

ABSTRACT
Populations of RNA viruses can spontaneously produce variants that differ in genome size, sequence, and biological activity. Defective variants that lack essential genes can nevertheless reproduce by coinfecting cells with viable virus, a process that interferes with virus growth. How such defective interfering particles (DIPs) change in abundance and biological activity within a virus population is not known. Here, a prototype RNA virus, vesicular stomatitis virus (VSV), was cultured for three passages on BHK host cells, and passages were subjected to Illumina sequencing. Reads from the initial population, when aligned to the full-length viral sequence (11,161 nucleotides [nt]), distributed uniformly across the genome. However, during passages two plateaus in read counts appeared toward the 5′ end of the negative-sense viral genome. Analysis by normalization and a simple sliding-window approach revealed plateau boundaries that suggested the emergence and enrichment of at least two truncated species having medium (∼5,900 nt) and short (∼4,000 nt) genomes. Relative measures of full-length and truncated species based on read counts were validated by quantitative reverse transcription-PCR (qRT-PCR). Limit-of-detection analysis suggests that deep sequencing can be more sensitive than complementary measures for detecting and quantifying defective particles in a population. Further, particle counts from transmission electron microscopy, coupled with infectivity assays, linked the rise in smaller genomes with an increase in truncated particles and interference activity. In summary, variation in deep sequencing coverage simultaneously shows the size, location, and relative level of truncated-genome variants, revealing a level of population heterogeneity that is masked by other measures of viral genomes and particles.
IMPORTANCE We show how deep sequencing can be used to characterize the emergence, diversity, and relative abundance of truncated virus variants in virus populations. Adaptation of this approach to natural isolates may elucidate factors that influence the stability and persistence of virus populations in nature.
INTRODUCTION
Defective interfering particles (DIPs) are truncated forms of viruses that emerge during viral infections. DIPs cannot replicate on their own, but upon coinfection with intact virus, they can divert the viral replication machinery to make defective genomes and particles, ultimately at the expense of intact virus production (1, 2). In culture, DIPs have been observed for most classes of DNA and RNA viruses (2–4). In nature, DIPs have been discovered in an outbreak of influenza A virus in chickens (5) and in West Nile virus isolates from avian hosts (6). In human hosts, clinical isolates from patients infected with dengue virus (7) and influenza A virus (8) provide evidence that DIPs and their genomes can be transmitted between patients.
DIPs have been characterized by their genome length and sequences (9–13), their particle sizes and morphologies (14–18), and their biological activities (19, 20). Few studies have combined genome, particle, and activity measurements of mixed DIP and virus populations (21, 22), though such measures can be useful for guiding computational models that aim to elucidate how such populations may grow, change, and be transmitted (23–26). To advance models that not only elucidate but also enable forecasting of how virus populations grow and spread, new measures that facilitate the rapid quantitative characterization of viruses and their DIPs will need to be developed.
High-throughput (deep) sequencing has been widely used to characterize the diversity of virus variants in natural infections as well as their sequence level adaptations to antiviral drugs (27–35). Deep sequencing has also revealed central roles for DIPs derived from influenza A, Sendai, measles, and parainfluenza 5 viruses in binding to the cellular pathogen recognition receptor retinoic acid-inducible gene I (RIG-I) as an initial step toward establishing an antiviral state within the host cell (36–38). In deep sequencing of viral RNA, the samples of genomes or subgenomes are reverse transcribed, amplified, and sequenced. When these reads are aligned to a reference genome, their match to a region on the reference genome (coverage) can approximate the level of that region in the analyzed genome population (37). In this way, deep sequencing can potentially be used to identify genome or subgenomic sizes, sequences, and relative levels within a biological sample.
Here we employed Illumina sequencing to track the emergence and enrichment of truncated genomic templates during serial transfer culture of the nonsegmented negative-sense RNA virus vesicular stomatitis virus (VSV), a prototypical virus for the study of DIPs (2, 20, 23, 39). Extents of template coverage by deep sequencing revealed changing relative levels of genomic and subgenomic templates that correlated with the emergence DIPs. Moreover, deep sequencing of the populations revealed variation in the genome position, size, and relative levels of subgenomic species, features of the virus populations that are not readily detected by quantitative reverse transcription-PCR (qRT-PCR) or particle size measures.
MATERIALS AND METHODS
Cell culture.
Baby hamster kidney (BHK-21) cells were maintained in 10% fetal bovine serum (FBS; Atlanta Biologicals)–1% Glutamax I (Gibco) in Eagle's minimum essential medium (MEM; CellGro). Cells were passaged every 2 or 3 days when they reached confluence. For virus passaging infections, cells were plated in a T-75 flask, grown for 1 day, and then infected.
Preparation of viral passages at fixed MOI.
Viral passages were prepared by serial infection of BHK-21 cells at fixed multiplicity of infection (MOI) (20). For virus passaging, we used the N1 virus arrangement (40), which has all genes from the San Juan isolate of the Indiana vesicular stomatitis virus (VSV) strain except the G protein, which originated from the Orsay isolate of the Indiana strain. Sequencing of our strain revealed eight fixed mutations, which were likely accumulated during passaging of the original strain. Viral infections were performed in 2% FBS (50 ml; Atlanta Biologicals)–1% GlutaMAX (5 ml; LifeTechnologies/Invitrogen) in MEM (500 ml; Gibco). BHK-21 cells were grown to 90% confluence and infected at an MOI of 10. Virus was adsorbed for 1 h at 37°C. Cells were rinsed with Dulbecco's phosphate-buffered saline (DPBS; Gibco), and 10 ml medium was added. The viral infection was allowed to progress for 24 h, and the supernatant containing virus particles was centrifuged (300 × g) to remove cell debris. Samples were stored at −80°C until analysis. Based on a preliminary passaging experiment, viral yields were sharply reduced after three passages, which is within the range reported in the literature (23, 41). After the first MOI of 10 passage (P0), the populations were named P1 and then P2 and then P3. The virus titer after each passage was measured, and samples were diluted appropriately to sustain an MOI of 10.
Interference assay.
DIP-mediated interference of infection in samples was quantified by the yield reduction assay (20). Prior to all infections, the cells were counted, enabling control of the MOI and quantification of virus and DIP yields on a per-cell basis. Serial dilutions of samples (1:10 or 1:2) were performed and then used to initiate infections. After 1 h of adsorption, stock virus was added at an MOI of 20, allowed to adsorb for 1 h, rinsed with DPBS, and replaced with MEM containing 2% FBS. Infections were incubated for 24 h and stored at −80°C until quantification by plaque assay. The plaque assay results were used to calculate the interfering unit concentration as previously described (20).
