

ABSTRACT
One role of mRNA cap guanine-N-7 (G-N-7) methylation is to facilitate the efficient translation of mRNA. The role of mRNA cap ribose 2′-O methylation is enigmatic, although recent work has implicated this as a signature to avoid detection of RNA by the innate immune system (S. Daffis, K. J. Szretter, J. Schriewer, J. Q. Li, S. Youn, J. Errett, T. Y. Lin, S. Schneller, R. Zust, H. P. Dong, V. Thiel, G. C. Sen, V. Fensterl, W. B. Klimstra, T. C. Pierson, R. M. Buller, M. Gale, P. Y. Shi, M. S. Diamond, Nature 468:452-456, 2010, doi:10.1038/nature09489). Working with vesicular stomatitis virus (VSV), we previously showed that a panel of recombinant VSVs carrying mutations at a predicted methyltransferase catalytic site (rVSV-K1651A, -D1762A, and -E1833Q) or S-adenosylmethionine (SAM) binding site (rVSV-G1670A, -G1672A, and -G4A) were defective in cap methylation and were also attenuated for growth in cell culture. Here, we analyzed the virulence of these recombinants in mice. We found that rVSV-K1651A, -D1762A, and -E1833Q, which are defective in both G-N-7 and 2′-O methylation, were highly attenuated in mice. All three viruses elicited a high level of neutralizing antibody and provided full protection against challenge with the virulent VSV. In contrast, mice inoculated with rVSV-G1670A and -G1672A, which are defective only in G-N-7 methylation, were attenuated in vivo yet retained a low level of virulence. rVSV-G4A, which is completely defective in both G-N-7 and 2′-O methylation, also exhibited low virulence in mice despite the fact that productive viral replication was not detected in lung and brain. Taken together, our results suggest that abrogation of viral mRNA cap methylation can serve as an approach to attenuate VSV, and perhaps other nonsegmented negative-strand RNA viruses, for potential application as vaccines and viral vectors.
IMPORTANCE Nonsegmented negative-sense (NNS) RNA viruses include a wide range of significant human, animal, and plant pathogens. For many of these viruses, there are no vaccines or antiviral drugs available. mRNA cap methylation is essential for mRNA stability and efficient translation. Our current understanding of mRNA modifications of NNS RNA viruses comes largely from studies of vesicular stomatitis virus (VSV). In this study, we showed that recombinant VSVs (rVSVs) defective in mRNA cap methylation were attenuated in vitro and in vivo. In addition, these methyltransferase (MTase)-defective rVSVs triggered high levels of antibody responses and provided complete protection against VSV infection. Thus, this study will not only contribute to our understanding of the role of mRNA cap MTase in viral pathogenesis but also facilitate the development of new live attenuated vaccines for VSV, and perhaps other NNS RNA viruses, by inhibiting viral mRNA cap methylation.
INTRODUCTION
Nonsegmented negative-sense (NNS) RNA viruses include a wide range of significant human, animal, and plant pathogens. The NNS RNA viruses are classified into four families: the Rhabdoviridae, exemplified by rabies virus and vesicular stomatitis virus (VSV); the Filoviridae, exemplified by Ebola and Marburg viruses; the Bornaviridae, exemplified by Borna disease virus; and the Paramyxoviridae, exemplified by human respiratory syncytial virus (hRSV), human metapneumovirus (hMPV), human parainfluenza virus type 3 (hPIV3), and measles, mumps, Nipah, and Hendra viruses. For many of these viruses, there are no vaccines available, and there are no antiviral drugs with proven efficacy against this entire class of viruses. With the exception of the Bornaviridae, the mammalian NNS RNA viruses replicate within the host cell cytoplasm and do not have access to the host mRNA capping machinery. Instead, the viruses have evolved their own mRNA capping machinery. Specifically, the large polymerase protein (L) is a 220- to 240-kDa multifunctional protein that possesses nucleotide polymerization (1–3) and mRNA modification activities, including cap addition (4–6), cap methylation (7–11), and polyadenylation (12–15). The enzymatic activities have been mapped within L to motifs that are highly conserved among all NNS RNA viruses (16). Specifically, a GDN motif, present within conserved region III (CR-III) is essential for nucleotide polymerization (1). CR-V contains a GXXT[n]HR motif that is required for mRNA capping catalyzed by a GDP:polyribonucelotidyltransferase (PRNTase) (4–6), and CR-VI contains a methyltransferase (MTase) motif (KDKE) that catalyzes both guanine-N-7 (G-N-7) and ribose 2′-O (2′-O) methylation (6, 9, 11, 17). The molecular architecture of the VSV L polymerase protein (18) shows that a core ring-like domain contains the RNA-dependent RNA polymerase (RdRp) and an appendage of three globular domains contains the cap-forming machinery.
Our understanding of mRNA modifications of NNS RNA viruses comes largely from studies of VSV. In response to a specific promoter element provided by the genomic leader region, the polymerase initiates mRNA synthesis at the first gene start sequence (19, 20). The nascent mRNA is capped through an unconventional mechanism in which the GDP:PRNTase of L transfers a monophosphate RNA onto a GDP acceptor through a covalent protein-RNA intermediate (4–6). Following cap formation, VSV mRNAs are sequentially methylated at the ribose 2′-O position and G-N-7 position (8, 21), an order which is opposite that of other known methylation reactions. Unlike traditional cap-forming enzymes (22), the VSV capping and methylation machinery require cis-acting signals in the RNA (8, 23–25) and a minimal nascent RNA chain length of 31 nucleotides (nt) (26). In response to a gene end sequence, L polyadenylates and terminates mRNA synthesis by the programmed stuttering of polymerase on a U7 tract (27).
mRNA 5′ cap structures are essential for mRNA stability and efficient translation (28). Guanine-N-7 (G-N-7) methylation of the mRNA cap structure is required for recognition of the cap by the rate-limiting factor for translation initiation, eIF-4E (28–30). Although the precise mechanism by which VSV mRNAs are translated is not fully understood, they utilize a variation of the canonical cap-dependent translational pathway that is hypersensitive to the lack of a ribosomal protein, rpL40 (30–34). In infected cells, host mRNA translation is rapidly inhibited through suppression of the intracellular pools of eIF-4E by a manipulation of the phosphorylation status of the 4E binding protein (4E-BP1) (35). Nevertheless, in vitro experiments have shown that G-N-7 cap methylation facilitates translation of VSV mRNA (30, 31, 33, 34). The biological function of mRNA cap 2′-O methylation, however, remains less well understood. Recent work with West Nile virus (WNV), vaccinia virus, mouse hepatitis virus, and human coronavirus strain 229E showed that 2′-O methylation of viral RNA cap structure functions to prevent the detection of viral RNA by the innate immune system (36–38, 57). From this perspective, 2′-O-methylation provides a molecular signature for the discrimination of self and nonself mRNA.
Previously, we characterized recombinant MTase-defective VSV that contain substitutions to a predicted MTase catalytic motif, KDKE, or a S-adenosyl-l-methionine (SAM) binding site (9, 17, 21). Those VSVs can be classified into three groups: viruses completely defective in both G-N-7 and 2′-O methylation (rVSV-K1651A, -D1762A, -K1795A, -E1833Q, and -G4A), those defective in G-N-7 but not 2′-O MTase (rVSV-G1670A and -G1672A), and those that require elevated SAM concentrations to permit full methylation (rVSV-G1674A and -Y1835A). With the exception of rVSV-G1674A and -Y1835A, all MTase-defective VSVs were attenuated in cell culture, as judged by diminished viral plaque size, reduced infectious viral progeny release (in single-step growth curves), and decreased levels of viral RNA and protein synthesis (9, 17, 21). However, whether these MTase-defective VSVs are attenuated in vivo is not known.
In this study, we examined the impact of mRNA cap methylation status on VSV pathogenicity in mice. We found that viruses with mutations in the predicted MTase catalytic site were highly attenuated and that viruses with substitutions in the SAM binding site that led to defects in G-N-7 but not 2′-O methylation retained low virulence. Each of those recombinants stimulated high levels of VSV-specific antibody and provided full protection against challenge with wild-type virus, demonstrating that such methylation-defective viruses might be good vaccine candidates. Remarkably, not all mutations that blocked methylation were highly attenuated, since rVSV-G4A, which contains 4 amino acid substitutions in the predicted SAM binding site, retained low virulence in mice despite the fact that it is completely defective in G-N-7 and 2′-O methylation and lacks evidence of productive viral replication in vivo. Since the transcripts generated by this virus in vitro lack both G-N-7 and ribose 2′-O methyl groups, we anticipated that this virus would be exquisitely sensitive to control by the innate immune system. The molecular mechanism of the virulence associated with this virus will likely be of interest to determine and may help further our understanding of the innate immune control of virus infection.
MATERIALS AND METHODS
Recombinant VSV.
Wild-type recombinant VSV (rVSV) and MTase-defective VSVs (rVSV-K1651A, -D1762A, -E1833Q, -G1670A, -G1672A, -G4A, and -G1674A) were recovered from an infectious clone of VSV Indiana strain as described previously (9, 21). All recombinant VSVs were grown on BHK-21 cells in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Carlsbad, CA) containing 2% fetal bovine serum (FBS; Invitrogen). Virus titers were determined by plaque assay in Vero cells as previously described (9, 21).
Transcription of viral RNA in vitro and scintillation counting.
Viral RNA was synthesized in vitro by using 10 μg of purified virus (rVSV and rVSV-G4A) as described previously (9, 21). Reactions were performed in the presence of 1 mM ATP, 0.5 mM CTP, GTP, and UTP, and 15 μCi [3H]SAM) (85 Ci/mmol; PerkinElmer, Billerica, MA), and 25% (vol/vol) rabbit reticulocyte lysate. Where indicated, reaction mixtures were supplemented with 1 mM S-adenosylhomocysteine (SAH). After incubation at 30°C for 5 h, RNA was purified with an RNeasy minikit (Qiagen, Valencia, CA). Aliquots of purified RNA were mixed with 4 ml of ReadySafe scintillation mixture (Beckman Coulter, Brea, CA), and the number of disintegrations per minute (dpm) was measured by scintillation counting using a 1414 series counter (PerkinElmer).
Replication of VSV in different cell lines.
Confluent BHK-21, Vero, or HEp-2 cells in 35-mm dishes were infected by rVSV or rVSV-G4A at a multiplicity of infection (MOI) of 0.1. After 1 h of adsorption, the inoculum was removed, the cells were washed three times with DMEM, fresh DMEM (supplemented with 2% fetal bovine serum) was added, and infected cells were incubated at 37°C. After 24 h, supernatants were harvested, and virus titers were determined by plaque assay on Vero cells.
Analysis of viral mRNA cap methylation in cell culture.
