

ABSTRACT
Endogenous retroviruses (ERVs) represent ancestral sequences of modern retroviruses or their extinct relatives. The majority of ERVs cluster alongside exogenous retroviruses into two main groups based on phylogenetic analyses of the reverse transcriptase (RT) enzyme. Class I includes gammaretroviruses, and class II includes lentiviruses and alpha-, beta-, and deltaretroviruses. However, analyses of the transmembrane subunit (TM) of the envelope glycoprotein (env) gene result in a different topology for some retroviruses, suggesting recombination events in which heterologous env sequences have been acquired. We previously demonstrated that the TM sequences of five of the six genera of orthoretroviruses can be divided into three types, each of which infects a distinct set of vertebrate classes. Moreover, these classes do not always overlap the host range of the associated RT classes. Thus, recombination resulting in acquisition of a heterologous env gene could in theory facilitate cross-species transmissions across vertebrate classes, for example, from mammals to reptiles. Here we characterized a family of class II avian ERVs, “TgERV-F,” that acquired a mammalian gammaretroviral env sequence. Although TgERV-F clusters near a sister clade to alpharetroviruses, its genome also has some features of betaretroviruses. We offer evidence that this unusual recombinant has circulated among several avian orders and may still have infectious members. In addition to documenting the infection of a nongalliform avian species by a mammalian retrovirus, TgERV-F also underscores the importance of env sequences in reconstructing phylogenies and supports a possible role for env swapping in allowing cross-species transmissions across wide taxonomic distances.
IMPORTANCE Retroviruses can sometimes acquire an envelope gene (env) from a distantly related retrovirus. Since env is a key determinant of host range, such an event affects the host range of the recombinant virus and can lead to the creation of novel retroviral lineages. Retroviruses insert viral DNA into the host DNA during infection, and therefore vertebrate genomes contain a “fossil record” of endogenous retroviral sequences thought to represent past infections of germ cells. We examined endogenous retroviral sequences in avian genomes for evidence of recombination events involving env. Although cross-species transmissions of retroviruses between vertebrate classes (from mammals to birds, for example) are thought to be rare, we here characterized a group of avian retroviruses that acquired an env sequence from a mammalian retrovirus. We offer evidence that this unusual recombinant circulated among songbirds 2 to 4 million years ago and has remained active into the recent past.
INTRODUCTION
Retroviruses are nearly unique in having left an abundance of “fossilized” viral sequences in the genomes of vertebrate species. Such sequences are known as endogenous retroviruses (ERVs) and are thought to be the outcomes of germ cell infections by ancient retroviruses. Previous analysis of the ERV “fossil record” had suggested that transmission of retroviruses between vertebrate classes has been rare (1). However, the addition of genomic sequences from an increasing number of nonmammalian species to the publicly available databases allows a greater breadth of ERV sampling, offering fresh insights into the dynamics of cross-species transmission events. In particular, the sequencing of the zebra finch genome (Passeriformes order) (2), along with the ever growing amount of genomic sequence from birds available in the databases, provides a glimpse into the evolutionary dynamics of avian retroviruses within the context of the highly speciated clade of neoaves (modern birds). Although the zebra finch genome, like that of the chicken, carries a much lower mobile element content than mammalian genomes (∼8%, compared to ∼45% for mammals), zebra finches have approximately three times the number of ERV-related sequences as chickens, a percentage comparable to that of humans (3).
The ability of a virus to replicate in a given host is subject to the interplay of various cellular and viral factors. However, the first step in infection is successfully mediating entry into the target cell. The envelope glycoprotein (Env) at the surface of a virion is a primary determinant of access and thus of host range, allowing entry into cells that express receptors recognized by it. Retroviral Env proteins consist of two subunits, SU (the surface-exposed, receptor binding subunit) and TM (the transmembrane fusion subunit). While SU is the most variable region of the genome, TM is highly conserved and can be aligned across a wide variety of retroviruses (4). The majority of retroviral envelope glycoproteins can be divided into those with Env subunits that are covalently associated (gamma-type) and those with noncovalently associated subunits (beta-type), and the two groups can be readily distinguished by sequence motifs in TM (5, 6) (Fig. 1). A variant of the covalent, gamma-type Env is that of alpharetroviruses, which has an internal fusion peptide flanked by a pair of cysteines.
FIG 1.
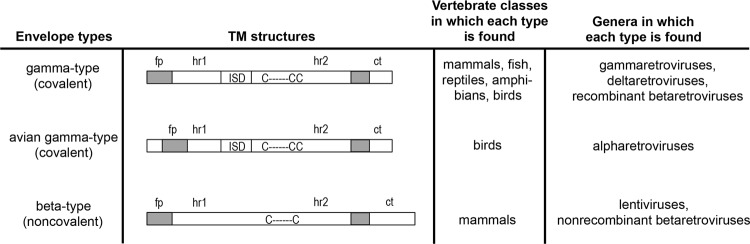
Envelope types found among the Orthoretroviridae. For each type, the TM domain is depicted, along with its distribution among vertebrate classes and retroviral genera. fp, fusion peptide; hr1 and hr2, heptad repeats 1 and 2; ISD, immunosuppressive domain; ct, cytoplasmic tail. “C- - - - - -C” and “C- - - - - -CC” represent the two- and three-cysteine motifs of beta-type and gamma-type envelopes, respectively.
