 20 min with [γ-32P]ATP in the presence of 100 nM DG2. Samples were subjected to SDS-PAGE and transferred onto nitrocellulose membrane, followed by Western blotting and autoradiography.
To investigate whether S6K1 can phosphorylate eIF3 in vitro, we performed a kinase assay using recombinant S6K1 lacking the autoinhibitory domain and eIF3 purified from rabbit reticulocyte lysate. First, in the absence of additional factors, autophosphorylation of S6K1 occurred (Fig. 4B, lane 1). Addition of purified eIF3 to S6K1 induced the appearance of two major bands. One corresponds to the molecular masses (110 to 120 kDa) of subunit 3b, 3c, or 3a; the last subunit is prone to degradation and therefore fragmented in the RRL-purified eIF3 complex (28). The other band (44 to 47 kDa) might correspond to either 3e, 3f, 3g, or 3h subunit. The 44- to 47-kDa band disappeared upon the addition of the S6K inhibitor, DG2, in contrast to the 110- to 120-kDa band (Fig. 4B). This suggests a greater accessibility of the 44- to 47-kDa protein. These data show that S6K1 phosphorylates eIF3 in vitro at least on two subunits, because each 32P signal on the autoradiography corresponds to the phosphorylation of one or several eIF3 subunits closely migrating on SDS-PAGE.
We next sought to determine whether the inhibition of S6K impairs the stimulatory activity of Paip1 on translation. In vivo translation assays were performed after rapamycin or DG2 treatment. DNA vectors expressing HA-tagged Paip1 p65 under the control of the Tet-off promoter were transfected into HeLa cells, along with constructs expressing the Renilla luciferase and the tTA (19). The relative induction of luciferase activity mediated by Paip1 was determined by calculating the ratio of Renilla luciferase activity between induced (no Dox) and repressed (with Dox) expression of HA-tagged Paip1. Paip1 stimulatory activity was reduced about 35% in rapamycin- and DG2-treated compared to nontreated cells (Fig. 5A). Rapamycin- or DG2-mediated S6 dephosphorylation, which was confirmed by Western blotting (Fig. 5B) did not affect Paip1 expression. These data indicate that the Paip1 stimulatory effect requires S6K activity. We also investigated Paip1-mediated translational control in response to serum and/or amino acids availability (Fig. 5C). Amino acid withdrawal induced about 25% decrease of Paip1 stimulatory activity, whereas serum deprivation had no effect. This is consistent with the significant S6 dephosphorylation observed upon amino acid deprivation compared to serum deprivation (Fig. 5D).
FIG 5.

S6K controls Paip1 activity in vivo. (A) Paip1-dependent translation stimulation in vivo. Cells were either mock transfected (Ctrl) or transfected with the pTet-HA-Paip1 p65 plasmid (HA-Paip1), together with constructs expressing the Renilla luciferase and the tTA. Cells were placed in medium containing 300 ng of Dox/ml, 20 nM rapamycin (Rapa.), and/or 20 μM DG2 or left untreated. Renilla luciferase activity was quantified in HeLa cell extracts. Relative induction of the luciferase reporter was determined by calculating the ratio of Renilla luciferase activity between induced (no Dox) and repressed (with Dox) expression of the HA-tagged Paip1 p65. Error bars denote the standard errors of the mean for three independent experiments. (B) Extracts from panel A were subjected to SDS-PAGE and Western blotting with the indicated antibodies. (C) Cells were transfected as in panel A and placed in serum-free medium containing 300 ng of Dox/ml or left untreated for 24 h. Cell media were then supplemented with 10% serum (+serum), replaced by HBSS (−AA), or left unchanged in the presence or absence of doxycycline for an additional 8 h before harvesting. (D) Extracts from panel C were subjected to SDS-PAGE and Western blotting with the indicated antibodies.
DISCUSSION
We show here that amino acids regulate eIF3 binding to Paip1 via mTOR and S6Ks. S6Ks promote the stability of Paip1-eIF3 complex and eIF3f/p47 binds S6K1 within the eIF3 complex. We also show that S6K1 phosphorylates eIF3 in vitro. These data demonstrate that eIF3 is a new translation target of the mTOR/S6K pathway.