Confluent BHK-21, Vero cells, or HEp-2 in 35-mm dishes were infected by rVSV or G4A at an MOI of 0.1. After 24 h postinfection, total RNA was isolated with an RNeasy minikit (Qiagen). Subsequently, poly(A)-containing RNA was isolated from total RNA using a Dynabeads mRNA isolation kit (Invitrogen) according to the manufacturer's recommendations. To examine G-N-7 methylation, equal amounts of viral RNA were incubated with 10 units of vaccinia virus G-N-7 MTase supplied with the m7G capping system (ScriptCap) in the presence of 15 μCi [3H]SAM (85 Ci/mmol; PerkinElmer). To examine 2′-O methylation, viral RNA was incubated with 10 units of vaccinia virus G-N-7 MTase in the presence of 100 μM cold SAM, and RNA was purified and further incubated with vaccinia virus 2′-O MTase (VP39). Finally, RNA was purified with an RNeasy minikit (Qiagen), and the methylation of the mRNA cap structure was measured by determining 3H incorporation using a 1414 series scintillation counter (PerkinElmer).
Animal experiments. (i) Animal experiment 1 (pathogenicity).
Seventy-two 4-week-old specific-pathogen-free female BALB/c mice (Charles River Laboratories, Wilmington, MA) were randomly divided into nine groups (8 mice per group). These mice were housed within ULAR facilities of The Ohio State University under approved Institutional Laboratory Animal Care and Use Committee (IACUC) guidelines. Each inoculation group was separately housed in rodent cages under biosafety level 2 (BSL-2) conditions. Mice in group 1 were inoculated with rVSV and served as a virulent control. Mice in groups 2 to 8 were inoculated with seven different MTase-defective VSVs (rVSV-K1651A, -D1762A, -E1833Q, -G1670A, -G1672A, -G1674A, and -G4A). Mice in group 9 were inoculated with DMEM and served as uninfected controls (the normal control). Each mouse was inoculated intranasally at a dose of 5 × 106 PFU in a volume of 50 μl. After inoculation, the animals were evaluated twice every day for mortality and the presence of any symptoms of VSV infection. The severity of clinical signs associated with VSV infection was scored based on the following criteria: grade 3 (severe) was characterized by ruffled fur, hyperexcitability, tremors, circling, and paralysis; grade 2 (moderate) was characterized by ruffled fur with neurological symptoms such as circling; grade 1 (mild) was characterized by ruffled fur but no neurological symptoms; grade 0 was defined as no symptoms. The body weight of each mouse was monitored on a daily basis. At day 7 postinoculation, 4 mice from each group were euthanized. At day 14 postinoculation, the remaining 4 mice from each group were euthanized. The brain, lungs, and spleen from each mouse were collected for both virus isolation and histological analysis as described below.
(ii) Animal experiment 2 (pathogenicity).
Forty 4-week-old specific-pathogen-free female BALB/c mice were randomly divided into 5 groups (8 mice per group). Mice in group 1 were inoculated with DMEM and served as the uninfected control. Mice in group 2 were inoculated with rVSV and served as the virulent control. Mice in groups 3 to 5 were inoculated with MTase-defective rVSV-K1651A, -G1670A, and -G4A, respectively. All mice were inoculated intranasally at a dose of 5 × 106 PFU per mouse and were evaluated as described for animal experiment 1 except that euthanasia was performed at days 3 and 5 postinoculation.
(iii) Animal experiment 3 (pathogenicity).
Twenty-four 4-week-old specific-pathogen-free female BALB/c mice were randomly divided into 3 groups (8 mice per group). Mice in groups 1 to 3 were inoculated with DMEM, rVSV, and rVSV-G4A, respectively. All mice were inoculated intranasally at a dose of 5 × 106 PFU per mouse and were evaluated as described for animal experiment 1 except that euthanasia was performed at days 1 and 2 postinoculation.
(iv) Animal experiment 4 (LD50).
The 50% lethal doses (LD50) of selected recombinant VSVs (rVSV, rVSV-G1674A, rVSV-G4A, rVSV-G1762A, and rVSV-K1651A) were determined. Briefly 4-week-old specific-pathogen-free female BALB/c mice were inoculated with four doses of each recombinant virus (105, 106, 107, and 108 PFU per mouse, 5 mice per group). Animals were observed every 12 h until 14 days postinoculation. The 50% lethal dose (LD50) for each virus was calculated by the method of Reed and Muench (39).
(v) Animal experiment 5 (immunogenicity).
Twenty-eight 4-week-old specific-pathogen-free female BALB/c mice were randomly divided into seven groups (4 mice per group). Mice in groups 1 to 5 were immunized with MTase-defective rVSV-K1651A, -D1762A, -E1833Q, -G1670A, and -G1672A, respectively. Mice in group 6 were inoculated with DMEM and served as the challenged control. Mice in group 7 were inoculated with DMEM and served as the uninfected controls (normal controls). All mice were immunized intranasally at a dose of 5 × 106 PFU per mouse. After immunization, the animals were evaluated daily for body weight, mortality, and the presence of any symptoms of VSV infection. Blood samples were collected from each mouse weekly by facial vein bleed, and serum was isolated for antibody detection. At week 4 postimmunization, mice in groups 1 to 6 were challenged with rVSV at a dose of 5 × 106 PFU per mouse. After challenge, the animals were evaluated twice every day for mortality and the presence of any symptoms of VSV infection. The body weight for each mouse was monitored on a daily basis. At day 7 postchallenge, all mice from each group were euthanized. The brain, lungs, and spleen from each mouse were collected for virus isolation and histological evaluation.
(vi) Animal experiment 6 (immunogenicity).
Experimental design for experiment 6 was essentially identical to that for experiment 3 with the exception that a booster immunization was given. Briefly, 28 4-week-old specific-pathogen-free (SPF) female BALB/c mice were randomly divided into seven groups (4 mice per group). Mice in groups 1 to 6 were immunized intranasally at a dose of 5 × 106 PFU with rVSV-K1651A, -E1833Q, -D1762A, -G1670A, -G1672A, and -G4A, respectively. At week 2 after primary immunization, mice were given boosters intranasally with 5 × 106 PFU of the corresponding virus. At week 3 after booster immunization, mice in groups 1 to 6 were challenged with rVSV at a dose of 5 × 106 PFU per mouse. At day 7 postchallenge, all mice were euthanized, and brains and lungs were collected for virological and pathological evaluation.
Determination of virus burden in lungs and brains.
A brain hemisphere and right lung from each mouse were homogenized in phosphate-buffered saline (PBS) solution, and the presence of infectious virus was determined by plaque assay in Vero cells as described previously (9, 21). The presence of viral RNA in brain and lung tissues was determined by reverse transcription-PCR (RT-PCR). Briefly, total RNA was extracted from lungs and brain using an RNeasy minikit (Qiagen) according to the manufacturer's instructions. Two primers (5′-ATGTCTGTTACAGTCAAGAG-3′ and 5′-TCATTTGTCAAATTCTGAC-3′) were designed to target the VSV N gene, ranging from position 64 to position 1332 (numbering refers to the complete VSV Indiana strain genome sequence). RT-PCR was performed using a one-step RT-PCR kit (Qiagen). The amplified products were analyzed on a 1% agarose gel.
Quantification of beta interferon (IFN-β) mRNA by real-time RT-PCR.
Total RNA was extracted from lung or brain tissues using the Qiagen RNeasy kit (Qiagen). The RNA was treated with DNase I (Ambion), and first-strand cDNA was synthesized with Superscript III reverse transcriptase (Invitrogen) using oligo(dT) primers (Promega). Real-time PCR was performed in 20-μl reaction mixtures with primers for mouse IFN-β (forward, 5′-CTC CAG CTC CAA GAA AGG AC-3′; reverse, 5′-TGG CAA AGG CAG TGT AAC TC-3′). The mRNA levels of IFN-β were determined using SYBR green master mix (Applied Biosystems). The PCR conditions and cycles were as follows: initial DNA denaturation for 10 min at 95°C, followed by 45 cycles at 95°C for 10 s, followed by an annealing step at 58°C for 10 s, extension at 72°C for 10 s, and then signal detection 81°C for 2 s. A plasmid containing the mouse IFN-β sequence was used as a standard.
RT-PCR and sequencing.
To determine whether MTase-defective VSVs were genetically stable in mice, viral RNA was isolated from lungs and brain. The entire L gene of each MTase-defective VSV was amplified by one-step RT-PCR using four overlapping fragments. In addition, the full-length genome of rVSV-G4A isolated from lung or brain was also amplified. The PCR products were purified and sequenced at The Ohio State University Plant Microbe Genetics Facility to confirm the presence of a mutation(s) in the MTase region.
Detection of antibody by ELISA.
Ninety-six-well plates were first coated with 50 μl of highly purified VSV (30 μg/ml, in 50 mM Na2CO3 buffer, pH 9.6) per well at 4°C overnight. Subsequently, individual serum samples were tested for VSV-specific antibody on antigen-coated plates. Briefly, serum samples were 2-fold serially diluted and added to VSV-coated wells. After incubation at room temperature for 1 h, the plates were washed five times with phosphate-buffered saline (PBS)–Tween (0.05%), followed by incubation with 50 μl of goat anti-mouse IgG horseradish peroxidase (HRP)-conjugated secondary antibodies (Sigma) at a dilution of 1:80,000 for 1 h. Plates were washed and developed with 75 μl of 3,3′,5,5′-tetramethylbenzidine (TMB), and the optical density (OD) at 450 nm was determined using an enzyme-linked immunosorbent assay (ELISA) plate reader. Endpoint titers were determined as the reciprocal of the highest dilution that had an absorbance value greater than the background level (DMEM control). Antibody titers were calculated by the geometric mean titers (GMT).
Histology.
Half of the tissues from each experiment were preserved in 4% (vol/vol) phosphate-buffered paraformaldehyde. After fixation, the brain was removed from the skull. A horizontal brain section containing the olfactory bulb, forebrain, cerebrum, hippocampus, and cerebellum was recovered. Fixed tissues were embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin-eosin (HE) for the examination of histological changes by light microscopy. The severity of lung histological change was scored based on the following criteria: grade 3 (severe), prominent lymphoid infiltrates around bronchovascular bundles and venules with extension into interlobular septa and interstitium; grade 2 (moderate), lymphoid infiltrates present around bronchovascular bundles and venules without extension into interlobular septa and interstitium; grade 1 (mild), minimal lymphoplasmacytic infiltrates involving occasional bronchovascular bundles and venules; grade 0, no lesions. The severity of brain histological change was scored according to the following criteria: grade 3 (severe), focally extensive and/or multifocal mononuclear inflammatory cell infiltrates around bronchi and blood vessels in the parenchyma (perivascular cuffs) with extension into surrounding neuropil, and evidence of gliosis (i.e., parenchymal reaction); grade 2 (moderate), two or three foci of limited infiltration; grade 1 (mild), one infiltrate or parenchymal reaction; grade 0, no lesions. The histological slides were examined independently by three pathologists.
Quantitative and statistical analyses.
Quantitative analysis was performed either by densitometric scanning of autoradiographs or by using a phosphorimager (Typhoon; GE Healthcare) and ImageQuant TL software (GE Healthcare, Piscataway, NJ). Statistical analysis was performed by one-way multiple comparisons using SPSS 8.0 statistical analysis software (SPSS Inc., Chicago, IL). A P value of <0.05 was considered statistically significant.
RESULTS
MTase-defective VSVs exhibited a variable degree of attenuation in mice.