We previously performed an exhaustive survey of TM sequences from the genomes of 78 vertebrate species and found distinct host range patterns for covalent (gamma-type) and noncovalent (beta-type) endogenous retroviral sequences (Fig. 1). Sequences representing the noncovalent Env type (typical of betaretroviruses and lentiviruses) were found only in mammals, while covalent type sequences (typical of gammaretroviruses) were found among species representing five vertebrate classes (5).
Retroviruses are most commonly analyzed according to phylogenetic relationships of the highly conserved reverse transcriptase (RT) region of pol (7–11), rather than env. In phylogenetic analyses of the RT region, ERVs cluster into three broad classes that include the known exogenous retroviruses: class I includes gamma- and epsilonretroviruses; class II, beta-, delta-, and alpharetroviruses and lentiviruses; and class III, spumaretroviruses (10).
A confounding factor in classifying ERVs is that pol and env can have separate evolutionary histories. Class II retroviruses, in particular, can be found with any of the three described Env protein types (6). The Betaretrovirus genus, for example, is split between members having a beta-type (noncovalent) Env protein and those having a gamma-type (covalent) Env protein. The first group includes mouse mammary tumor virus (MMTV), Jaagsiekte sheep retrovirus (JSRV), enzootic nasal tumor virus (ENTV), and the human ERV-K(HML-2) group, as well as many betaretrovirus-like ERVs. The second group includes the type D betaretroviruses, typified by Mason-Pfizer monkey virus (MPMV), which resulted from a recombination event in which an ancestral retrovirus acquired a gammaretroviral envelope gene (12). The recombinant origin of the latter group is reflected in incongruent topologies between phylogenetic trees based on pol and those based on TM (4). Thus, while pol sequences from both groups of betaretroviruses cluster together, TM sequences from the group that includes the type D betaretroviruses cluster with gammaretroviral sequences. Incongruent topologies also characterize deltaretroviruses and alpharetroviruses, implying recombinant origins for these class II genera as well (4). In contrast, pol and TM clustering is congruent in the case of the MMTV-containing group of betaretroviruses and the lentiviruses, suggesting that beta-type env (noncovalent) is the “natural” type for class II retroviruses.
The best-characterized class II retroviruses associated with birds are members of the Alpharetrovirus genus, typified by avian leukosis virus (ALV), found in the genomes of galliform birds. Interestingly, previous research focusing on pol sequences has shown that class II ERVs are found in the genomes of numerous additional avian species and moreover that many of these sequences cluster outside the Alpharetrovirus genus (8, 13). Since beta-type env sequences have been found only in mammalian genomes, this raises the question of which env types associate with avian class II ERVs, particularly those ERVs that do not belong to the Alpharetrovirus genus.
Here we describe a group of class II ERVs in the genome of zebra finches that forms a clade separate from that for alpharetroviruses. These elements display an intriguing mix of betaretroviral and alpharetroviral features and have acquired a gammaretroviral env sequence that is typical of mammals, suggesting an interclass transmission event. Moreover, the presence of highly similar sequences in the genomes of representatives of several additional avian species suggests that, despite the challenges a virus must overcome in order to adapt to a species of another class, this unusual recombinant was able to circulate among various avian species.
MATERIALS AND METHODS
Data mining.
Build 1.1 of the Taeniopygia guttata (zebra finch) genome, as well as sequence data for bird species in the National Center for Biotechnology Information (NCBI) databases, was screened for class II ERVs using as query sequences class II RT regions from a variety of retroviruses, employing the tBLASTn algorithm (14). Reading frames for putative pol sequences were examined for the presence of the typical class II YMDD motif (11, 13) and confirmed by their recovery of previously characterized class II retroviral pol genes when used as BLAST queries themselves. Regions downstream from the pol genes were translated in all three reading frames and examined by eye for sequences typical of retroviral envelope glycoproteins. Putative env sequences were confirmed by their recovery of known retroviral env genes in BLAST searches. Flanking long terminal repeats (LTRs) were discerned by constructing DNA self-matrices with a 12- to 15-kb window centered on the identified pol and env regions, in the DNA Strider program, and examining any flanking repeated regions for the canonical features of LTRs. Putative gag and pro sequences were confirmed in the same manner as for pol and env sequences. The TgERV-F consensus sequence was assembled based on alignments made in ClustalW from full-length sequences, using the Consensus program (coot.embl.de/Alignment/consensus).
Sequence analysis.