RNA extraction.
Viral RNA was collected using the Qiagen Viral RNA extraction kit according to the manufacturer's instructions, except that carrier RNA was not added to the lysis buffer. Carrier RNA would interfere with downstream sequencing. The VSV-associated RNAs were extracted from a full lineage including two technical replicates on supernatant from P3 (P0, P1, P2, P3, and P3b).
Construction of RNA-Seq libraries.
Viral RNA was submitted to the University of Wisconsin—Madison Biotechnology Center for quality control testing (Qubit Fluorometer and Agilent 2100 BioAnalyzer for quantity and integrity verification, respectively) and transcriptome sequencing (RNA-Seq) library preparation. Each library was generated using Illumina's guide, Preparing Samples for Sequencing of mRNA (Rev. A), and the Illumina mRNA-seq 8 sample preparation kit (Illumina Inc., San Diego, CA, USA) with minimal modification. To create the libraries, each viral RNA sample was fragmented into small pieces using divalent cations under elevated temperature and ethanol precipitated. Each mRNA pellet was suspended in 11.1 μl RNase-free water. Double-stranded cDNAs were synthesized using SuperScript II (Invitrogen, Carlsbad, CA, USA) and random primers for first-strand cDNA synthesis followed by second-strand synthesis using DNA Polymerase I and RNase H for removal of mRNA. Double-stranded cDNA was purified using QIAquick PCR Purification columns (Qiagen, Valencia, CA, USA). cDNAs were end repaired by T4 DNA polymerase and Klenow DNA Polymerase and phosphorylated by T4 polynucleotide kinase. The blunt-ended cDNA was purified using QIAquick PCR Purification columns. The cDNA products were incubated with Klenow DNA Polymerase to add an A base (adenine) to the 3′ end of the blunt phosphorylated DNA fragments and then purified using MinElute PCR Purification columns (Qiagen). DNA fragments were ligated to Illumina adapters, which have a single T base (thymine) overhang at their 3′ end. The adapter-ligated products were purified using MinElute PCR Purification columns. Products of the ligation reaction were purified by gel electrophoresis using 2% SizeSelect agarose gels (Invitrogen, Carlsbad, CA, USA) targeting 225- and 300-bp fragments. cDNA products were PCR amplified with Phusion DNA Polymerase using Illumina's genomic DNA primer set and purified using QIAquick PCR Purification columns. Quality and quantity of the finished libraries were assessed using an Agilent DNA HS series chip assay (Agilent Technologies, Santa Clara, CA, USA) and QuantIT PicoGreen dsDNA kit (Invitrogen, Carlsbad, CA, USA), respectively, and libraries were standardized to 10 μM. Cluster generation was performed using standard Cluster kits (v2) and the Illumina Cluster Station, with each sample placed in a single lane. A single-read, 36-bp run was performed, using standard 36-bp sequencing by synthesis (SBS) kits (v3) and SCS 2.4 software, on an Illumina Genome Analyzer IIx. Images were analyzed using the standard Illumina Pipeline, version 1.4.
Sequence alignment and P0 consensus determination.
Sequence alignment and de novo sequence identification were performed in the CLCBio Genomics Workbench using default settings. To generate a P0 consensus sequence, the reads from P0 were initially aligned to a published VSV sequence (NCBI Reference Sequence NC_001560.1). We extracted the consensus of P0 and used it as a reference to determine coverage in P0, P1, P2, P3, and P3b. The second alignment had an increased number of reads compared to the first.
Quantification and analysis of coverage data.
The sequence coverage was extracted from CLCBio, and analysis was performed in MATLAB. The coverage in each passage was normalized by dividing the coverage at each base by its corresponding coverage in P0. Following normalization, locations of plateau boundaries were estimated by defining local mean coverage, systematically applying these measures across the genome, and identifying where significant changes occurred. More specifically, 400-nucleotide-long windows, centered at every 5 nucleotides along the genome, were expanded toward the 3′ or 5′ end of the genome by 10 nucleotides. Mean coverages for each window before and after expansion were compared by the t test. When the means were found to be significantly different (α = 10−11), the added bases were recorded as potential plateau boundaries. The frequency of bases recorded as potential boundaries was plotted as a histogram with a bin size of 100 nucleotides. The bases identified as boundary more than 100 times were chosen as plateau boundaries. After plateau locations were determined, we calculated the averages and standard deviations over the entire range of each plateau and used those values to determine population fractions. Confidence intervals for population fractions were calculated using propagation of uncertainty.
qRT-PCR.
Viral RNA was extracted from cell culture supernatant using the QIAAmp Viral RNA Minikit (Qiagen) according to the manufacturer's instructions. Reverse transcription was performed with VSV genome-specific primer (see below) using the GoScript reverse transcriptase (Promega, Madison, WI, USA) according to the manufacturer's instructions. qPCR was performed with a forward primer and reverse primer (see below) from RT reaction using the SsoFast Supermix (Bio-Rad, Hercules, CA, USA) on a C1000 thermal cycler (Bio-Rad) according to the manufacturer's instructions with an annealing temperature of 62.5°C for all primer sets. A purified plasmid containing the VSV genome was used as a known standard to determine the numbers of copies per ml. Primers and the reverse transcription protocol were optimized to determine the linear range of detection using serial dilutions of extracted viral genomes. Linearity at the determined detection range was preserved for all four virus populations (P0 to P3), showing specificity of primers to full-length negative-sense genomes. Test samples were diluted when necessary to be within the linear range of detection. The primer sequences for full-length genomes spanned the NP gene junction with sequences CAGTCATGTCACTGCAAGGCCTAA and TACCGCCTGATCCAGACGAGAATA. The medium-length genomes were detected with primers TGCTGTCTCTGAGGTGGAATTGCT and AAGCTGGGAACCCTAATCCTGCAT. The short genomes were detected in the L region of the genome with CAGCCTTGGGAAAGGGAAAGCAAA and AACCTATGAAGGGCAGACCCTGTT. All primers were synthesized by Integrated DNA Technologies (Coralville, IA, USA) and were resuspended in RNase-free water.
Particle quantification by TEM.