To determine whether MTase-defective VSVs were attenuated in an in vivo pathogenesis model, we inoculated purified virus into BALB/c mice and monitored body weight changes and scored clinical symptoms. Mice infected with rVSV lost 3 to 5 g of body weight over a 7-day period, and 2/8 mice died (Fig. 1). All mice in this group exhibited severe clinical symptoms of VSV infection, including ruffled fur, hyperexcitability, tremors, circling, and paralysis. The average score of clinical symptoms in rVSV group was 3.0 (severe) (Table 1). In contrast, mice inoculated with rVSV-K1651A, -D1762A, and -E1833Q exhibited no clinical signs of VSV infection, including weight loss (score 0, no symptoms). The average body weight of mice inoculated with these recombinants (data for rVSV-D1762A not shown) was indistinguishable from that of the uninfected controls (P > 0.05) during the entire experimental period (Fig. 1). Mice inoculated with rVSV-G4A, -G1670A, and -G1672A showed moderate weight loss (2 to 3 g during days 3 to 7 postinoculation). The average body weights of mice in the rVSV-G4A and -G1670A groups were indistinguishable (P > 0.05) by day 14 but were lower than those of the uninfected controls (P < 0.05). During days 3 to 5 postinoculation, mice inoculated with rVSV-G1672A had significantly less weight loss than those in the rVSV-G4A and -G1670A groups (P < 0.05). In addition, mice in the rVSV-G4A group had a faster weight recovery than those in the rVSV-G1670A and rVSV-G1672A groups (Fig. 1). A few mice inoculated with these recombinants showed mild illnesses, such as ruffled coats for 2 to 3 days, but recovered quickly. None of the mice receiving rVSV-G1670A or rVSV-G1672A had neurological symptoms or died. The average score for clinical symptoms in these groups was 1.0 (mild) (Table 1). However, some mice in rVSV-G4A groups exhibited mild neurological symptoms, with an average score of 1.375, which was not significantly different from that of mice challenged with rVSV-G1670A and rVSV-G1672A (P > 0.05) (Table 1). Mice inoculated with rVSV-G1674A exhibited body weight loss similar to that of the rVSV group (P > 0.05). In addition, mice in rVSV-G1674A groups showed significant clinical signs of VSV infections, including ruffled fur and neurological disorders, although the severity was diminished compared to that seen with rVSV. Taken together, these data demonstrated that (i) rVSV-K1651A, -D1762A, and -E1833Q are avirulent in mice; (ii) rVSV-G4A, -G1670A, and G1672A retain low virulence; and (iii) rVSV-G1674A remains virulent in mice.
FIG 1.

Dynamics of mouse body weight change after VSV infection. Four-week-old SPF female BALB/c mice (8 mice per group) were inoculated with each MTase-defective VSVs intranasally at a dose of 5×106 PFU. At day 7 postinoculation, 4 mice from each group were euthanized. The body weight of the remaining 4 mice in each group was monitored until day 14 postinoculation. The average body weight for each group is shown.
TABLE 1.
Pathogenicity of MTase-defective VSVs in mice
| Groupa | No. of sick mice/total (score)b |
Histology scorec |
No. of mice (n = 4) with viral RNA in: |
Viral titer at day 7 (PFU/g)d |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Lung |
Brain |
Lung |
Brain |
|||||||||
| Day 7 | Day 14 | Day 7 | Day 14 | Day 7 | Day 14 | Day 7 | Day 14 | Day 7 | Day 14 | Lung | Brain | |
| DMEM | 0/8 (0) | 0/4 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| rVSV | 8/8 (3.0 A) | 1/4 | 3.0 A | 2.5 A | 2.5 A | 0 | 4 | 4 | 3 | 2 | 0 | 2.6 × 105 |
| rVSV-K1651A | 0/8 (0) | 0/4 | 2.0 B | 1.5 B | 0 | 0 | 3 | 0 | 0 | 1 | 0 | 0 |
| rVSV-E1833Q | 0/8 (0) | 0/4 | 1.0 C | 0.5 C | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 0 |
| rVSV-G1670A | 6/8 (1.0 C) | 0/4 | 2.0 B | 1.5 B | 0.5 B | 0 | 3 | 0 | 0 | 0 | 0 | 0 |
| rVSV-G1672A | 5/8 (1.0 C) | 0/4 | 2.0 B | 1.0 B | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 0 |
| rVSV-G1674A | 8/8 (2.25 B) | 0/4 | 3.0 A | 2.0 A | 0.5 B | 0 | 4 | 0 | 1 | 1 | 0 | 0 |
| rVSV-G4A | 8/8 (1.375 C) | 0/4 | 3.0 A | 2.0 A | 0.5 B | 0 | 4 | 0 | 2 | 1 | 0 | 0 |
Each group was infected with the indicated virus or administered DMEM as a control. Each group contained 8 mice.
The severity of VSV symptoms at day 7 was scored as grade 3 (severe), grade 2 (moderate), grade 1 (mild), or grade 0 (no symptoms). The average score for each group is shown. Values within a column followed by the same letter are significantly different (P < 0.05).
The severity of histological change was scored based on the extent of lymphoid infiltrates (lung) or inflammatory cell infiltrates and gliosis (brain) as described in Materials and Methods: grade 3 (severe), grade 2 (moderate), grade 1 (mild), and grade 0 (no lesion). The average scores for four mice are shown. Values within a column followed by different letters are significantly different (P < 0.05).
Viral titer was determined by plaque assay. 3 out of 4 mice in rVSV group had average titer of 2.6 × 105 in brains at day 5 postinfection. All other groups did not have detectable infectious VSV. “0” indicates that the viral titer was undetectable by plaque assay.
MTase-defective VSV had diminished pathological changes in lung and brain.
To examine pathological changes, four out of eight mice from each group were euthanized at day 7 postinoculation. Lungs, spleen, and brain from each mouse were collected for histological examination and antigen detection. At the gross level, lung tissues from rVSV- and rVSV-G1674A-infected mice appeared to be dark red, a sign of inflammation, whereas lungs from other MTase-defective VSV groups as well as the uninfected control group were pink. The spleen samples from rVSV- and rVSV-G1674A-infected mice were enlarged compared to those from the uninfected control group. As for brain tissues, no significant gross pathological changes were observed in all infected groups compared with the uninfected control. Following euthanization of the remaining 4 mice in each group at day 14 postinoculation, no significant gross pathological changes were found in lungs, spleens, and brains in comparison to the uninfected controls.
To evaluate the histopathological changes in the brain and lung tissues, samples were fixed and embedded in paraffin and sectioned, followed by hematoxylin-eosin staining. At day 7 postinfection, pulmonary tissues from the rVSV group showed severe histological changes characterized by prominent mononuclear cell infiltrates (lymphocytes and macrophages) around bronchi and bronchiole vascular bundles and venules (Fig. 2 and Table 1). This inflammatory response extended into interlobular septa and the interstitium, resulting in variably severe interstitial pneumonia and mononuclear cell alveolitis (Fig. 2). Pulmonary tissue sections from mice in the rVSV-K1651A, -G1670A, -G1672A, -G4A, and -G1674A groups had moderate lymphocyte and monocyte cellular infiltrates, while mice receiving rVSV-E1833Q exhibited only mild inflammatory cell changes in the lungs (Fig. 2 and Table 1). At day 14 postinfection, the pulmonary infiltrates were diminished and were scored as moderate. In contrast, lung tissue sections from all MTase-defective VSV groups exhibited either no or mild lymphoid infiltrates (Table 1). No inflammation was observed in lung tissues from the uninfected control group. The extent and severity of brain inflammatory cell infiltrates varied by inoculation group. By day 7 postinfection, brain sections from the rVSV group showed moderate histological changes characterized by multifocal meningeal mononuclear cell infiltrates with formation of perivascular cuffs in the parenchyma and evidence of inflammatory cells extending into the neuropil (Fig. 3 and Table 1). Brain tissues from the rVSV-G1670A-, rVSV-G1674A-, and rVSV-G4A-infected mice had mild inflammatory changes, whereas those from rVSV-K1651A-, rVSV-E1833Q-, and rVSV-G1672A-infected mice had no apparent histological change (Table 1). By day 14 postinfection, no histological change was found in brain tissues from all groups. Taken together, these data demonstrated that (i) infection with rVSV causes moderate inflammatory changes in both lung and brain tissues; (ii) infection with rVSV-K1651A, -E1833Q, and -G1672A causes mild histological changes in the lungs but not the brain; and (iii) infection with rVSV-G1670A, -G4A, and -G1674A causes moderate histological changes in the lungs and mild changes in brains.
FIG 2.
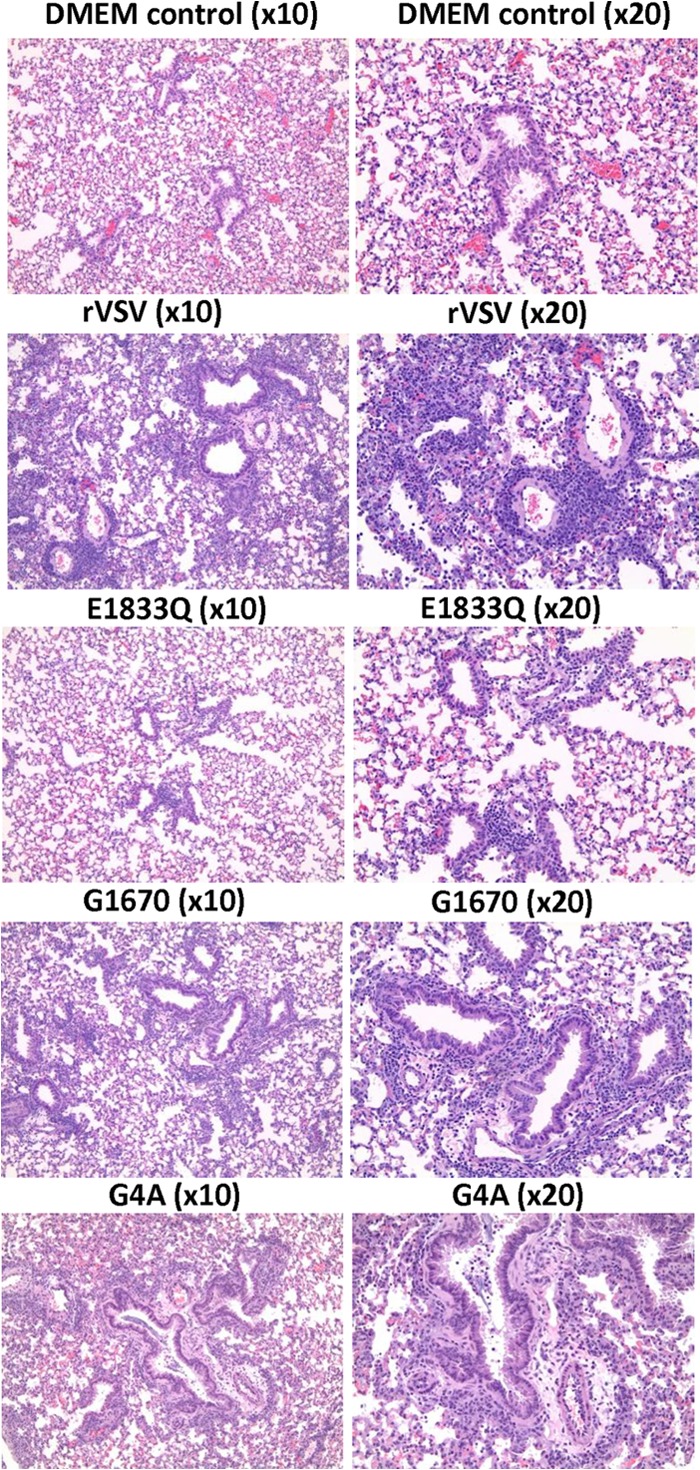
Histopathological changes in lungs after VSV infection. Four-week-old SPF female BALB/c mice were inoculated with each MTase-defective VSVs intranasally at a dose of 5 × 106 PFU. Mice from each group were euthanized at day 7 postinoculation. The right lung from each mouse was fixed in 4% (vol/vol) phosphate-buffered paraformaldehyde, embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin-eosin (HE). Detailed criteria for evaluation of lung histological change are described in Materials and Methods. Representative lung histology from each group is shown. DMEM control, grade 0 (no lesion); rVSV, grade 3 (severe); rVSV-E1833Q, grade 1 (mild); rVSV-G1670A, grade 2 (moderate); and rVSV-G4A, grade 3 (severe).