We previously reported on the Paip1-eIF3 interaction and showed that Paip1 directly binds to the eIF3g/p44 subunit (19). However, eIF3g phosphorylation on Thr 41 and Ser 42 is not controlled by amino acids. This suggests that Paip1-eIF3 interaction requires several eIF3 subunits and that phosphorylation of a subunit(s) other than eIF3g effects Paip1 binding. Optimal folding of eIF3g is most likely achieved only when it is incorporated into eIF3. Thus, it is conceivable that phosphorylation of other subunits would affect the global conformation or surface charge of the eIF3 complex. Recent data from eIF3 in vitro reconstitution experiments indicate that the octameric eIF3 subcomplex composed of subunits a, c, e, f, h, k, l, and m is highly stable (29). This complex associates with the “b,g,i” subcomplex to form a dodecameric complex lacking the j subunit. The recombinant octameric eIF3 structure strikingly resembles the native eIF3 (30). It is thus possible that 3f serves as a docking site for S6K to phosphorylate another eIF3 subunit. eIF3f is tightly associated with the octameric eIF3 complex containing eIF3a and 3c, the most phosphorylated subunits of eIF3 (17, 20). In addition, eIF3f is located in the left arm and leg of the octameric structure and binds directly to j subunit and the b,g,i subcomplex. Thus, S6K might also regulate the assembly of eIF3 subcomplexes through phosphorylation.
S6Ks modulate translation initiation through phosphorylation of multiple targets. S6Ks phosphorylate ribosomal protein S6 (RPS6) on Ser 235/236 and 240/244. However, genetic ablation of S6 phosphorylation modestly affects global translation (31, 32). S6Ks also regulate eIF4A activity through the phosphorylation of PDCD4 (33). One of the best-characterized targets of S6Ks is eIF4B whose activity and binding to eIF3 is regulated by phosphorylation on Ser422 in an RXRXXS/T consensus motif common to RSK, AKT, and S6K. Our data strongly suggest that S6Ks could also target eIF3. Whereas phosphorylation on Ser235/236 of RPS6 or Ser422 of eIF4B is readily detected in S6K1/2 DKO MEFs due to the activity of p90-RSK, phosphorylation of Ser240/244 on RPS6 is abrogated (31). This raises the question whether the S6K recognition motif has been properly defined and whether important S6K-specific substrates remain to be discovered. We found a putative S6K recognition site in eIF3c/p110 on residue Ser711 but could not detect any changes in phosphorylation by mass spectrometry in response to amino acids deprivation (data not shown). An S6K recognition site was described in eIF3h/p40 from Arabidopsis thaliana (34), but in HeLa cells, we could not detect changes in its phosphorylation by 2D electrophoresis after prolonged amino acid deprivation or mTOR inhibitor treatment (data not shown).
Thus far, the eIF3 phosphorylation sites in mammals (17) have not been shown to have functional significance for any of the subunits, with the exception of the noncore eIF3h subunit, for which its oncogenic potential requires phosphorylation (18). One possible reason is the difficulty in replacing the endogenous eIF3 subunit by a nonphosphorylatable mutant at an isostoichiometric ratio in mammals. In Saccharomyces cerevisiae, phosphoresidues were described in eIF3b/Prt1 and eIF3c/Nip1, and casein kinase II can phosphorylate eIF3c/Nip1. Nevertheless, a difference in eIF3c/Nip1 binding with eIF3 subunits or initiation factors associated with the expression of eIF3c/Nip1 phosphomutant was not reported, but a mild slow-growth phenotype was described previously (35). Casein kinase II was shown to phosphorylate mammalian eIF3 in vitro similarly to RSK and protein kinase C (36) but was not associated with any specific function or change in affinity for binding partners.
eIF3 phosphorylation by S6Ks might also regulate the association of other factors with eIF3. Endogenous eIF4B was shown to exhibit enhanced association with eIF3 upon phosphorylation by RSK or S6K, which might be due to the increased phosphorylation of eIF3 (37). In addition, eIF4G binding to eIF3 could also behave in a similar manner, since the complex is stabilized by insulin treatment and is sensitive to rapamycin (14). The use S6K-specific inhibitors such as DG2 or PF-4708671 (38) shed light on the specific role of S6Ks in those mechanisms compared to mTOR inhibitors, which abolished both mTOR and S6K activity.