Sequences extracted from the in silico searches were translated in all three reading frames and compared to amino acid alignments of previously characterized, published sequences to reconstruct putative open reading frames (ORFs). Multiple alignments of the adjusted sequences were then performed using the ClustalW program (15) in MEGA 4.0 (16) or GENEIOUS v. 6.0.4 (Biomatters, Auckland, New Zealand). Alignments of the RT region of pol spanned 295 amino acids and included the conserved motifs characterized by Xiong and Eickbush (17). Alignments of TM sequences of env always included the cysteine region and spanned 150 to 200 amino acids, excluding the cytoplasmic domain, which can vary widely in sequence even within genera (4).
Phylogenetic analysis.
Analyses using the neighbor-joining method (18) and determination of average pairwise genetic distances were performed in MEGA4.0 and GENEIOUS v. 6.0.4, using the ClustalW algorithm. As ERV sequences often represent degraded, repetitive elements that contain many indels, positions containing missing data or alignment gaps were eliminated in a pairwise manner only, using the p-distance model, instead of by the standard means of stripping from the analysis all regions containing one or more gaps. Bootstrap values, when indicated, were inferred from 1,000 replicates.
RESULTS
In order to investigate the types of env genes associated with class II avian ERVs, we screened avian genomic sequences in the NCBI databases for class II RT sequences, using RT regions from a variety of class II retroviruses as query sequences. We then examined the region downstream from all RT hits for the presence of env genes and found an element in the genome of Taeniopygia guttata (zebra finch, Passeriform order) that was associated with a gammaretroviral env sequence. Using this pol-env sequence as a query to screen the zebra finch genome, we were able to recover 14 near-full-length proviruses flanked by LTRs in the genome, each having distinct pairs of matching target site duplications (TSDs) and thus representing unique insertions (Table 1). Nine of the sequences were found in build 1.1 of the zebra finch genome, while the remaining five sequences were found in a screen of the high-throughput genomic sequence (htgs) database. The sequences exhibited 95% identity at the nucleotide level (as measured by average pairwise distance) across the env gene, 91% across the pol gene, and 86% across the gag gene, which is the least conserved gene among the 14 sequences. We then generated a consensus provirus (see Fig. S1 in the supplemental material) from an alignment of the 14 sequences.
TABLE 1.
Full-length TgERV-F proviruses flanked by unique, intact target site duplications
| Accession no., position | Structure | Estimated insertion time (million yr) based on LTR differences | TSD |
|---|---|---|---|
| NW_002198511.1, 789669–798230 | Δgag-Δpro-pol-env | <0.7 | TTAAAG |
| AC188309.1, 31532–39714 | Δgag-pro-pol-env | <0.7 | GTCGGC |
| AC199447, c108254–c100074 | Δgag-pro-pol-env | <0.7 | CAGGTG |
| NW_002198276.1, c598839–c587085 | Δgag-pro-Δpol-Δgag-pro-Δpol-env | 0.7–1.4 | CCAGGG |
| NW_002210925.1, 5145–14422 | gag-pro-Δpol-env | 0.7–1.4 | GGCCCC |
| NW_002218503.1, c12645–c579 | Δgag-pro-Δpol-env | 0.7–1.4 | *ACCCT |
| AC188375.1, 59415–67585 | Δgag-pro-Δpol-env | 1.1–2.1 | GACACT |
| AC192433.2, c67750–c59570 | Δgag-pro-Δpol-Δenv | 1.1–2.1 | GTGTCC |
| AC199447, 73654–81822 | Δgag-pro-pol-env | 1.1–2.1 | ACAATG |
| NW_002198510.1, 914525–924332 | Δgag-pro-Δpol-pro-pol-Δenv | 1.1–2.2 | GGAATG |
| NW_002234469, 2805393–2813547 | Δgag-pro-Δpol-Δenv | 1.8–3.5 | AGAGTG |
| NW_002198918.1, 68352–77921 | Δgag-pro-Δpol-Δenv | 1.8–3.6 | GTATGa |
| NW_002234472.1, 7877009–7885717 | Δgag-pro-Δpol-Δenv | 1.9–3.6 | AACTAG |
| NW_002204614.1, 79242–90654 | Δgag-pro-Δpol-Δenv-Δpol-Δ-env | 2.1–4.2 | CCAGTC |
Five-base-pair TSD that may have resulted from a single nucleotide deletion.
A search of the whole-genome shotgun (wgs) database of the NCBI confirmed the presence of contiguous pol and env sequences with high similarity to the zebra finch sequences in the genomes of three additional passerine species: the white-throated sparrow (Zonotrichia albicollis), the ground tit (Pseudopodoces humilis), and the medium ground finch (Geospiza fortis) (see Table S1 in the supplemental material). However, due to the short sequence length and incomplete genome coverage of the wgs database entries, we were unable to confirm any full-length proviruses in these species. Interestingly, no related sequences were found in galliform species.
Features of the proviral genome and predicted proteins.