FIG 3.
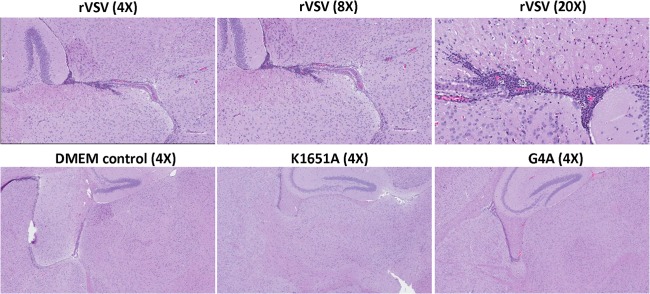
Histopathological changes in brain after VSV infection. Mice were euthanized at day 7 postinoculation. A horizontal brain section containing the olfactory bulb, forebrain, cerebrum, hippocampus, and cerebellum was fixed in 4% (vol/vol) phosphate-buffered paraformaldehyde, followed by HE straining. Detailed criteria for evaluation of brain histological change are described in Materials and Methods. DMEM control, grade 0 (no lesion); rVSV-K1651A, grade 1 (mild); rVSV-G4A, grade 1 (mild); and rVSV, grade 3 (severe).
Disparate levels of virus burden in the lungs and brains of mice inoculated with MTase-defective VSV.
Since the above-described analysis revealed disparate effects in brain and lung pathology following infection of mice with the MTase-defective viruses, we examined the amount of virus present in the lung and brains by plaque assay (Table 1). At 7 days postinoculation, 3 out of 4 mice in the rVSV group had virus in the brain (average titer of 2.6 × 105), whereas no infectious virus particles were detected in all other groups. At 14 days postinoculation, no virus was detected in any brain or lung tissue.
Since we were unable to detect any infectious virus particles at both days 7 and 14 postinoculation in MTase-defective VSV groups, we next tested for viral RNA by RT-PCR. Briefly, after extraction of total RNAs from equal amounts of lung and brain tissue, a 1,268-bp fragment spanning the VSV N gene was amplified by RT-PCR and subsequently analyzed by agarose gel electrophoresis. The presence of the viral N gene was detected in most lung samples from mice infected with MTase-defective virus at day 7 postinoculation; however, the amount of the RT-PCR product was lower than that obtained for the rVSV-infected mice (Fig. 4). Similar observations were obtained with brain samples. While 3 out of 4 brain samples from rVSV-infected mice at day 7 postinoculation were positive for viral RNA, no viral RNA was detected in most brain samples from mice infected by MTase-defective VSVs (Table 1; Fig. 4). At day 7, 2 out of 4 mice in the rVSV-G4A challenge group had detectable viral RNA in brain, whereas no viral RNA was detected in rVSV-G1670A- and rVSV-G1672A-infected animals (Table 1). By day 14, lung samples from the mice infected with MTase-defective VSV were negative for viral RNA, whereas those from the rVSV group remained positive. Typically, viral RNA levels in the brain were significantly less than those in lung tissues (Table 1; Fig. 4).
FIG 4.

Detection of viral RNA in lung and brain by RT-PCR. Equal amounts (0.5 g) of lung and brain tissues were homogenized in 0.5 ml of DMEM. Total RNA was extracted from each sample, followed by RT-PCR to amplify the VSV N gene. The amplified products were analyzed on a 1% agarose gel. M indicates DNA marker; lanes 1 to 4 indicate the numbers of mice.
One possibility underlying our inability to detect infectious virus in most lung and brain samples from mice infected with MTase-defective VSVs is that the virus had already been cleared by day 7 postinoculation. To determine whether VSV was present in the lung and brain tissues from infected mice at earlier time points, we inoculated mice with rVSV, rVSV-K1651A, rVSV-G1670A, and rVSV-G4A and monitored them as described above except that the euthanization time points were day 3 and day 5 postinoculation (Table 2). By day 3 postinoculation, 6.7 × 105, 1.1 × 103, 3.9 × 104, and 2.1 × 101 PFU/g of viruses were detected in lung samples from mice infected with rVSV, rVSV-K1651A, rVSV-G1670A, and rVSV-G4A, respectively. By day 5 postinoculation, virus titers were diminished to 1.2 ×103 and 6.3 ×101 PFU/g for rVSV and rVSV-G1670A groups, respectively, and below the limit of detection for rVSV-K1651A and rVSV-G4A groups. All brain samples from mice in the rVSV group also had high viral burdens at day 3 postinoculation, with an average titer of 8.0 × 105 PFU/g. In comparison, the brains of mice inoculated with rVSV-G1670A contained 1.5 × 104 PFU/g (P < 0.05). By day 5 postinoculation, the virus load in brain samples from the rVSV group was reduced to 1.1 ×104 PFU/g, whereas virus was completely cleared in the rVSV-G1670A group. No virus was detected in brain samples from mice infected by rVSV-K1651A and rVSV-G4A at day 3 or 5 postinoculation. These data demonstrated that MTase-defective VSVs created much lower viral burdens in both lungs and brains than rVSV. Specifically, viral titer in brain and lung was ranked as follows: rVSV > rVSV-G1670A > rVSV-K1651A > rVSV-G4A. These results illustrate a reduced ability of MTase-defective VSVs to replicate and spread in tissues of mice.
TABLE 2.
Viral titers in lungs and brain of mice at days 3 and 5 postinfection
| Groupa | No. of sick mice/total (score)b |
Viral titer (no. of positive mice) inc: |
||||
|---|---|---|---|---|---|---|
| Lung |
Brain |
|||||
| Day 3 | Day 5 | Day 3 | Day 5 | Day 3 | Day 5 | |
| DMEM | 0/8 (0) | 0/4 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| rVSV | 8/8 (3.0 A) | 4/4 (3.0 A) | 6.7 × 105 (4) | 1.2 × 103 (4) | 8.0 × 105 (4) | 1.1 × 104 (4) |
| rVSV-K1651A | 0/8 (0) | 0/4 (0) | 1.1 × 103 (3) | 0 (0) | 0 (0) | 0 (0) |
| rVSV-G1670A | 8/8 (1.0 C) | 2/4 (1.0 B) | 3.9 × 104 (4) | 6.3 × 101 (1) | 1.5 × 104 (4) | 0 (0) |
| rVSV-G4A | 8/8 (1.5 B) | 4/4 (1.25 B) | 2.1 × 101 (1) | 0 (0) | 0 (0) | 0 (0) |
Each group was infected with the indicated virus or administered DMEM as a control. Each group contained 8 mice.
The severity of VSV symptoms at days 3 and 5 was scored as grade 3 (severe), grade 2 (moderate), grade 1 (mild), or grade 0 (no symptoms). The average score for each group is shown. Values within a column followed by different letters are significantly different (P < 0.05).
Viral titer (PFU/g) was determined by plaque assay. The average titers from four mice are shown. A value of 0 indicates that the viral titer was undetectable by plaque assay.
rVSV-G4A induced body weight changes and clinical symptoms that were comparable in severity to those seen in rVSV-G1670A- and rVSV-G1672A-infected mice, despite the fact that no infectious rVSV-G4A virus was detected in lungs or brains at days 3 and 5 postinoculation (Table 2). Thus, we performed an additional animal experiment in which mice were euthanized at days 1 and 2 postinoculation. As shown in Table 3, all of the mice in the rVSV group had 105 to 106 PFU/g of virus in lungs and brain, whereas only 1 of 4 mice in the rVSV-G4A group had a low titer at days 1 and 2 postinoculation. The plaque size of rVSV-G4A isolated from lung or brain (designated rVSV-G4A-M) was indistinguishable from that of the input rVSV-G4A (Fig. 5A). To further characterize rVSV-G4A-M, virus-positive brain and lung homogenates were inoculated into BHK-21 cells. The rVSV-G4A-M was purified by sucrose ultracentrifugation as previously described (22, 25). To examine the methylation status of rVSV-G4A-M, in vitro transcription reactions were performed in the presence of 20 μM S-adenosyl-l-[3H]methionine (SAM) as previously described (22, 25), and the amount of [3H]SAM incorporated into the mRNA was measured by scintillation counting (Fig. 5B). The level of incorporation of [3H]SAM into RNA by rVSV-G4A-M was not significantly different from that by rVSV-G4A and the background control (rVSV + SAH) (P > 0.05), suggesting that mRNAs synthesized by rVSV-G4A-M and rVSV-G4A were completely defective in both G-N-7 and 2′-O methylation.
TABLE 3.
Viral titers in lungs and brain at days 1 and 2 postinfection
| Groupa | No. of sick mice/total (score)b |
Viral titer (no. of positive mice)c in: |
||||
|---|---|---|---|---|---|---|
| Lung |
Brain |
|||||
| Day 1 | Day 2 | Day 1 | Day 2 | Day 1 | Day 2 | |
| DMEM | 0/8 (0) | 0/4 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| rVSV | 8/8 (2.0 A) | 4/4 (3.0 A) | 1.2 × 106 (4) | 5.5 × 105 (4) | 9.6 × 105 (4) | 6.6 × 105 (4) |
| rVSV-G4A | 8/8 (0.5 B) | 4/4 (1.0 B) | 2.7 × 102 (1) | 5 (1) | 2.3 × 101 (1) | 0 (0) |
Each group was infected with the indicated virus or administered DMEM as a control. Each group contained 8 mice.
The severity of VSV symptoms at days 1 and 2 was scored as grade 3 (severe), grade 2 (moderate), grade 1 (mild), or grade 0 (no symptoms). The average score for each group is shown. Values within a column followed by different letters are significantly different (P < 0.05).
Viral titer (PFU/g) was determined by plaque assay. The average titers from four mice are shown. A value of 0 indicates that the viral titer was undetectable by plaque assay.
FIG 5.