The virus samples were mixed with a NIST Traceable size standard (115 nm in diameter, 109 particles/ml) in a 1:1 ratio. The prepared mixture was diluted 1:1 with methylamine tungstate (Nanoprobes), and a drop was loaded on a Pioloform (polyvinyl butyral)-coated grid. The sample was then imaged using a transmission electron microscope (TEM; CM120; Philips). For each sample, 15 TEM images were acquired, and VSV particles and microspheres were counted and averaged (approximately 100 particles per sample per grid). The VSV particle concentration was estimated relative to the concentration of NIST Traceable size standard beads.
LOD calculations.
The statistical limit of detection (LOD) for genome deletion mutants was determined by measuring the mean (μ) and standard deviation (σ) across a given window size and then solving for LOD in the equation
at a confidence limit α of 0.001 and n = (window size − 1). All coverages were extracted from P1 raw coverage or coverage normalized to P0.
The empirical limit of detection of genome deletion mutants was calculated by comparing fractions measured by sequencing to fractions measured by all complementary measures. A trendline was fitted through all data points to determine slope and intercept. The limit of detection was calculated by determining the x-intercept (predicted sequence measure when other measures are zero).
Nucleotide sequence accession numbers.
The GenBank accession numbers for genome sequences determined in this study are KF935251 for the passage 0 (P0) virus and KF935252 for the de novo defective interfering particle (DIP) from passage 3 (P3).
RESULTS
Deep sequencing can detect differential coverage profiles in related populations.
Vesicular stomatitis virus was cultured at a multiplicity of infection (MOI) of 10 PFU/cell on baby hamster kidney cells (BHK-21) for three passages (P1, P2, and P3), starting with an initial population (P0), as shown in Fig. 1A. RNA extracted from supernatants of each passage was sequenced using Illumina technology with 35-bp-long reads. All samples contained reads that mapped to the initial passage zero (P0) VSV consensus sequence (Table 1). Unmapped reads could not be directly attributed to the host (Syrian golden hamster) for which a genomic sequence is not yet available. However, de novo alignment on all reads from passages showed multiple contigs that aligned to rRNA of other species, including Chinese hamster (data not shown).
FIG 1.

Experimental setup for serial passage culture of VSV and raw coverage data. (A) VSV was passaged at an MOI of 10 for three passages to promote amplification of defective interfering particles. (B) Deep sequencing coverage (y axis) across the viral genome (x axis) is plotted for each passage. The genome is from 3′ to 5′, with base 1 representing the first base on the 3′ end. The coverage profile of the P3 sequencing replicate is plotted as a gray line.
TABLE 1.
Read mapping statistics
| Reads | Passage |
|||||
|---|---|---|---|---|---|---|
| P0 initiala | P0b | P1 | P2 | P3 | P3b | |
| Total no. | 913,387 | 913,387 | 1,044,166 | 1,624,786 | 1,706,897 | 1,726,404 |
| No. mapped | 508,249 | 513,612 | 106,302 | 713,200 | 35,581 | 30,877 |
| % Mapped | 55.64 | 56.23 | 10.18 | 43.90 | 2.08 | 1.79 |
P0 aligned to the VSV reference genome.
P0 aligned to the consensus of P0 initial.
In our initial alignment of P0 to the VSV Indiana reference, 55.6% (508,249 of 913,387) of the total reads were mapped to the viral genome. When the extracted consensus of this alignment was used as the reference, the mapped reads increased to 56.2%. For each passage, total reads of 9 × 105 to 1.7 × 106 were obtained, with a maximum of 56% and a minimum of 1.8% for later passages mapping to the P0 consensus. Details of the mapping by passage are in Table 1.
Base-by-base coverages for each of the 11,161 nucleotide positions of the VSV genome mapped to the P0 consensus, as shown in Fig. 1B, and the coverage for P0 exhibited a relatively flat profile across the VSV genome. A sharp drop in coverage of P0 between bases 1622 and 1625 is attributed to zero coverage at those four bases. In passage 2, different plateaus of coverage appeared, with 5- to 10-fold-lower coverage toward the 3′ end than toward the 5′ end. By passage 3, these differences were enhanced with 100-fold differences in coverage between the lowest and highest plateaus. The coverage profile of the technical replicates of P3 showed a good agreement, highlighting the reproducibility of sequencing.
Normalizing coverage to the initial population coverage reduced the noise in coverage profiles.
To test whether fluctuations in coverage at the base level reflected sequence-dependent biases, we normalized the number of reads at each base in P1 by its corresponding value in P0, as shown in Fig. 2A. As an unbiased control, we also normalized P1 to shifted P0, where P0 coverage was offset by a fixed number of nucleotide positions prior to normalization. Normalization of P1 by P0 (offset = 0) significantly reduced the bias (Fig. 2A), while increasing the offset produced similar fluctuations when P1 or P0 was normalized by P0 (offset = 100 nt), suggesting coverage bias due to sequence similarity between P1 and P0.
FIG 2.

Normalization by P0 coverage reduces variability. P1 coverage values are divided into P0 coverages at the corresponding positions shifted by an offset. (A) Normalized P0 (black curve) and P1 (gray curve) coverages with increasing offset from top to bottom. The x axis represents the nucleotide positions on the VSV genome. (B) As the reference P0 coverages are shifted by a higher offset, the noise in data increases, which can be seen in the increase of coefficient of variance (COV; calculated as the standard deviation of normalized coverages divided by the coverage mean across genome) of P1. The COV approaches its maximum after about 30 bases.
Coverage analysis reveals two distinct genome deletion mutants in the population.
Different coverage plateaus suggested the formation and enrichment of truncated genomes associated with defective particles in the virus population. Boundaries of the plateaus, estimated as described in Materials and Methods, highlight a 100-nucleotide-long window centered at position 5250, a second window 100 nucleotides long around position 6450, and finally a 200-nucleotide-long window centered at position 7200 (Fig. 3A). The boundaries at positions 5250 and 7200 occurred in both P2 and P3. However, the boundary at 6450 appeared only in P2. Therefore, we focused on conserved boundaries at positions 5250 and 7200, corresponding to defective genomes with lengths of 5,900 and 4,000 nucleotides (Fig. 3C). Analysis of the coverage profile from P1 showed no detectable changes in coverage.
FIG 3.

Determination of plateau boundaries and population fractions. (A) Frequency data showing bases recorded as boundaries using the algorithm described in Materials and Methods. The horizontal line is at 100 counts. (B) Normalized coverage data for P3. (C) Representation of VSV genome and genome deletion mutants that contribute to coverage levels.
De novo read assembly suggests a genome deletion mutant.