Regulation of Paip1-eIF3 interaction by mTOR/S6K signaling adds an extra layer of complexity to the translational control of this pathway, leading to a better synchronization of translation initiation. mRNA circularization is brought about by bridging the 5′ and the 3′ end through eIF4G/PABP interaction (2). The stabilized association of Paip1 with eIF3 after mTOR/S6K activation reinforces the closed model. Through the regulation of eIF4B, PDCD4, CBP80, RPS6 and others, S6Ks appears to fine-tune translation, by synchronizing the rhythm of multiple components of the translational machinery. Regulation of eIF3 by S6Ks brings a supplemental instrument to the orchestra. Nevertheless, detailed functions of eIF3 and its phosphorylation remain to be fully understood in order to improve the sound of this complex symphony.
ACKNOWLEDGMENTS
We thank C. Lister for exceptional technical assistance, Y. Svitkin for suggestions and experimental assistance, and C. Bousquet, J. Guillermet-Guibert, and C. S. Fraser for comments and careful reading of the manuscript. We thank N. Saint-Laurent and F. Lopez of the Groupe Protéomique CRCT for 2D electrophoresis.
Y.M. was recipient of a postdoctoral fellowship from la Fondation de France, and B.F. was supported by a Ph.D. program from CNRS/Région Midi-Pyrénées. This study was supported by Canadian Institute of Health Research grant MOP-7214 to N.S., grants from INSERM-Université Paul Sabatier and from La LIGUE (Comités de Hautes-Pyrénées et de Lot-et-Garonne and Equipes Labelisées programs) to S.P., and grants from Région Midi-Pyrénées and Europe to B.M.
Footnotes
Published ahead of print 6 January 2014
REFERENCES
- 1.Holcik M, Sonenberg N. 2005. Translational control in stress and apoptosis. Nat. Rev. Mol. Cell. Biol. 6:318–327. 10.1038/nrm1618 [DOI] [PubMed] [Google Scholar]
- 2.Sonenberg N, Hinnebusch AG. 2009. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136:731–745. 10.1016/j.cell.2009.01.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Laplante M, Sabatini DM. 2012. mTOR signaling in growth control and disease. Cell 149:274–293. 10.1016/j.cell.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma XM, Blenis J. 2009. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell. Biol. 10:307–318. 10.1038/nrm2672 [DOI] [PubMed] [Google Scholar]
- 5.Kimball SR, Jefferson LS. 2000. Regulation of translation initiation in mammalian cells by amino acids, p 561–579 In Sonenberg N, Hershey JWB, Mathews MB. (ed), Translational control of gene expression, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 6.Sengupta S, Peterson TR, Sabatini DM. 2010. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol. Cell 40:310–322. 10.1016/j.molcel.2010.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dodd KM, Tee AR. 2012. Leucine and mTORC1: a complex relationship. Am. J. Physiol. Endocrinol. Metab. 302:E1329–E1342. 10.1152/ajpendo.00525.2011 [DOI] [PubMed] [Google Scholar]
- 8.Jastrzebski K, Hannan KM, Tchoubrieva EB, Hannan RD, Pearson RB. 2007. Coordinate regulation of ribosome biogenesis and function by the ribosomal protein S6 kinase, a key mediator of mTOR function. Growth Factors 25:209–226. 10.1080/08977190701779101 [DOI] [PubMed] [Google Scholar]
- 9.Shahbazian D, Parsyan A, Petroulakis E, Hershey J, Sonenberg N. 2010. eIF4B controls survival and proliferation and is regulated by proto-oncogenic signaling pathways. Cell Cycle 9:4106–4109. 10.4161/cc.9.20.13630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dorrello NV, Peschiaroli A, Guardavaccaro D, Colburn NH, Sherman NE, Pagano M. 2006. S6K1- and βTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science 314:467–471. 10.1126/science.1130276 [DOI] [PubMed] [Google Scholar]