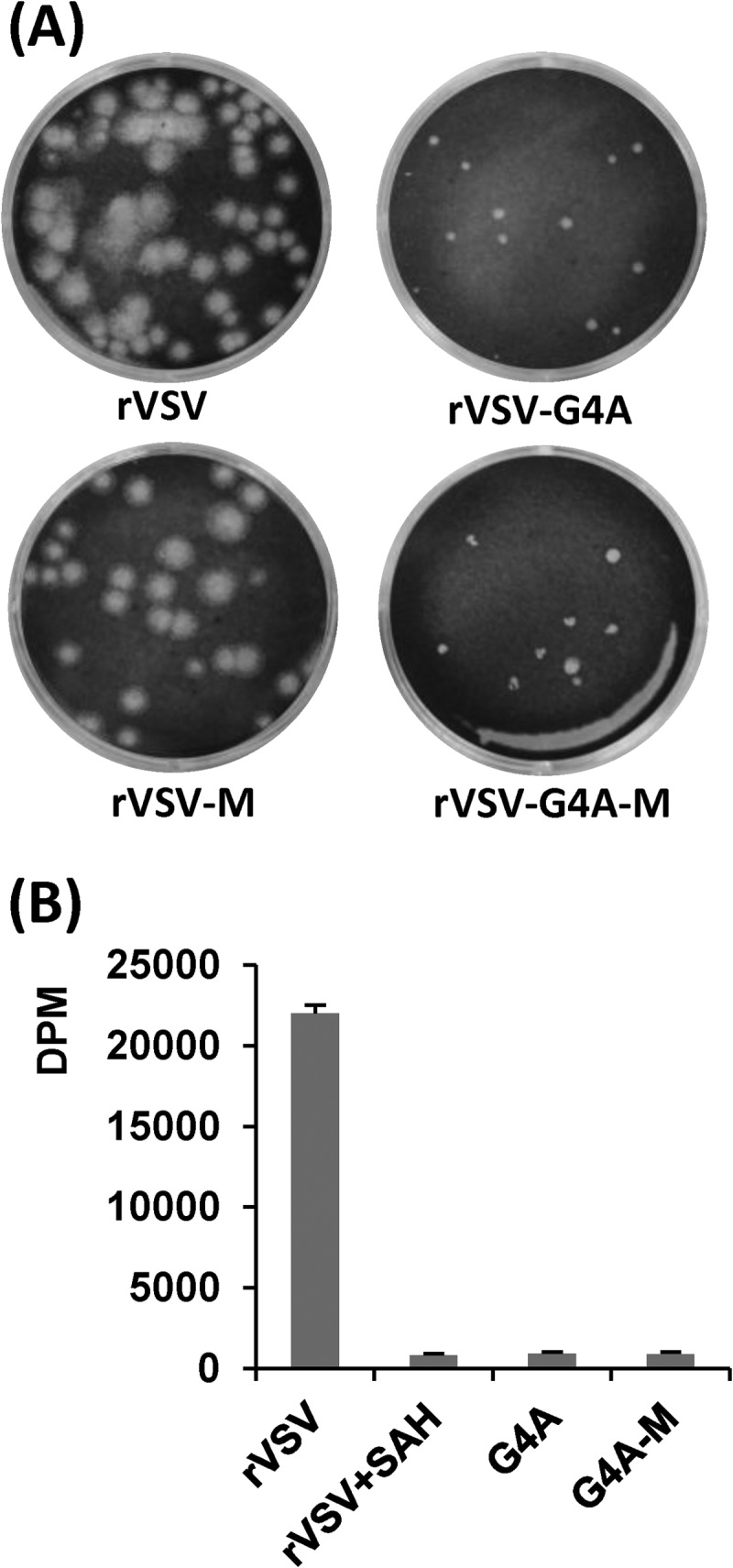
Phenotype of rVSV-G4A isolated from mice. (A) Plaque morphology of rVSV-G4A on Vero cells. rVSV-G4A-M, virus isolated from lungs of rVSV-G4A-infected mice; rVSV-M, virus isolated from lungs of rVSV-infected mice. Plaques of rVSV and rVSV-M were developed after 24 h; those of rVSV-G4A, and rVSV-G4A-M were developed after 96 h. (B) [3H]SAM incorporation monitored by scintillation counting. Viral RNA was synthesized in vitro by using 10 μg of purified virus. The amount of [3H]SAM incorporated into the RNA was determined by scintillation counting, and the number of dpm was normalized to the amount of RNA synthesized. Three independent experiments were used to generate the graph.
To confirm that MTase-defective VSVs retained the designed mutations after infection, we extracted total RNAs from the lung and brain tissues of each mouse, amplified them by RT-PCR, and sequenced the L gene. In case of rVSV-G4A-M, the entire genome was sequenced. The results showed that all MTase-defective VSVs recovered from infected mice contained the expected mutations in MTase region, indicating that these viral mutants are genetically stable in mice. No other changes were detected in any of the MTase-defective VSVs.
One possible reason for the low level of rVSV-G4A replication in vivo is that rVSV-G4A induces a strong type 1 interferon response. To address this possibility, we determined the levels of IFN-β mRNA in lungs and brains from mice infected with rVSV, rVSV-K1651A, rVSV-G4A, and rVSV-G1670A at day 3 postinfection. As shown in Fig. 6, there is no significant difference in IFN-β mRNA levels in lung samples among the groups (P > 0.05). However, IFN-β mRNA levels in brain samples of rVSV-G4A-infected mice were significantly lower than levels in the other challenge groups (P < 0.05). Therefore, it is unlikely that cytokine responses contribute to the low level of G4A replication in vivo.
FIG 6.
Quantification of IFN-β mRNA by real-time RT-PCR. Mice were infected with rVSV, rVSV-G4A, rVSV-K1651A, and rVSV-G1670A at dose of 5 × 106 PFU/mouse. At days 3 postinfection, lung and brain tissues were collected. Total RNA was extracted from lung or brain tissues and was treated with DNase I (Ambion). The IFN-β mRNA in each tissue was quantified by real-time RT-PCR. The average amounts of IFN-β mRNA from 5 mice for each group are shown.
Next, we determined whether defects in G4A replication are cell type specific. Briefly, Vero, BHK-21, and HEp-2 cells were infected with rVSV and rVSV-G4A, and virus titers in supernatants were determined by plaque assay. As shown in Fig. 7A, rVSV-G4A replicated much better in Vero and BHK-21 cells than in HEp-2 cells. The ratios of viral titer between rVSV and rVSV-G4A in BHK-21, Vero, and HEp-2 cells are 63:1, 126:1, and 3,162:1, respectively, suggesting that rVSV-G4A is a host range mutant.
FIG 7.

Replication and methylation of VSV in different cell lines. (A) Replication of rVSV-G4A is cell type dependent. Confluent BHK-21, Vero, or HEp-2 cells were infected with rVSV or G4A at an MOI of 0.1. After 24 h, supernatants were harvested, and virus titers were determined by plaque assay. (B) rVSV-G4A is defective in G-N-7 methylation in virus-infected cells. Confluent BHK-21, Vero cells, or HEp-2 in 35-mm dishes were infected by rVSV or G4A at an MOI of 0.1. After 24 h postinfection, poly(A)-containing viral mRNA was isolated. Equal amounts of viral RNA were incubated with 10 units of vaccinia virus G-N-7 MTase supplied with the m7G capping system (ScriptCap) in the presence of 15 μCi [3H]SAM (85 Ci/mmol; PerkinElmer). The G-N-7 methylation was measured by 3H incorporation using a 1414 series scintillation counter (PerkinElmer). (C) rVSV-G4A is defective in 2′-O methylation in virus-infected cells. Poly(A)-containing viral mRNA was incubated with 10 units of vaccinia virus G-N-7 MTase in the presence of 100 μM cold SAM. RNA was purified and incubated with vaccinia virus 2′-O MTase (VP39) in the presence of 15 μCi [3H]SAM. The 2′-O methylation was measured by 3H incorporation using a 1414 series scintillation counter.
We also determined whether G4A methylation defects are cell type specific. Briefly, Vero, BHK-21, and HEp-2 cells were infected with rVSV or rVSV-G4A, and viral mRNAs for each recombinant were harvested. Equal amounts of viral mRNA from each VSV mutant were trans-methylated by vaccinia virus mRNA cap G-N-7 MTase in the presence of [3H]SAM, and the extent of methylation was determined by scintillation counting. As shown in Fig. 7B, rVSV mRNAs isolated in Vero, BHK-21, and HEp-2 cells were not methylated by vaccinia virus G-N-7 MTase, which is consistent with the fact that rVSV produces fully methylated mRNA. In contrast, rVSV-G4A mRNAs were efficiently methylated by vaccinia virus G-N-7 MTase. There was no significant difference in the levels of G-N-7 methylation of viral mRNA isolated from Vero, BHK-21, and HEp-2 cells (P > 0.05). Similarly, mRNAs of rVSV-G4A but not rVSV were efficiently methylated by vaccinia virus 2′-O MTase, VP39 (Fig. 7C), suggesting that G4A was equally defective in 2′-O methylation in all three cell lines (P > 0.05). Together, these results suggest that rVSV-G4A produced unmethylated mRNA in each cell line tested and that differences in methylation do not account for the different levels of G4A replication.
The above results showed that MTase-defective VSVs can be classified into four pathotypes in mice. (i) MTase-defective VSVs carrying mutations in the MTase catalytic site, i.e., rVSV-K1651A, rVSV-D1762A, and rVSV-E1833Q, were highly attenuated in mice. These recombinant viruses did not cause any detectable weight loss or clinical signs or any pathological changes in lung and brain. In addition, no infectious virus particles were detected in lung and brain tissues after 3 days postinfection. (ii) MTase-defective VSVs carrying mutations in the SAM binding site, rVSV-G1670A and rVSV-G1672A, were moderately attenuated in mice. These two recombinant viruses caused only mild clinical signs and moderate histopathological changes in lungs, and viruses were detected at early time points (up to 5 days) after inoculation but were cleared quickly. (iii) rVSV-G4A retains low virulence, although no infectious virus was detected in lung and brain tissues except day 1 postinoculation. (iv) Recombinant G1674A was still pathogenic for mice but appeared to be less virulent than rVSV. This recombinant virus led to significant weight loss and moderate clinical signs and histopathological changes in lung and brain and replicated to high titers in lung and brain tissues.
LD50s of selected MTase-defective VSVs.
To further verify the differences in virulence between the MTase defective viruses, the LD50s for selected mutants (rVSV, rVSV-G1674A, rVSV-G4A, rVSV-G1670A, and rVSV-K1651A) were determined (Table 4). Briefly, mice were inoculated with four doses of each recombinant VSV (105, 106, 107, and 108 PFU per mouse, 5 mice per group). Under our experimental conditions, the LD50s for rVSV and rVSV-G1674A were 2.37 × 106 and 2.37 × 107, respectively. However, no mortality was observed in rVSV-G4A, rVSV-G1670A, and rVSV-K1651A groups at the dose of 108 PFU. Therefore, the LD50s for these mutants are above 108 PFU. However, it should be emphasized that mice inoculated with rVSV-G4A and rVSV-G1670A showed mild symptoms (such as ruffled fur) of VSV infection but recovered by day 14 postinfection. Mice inoculated with rVSV-K1651A did not show any symptoms at a dose of 108 PFU. Infection at higher doses was not feasible due to limitations in stock viral titers. These data further support the conclusion that rVSV-G1674A is virulent, rVSV-G4A and rVSV-G1670A retain low virulence, and rVSV-K1651A is highly attenuated.