Using de novo read assembly algorithms available in CLC Genomics Workbench (CLC bio, Denmark), 8 to 20 contigs were obtained at each passage. One of the contigs in P3 was a 6,025-nucleotide sequence containing the 5′ end of the VSV genome, including most of the L gene (Fig. 4). The 3′ end was complementary to the 5′ end across a 33-nucleotide-long section, as previously observed for other DI particles (12). This de novo sequence is likely a DI genome and corresponds to additional coverage in P2 and P3 as described above. However, we were unable to find by de novo alignment evidence for a ∼4,000-nt length species (short DIP) corresponding to the high coverage at the 3′ end.
FIG 4.
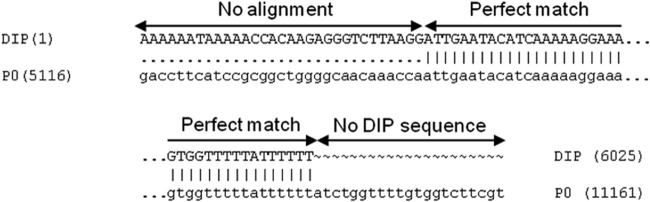
Genome deletion mutant detected by de novo alignment has complementary ends and similarity to full-length virus. The first base of the genome deletion lines up with base 5116 of the parental genome (P0). There is a short segment where there is no alignment, followed by a long stretch of perfect match. Finally, the DIP sequence aligns to the end but is truncated before the final base of the parent sequence.
Relative coverage levels were used to quantify population fractions of full-length and genome deletion mutants.
The emergence of different plateaus (Fig. 5A) is consistent with the enrichment of two truncated-genome populations, one of medium length (5,911 nucleotides) and the other of shorter length (3,961 nucleotides). Relative levels of each species were estimated by normalized average read coverages across each of three regions (bases 1 to 5250, 5251 to 7200, and 7201 to 11161), and this average was used to calculate population fractions. In Fig. 5B, the larger pie charts represent the fractions of full-length genomes (black) and truncated genomes (gray), and the smaller pie charts show how the truncated genomes were distributed between short genomes (light gray) and medium-length genomes (dark gray). Over the course of three passages, the fraction of species carrying full-length genomes declined as the species carrying the shorter length genome emerged to become the dominant species (Fig. 5B).
FIG 5.

Comparison of all quantification techniques. (A) Coverage values from deep sequencing were normalized to the corresponding base in P0, ignoring any zero values. The technical replicate of the P3 sample is shown as the gray curve. (B to E) Population fractions are calculated for each method. The black segment corresponds to full-size species, and the gray in the large pie represents all defective particles. In panels B and C, a second pie chart shows the fractions of medium and short genomes in the defective population of P2 and P3. Values reported are percentages of population ± standard deviations. (B) Fraction calculated from sequencing coverage. The standard deviation was calculated by coverage noise and propagation of uncertainty. (C) Fractions determined by qRT-PCR (n = 3). (D) Fraction calculated from TEM images (n = 2, ∼100 particles/grid). (E) Fractions calculated from PFU and interfering units measured by plaque and interference assays (n = 3).
Complementary measures agree with quantification by relative coverage.
We employed qRT-PCR, TEM, and activity assays (plaque and yield reduction assays) as complementary measures of how the virus populations changed with passage. A qRT-PCR assay was designed to measure the relative abundance for genomic species corresponding to each plateau region, with primer sets targeting the M mRNA region of the full-length genome, the medium-length species, and the L mRNA region associated with the shortest subgenomic species. To compare levels of truncated and full-length genomes estimated from sequencing with an independent measure, we normalized all qRT-PCR values to their measures in P0, which served as our base case pure population of full-length genomes. Figure 5C shows the distribution of the genome population measured by qRT-PCR. The qRT-PCR results were consistent with estimates from sequencing in P2 and P3. Measures of relative abundance for genomic species from the same locations in P1 did not show a significant change in levels between locations.
TEM of populations enabled visualization and approximation of relative levels of full-size and truncated virus particles. Here, truncated particles were treated as a single class, without attempting to distinguish between ones carrying medium-length or short genomes. The fraction of truncated VSV particles relative to full-size particles increased with passage (Fig. 5D), consistent with the overall trend observed by sequencing and qRT-PCR. However, the rate of enrichment of truncated particles appeared to lag behind the genomic measures, potentially reflecting the uncertainty in distinguishing particles that carry full-sized versus medium-length genomes.
Over the course of the three passages, levels of infectious virus declined more than 5,000-fold, suggesting that the emergence of DIPs interfered with infectious virus production. Measures of interference activity by the yield reduction assay indicated a significant rise in interfering units. These infection and interference results were combined to provide estimates of DIP fractions, which increased with passage, consistent with genome and particle measures (Fig. 5E).
The correlation between virus population distributions obtained by deep sequencing and genome, particle, and activity distributions highlights the accuracy of deep sequencing to quantify virus population distributions and their dynamics. Moreover, the similarity between genome and activity distributions suggests that all intact genomes are infectious while all defective genomes are interfering with infection. In order to test this, the absolute particle, genome, and activity levels were compared.
Quantification by relative coverage is limited by statistical and empirical measurement noise.
In contrast to what was observed with the P2 and P3 samples, we did not find evidence for different coverages in the P1 sample. Assuming normally distributed noise, we calculated a statistical lower detection limit of ∼1.5% of additional reads attributed to genome deletion mutants (Fig. 6A). As we increased the window size to the theoretical optimum value, we observed a correlated increase in the predicted number of plateaus, which we attributed to a higher detection sensitivity of deep sequencing. The empirical lower detection limit in this analysis was determined as a 10% representation of genome deletions (Fig. 6B), defined by the x-intercept and confidence intervals of a linear fit between sequencing and complementary measures. The discrepancy between statistical and empirical limits can be attributed to inherent measurement variation of the complementary measures (qRT-PCR, TEM, and biological activity), nonnormally distributed noise in sequencing data, or the gradual change (over tens to hundreds of bases) in coverage at the plateau boundaries. Based on this analysis, a conservative estimate of the limit of detection of deep sequencing is ∼10%. However, if one accepts a trade-off between specificity and sensitivity, deep sequencing can be used to detect genome deletion mutants at lower levels.
FIG 6.