A schematic of the consensus proviral sequence is shown in Fig. 2. The TSDs are 6 bp in length, in keeping with the integration mechanics of alpharetroviruses but not gammaretroviruses, which typically produce 4-bp TSDs (19–21). The length of the provirus is 8.16 kb, with the canonical CA dinucleotides at each 3′ end of the LTRs. The LTRs are 356 nucleotides (nt) in length. Only two of the elements contain a predicted promoter region with a canonical TATAA sequence, while this region is missing in the other proviruses. However, all of the proviruses contain a polyadenylation signal near the 3′ end of the R region. Four nucleotides downstream of the 5′ LTR is the primer binding site (PBS), consisting of a 17-nt region that is complementary to a portion of tRNAPhe, prompting us to name the element TgERV-F. Just upstream of the start of the 3′ LTR is a 14-nt polypurine tract (ppt).
FIG 2.

Genome structure of the TgERV-F provirus. Top, typical domains found in the consensus sequence (see the text for description); bottom, comparison of the genome structure of TgERV-F with those of alpha-, beta-, and gammaretroviruses.
The predicted Gag-coding region is 2.3 kb, and like Gag of alpharetroviruses (22), the protein lacks a myristylation signal at the N terminus. The matrix (MA) domain, at the N terminus of Gag, is followed by a proline-rich region (PRR) that contains a PPPY motif characteristic of L domains, which are regions that contribute to virus budding, possibly by interaction with the ESCRT machinery of the cell, as in HIV-1 (23). The position of the L domain at the C terminus of MA is common to members of the Alpha-, Beta-, and Gammaretrovirus genera, while in lentiviruses, it is found at the C terminus of Gag (24). Further downstream are the capsid (CA) and nucleocapsid (NC) domains. The NC domain contains two zinc finger (ZF) RNA binding domains with typical C-C-H-C motifs (25). The presence of two ZF domains is typical of beta- and alpharetroviruses but not gammaretroviruses, most of which have only one ZF domain in Gag (11). Immediately downstream of the second ZF domain is a second PRR of ∼95 amino acids that has no recognizable protein domains or similarity to any known protein. At the end of the PRR is a stretch of adenosine residues that likely serves as a frameshift site, leading into the protease gene (pro).
Notably, the predicted protease (PR)-coding region of TgERV-F is in a separate reading frame from both Gag and Pol, akin to the genome organization of betaretroviruses. Furthermore, the catalytic domain of PR is DTG, as in betaretroviruses, rather than the typical alpharetroviral catalytic domain, DSG. However, whereas betaretroviral PR carries a dUTPase at its N terminus, PR of TgERV-F lacks a discernible dUTPase. At the end of the PR reading frame is another stretch of adenosines and thymines that likely serves as a frameshift site leading into the pol gene.
The Pol-coding region of TgERV-F is ∼2.6 kb, and the predicted protein product contains the usual conserved regions of reverse transcriptase (17), an RNase H domain, and an integrase domain. The RT catalytic site (YMDD) is typical of class II ERVs (11, 13). The integrase region has a Zn binding domain, a catalytic domain, and a DNA binding domain.
While the predicted TgERV-F Pol protein has features of a class II ERV, the predicted Env protein is typical of mammalian gammaretroviral (class I) Env. The SU domain includes a CWLC motif, which has been shown in murine leukemia virus (MLV) to isomerize the intersubunit disulfide bond after binding of the receptor (26). The basic amino acid-rich furin recognition and cleavage site separating the SU and TM domains is unusual (RLHKR) but highly conserved among the 14 proviruses. The fusion peptide of TgERV-F is located at the N terminus of the TM subunit, whereas in alpharetroviral Env, it is located internally and flanked by cysteines (Fig. 1). There is a conserved immunosuppressive domain (ISD) and a CX6CC motif, typical of gammaretroviral and alpharetroviral, but not betaretroviral, TM proteins (4, 5).
Age of TgERV-F.
None of the TgERV-F elements has ORFs across all coding regions (Table 1); however, each of the genes (gag, pro, pol, and env) has an intact ORF in at least one provirus. Four of the 12 proviruses are nearly intact, with only one of the four reading frames disrupted. env is the best conserved of the coding regions, with eight of 14 TgERV-F proviruses having an ORF across this entire region. The Pol-coding region is intact in three of the proviruses. The most frequently mutated coding region is that of the gag gene, and only one provirus has an intact ORF across this region. The pro gene has an intact ORF in all but one provirus, but this is the shortest coding region, consisting of 342 nt.
Since the mechanism of reverse transcription ensures that LTRs are identical at the time of integration, a rough estimate of the time of insertion can be made by applying the neutral rate of substitution to the number of differences between the 5′ and 3′ LTRs (27). Among the 14 full-length TgERV-F proviruses, LTR pairs differ by zero to six nucleotides (of 356). Applying an estimated neutral substitution rate for the bird genome of 2 × 10−9 and 3.9 × 10−9 substitutions per site per million years (28) gives an estimated integration time of two to four million years ago for the oldest of the 14 TgERV-F proviruses (Table 1). Interestingly, three of the proviruses have LTR pairs that are identical or differ by only one mutation, implying integration into the germ line in the recent past. Thus, TgERV-F has likely been active in the population for at least several million years, with possible ongoing activity. However, our search also uncovered numerous degraded TgERV-F sequences without intact LTRs, suggesting a longer period of activity in the zebra finch genome.