TABLE 4.
LD50s of MTase-defective VSVs
| Group | LD50a | Phenotypeb |
|---|---|---|
| rVSV | 2.37 × 106 | Highly virulent |
| rVSV-G1674A | 2.37 × 107 | Virulent |
| rVSV-G4A | >108 | Attenuated, poorly virulent |
| rVSV-G1670A | >108 | Attenuated, poorly virulent |
| rVSV-K1651A | >108 | Highly attenuated, avirulent |
No mortality was observed in the rVSV-G4A, rVSV-G1670A, and rVSV-K1651A groups at the highest dose (108 PFU per mouse).
The phenotype is based on animal experiments 1 to 4, as described in the text.
Immunogenicity of MTase-defective VSVs in mice.
We next examined whether the limited replication of the MTase defective viruses in mice was sufficient to induce a protective immune response. To do this, 4 mice were immunized with 5 × 106 PFU of each MTase-defective VSV and challenged 4 weeks later with 5 × 106 PFU of rVSV. Protection was evaluated based on changes in the body weight, clinical symptoms, viral burdens in lungs and brains, and pathological changes in lungs and brains. Prior to challenge, mice vaccinated with rVSV-K1651A, rVSV-D1762A, and rVSV-E1833Q did not lose body weight or show clinical signs of infection (Fig. 8; Table 5). Mice vaccinated with rVSV-G1670A and rVSV-G1672A had only mild clinical signs (such as ruffled fur) at days 3 postvaccination but recovered at day 5. Mice vaccinated with these two mutant viruses lost 2 to 3 g of body weight at days 3 to 7 postvaccination, indicating that these two MTase-defective VSVs retained low virulence for mice (Fig. 8).
FIG 8.

Dynamics of mouse body weight change after primary immunization followed by virulent challenge. Four-week-old SPF female BALB/c mice were immunized with MTase-defective VSVs intranasally at a dose of 5 × 106 PFU per mouse. At week 4 postimmunization, mice were challenged with rVSV at a dose of 5 × 106 PFU per mouse. The body weight for each mouse was monitored every 3 days. The average body weight of four mice is shown. DMEMC, inoculated with DMEM and challenged with rVSV.
TABLE 5.
Immunogenicity of MTase-defective VSV in mice (primary immunization)
| Groupa | Log10 antibody GMT at week 3b | No. of sick mice | Histology score |
No. of mice with viral RNA in: |
Protection rated | ||
|---|---|---|---|---|---|---|---|
| Lungc | Brain | Lung | Brain | ||||
| DMEM | 0 | 0 | 0 | 0 | 0 | 0 | NA |
| DMEMC | 0 | 4 | 3.0 A | 3.0 | 4 | 4 | 0/4 |
| rVSV-K1651A | 5.31 | 0 | 1.0 B | 0 | 0 | 0 | 4/4 |
| rVSV-E1833Q | 5.01 | 0 | 1.0 B | 0 | 0 | 0 | 4/4 |
| rVSV-D1762A | 4.48 | 0 | 1.0 B | 0 | 0 | 1 | 4/4 |
| rVSV-G1670A | 5.04 | 0 | 1.0 B | 0 | 0 | 0 | 4/4 |
| rVSV-G1672A | 4.88 | 0 | 1.0 B | 0 | 0 | 0 | 4/4 |
Each group was infected with the indicated virus or administered DMEM as a control. Each group contained 4 mice. DMEMC, inoculated with DMEM and challenged with rVSV.
Antibody titer was determined by ELISA. The geometric mean titers (GMT) for four mice are shown.
Values followed by different letters are significantly different (P < 0.05).
Protection is based on the presence of VSV symptoms, mouse body weight change, and lung and brain histology after virulent challenge. There were 4 mice per group. NA, not applicable.
To evaluate whether MTase-defective VSVs can induce humoral immunity, serum samples were isolated from each mouse, serum IgG antibody response was determined by ELISA, and geometric mean titers (GMT) of VSV antibodies were calculated for each group of mice. Prior to inoculation, all mice were negative (GMT < 10) for VSV antibody (data not shown). Mice inoculated with MTase-defective VSVs triggered strong antibody responses (Fig. 9A; Table 5). At 1 week postvaccination, all mice inoculated with MTase-defective viruses had a high level of serum IgG, with an average GMT ranging from 1,023 to 25,704. The antibody level gradually increased during weeks 2 to 3 postinoculation. By 3 weeks postvaccination, the antibody GMT in mice infected with MTase-defective virus reached an average of 15,241 to 215,278. rVSV-G1670A triggered the highest antibody titer, whereas rVSV-D1762A induced the lowest antibody titer. There was no significant difference in the antibody titers among mice infected with rVSV-G1670A, rVSV-G1672A, rVSV-K1651A, and rVSV-E1833Q. Control mice inoculated with DMEM did not have any detectable anti-VSV antibody response during the experiment period.
FIG 9.

MTase-defective VSVs triggered high levels of antibody responses in mice. Blood samples were collected from each mouse weekly, and serum was isolated for antibody detection by ELISA. Antibody titers were calculated by the geometric mean titers (GMT). (A) Antibody titer after primary immunization. (B) Antibody titer after booster immunization.
Following challenge with virulent VSV, mice in the unimmunized group exhibited typical symptoms of infection, and their body weight dropped rapidly (Table 5). In contrast, mice immunized with MTase-defective VSVs did not exhibit any clinical sign or weight loss (Table 5; Fig. 8). After euthanization at day 7 postchallenge, lungs from the unimmunized but challenged group were dark red, whereas lungs from mice in MTase-defective VSVs-vaccinated groups were a normal pink color. Viral RNA load in lung and brain samples was determined by RT-PCR, which revealed a high level of viral RNA in the challenge control samples (data not shown). In contrast, levels of viral RNA were at the limit of detection in samples from mice immunized with MTase-defective viruses (Table 5). Histologic analyses showed that lung and brain tissues from mice vaccinated with MTase-defective VSVs had only mild histopathological changes following rVSV challenge, compared to moderate changes in unimmunized rVSV-challenged mice (Table 5). Taken together, these data demonstrated that a single-dose immunization with MTase-defective VSVs elicited a strong VSV-specific immunity and completely protected mice from virulent challenge.
We also evaluated the safety and immunogenicity following a booster immunization for each MTase-defective VSV. Briefly, 4-week-old mice were first immunized with 5 × 106 PFU of each MTase-defective VSV and then given a booster of the same MTase-defective VSV at the same dose at week 4 of primary immunization. After 3 weeks after booster immunization, mice were challenged with 5 × 106 PFU of rVSV. As shown in Fig. 10, booster immunization did not cause any weight loss or VSV infection symptoms, suggesting that these MTase-defective VSVs are safe for mice. In addition, the level of VSV-specific antibody further increased upon booster immunization (Fig. 9B). The antibody titer in the rVSV-K1651A group was significantly higher than those of other MTase-defective VSV groups (P < 0.05). After virulent challenge, mice in the challenge control group had severe weight loss and exhibited typical VSV symptoms (Fig. 10; Table 6). In contrast, none of mice in the vaccination groups had weight loss or any clinical signs of VSV infection, indicating that mice were protected from virulent challenge. Lastly, mice vaccinated with MTase-defective VSV did not have any gross pathological changes in lungs at day 7 postchallenge, whereas those from the challenge control group were dark red, indicative of inflammation. Moreover, neither infectious virus particles nor viral RNA was detected in lung and brain tissues from MTase-defective-VSV-immunized mice (Table 6). However, high viral RNA loads in lung and brain were detected in mice from the challenge control group. Furthermore, histological examination found that lung and brain tissues from the challenge control were moderately inflamed, whereas those from MTase-defective VSV-vaccinated groups only had mild inflammation (Table 6). Taken together, these data demonstrated that booster immunization of MTase-defective not only was safe for mice but also elicited strong VSV-specific immunity and completely protected mice from virulent challenge.
FIG 10.

Dynamics of mouse body weight change after booster immunization followed by virulent challenge. Four-week-old SPF female BALB/c mice were immunized with MTase-defective VSVs intranasally at a dose of 5 × 106 PFU per mouse. At week 2 after primary immunization, mice were boosted intranasally with 5 × 106 PFU of the corresponding virus. At week 3 after booster immunization, mice were challenged with rVSV at a dose of 5 × 106 PFU per mouse. The body weight for each mouse was monitored every 3 days. The average body weight of four mice is shown.
TABLE 6.
Immunogenicity of MTase-defective VSV in mice (booster immunization)
| Groupa | Log10 antibody GMT at week 5b | No. of sick mice | Histology score |
No. of mice with viral RNA in: |
Protection rated | ||
|---|---|---|---|---|---|---|---|
| Lung histologyc | Brain histology | Lung | Brain | ||||
| DMEM | 0 | 0/4 | 0 | 0 | 0 | 0 | NA |
| DMEMC | 0 | 4/4 | 3.0 A | 3.0 | 4 | 4 | 0/4 |
| rVSV-K1651A | 6.03 | 0/4 | 0.5 B | 0 | 0 | 0 | 4/4 |
| rVSV-E1833Q | 5.28 | 0/4 | 1.0 B | 0 | 0 | 0 | 4/4 |
| rVSV-D1762A | 5.35 | 0/4 | 1.0 B | 0 | 0 | 0 | 4/4 |
| rVSV-G1670A | 5.21 | 0/4 | 1.0 B | 0 | 0 | 0 | 4/4 |
| rVSV-G1672A | 5.43 | 0/4 | 1.0 B | 0 | 0 | 0 | 4/4 |
Each group was infected with the indicated virus or administered DMEM as a control. Each group contained 4 mice. DMEMC, inoculated with DMEM and challenged with rVSV.
Antibody titer was determined by ELISA. The geometric mean titers (GMT) for four mice are shown.
Values followed by different letters are significantly different (P < 0.05).
Protection is based on the presence of VSV symptoms, mouse body weight change, and lung and brain histology after virulent challenge. There were 4 mice per group. NA, not applicable.
DISCUSSION
In this study, we evaluated the impact of mRNA cap methylation in VSV pathogenesis in vivo. We found that the VSV mutants rVSV-K1651A, rVSV-D1762A, and rVSV-E1833Q, which have mutations in the MTase catalytic site and are defective in both G-N-7 and 2′-O methylation, were highly attenuated in mice. rVSV-G1670A and rVSV-G1672A, which have mutations in the SAM binding site and are defective in G-N-7 but not 2′-O methylation, retained low virulence in mice. In contrast, rVSV-G4A, a mutant producing unmethylated mRNA cap structures in vitro, induced weight loss and clinical signs that were indistinguishable from those observed in rVSV-G1670A and rVSV-G1672A infections despite the extremely low level of rVSV-G4A replication in vivo. Three major conclusions are apparent from our data: (i) inactivation of the predicted catalytic residues attenuates the virus to a greater extent in vivo than does inactivation of the predicted SAM binding site, (ii) the relationship between mRNA cap methylation and viral pathogenesis is not clear-cut in VSV infections, and (iii) some MTase-defective VSVs may serve as useful vaccine candidates.