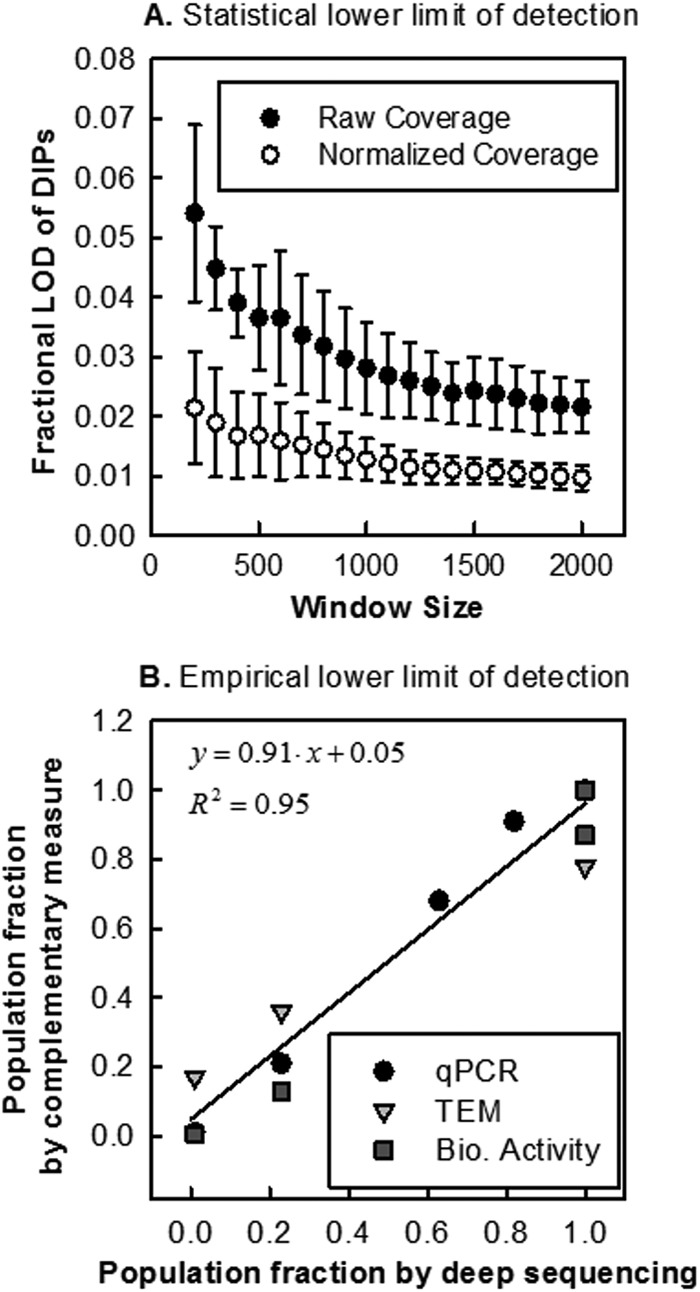
Limit of detection calculated two ways. (A) The statistical limit of detection was calculated by determining detection levels (x axis) at different window sizes (y axis) for average coverages. Noise measure was calculated based on raw and normalized P1 data. (B) Population fractions from sequencing (x axis) and complementary measures (y axis) were used to determine the lowest detectable level by sequencing by calculating the x-intercept for a best-fit line.
Absolute quantification of population shows correlation between genome structure and viral activity.
In addition to measuring relative levels of different species with a population passage, TEM and qRT-PCR measures allowed us to estimate the absolute particle and genome concentrations in passaged samples, while the plaque and interference assays provided infectious and interference measures. Concentrations of full-size particles, full-length genomes, and infectious units were within a factor of 10 from P0 to P2 (Fig. 7A), and defective virus particles, truncated genomes, and interference units were within a factor of 10 from P1 to P3, all with a maximum at P2 (Fig. 7B). The level of infectious particles followed an 8-fold-lower but parallel trend with full-length genome levels (Fig. 7A), and full-length particle levels dropped to a lesser extent across passages than viral genomes and infectious virus. This may suggest that some full-length particles contain complete or mostly complete genomes that are not infectious. For defective particles, there was greater agreement between particle, genome, and activity levels (Fig. 7B), evidence that most defective particles were able to interfere with infectious virus production. Interestingly, following their initial appearance and amplification, defective virus levels dropped from P2 to P3. Likewise, total virus particle levels dropped from P2 to P3 after being stable from P0 to P2 (Fig. 7C). These parallel drops in total and defective virus levels reflect that DIPs can interfere with production of infectious virus and thereby indirectly and adversely affect their own production.
FIG 7.
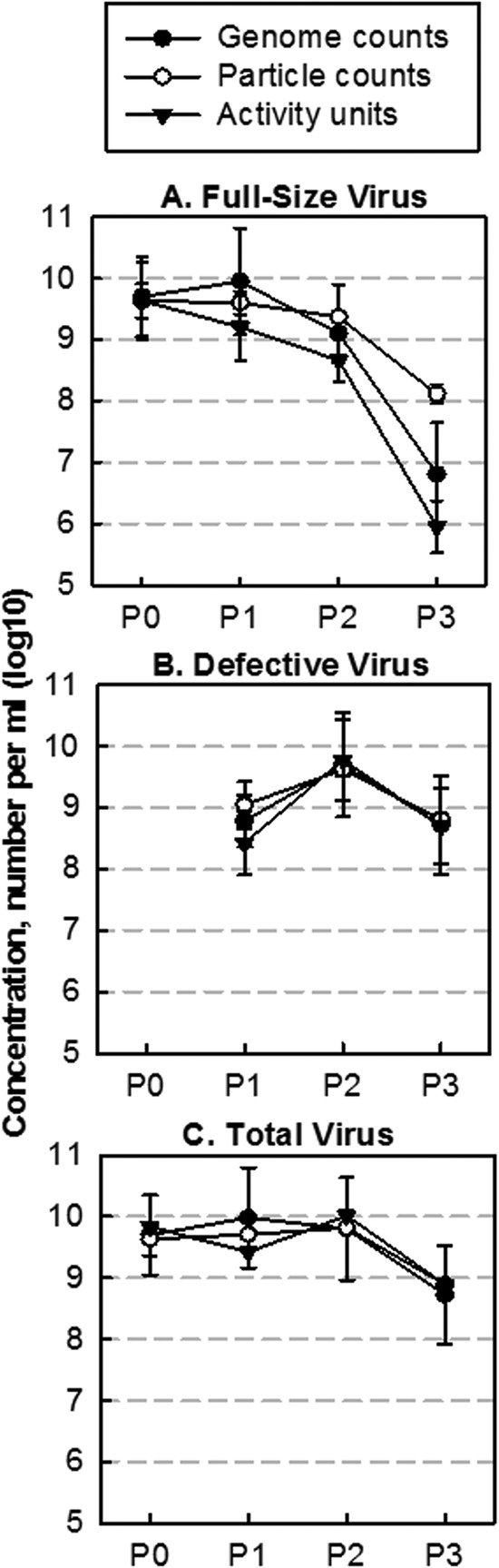
Absolute virus and DIP concentrations using different methods. Genomes (full circle) were measured by qRT-PCR, particles (empty circle) were measured by TEM, and activity (triangle) was determined by plaque assay or interference assay. (A) Full-length genomes, particles, and infectious units decline at a similar rate. (B) Defective genomes, particles, and interfering units follow a similar trend. (C) Total genomes, particles, and activity follow similar trends.