LTR pairs frequently undergo recombination events in which the coding region is deleted, resulting in a single “solo-LTR.” We found approximately 260 solo-LTR sequences matching TgERV-F in the sequenced genome, a ratio (ca. 18.5) that is consistent with that seen in various other ERV families, where solo-LTRs outnumber their full-length ancestors by 10 to 100 (39), with evidence for more recently integrated families having a ratio at the lower end of the range (30).
Phylogenetic analysis of TgERV-F.
In a previous study of class II retroviruses among avian species, a region of ∼801 nt spanning part of the pro gene and a portion of the reverse transcriptase (RT) region of the pol gene was PCR amplified from 38 taxa of birds (13). Phylogenetic analysis of the aligned sequences produced a tree that suggested the existence of several uncharacterized avian retroviral lineages, including one that clustered as a sister clade to ALV. In order to infer the relationship of the TgERV-F group to other known class II retroviruses, we aligned the analogous regions of the TgERV-F proviruses with the previously studied sequences, along with the corresponding regions of a selection of endogenous and exogenous class II retroviruses, to produce the neighbor-joining tree shown in Fig. 3. Despite the betaretrovirus-like features of its genome structure and PR catalytic site, the pro-pol region of TgERV-F places it firmly outside the betaretroviruses and closer to the ALV sister clade. This sister clade includes sequences isolated in the previous study from the hermit thrush (Catharus guttata) and blue tit (Parus caeruleus), both of which are members of the order Passeriformes, as well as the toucanette (order Piciformes) and the more distantly related black duck (order Anseriformes) (13). Although the original study sampled only for RT sequences, leaving the presence of the env sequence unconfirmed, this clade also includes the sequences that we recovered from several additional passerine species, i.e., the medium ground finch (Geospiza fortis), the ground tit (Pseudopodoces humilis), and the white-throated sparrow (Zonotrichia albicollis), all of which were fused to env genes that were highly related to that of TgERV-F (see Table S1 in the supplemental material). The presence of TgERV-F-like proviruses in several different passerine species suggests that TgERV-F is not an artifact of inbreeding in aviaries but has circulated in the wild.
FIG 3.

TgERV-F pro-pol clusters with a sister clade of ALV. A neighbor-joining consensus tree based on a nucleotide alignment of 801 nt spanning the pro and pol regions and rooted on human T cell leukemia virus (HTLV-1) as a class II outgroup is shown. TgERV-F is shown in bold. Asterisks mark species in which a TgERV-F-like env sequence was identified in this study. Bootstrap values, in percent, are shown for each node. The scale bar represents percent substitutions. Brackets to the right identify retroviral genera. Names and accession numbers of viruses used as reference sequences are in Table S2 in the supplemental material.
The SU region of the env gene is highly variable and unsuited to phylogenetic analyses, while the TM region is highly conserved (4). In order to infer the relationship of the TgERV-F env gene to other gamma-type env sequences, we aligned the TM regions of TgERV-F and a panel of gammaretroviruses and alpharetroviruses. As seen in Fig. 4, TgERV-F TM and the related passerine sequences cluster firmly with mammalian gammaretroviral sequences and separately from other known retroviruses of birds. The closest matching mammalian TM sequence found in the NCBI databases, that of the horseshoe bat, exhibited 69% identity and 88% similarity at the amino acid level. The TM sequence from reticuloendotheliosis viruses (REVs), which are recombinant gammaretroviruses that infect several species of waterfowl and game birds (31), clusters separately from that of TgERV-F, as does TM from chicken retrovirus 1 (ChiRV1) (32), a gammaretroviral-like ERV of chickens whose TM clusters with alpharetroviral TM. Thus, neither REVs nor ChiRV1 was a contributing partner in the recombination event that produced TgERV-F.
FIG 4.

TgERV-F TM clusters with gammaretrovirus sequences. A neighbor-joining tree based on an amino acid alignment of the TM-coding region and rooted on HTLV-1 is shown. TgERV-F is shown in bold. Asterisks mark species in which a TgERV-F-like pol sequence was identified in this study. The scale bar represents percent substitutions. Brackets to the right identify retroviral genera. Names and accession numbers of viruses used as reference sequences are in Table S2 in the supplemental material.
The region encoding Gag is less conserved than that encoding either Pol or Env, but BLAST searches with TgERV-F Gag returned ALV/RSV and EAV-HP as the closest hits, in keeping with the phylogeny of pro-pol.
DISCUSSION
The zebra finch genome has revealed a percentage of ERV sequences that is three times higher than that of the chicken genome (2). This higher load of ERVs in the zebra finch than in the chicken may be related to the intense period of speciation in the neoavian lineage (33). The context of intense speciation makes the ERV composition of the zebra finch particularly interesting for what it can reveal about the dynamics of cross-species transmissions and its role in the formation of novel retroviral lineages.