Impact of mRNA cap methylation status on VSV pathogenesis.
VSV infects a wide range of wild and domestic animals, such as cattle, horses, deer, and pigs, and the infection is characterized by vesicular lesions in the mouth, tongue, lips, gums, teats, and feet. Although VSV does not cause vesicular diseases in mice, the mouse represents an excellent small animal model to understand pathogenesis because VSV causes systemic infection and fatal encephalitis (40–43). After intranasal inoculation, VSV infects olfactory neurons in the nasal mucosa and subsequently enters the central nervous system (CNS) through the olfactory nerves (44, 45). The virus is then disseminated to other areas in the brain through retrograde and possibly anterograde transneuronal transport, ultimately causing an acute brain infection. The mortality, pathology, and viral burden in lung and brain tissues are dependent on the age and species of the mouse (40–42, 46).
Based on the pathogenesis in mice, MTase-defective VSVs can be classified into three pathotypes: highly attenuated, poorly virulent, and virulent. With the exception of rVSV-G4A, the pathotype of MTase-defective VSVs in mice is essentially consistent with the phenotypes observed in cell cultures. The VSV mutants rVSV-K1651A, rVSV-D1762A, and rVSV-E1833Q, which carry point mutations in the MTase catalytic site and are defective in both G-N-7 and 2′-O methylation, were highly attenuated in mice. This is consistent with our previous finding that these recombinants were highly attenuated in cell culture (9). Specifically, these mutant viruses had decreased virus titers (2 to 3 logs) and reduced levels of protein and RNA synthesis (by 30 to 50%) compared to rVSV. In contrast, mutations in the SAM binding site impact viral pathogenesis differently. VSV mutants specifically defective in G-N-7 but not 2′-O MTase, including rVSV-G1670A and rVSV-G1672A, retained low virulence. These findings suggest that VSV mutants with a defect only in G-N-7 methylation are insufficiently attenuated. In cell culture, these two mutants led to decreases in virus titer of only 1 to 2 log units, while the levels of viral protein and RNA synthesis were comparable to those of rVSV (21). Recombinant West Nile virus (WNV-K61A and -K182A) defective in 2′-O but not the G-N-7 MTase activity is also attenuated in vitro as well as in vivo (47–49). In this case, inhibition of 2′-O methylation is sufficient to attenuate WNV. As predicted, rVSV-G1674A remained virulent for mice, although the virulence was reduced compared to that of rVSV. This mutant was fully methylated in cap structure only at high SAM concentrations (200 μM), whereas rVSV requires only 20 μM SAM for full methylation (21). It replicated as efficiently as rVSV and had no defect in RNA and protein synthesis in cell culture.
Most striking in our study was the phenotype associated with infections by rVSV-G4A, a virus which is completely defective in both G-N-7 and 2′-O methylation. rVSV-G4A induced body weight losses and clinical signs that were comparable to those associated with rVSV-G1670A and rVSV-G1672A infections. However, unlike rVSV-G1670A and rVSV-G1672A, rVSV-G4A was highly attenuated for replication and/or spread in vivo. Reduction in brain and lung viral titers was not attributed to enhanced production of antiviral cytokines, such as IFN-β (Fig. 6). The phenotype (small plaque and no cap methylation) of rVSV-G4A-M isolated from mice was identical to that of the rVSV-G4A original stock (Fig. 5). Also, no additional mutations were identified in rVSV-G4A-M. In fact, rVSV-G4A is the most attenuated mutant in cell culture among MTase-defective VSVs (21). We also showed that rVSV-G4A is a host range mutant which had much poorer replication in HEp-2 cells than in BHK-21 and Vero cells. rVSV-G4A produced unmethylated mRNA in all three cell lines, suggesting that the differences in replication among cell lines do not reflect a compensatory cellular methylase modifying the viral mRNA. Also, it is unlikely that cytokine responses contribute to the low level replication of rVSV-G4A in vivo (Fig. 6). The molecular mechanisms underlying the apparent discrepancy between replication in cell culture and in vivo pathogenesis remain to be elucidated. One feature of the gene expression strategy of NNS RNA viruses is that the polymerase complex controls two distinct RNA synthetic events: genomic RNA replication and mRNA transcription (19, 50, 51). Previously, we showed that regulation of those activities is perturbed for rVSV-G4A such that genomic replication is enhanced 4-fold (21). Perhaps, the defects in mRNA cap methylation and the alternation of the polymerase function collectively contribute to the unique phenotype of G4A.
MTase-defective viruses as potential live vaccine candidates.
In this study, we demonstrated that ablating viral mRNA cap methylation is a means of attenuating VSV, and by extrapolation perhaps other NNS RNA viruses, for development of live attenuated vaccines. Our results showed that MTase-defective VSVs were able to induce high levels of VSV-specific antibody in mice and provided full protection against a virulent challenge with the VSV Indiana serotype. rVSV-K1651A, rVSV-D1762A, and rVSV-E1833Q are attractive vaccine candidates, since they not only are highly attenuated but also retain high immunogenicity. Although rVSV-G1670A and rVSV-G1672A retained low virulence for mice, their pathogenicity was significantly reduced compared to that of rVSV. The safety of using these two viruses as live vaccine candidates remains to be further investigated.
This study extends the approaches that have been employed to attenuate VSV, including gene rearrangement, M gene mutation, G protein cytoplasmic tail truncations, G protein deletions, and combinations thereof (52–55). We now add to this list mutations in the MTase domain of L, which we show can result in attenuated and immunogenic viruses. Since our sequence data indicate that the viruses isolated from lung and brain tissues retained the original mutation(s), reversion may be less of a problem than anticipated. Moreover, by combining mutations in this region of L, it may be possible to diminish further the chance of selecting virulent revertants. We do not understand the basis for the virulence of rVSV-G4A, which lacks the ability to methylate mRNA in vitro. Consequently, we cannot yet conclude that elimination of the MTase activities of L is an effective vaccine strategy. Resolving why some MTase-defective VSVs are highly attenuated whereas others are not may facilitate a strategy to develop viral vectors for vaccines, oncolytic therapy, and gene delivery.
Our studies on MTase-defective VSVs also shed light on developing live vaccine candidates for other NNS RNA viruses, particularly paramyxoviruses. Live attenuated vaccines are the most promising vaccine candidates for paramyxoviruses such as hMPV, RSV, and PIV3 (54, 56), which account for the majority of respiratory diseases in infants, children, and the elderly. However, it has been technically challenging to isolate viruses with low virulence but high immunogenicity. Introducing mutations in the MTase may provide a novel approach to generate live attenuated viruses for these viruses. Recently, a panel of MTase-defective hMPVs which showed only 0.5 to 1 log reductions in growth relative to the parent virus was generated (Y. Zhang and J. Li, unpublished data). These recombinants were highly attenuated and immunogenic in cotton rats. Thus, it is feasible to utilize MTase-defective viruses as vaccine candidates for paramyxoviruses.
Recently, the concept of using MTase-defective viruses as live vaccine candidates has also been demonstrated in flaviviruses such as Japanese encephalitis virus (JEV) and dengue virus (36, 58). In both cases, 2′-O methylation-defective flaviviruses were shown to be promising vaccine candidates. For example, 2′-O methylation-defective JEV was attenuated in mice, elicited robust humoral and cellular immune responses, and was genetically stable in vivo (58). A single immunization induced full protection against lethal challenge with JEV strains in mice. Similarly, dengue virus mutants lacking 2′-O MTase activity are attenuated in mice and rhesus monkeys and elicit a strong adaptive immune response (36).
Biological role of 2′-O methylation: discrimination of self and nonself mRNA?
While it is firmly established that G-N-7 methylation is essential for mRNA stability as well as efficient translation, the role(s) of ribose 2′-O methylation has proved more elusive. Recent studies on West Nile virus (WNV) suggest that the 2′-O methylation of the 5′ cap of viral RNA functions to evade innate host antiviral responses through escape of the suppression of interferon-stimulated genes, which encode tetratricopeptide repeats (IFIT) (38). A WNV MTase mutant (E218A), which is defective in 2′-O MTase activity, was attenuated in wild-type C57BL/6 mice but remained pathogenic in mice that lack a type I interferon (IFN) signaling pathway. The vaccinia virus mutant J3-K175R and the mouse hepatitis virus mutant MHV-D130A, both of which lack 2′-O MTase activity, also exhibited enhanced sensitivities to the antiviral actions of IFN mediated by IFIT proteins (38). Similarly, 2′-O methylation of mouse and human coronavirus RNA seems to facilitate evasion from detection by the cytoplasmic RNA sensor Mda5 (57). Those studies suggest that 2′-O methylation of viral RNA provides a molecular signature for the discrimination of self and nonself mRNA. The finding here that VSV-G4A produces unmethylated mRNA yet remains as virulent as VSV-G1670A and VSV-G1672A underscores the idea that the recognition of non-2′-O methylated mRNA is only one aspect of the immune response to virus infection.
In summary, the present study highlights a major gap in our understanding of the impact of mRNA cap methylation in VSV pathogenesis. Our work suggests that abrogation of viral mRNA cap methylation can serve as a novel approach to attenuate VSV, and perhaps other NNS RNA viruses, for use as vaccines and viral vectors. It will be of interest to determine the role of mRNA cap methylation in innate immunity to NNS RNA viruses.
ACKNOWLEDGMENTS
This study was supported in part by grants from the NIH (AI090060) and the USDA Agriculture and Food Research Initiative (2010-65119-20602) and a pilot grant from the OSU Center for Clinical and Translational Science (CCTS) to J.L. and by a grant from NIH (AI059371) to S.P.J.W. Y.Z. is a fellow of the Center for RNA Biology at OSU.
We thank members of Jianrong Li's laboratory for critical review of the manuscript.