Other studies (27, 32, 42) have observed a correlation between absolute measure and mapped reads, but when we compared our mapped reads with other absolute measures, we did not see a correlation (not shown). Therefore, we were unable to use mapped reads as an absolute measure of genomes in the population.
DISCUSSION
Deep sequencing can reveal features of virus populations that are currently masked by other measures. Here, sequencing reads aligned to the viral genome exhibited plateaus in coverage that spanned thousands of bases, providing novel evidence for the simultaneous emergence and enrichment of at least two distinct truncated-genome variants of VSV. Previous studies have correlated deep sequencing with measures by qRT-PCR and microarrays (27, 32, 43–46). Based on the observed plateau locations, we selected primers for qRT-PCR measures that enabled us to validate the estimated levels of the two variants, relative to the full-length wild type, in the population. Further, analysis of TEM images confirmed a coexistence of truncated and full-sized particles, corresponding to truncated and full-sized genomes, but TEM estimates were not of sufficiently high resolution to distinguish more than one class of truncated particles. In addition, measures of biological activity demonstrated the presence of both viable virus and defective interfering activity, consistent with the presence of particles containing full-length and truncated genomes, respectively. Such population level measures cannot currently resolve whether interference arises from one or more distinct classes of truncated genomes.
The normalization of the coverage profile to that of initial population (P0) reduced the noise and allowed for the identification of genomic subpopulations and the estimation of population distributions. The good correlation between virus population distributions by sequencing and by other independent methods highlights that deep sequencing can accurately quantify the relative levels of genomic variants. We were unable to detect the presence of DIPs in P1 by deep sequencing, while TEM and interference assays suggested their presence in P1 at a low level. Based on these other measures, we determined that the lower detection limit of deep sequencing to distinguish between two genome subpopulations (intact and defective genomes) was about 10%. In short, genome deletion mutants must comprise at least 10% of the population in order to be detectable based on sequence coverage.
The coverage changes between observed plateaus were not discrete: the change occurred gradually over an ∼200-nt stretch of the genome. This gradual change may be due to the presence of multiple genome lengths of similar size, as observed in poliovirus defective interfering particles (47, 48). The existence of a distribution of lengths suggests that multiple defective particles may arise in culture and become simultaneously enriched.
Differential sequencing coverage levels, compared to other methods, allowed us to estimate an empirical limit of detection of ∼10% for truncated genomes in a sample. The theoretical limit of detection was a function of window size in the plateau-finding algorithm. For larger window sizes, the detection limit decreased, and new peaks emerged, defining potential plateau boundaries associated with additional genome deletion mutants in the population. Sampling with larger window sizes may enable deep sequencing measures to detect variants at lower levels, but this approach will need to be validated by more-sensitive measurements than those currently employed.
It is important to distinguish that the relatively high detection limit applies to overlapping genome sequences, while unique sequences in a population can be identified at even lower fractions, as we observed full-length genomic segments in P3 at fractions of ∼1%. The quantitative levels of DIPs in natural samples have not been reported, but genome deletion DIPs may be present at levels of <10%, emphasizing the need for other quantification methods. Methods of detection of DIPs in natural populations include sequencing of promoters or other conserved sequences (8), but these have not provided quantitative population information. We have shown that differences in sequencing coverage for overlapping segments can reduce the effect of sequencing noise to provide more-quantitative estimates of population fractions and population changes during passaging.
We observed good agreement between biological activity and physical particle measurements for early passages and greater disagreement in later passages. However, the total particle counts produced were relatively stable through P0, P1, and P2, lending support to the idea that the total particle production (viable and nonviable) is limited by a finite pool of host cellular resources (49, 50). Future measures of intracellular viral and DIP components, combined with mechanistic modeling (50), will enable more-detailed assessments of resource use during coinfections. The drop in total particle production seen in P3 can be attributed to the ability of DIPs at high intracellular levels to interfere with their own replication (23).
While deep sequencing has been used primarily to detect and identify viral genome sequences, its ability to quantify the population distributions has been in part limited because of the noise in the coverage profiles. Here, we observed a similarity in the noise patterns of related samples and found that normalizing coverage measurements to a common ancestor reduced much of the sequence-dependent noise, allowing for unbiased species identification and more-precise quantification. Such a normalization strategy can be applied to reduce the noise in chronologically linked biological samples, which may aid in the quantitative analysis of the population dynamics.
ACKNOWLEDGMENTS
We thank Ming Yuan (University of Wisconsin—Madison) for helpful discussions. We thank the University of Wisconsin DNA Sequencing Facility (UWBC) for their help in preparing and analyzing sequencing samples. We also thank the University of Wisconsin Medical School Electron Microscope Facility.
We are grateful for support from the National Institutes of Health (AI071197). C.T. acknowledges support from the National Library of Medicine (training grant 5T15LM007359).
We declare that we have no conflicts of interest.