- 11.Raught B, Peiretti F, Gingras AC, Livingstone M, Shahbazian D, Mayeur GL, Polakiewicz RD, Sonenberg N, Hershey JW. 2004. Phosphorylation of eucaryotic translation initiation factor 4B Ser422 is modulated by S6 kinases. EMBO J. 23:1761–1769. 10.1038/sj.emboj.7600193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang L, Gout I, Proud CG. 2001. Cross-talk between the ERK and p70 S6 kinase (S6K) signaling pathways. MEK-dependent activation of S6K2 in cardiomyocytes. J. Biol. Chem. 276:32670–32677. 10.1074/jbc.M102776200 [DOI] [PubMed] [Google Scholar]
- 13.Csibi A, Cornille K, Leibovitch MP, Poupon A, Tintignac LA, Sanchez AM, Leibovitch SA. 2010. The translation regulatory subunit eIF3f controls the kinase-dependent mTOR signaling required for muscle differentiation and hypertrophy in mouse. PLoS One 5:e8994. 10.1371/journal.pone.0008994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harris TE, Chi A, Shabanowitz J, Hunt DF, Rhoads RE, Lawrence JC., Jr 2006. mTOR-dependent stimulation of the association of eIF4G and eIF3 by insulin. EMBO J. 25:1659–1668. 10.1038/sj.emboj.7601047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holz MK, Ballif BA, Gygi SP, Blenis J. 2005. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell 123:569–580. 10.1016/j.cell.2005.10.024 [DOI] [PubMed] [Google Scholar]
- 16.Hinnebusch AG. 2006. eIF3: a versatile scaffold for translation initiation complexes. Trends Biochem. Sci. 31:553–562. 10.1016/j.tibs.2006.08.005 [DOI] [PubMed] [Google Scholar]
- 17.Damoc E, Fraser CS, Zhou M, Videler H, Mayeur GL, Hershey JW, Doudna JA, Robinson CV, Leary JA. 2007. Structural characterization of the human eukaryotic initiation factor 3 protein complex by mass spectrometry. Mol. Cell. Proteomics 6:1135–1146. 10.1074/mcp.M600399-MCP200 [DOI] [PubMed] [Google Scholar]
- 18.Zhang L, Smit-McBride Z, Pan X, Rheinhardt J, Hershey JW. 2008. An oncogenic role for the phosphorylated h-subunit of human translation initiation factor eIF3. J. Biol. Chem. 283:24047–24060. 10.1074/jbc.M800956200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martineau Y, Derry MC, Wang X, Yanagiya A, Berlanga JJ, Shyu AB, Imataka H, Gehring K, Sonenberg N. 2008. Poly(A)-binding protein-interacting protein 1 binds to eukaryotic translation initiation factor 3 to stimulate translation. Mol. Cell. Biol. 28:6658–6667. 10.1128/MCB.00738-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou M, Sandercock AM, Fraser CS, Ridlova G, Stephens E, Schenauer MR, Yokoi-Fong T, Barsky D, Leary JA, Hershey JW, Doudna JA, Robinson CV. 2008. Special feature: mass spectrometry reveals modularity and a complete subunit interaction map of the eukaryotic translation factor eIF3. Proc. Natl. Acad. Sci. U. S. A. 105:18139–18144. 10.1073/pnas.0801313105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Craig AW, Haghighat A, Yu AT, Sonenberg N. 1998. Interaction of polyadenylate-binding protein with the eIF4G homologue PAIP enhances translation. Nature 392:520–523. 10.1038/33198 [DOI] [PubMed] [Google Scholar]
- 22.Alain T, Lun X, Martineau Y, Sean P, Pulendran B, Petroulakis E, Zemp FJ, Lemay CG, Roy D, Bell JC, Thomas G, Kozma SC, Forsyth PA, Costa-Mattioli M, Sonenberg N. 2010. Vesicular stomatitis virus oncolysis is potentiated by impairing mTORC1-dependent type I IFN production. Proc. Natl. Acad. Sci. U. S. A. 107:1576–1581. 10.1073/pnas.0912344107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dowling RJ, Topisirovic I, Alain T, Bidinosti M, Fonseca BD, Petroulakis E, Wang X, Larsson O, Selvaraj A, Liu Y, Kozma SC, Thomas G, Sonenberg N. 2010. mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science 328:1172–1176. 