We have described a zebra finch ERV group, TgERV-F, for which we found 14 near-full-length insertions. TgERV-F is unique in several attributes. First, it is the only known class II/gammaretroviral recombinant in an avian genome. Other such recombinants have been described in mammals, including the exogenous and endogenous primate type D betaretroviruses (e.g., MPMV) (12, 29, 34), a recently described bat ERV (35), and endogenous intracisternal A particles (IAPs), which have recombined with gammaretroviral env, in the genomes of the shrew and guinea pig (36). The only previously characterized class II/gammaretroviral recombinants found outside mammals are two related ERVs of pythons (PyERV) (37). Second, TgERV-F demonstrates a unique mix of alpha-, beta-, and gammaretroviral features. It possesses betaretrovirus-like features of genome organization, such as −1 frameshifts between Gag, Pro, and Pol and a DTG active site in PR, yet like alpharetroviruses, it lacks a myristylation signal on Gag and a dUTPase, and its RT clusters more closely with alpharetroviruses than with betaretroviruses.
Of further interest is the possession by TgERV-F of a gammaretroviral env that appears of be of mammalian origin yet whose sequence does not closely match the env sequence of any of the previously characterized gammaretroviruses in birds, such as ChiRV1 and REVs, indicating a separate transmission event. Interestingly, recent work has suggested that REVs were accidentally introduced into birds during experiments with malarial parasites and so do not represent natural infections and are far too recent to be represented as avian ERVs, although related ERVs can be found in some mammals (31).
Furthermore, while other class II recombinants involving betaretroviruses with gammaretroviral env have been described, TgERV-F is the first characterized alpharetrovirus-like element that has recombined with a gammaretroviral env. Intriguingly, env sequences that cluster with alpharetroviruses do exist in the zebra finch genome (Fig. 4, Tg alpha), but these are found with pro-pol sequences that are more closely related to those of betaretroviruses (8).
Lastly, phylogenetic analysis of gammaretroviral ERVs among various vertebrate classes indicates that interclass transmission is very rare (1), in keeping with the many layers of hurdles presented by a genetically distant potential host, yet TgERV-F was apparently able to establish an ongoing, productive infection in the zebra finch genome over a span of several million years. Indeed, our analysis of LTR divergence and ORF intactness suggests that TgERV-F has been active in the zebra finch genome for at least 2 to 4 million years, and likely much longer, based on the presence of highly degraded sequences. Moreover, three TgERV-F proviruses have LTRs with zero or one mismatches between them, suggesting recent germ line infection.
Even more remarkable, given the probable mammalian origins of its env, TgERV-F has been able to circulate among various avian species. RT sequences with greater than 90% similarity to TgERV-F are found in several other passeriform (songbird) species, a piciform (woodpecker and other arboreal) species, and an anseriform (waterfowl) species. Additionally, mammalian gammaretroviral env sequences whose TMs cluster with that of TgERV-F were confirmed downstream from the RT sequences in three additional passerine species: the medium ground finch, ground tit, and white-throated sparrow. Although there are not enough data from these other species to verify if any of the insertions are or are not orthologous, Anseriformes are estimated to have diverged from Passeriformes 90 to 100 million years ago (38), strongly suggesting that the appearance of TgERV-F in both orders represents one or more cross-species transmission events.
Based on the wider species distribution of the gamma-type than of the beta-type env (Fig. 1) (5), acquisition of a gamma-type env could in theory afford a virus access to a new host environment, significantly different from that to which it is adapted. If the virus is able to adapt to this new environment, its evolutionary trajectory can be greatly affected, especially if it is able to circulate further among related species of the new class. TgERV-F may offer one such example of a cross-class transmission that was able to adapt and even gain access to other avian species, emphasizing the important role of env in generating novel lineages. As more avian ERVs and their env sequences become available, a fuller picture of the role of env recombination in adaptation, cross-species transmissions, and the formation of retroviral novelty will emerge.
Supplementary Material
ACKNOWLEDGMENTS
We thank Ted Diehl for helpful discussion and comments on the manuscript.
This work was supported by research grant R37CA089441 from the National Cancer Institute and NIH grant AI095092 (W.E.J.). J.M.C. was a Research Professor of the American Cancer Society.
Footnotes
Published ahead of print 18 December 2103
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02863-13.