Footnotes
Published ahead of print 18 December 2013
REFERENCES
- 1.Von Magnus P. 1954. Incomplete forms of influenza virus. Adv. Virus Res. 2:59–79 [DOI] [PubMed] [Google Scholar]
- 2.Huang AS, Baltimore D. 1970. Defective viral particles and viral disease processes. Nature 226:325–327. 10.1038/226325a0 [DOI] [PubMed] [Google Scholar]
- 3.Lazzarini RA, Keene JD, Schubert M. 1981. The origins of defective interfering particles of the negative-strand RNA viruses. Cell 26:145–154. 10.1016/0092-8674(81)90298-1 [DOI] [PubMed] [Google Scholar]
- 4.Roux L, Simon AE, Holland JJ. 1991. Effects of defective interfering viruses on virus replication and pathogenesis in vitro and in vivo. Adv. Virus Res. 40:181–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bean WJ, Kawaoka Y, Wood JM, Pearson JE, Webster RG. 1985. Characterization of virulent and avirulent A/chicken/Pennsylvania/83 influenza A viruses: potential role of defective interfering RNAs in nature. J. Virol. 54:151–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pesko KN, Fitzpatrick KA, Ryan EM, Shi P-Y, Zhang B, Lennon NJ, Newman RM, Henn MR, Ebel GD. 2012. Internally deleted WNV genomes isolated from exotic birds in New Mexico: function in cells, mosquitoes, and mice. Virology 427:10–17. 10.1016/j.virol.2012.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li D, Lott WB, Lowry K, Jones A, Thu HM, Aaskov J. 2011. Defective interfering viral particles in acute dengue infections. PLoS One 6:e19447. 10.1371/journal.pone.0019447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saira K, Lin X, DePasse JV, Halpin R, Twaddle A, Stockwell T, Angus B, Cozzi-Lepri A, Delfino M, Dugan V, Dwyer DE, Freiberg M, Horban A, Losso M, Lynfield R, Wentworth DN, Holmes EC, Davey R, Wentworth DE, Ghedin E. 2013. Sequence analysis of in vivo defective-interfering (DI)-like RNA of influenza A H1N1 pandemic virus. J. Virol. 87:8064–8074. 10.1128/JVI.00240-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Epstein DA, Herman RC, Chien I, Lazzarini RA. 1980. Defective interfering particle generated by internal deletion of the vesicular stomatitis virus genome. J. Virol. 33:818–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Re GG, Gupta KC, Kingsbury DW. 1983. Genomic and copy-back 3′ termini in Sendai virus defective interfering RNA species. J. Virol. 45:659–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davis AR, Hiti AL, Nayak DP. 1980. Influenza defective interfering viral RNA is formed by internal deletion of genomic RNA. Biochemistry 77:215–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meier E, Harmison GG, Keene JD, Schubert M. 1984. Sites of copy choice replication involved in generation of vesicular stomatitis virus defective-interfering particle RNAs. J. Virol. 51:515–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.O'Hara PJ, Horodyski FM, Nichol ST, Holland JJ. 1984. Vesicular stomatitis virus mutants resistant to defective-interfering particles accumulate stable 5′-terminal and fewer 3′-terminal mutations in a stepwise manner. J. Virol. 49:793–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang AS, Greenawalt JW, Wagner RR. 1966. Defective T particles of vesicular stomatitis virus I. Preparation, morphology, and some biologic properties. Virology 30:161–172 [DOI] [PubMed] [Google Scholar]
- 15.Reichmann ME, Pringle CR, Follett EAC. 1971. Defective particles in BHK cells infected with temperature-sensitive mutants of vesicular stomatitis virus. J. Virol. 8:154–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hartford SL. 1975. Physical properties of New Jersey serotype of vesicular stomatitis virus and its defective particles. Proc. Natl. Acad. Sci. U. S. A. 72:1202–1205. 10.1073/pnas.72.3.1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nonoyama M, Watanabe Y, Graham AF. 1970. Defective virions of reovirus. J. Virol. 6:226–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petric M. 1970. Vesicular stomatitis virus—a new interfering particle, intracellular structures and virus-specific RNA. Virology 41:615–630. 10.1016/0042-6822(70)90427-7 [DOI] [PubMed] [Google Scholar]
- 19.Bellett AJD, Cooper PD. 1959. Some properties of the transmissible interfering component of vesicular stomatitis virus preparations. Microbiology 21:498–509 [DOI] [PubMed] [Google Scholar]
- 20.Stauffer Thompson KA, Rempala GA, Yin J. 2009. Multiple-hit inhibition of infection by defective interfering particles. J. Gen. Virol. 90:888–899. 10.1099/vir.0.005249-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Janda JM, Davis AR, Nayak DP, De BK. 1979. Diversity and generation of defective interfering influenza virus particles. Virology 95:48–58. 10.1016/0042-6822(79)90400-8 [DOI] [PubMed] [Google Scholar]
- 22.Marcus PI, Sekellick MJ. 1974. Cell killing by viruses. I. Comparison of cell-killing, plaque-forming, and defective-interfering particles of vesicular stomatitis virus. Virology 57:321–338 [DOI] [PubMed] [Google Scholar]
- 23.Thompson K, Yin J. 2010. Population dynamics of an RNA virus and its defective interfering particles in passage cultures. Virol. J. 7:257. 10.1186/1743-422X-7-257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kirkwood TB, Bangham CR. 1994. Cycles, chaos, and evolution in virus cultures: a model of defective interfering particles. Proc. Natl. Acad. Sci. U. S. A. 91:8685–8689. 10.1073/pnas.91.18.8685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zwart M, Pijlman G, Sardanyés J, Duarte J, Januário C, Elena S. 2013. Complex dynamics of defective interfering baculoviruses during serial passage in insect cells. J. Biol. Phys. 39:327–342. 10.1007/s10867-013-9317-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ke R, Aaskov J, Holmes EC, Lloyd-Smith JO. 2013. Phylodynamic analysis of the emergence and epidemiological impact of transmissible defective dengue viruses. PLoS Pathog. 9:e1003193. 10.1371/journal.ppat.1003193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsibris AMN, Korber B, Arnaout R, Russ C, Lo C-C, Leitner T, Gaschen B, Theiler J, Paredes R, Su Z, Hughes MD, Gulick RM, Greaves W, Coakley E, Flexner C, Nusbaum C, Kuritzkes DR. 2009. Quantitative deep sequencing reveals dynamic HIV-1 escape and large population shifts during CCR5 antagonist therapy in vivo. PLoS One 4:e5683. 