10.1126/science.1187532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mamane Y, Petroulakis E, Martineau Y, Sato TA, Larsson O, Rajasekhar VK, Sonenberg N. 2007. Epigenetic activation of a subset of mRNAs by eIF4E explains its effects on cell proliferation. PLoS One 2:e242. 10.1371/journal.pone.0000242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM. 2008. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320:1496–1501. 10.1126/science.1157535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, Shokat KM. 2009. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 7:e38. 10.1371/journal.pbio.1000038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Okuzumi T, Fiedler D, Zhang C, Gray DC, Aizenstein B, Hoffman R, Shokat KM. 2009. Inhibitor hijacking of Akt activation. Nat. Chem. Biol. 5:484–493. 10.1038/nchembio.183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pisarev AV, Unbehaun A, Hellen CU, Pestova TV. 2007. Assembly and analysis of eukaryotic translation initiation complexes. Methods Enzymol. 430:147–177. 10.1016/S0076-6879(07)30007-4 [DOI] [PubMed] [Google Scholar]
- 29.Sun CM, Todorovic A, Querol-Audi J, Bai Y, Villa N, Snyder M, Ashchyan J, Lewis CS, Hartland A, Gradia S, Fraser CS, Doudna JA, Nogales E, Cate JHD. 2011. Functional reconstitution of human eukaryotic translation initiation factor 3 (eIF3). Proc. Natl. Acad. Sci. U. S. A. 108:20473–20478. 10.1073/pnas.1116821108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Siridechadilok B, Fraser CS, Hall RJ, Doudna JA, Nogales E. 2005. Structural roles for human translation factor eIF3 in initiation of protein synthesis. Science 310:1513–1515. 10.1126/science.1118977 [DOI] [PubMed] [Google Scholar]
- 31.Pende M, Um SH, Mieulet V, Sticker M, Goss VL, Mestan J, Mueller M, Fumagalli S, Kozma SC, Thomas G. 2004. S6K1−/−/S6K2−/− mice exhibit perinatal lethality and rapamycin-sensitive 5′-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol. Cell. Biol. 24:3112–3124. 10.1128/MCB.24.8.3112-3124.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ruvinsky I, Sharon N, Lerer T, Cohen H, Stolovich-Rain M, Nir T, Dor Y, Zisman P, Meyuhas O. 2005. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev. 19:2199–2211. 10.1101/gad.351605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schmid T, Jansen AP, Baker AR, Hegamyer G, Hagan JP, Colburn NH. 2008. Translation inhibitor Pdcd4 is targeted for degradation during tumor promotion. Cancer Res. 68:1254–1260. 10.1158/0008-5472.CAN-07-1719 [DOI] [PubMed] [Google Scholar]
- 34.Schepetilnikov M, Dimitrova M, Mancera-Martinez E, Geldreich A, Keller M, Ryabova LA. 2013. TOR and S6K1 promote translation reinitiation of uORF-containing mRNAs via phosphorylation of eIF3h. EMBO J. 32:1087–1102. 10.1038/emboj.2013.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Farley AR, Powell DW, Weaver CM, Jennings JL, Link AJ. 2011. Assessing the components of the eIF3 complex and their phosphorylation status. J. Proteome Res. 10:1481–1494. 10.1021/pr100877m [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tuazon PT, Merrick WC, Traugh JA. 1989. Comparative analysis of phosphorylation of translational initiation and elongation factors by seven protein kinases. J. Biol. Chem. 264:2773–2777 [PubMed] [Google Scholar]
- 37.Shahbazian D, Roux PP, Mieulet V, Cohen MS, Raught B, Taunton J, Hershey JW, Blenis J, Pende M, Sonenberg N. 2006. The mTOR/PI3K and MAPK pathways converge on eIF4B to control its phosphorylation and activity. EMBO J. 25:2781–2791. 10.1038/sj.emboj.7601166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pearce LR, Alton GR, Richter DT, Kath JC, Lingardo L, Chapman J, Hwang C, Alessi DR. 2010. Characterization of PF-4708671, a novel and highly specific inhibitor of p70 ribosomal S6 kinase (S6K1). Biochem. J. 431:245–255. 10.1042/BJ20101024 [DOI] [PubMed] [Google Scholar]