REFERENCES
- 1.Martin J, Herniou E, Cook J, O'Neill RW, Tristem M. 1999. Interclass transmission and phyletic host tracking in murine leukemia virus-related retroviruses. J. Virol. 73:2442–2449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Warren WC, Clayton DF, Ellegren H, Arnold AP, Hillier LW, Künstner A, Searle S, White S, Vilella AJ, Fairley S, Heger A, Kong L, Ponting CP, Jarvis ED, Mello CV, Minx P, Lovell P, Velho TAF, Ferris M, Balakrishnan CN, Sinha S, Blatti C, London SE, Li Y, Lin Y-C, George J, Sweedler J, Southey B, Gunaratne P, Watson M, Nam K, Backström N, Smeds L, Nabholz B, Itoh Y, Whitney O, Pfenning AR, Howard J, Völker M, Skinner BM, Griffin DK, Ye L, McLaren WM, Flicek P, Quesada V, Velasco G, Lopez-Otin C, Puente XS, Olender T, Lancet D, Smit AFA, Hubley R, Konkel MK, Walker JA, Batzer MA, et al. 2010. The genome of a songbird. Nature 464:757–762. 10.1038/nature08819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann N, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, et al. 2001. Initial sequencing and analysis of the human genome. Nature 409:860–921. 10.1038/35057062 [DOI] [PubMed] [Google Scholar]
- 4.Benit L, Dessen P, Heidmann T. 2001. Identification, phylogeny, and evolution of retroviral elements based on their envelope genes. J. Virol. 75:11709–11719. 10.1128/JVI.75.23.11709-11719.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Henzy JE, Coffin JM. 2013. Betaretroviral envelope subunits are noncovalently associated and restricted to the mammalian class. J. Virol. 87:1937–1946. 10.1128/JVI.01442-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Henzy JE, Johnson WE. 2013. Pushing the endogenous envelope. Philos. Trans. R. Soc. Lond. B Biol. Sci. 368:20120506. 10.1098/rstb.2012.0506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Basta HA, Cleveland SB, Clinton RA, Dimitrov AG, McClure MA. 2009. Evolution of teleost fish retroviruses: characterization of new retroviruses with cellular genes J. Virol. 83:10152–10162. 10.1128/JVI.02546-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bolisetty M, Blomberg J, Benachenhou F, Sperber G, Beemon K. 2012. Unexpected diversity and expression of avian endogenous retroviruses. mBio 3(5):e00344–12. 10.1128/mBio.00344-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cordonnier A, Casella JF, Heidmann T. 1995. Isolation of novel human endogenous retrovirus-like elements with foamy virus-related pol sequence. J. Virol. 69:5890–5897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gifford R, Tristem M. 2003. The evolution, distribution and diversity of endogenous retroviruses. Virus Genes 26:291–315. 10.1023/A:1024455415443 [DOI] [PubMed] [Google Scholar]
- 11.Jern P, Sperber GO, Blomberg J. 2005. Use of endogenous retroviral sequences (ERVs) and structural markers for retroviral phylogenetic inference and taxonomy. Retrovirology 2:50. 10.1186/1742-4690-2-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sonigo P, Barker C, Hunter E, Wain-Hobson S. 1986. Nucleotide sequence of Mason-Pfizer monkey virus: an immunosuppressive D-type retrovirus. Cell 45:375–385. 10.1016/0092-8674(86)90323-5 [DOI] [PubMed] [Google Scholar]
- 13.Gifford R, Kabat P, Martin J, Lynch C, Tristem M. 2005. Evolution and distribution of class II-related endogenous retroviruses. J. Virol. 79:6478–6486. 10.1128/JVI.79.10.6478-6486.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410. 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- 15.Thompson JD, Gibson TJ, Higgins DG. 2002. Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinformatics Chapter 2:Unit 2.3. [DOI] [PubMed] [Google Scholar]
- 16.Tamura K, Dudley J, Nei M, Kumar S. 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24:1596–1599. 10.1093/molbev/msm092 [DOI] [PubMed] [Google Scholar]
- 17.Xiong Y, Eickbush TH. 1990. Origin and evolution of retroelements based upon their reverse transcriptase sequences. EMBO J. 9:3353–3362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saitou N, Nei M. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4:406–425 [DOI] [PubMed] [Google Scholar]
- 19.Derse D, Crise B, Li Y, Princler G, Lum N, Stewart C, McGrath CF, Hughes SH, Munroe DJ, Wu X. 2007. Human T-cell leukemia virus type 1 integration target sites in the human genome: comparison with those of other retroviruses. J. Virol. 81:6731–6741. 10.1128/JVI.02752-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holman AG, Coffin JM. 2005. Symmetrical base preferences surrounding HIV-1, avian sarcoma/leukosis virus, and murine leukemia virus integration sites. Proc. Natl. Acad. Sci. U. S. A. 102:6103–6107. 10.1073/pnas.0501646102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lewinski MK, Yamashita M, Emerman M, Ciuffi A, Marshall H, Crawford G, Collins F, Shinn P, Leipzig J, Hannenhalli S, Berry CC, Ecker JR, Bushman FD. 2006. Retroviral DNA integration: viral and cellular determinants of target-site selection. PLoS Pathog. 2:e60. 10.1371/journal.ppat.0020060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palmiter RD, Gagnon J, Vogt VM, Ripley S, Eisenman RN. 1978. The NH2-terminal sequence of the avian oncovirus gag precursor polyprotein (Pr76gag). Virology 91:423–433. 10.1016/0042-6822(78)90388-4 [DOI] [PubMed] [Google Scholar]