10.1371/journal.pone.0005683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bimber BN, Dudley DM, Lauck M, Becker EA, Chin EN, Lank SM, Grunenwald HL, Caruccio NC, Maffitt M, Wilson NA, Reed JS, Sosman JM, Tarosso LF, Sanabani S, Kallas EG, Hughes AL, O'Connor DH. 2010. Whole-genome characterization of human and simian immunodeficiency virus intrahost diversity by ultradeep pyrosequencing. J. Virol. 84:12087–12092. 10.1128/JVI.01378-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nasu A, Marusawa H, Ueda Y, Nishijima N, Takahashi K, Osaki Y, Yamashita Y, Inokuma T, Tamada T, Fujiwara T, Sato F, Shimizu K, Chiba T. 2011. Genetic heterogeneity of hepatitis C virus in association with antiviral therapy determined by ultra-deep sequencing. PLoS One 6:e24907. 10.1371/journal.pone.0024907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Verbinnen T, Van Marck H, Vandenbroucke I, Vijgen L, Claes M, Lin T-I, Simmen K, Neyts J, Fanning G, Lenz O. 2010. Tracking the evolution of multiple in vitro hepatitis C virus replicon variants under protease inhibitor selection pressure by 454 deep sequencing. J. Virol. 84:11124–11133. 10.1128/JVI.01217-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuroda M, Katano H, Nakajima N, Tobiume M, Ainai A, Sekizuka T, Hasegawa H, Tashiro M, Sasaki Y, Arakawa Y, Hata S, Watanabe M, Sata T. 2010. Characterization of quasispecies of pandemic 2009 influenza A virus (A/H1N1/2009) by de novo sequencing using a next-generation DNA sequencer. PLoS One 5:e10256. 10.1371/journal.pone.0010256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Greninger AL, Chen EC, Sittler T, Scheinerman A, Roubinian N, Yu G, Kim E, Pillai DR, Guyard C, Mazzulli T, Isa P, Arias CF, Hackett J, Schochetman G, Miller S, Tang P, Chiu CY. 2010. A metagenomic analysis of pandemic influenza A (2009 H1N1) infection in patients from North America. PLoS One 5:e13381. 10.1371/journal.pone.0013381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Görzer I, Guelly C, Trajanoski S, Puchhammer-Stöckl E. 2010. Deep sequencing reveals highly complex dynamics of human cytomegalovirus genotypes in transplant patients over time. J. Virol. 84:7195–7203. 10.1128/JVI.00475-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fischer W, Ganusov VV, Giorgi EE, Hraber PT, Keele BF, Leitner T, Han CS, Gleasner CD, Green L, Lo C-C, Nag A, Wallstrom TC, Wang S, McMichael AJ, Haynes BF, Hahn BH, Perelson AS, Borrow P, Shaw GM, Bhattacharya T, Korber BT. 2010. Transmission of single HIV-1 genomes and dynamics of early immune escape revealed by ultra-deep sequencing. PLoS One 5:e12303. 10.1371/journal.pone.0012303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wright CF, Morelli MJ, Thébaud G, Knowles NJ, Herzyk P, Paton DJ, Haydon DT, King DP. 2011. Beyond the consensus: dissecting within-host viral population diversity of foot-and-mouth disease virus by using next-generation genome sequencing. J. Virol. 85:2266–2275. 10.1128/JVI.01396-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Komarova AV, Combredet C, Sismeiro O, Dillies M-A, Jagla B, Sanchez David RY, Vabret N, Coppee J-Y, Vidalain P-O, Tangy F. 2013. Identification of RNA partners of viral proteins in infected cells. RNA Biol. 10:944–957. 10.4161/rna.24453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Killip MJ, Young DF, Gatherer D, Ross CS, Short JAL, Davison AJ, Goodbourn S, Randall RE. 2013. Deep sequencing analysis of defective genomes of parainfluenza virus 5 and their role in interferon induction. J. Virol. 87:4798–4807. 10.1128/JVI.03383-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baum A, Sachidanandam R, García-Sastre A. 2010. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc. Natl. Acad. Sci. U. S. A. 107:16303–16308. 10.1073/pnas.1005077107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holland JJ. 1987. Defective interfering rhabdoviruses, p 297–360 In Wagner RR. (ed), The rhabdoviruses. Springer, New York, NY [Google Scholar]
- 40.Wertz GW, Perepelitsa VP, Ball L. 1998. Gene rearrangement attenuates expression and lethality of a nonsegmented negative strand RNA virus. Proc. Natl. Acad. Sci. U. S. A. 95:3501–3506. 10.1073/pnas.95.7.3501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stampfer M, Baltimore D, Huang AS. 1971. Absence of interference during high-multiplicity infection by clonally purified vesicular stomatitis virus. J. Virol. 7:409–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mortazavi A, Williams BA, Mccue K, Schaeffer L, Wold B. 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5:1–8. 10.1038/nmeth1153 [DOI] [PubMed] [Google Scholar]
- 43.Bloom JS, Khan Z, Kruglyak L, Singh M, Caudy AA. 2009. Measuring differential gene expression by short read sequencing: quantitative comparison to 2-channel gene expression microarrays. BMC Genomics 10:221. 10.1186/1471-2164-10-221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Willenbrock H, Salomon J, Søkilde R, Barken KB, Hansen TN, Nielsen FC, Møller S, Litman T. 2009. Quantitative miRNA expression analysis: comparing microarrays with next-generation sequencing. RNA 15:2028–2034. 10.1261/rna.1699809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Camarena L, Bruno V, Euskirchen G, Poggio S, Snyder M. 2010. Molecular mechanisms of ethanol-induced pathogenesis revealed by RNA-sequencing. PLoS Pathog. 6:e1000834. 10.1371/journal.ppat.1000834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oliver HF, Orsi RH, Ponnala L, Keich U, Wang W, Sun Q, Cartinhour SW, Filiatrault MJ, Wiedmann M, Boor KJ. 2009. Deep RNA sequencing of L. monocytogenes reveals overlapping and extensive stationary phase and sigma B-dependent transcriptomes, including multiple highly transcribed noncoding RNAs. BMC Genomics 10:641. 10.1186/1471-2164-10-641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cole CN, Smoler D, Wimmer E, Baltimore D. 1971. Defective interfering particles of poliovirus. I. Isolation and physical properties. J. Virol. 7:478–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kuge S, Saito I, Nomoto A. 1986. Primary structure of poliovirus defective-interfering particle genomes and possible generation mechanisms of the particles. J. Mol. Biol. 192:473–487. 10.1016/0022-2836(86)90270-6 [DOI] [PubMed] [Google Scholar]
- 49.Turner PE, Chao L. 1999. Prisoner's dilemma in an RNA virus. Nature 398:441–443. 10.1038/18913 [DOI] [PubMed] [Google Scholar]
- 50.Lim K, Lang T, Lam V, Yin J. 2006. Model-based design of growth-attenuated viruses. PLoS Comput. Biol. 2(9):e116. 10.1371/journal.pcbi.0020116 [DOI] [PMC free article] [PubMed] [Google Scholar]