- 23.Strack B, Calistri A, Craig S, Popova E, Göttlinger HG. 2003. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell 114:689–699. 10.1016/S0092-8674(03)00653-6 [DOI] [PubMed] [Google Scholar]
- 24.Freed EO. 2002. Viral late domains. J. Virol. 76:4679–4687. 10.1128/JVI.76.10.4679-4687.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chance MR, Sagi I, Wirt MD, Frisbie SM, Scheuring E, Chen E, Bess JW, Jr, Henderson LE, Arthur LO, South TL. 1992. Extended X-ray absorption fine structure studies of a retrovirus: equine infectious anemia virus cysteine arrays are coordinated to zinc. Proc. Natl. Acad. Sci. U. S. A. 89:10041–10045. 10.1073/pnas.89.21.10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pinter A, Kopelman R, Li Z, Kayman SC, Sanders DA. 1997. Localization of the labile disulfide bond between SU and TM of the murine leukemia virus envelope protein complex to a highly conserved CWLC motif in SU that resembles the active-site sequence of thiol-disulfide exchange enzymes. J. Virol. 71:8073–8077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson WE, Coffin JM. 1999. Constructing primate phylogenies from ancient retrovirus sequences. Proc. Natl. Acad. Sci. U. S. A. 96:10254–10260. 10.1073/pnas.96.18.10254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Axelsson E, Smith NGC, Sundström H, Berlin S, Ellegren H. 2004. Male-biased mutation rate and divergence in autosomal, z-linked and w-linked introns of chicken and Turkey. Mol. Biol. Evol. 21:1538–1547. 10.1093/molbev/msh157 [DOI] [PubMed] [Google Scholar]
- 29.Kato S, Matsuo K, Nishimura N, Takahashi N, Takano T. 1987. The entire nucleotide sequence of baboon endogenous virus DNA: a chimeric genome structure of murine type C and simian type D retroviruses. Jpn. J. Genet. 62:127–137. 10.1266/jjg.62.127 [DOI] [Google Scholar]
- 30.Nellåker C, Keane TM, Yalcin B, Wong K, Agam A, Belgard TG, Flint J, Adams DJ, Frankel WN, Ponting CP. 2012. The genomic landscape shaped by selection on transposable elements across 18 mouse strains. Genome Biol. 13:R45. 10.1186/gb-2012-13-6-r45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Niewiadomska AM, Gifford RJ. 2013. The extraordinary evolutionary history of the reticuloendotheliosis viruses. PLoS Biol. 11:e1001642. 10.1371/journal.pbio.1001642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Borysenko L, Stepanets V, Rynditch AV. 2008. Molecular characterization of full-length MLV-related endogenous retrovirus ChiRV1 from the chicken, Gallus gallus. Virology 376:199–204. 10.1016/j.virol.2008.03.006 [DOI] [PubMed] [Google Scholar]
- 33.Suh A, Paus M, Kiefmann M, Churakov G, Franke FA, Brosius J, Kriegs JO, Schmitz J. 2011. Mesozoic retroposons reveal parrots as the closest living relatives of passerine birds. Nat. Commun. 2:443. 10.1038/ncomms1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van der Kuyl AC, Mang R, Dekker JT, Goudsmit J. 1997. Complete nucleotide sequence of simian endogenous type D retrovirus with intact genome organization: evidence for ancestry to simian retrovirus and baboon endogenous virus. J. Virol. 71:3666–3676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hayward JA, Tachedjian M, Cui J, Field H, Holmes EC, Wang L-F, Tachedjian G. 2013. Identification of diverse full-length endogenous betaretroviruses in megabats and microbats. Retrovirology 10:35. 10.1186/1742-4690-10-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Magiorkinis G, Gifford RJ, Katzourakis A, De Ranter J, Belshaw R. 2012. Env-less endogenous retroviruses are genomic superspreaders. Proc. Natl. Acad. Sci. U. S. A. 109:7385–7390. 10.1073/pnas.1200913109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huder JB, Böni J, Hatt J-M, Soldati G, Lutz H, Schüpbach J. 2002. Identification and characterization of two closely related unclassifiable endogenous retroviruses in pythons (Python molurus and Python curtus). J. Virol. 76:7607–7615. 10.1128/JVI.76.15.7607-7615.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hackett SJ, Kimball RT, Reddy S, Bowie RCK, Braun EL, Braun MJ, Chojnowski JL, Cox WA, Han K-L, Harshman J, Huddleston CJ, Marks BD, Miglia KJ, Moore WS, Sheldon FH, Steadman DW, Witt CC, Yuri T. 2008. A phylogenomic study of birds reveals their evolutionary history. Science 320:1763–1768. 10.1126/science.1157704 [DOI] [PubMed] [Google Scholar]
- 39.Stoye JP. 2001. Endogenous retroviruses: still active after all these years? Curr. Biol. 11:R914–R916. 10.1016/S0960-9822(01)00553-X [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.