Footnotes
Published ahead of print 26 December 2013
REFERENCES
- 1.Sleat DE, Banerjee AK. 1993. Transcriptional activity and mutational analysis of recombinant vesicular stomatitis virus RNA polymerase. J. Virol. 67:1334–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Emerson SU, Yu YH. 1975. Both NS and L proteins are required for in vitro RNA synthesis by vesicular stomatitis virus. J. Virol. 15:1348–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Emerson SU, Wagner RR. 1973. L protein requirement for in vitro RNA synthesis by vesicular stomatitis virus. J. Virol. 12:1325–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ogino T, Yadav SP, Banerjee AK. 2010. Histidine-mediated RNA transfer to GDP for unique mRNA capping by vesicular stomatitis virus RNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 107:3463–3468. 10.1073/pnas.0913083107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ogino T, Banerjee AK. 2007. Unconventional mechanism of mRNA capping by the RNA-dependent RNA polymerase of vesicular stomatitis virus. Mol. Cell 25:85–97. 10.1016/j.molcel.2006.11.013 [DOI] [PubMed] [Google Scholar]
- 6.Li J, Rahmeh A, Morelli M, Whelan SPJ. 2008. A conserved motif in region V of the large polymerase proteins of nonsegmented negative-sense RNA viruses that is essential for mRNA capping. J. Virol. 82:775–784. 10.1128/JVI.02107-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Testa D, Banerjee AK. 1977. Two methyltransferase activities in the purified virions of vesicular stomatitis virus. J. Virol. 24:786–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rahmeh AA, Li J, Kranzusch PJ, Whelan SPJ. 2009. Ribose 2 ′-O methylation of the vesicular stomatitis virus mRNA cap precedes and facilitates subsequent guanine-N-7 methylation by the large polymerase protein. J. Virol. 83:11043–11050. 10.1128/JVI.01426-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li J, Fontaine-Rodriguez EC, Whelan SPJ. 2005. Amino acid residues within conserved domain VI of the vesicular stomatitis virus large polymerase protein essential for mRNA cap methyltransferase activity. J. Virol. 79:13373–13384. 10.1128/JVI.79.21.13373-13384.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hercyk N, Horikami SM, Moyer SA. 1988. The vesicular stomatitis virus L protein possesses the mRNA methyltransferase activities. Virology 163:222–225. 10.1016/0042-6822(88)90253-X [DOI] [PubMed] [Google Scholar]
- 11.Grdzelishvili VZ, Smallwood S, Tower D, Hall RL, Hunt DM, Moyer SA. 2005. A single amino acid change in the L-polymerase protein of vesicular stomatitis virus completely abolishes viral mRNA cap methylation. J. Virol. 79:7327–7337. 10.1128/JVI.79.12.7327-7337.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li J, Rahmeh A, Brusic V, Whelan SPJ. 2009. Opposing effects of inhibiting cap addition and cap methylation on polyadenylation during vesicular stomatitis virus mRNA synthesis. J. Virol. 83:1930–1940. 10.1128/JVI.02162-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hunt DM, Mehta R, Hutchinson KL. 1988. The L protein of vesicular stomatitis virus modulates the response of the polyadenylic acid polymerase to S-adenosylhomocysteine. J. Gen. Virol. 69:2555–2561. 10.1099/0022-1317-69-10-2555 [DOI] [PubMed] [Google Scholar]
- 14.Galloway SE, Wertz GW. 2008. S-adenosyl homocysteine-induced hyperpolyadenylation of vesicular stomatitis virus mRNA requires the methyltransferase activity of L protein. J. Virol. 82:12280–12290. 10.1128/JVI.01225-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Banerjee AK, Rhodes DP. 1973. In vitro synthesis of RNA that contains polyadenylate by virion associated RNA polymerase of vesicular stomatitis virus. Proc. Natl. Acad. Sci. U. S. A. 70:3566–3570. 10.1073/pnas.70.12.3566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poch O, Blumberg BM, Bougueleret L, Tordo N. 1990. Sequence comparison of five polymerases (L proteins) of unsegmented negative-strand RNA viruses: theoretical assignment of functional domains. J. Gen. Virol. 71:1153–1162. 10.1099/0022-1317-71-5-1153 [DOI] [PubMed] [Google Scholar]
- 17.Zhang XD, Wei YW, Ma YM, Hu SH, Li J. 2010. Identification of aromatic amino acid residues in conserved region VI of the large polymerase of vesicular stomatitis virus is essential for both guanine-N-7 and ribose 2 ′-O methyltransferases. Virology 408:241–252. 10.1016/j.virol.2010.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rahmeh AA, Schenk AD, Danek EI, Kranzusch PJ, Liang B, Walz T, Whelan SPJ. 2010. Molecular architecture of the vesicular stomatitis virus RNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 107:20075–20080. 10.1073/pnas.1013559107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whelan SPJ, Wertz GW. 2002. Transcription and replication initiate at separate sites on the vesicular stomatitis virus genome. Proc. Natl. Acad. Sci. U. S. A. 99:9178–9183. 10.1073/pnas.152155599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Whelan SP, Wertz GW. 1999. Regulation of RNA synthesis by the genomic termini of vesicular stomatitis virus: identification of distinct sequences essential for transcription but not replication. J. Virol. 73:297–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li J, Wang JT, Whelan SPJ. 2006. A unique strategy for mRNA cap methylation used by vesicular stomatitis virus. Proc. Natl. Acad. Sci. U. S. A. 103:8493–8498. 10.1073/pnas.0509821103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hagler J, Shuman S. 1992. A freeze-frame view of eukaryotic transcription during elongation and capping of nascent mRNA. Science 255:983–986. 10.1126/science.1546295 [DOI] [PubMed] [Google Scholar]
- 23.Wang JT, McElvain LE, Whelan SP. 2007. Vesicular stomatitis virus mRNA capping machinery requires specific cis-acting signals in the RNA. J. Virol. 81:11499–11506. 10.1128/JVI.01057-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stillman EA, Whitt MA. 1999. Transcript initiation and 5′-end modifications are separable events during vesicular stomatitis virus transcription. J. Virol. 73:7199–7209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stillman EA, Whitt MA. 1997. Mutational analyses of the intergenic dinucleotide and the transcriptional start sequence of vesicular stomatitis virus (VSV) define sequences required for efficient termination and initiation of VSV transcripts. J. Virol. 71:2127–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tekes G, Rahmeh AA, Whelan SP. 2011. A freeze frame view of vesicular stomatitis virus transcription defines a minimal length of RNA for 5′ processing. PLoS Pathog. 7:e1002073. 10.1371/journal.ppat.1002073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barr JN, Whelan SPJ, Wertz GW. 2002. Transcriptional control of the RNA-dependent RNA polymerase of vesicular stomatitis virus. BBA Gene Struct. Expr. 1577:337–353. 10.1016/S0167-4781(02)00462-1 [DOI] [PubMed] [Google Scholar]
- 28.Furuichi Y, Shatkin AJ. 2000. Viral and cellular mRNA capping: past and prospects. Adv. Virus Res. 55:135–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muthukrishnan S, Both GW, Furuichi Y, Shatkin AJ. 1975. 5′-terminal 7-methylguanosine in eukaryotic mRNA is required for translation. Nature 255:33–37. 10.1038/255033a0 [DOI] [PubMed] [Google Scholar]
- 30.Both GW, Banerjee AK, Shatkin AJ. 1975. Methylation-dependent translation of viral messenger RNAs in vitro. Proc. Natl. Acad. Sci. U. S. A. 72:1189–1193. 10.1073/pnas.72.3.1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mudd JA, Summers DF. 1970. Protein synthesis in vesicular stomatitis virus-infected HeLa cells. Virology 42:328–340. 10.1016/0042-6822(70)90277-1 [DOI] [PubMed] [Google Scholar]
- 32.Makarow M, Nevalainen LT, Kaariainen L. 1986. Expression of the RNA genome of an animal virus in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U. S. A. 83:8117–8121. 10.1073/pnas.83.21.8117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lodish HF, Porter M. 1980. Translational control of protein synthesis after infection by vesicular stomatitis virus. J. Virol. 36:719–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dratewka-Kos E, Kiss I, Lucas-Lenard J, Mehta HB, Woodley CL, Wahba AJ. 1984. Catalytic utilization of eIF-2 and mRNA binding proteins are limiting in lysates from vesicular stomatitis virus infected L cells. Biochemistry 23:6184–6190. 10.1021/bi00320a045 [DOI] [PubMed] [Google Scholar]
- 35.Connor JH, Lyles DS. 2002. Vesicular stomatitis virus infection alters the eIF4F translation initiation complex and causes dephosphorylation of the eIF4E binding protein 4E-BP1. J. Virol. 76:10177–10187. 10.1128/JVI.76.20.10177-10187.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zust R, Dong H, Li XF, Chang DC, Zhang B, Balakrishnan T, Toh YX, Jiang T, Li SH, Deng YQ, Ellis BR, Ellis EM, Poidinger M, Zolezzi F, Qin CF, Shi PY, Fink K. 2013. Rational design of a live attenuated dengue vaccine: 2′-O-methyltransferase mutants are highly attenuated and immunogenic in mice and macaques. PLoS Pathog. 9:e1003521. 10.1371/journal.ppat.1003521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Garcia-Sastre A. 2011. 2 methylate or not 2 methylate: viral evasion of the type I interferon response. Nat. Immunol. 12:114–115. 10.1038/ni0211-114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Daffis S, Szretter KJ, Schriewer J, Li JQ, Youn S, Errett J, Lin TY, Schneller S, Zust R, Dong HP, Thiel V, Sen GC, Fensterl V, Klimstra WB, Pierson TC, Buller RM, Gale M, Shi PY, Diamond MS. 2010. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature 468:452–456. 10.1038/nature09489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reed LJ, Muench H. 1938. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 27:493–497 [Google Scholar]
- 40.Sabin AB, Olitsky PK. 1938. Influence of host factors on neuroinvasiveness of vesicular stomatitis virus. III. Effect of age and pathway of infection on the character and localization of lesions in the central nervous system. J. Exp. Med. 67:201–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sabin AB, Olitsky PK. 1938. Influence of host factors on neuroinvasiveness of vesicular stomatitis virus. IV. Variations in neuroinvasiveness in different species. J. Exp. Med. 67:229–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sabin AB, Olitsky PK. 1937. Influence of host factors on neuroinvasiveness of vesicular stomatitis virus. I. Effect of age on the invasion of the brain by virus instilled in the nose. J. Exp. Med. 66:15–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Slavin HB, Hale HW, Berry GP. 1946. Passive protection of the central nervous system of mice against viruses that pursue the pathway of the olfactory nerves after intranasal instillation—vesicular stomatitis and St. Louis encephalitis. J. Immunol. 54:179–188 [PubMed] [Google Scholar]
- 44.Plakhov IV, Arlund EE, Aoki C, Reiss CS. 1995. The earliest events in vesicular stomatitis virus infection of the murine olfactory neuroepithelium and entry of the central nervous system. Virology 209:257–262. 10.1006/viro.1995.1252 [DOI] [PubMed] [Google Scholar]
- 45.Huneycutt BS, Plakhov IV, Shusterman Z, Bartido SM, Huang A, Reiss CS, Aoki C. 1994. Distribution of vesicular stomatitis virus proteins in the brains of BALB/c mice following intranasal inoculation: an immunohistochemical analysis. Brain Res. 635:81–95. 10.1016/0006-8993(94)91426-5 [DOI] [PubMed] [Google Scholar]
- 46.Cornish TE, Stallknecht DE, Brown CC, Seal BS, Howerth EW. 2001. Pathogenesis of experimental vesicular stomatitis virus (New Jersey serotype) infection in the deer mouse (Peromyscus maniculatus). Vet. Pathol. 38:396–406. 10.1354/vp.38-4-396 [DOI] [PubMed] [Google Scholar]
- 47.Zhou YS, Ray D, Zhao YW, Dong HP, Ren SP, Li Z, Guo Y, Bernard KA, Shi PY, Li HM. 2007. Structure and function of flavivirus NS5 methyltransferase. J. Virol. 81:3891–3903. 10.1128/JVI.02704-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ray D, Shah A, Tilgner M, Guo Y, Zhao YW, Dong HP, Deas TS, Zhou YS, Li HM, Shi PY. 2006. West Nile virus 5′-cap structure is formed by sequential guanine N-7 and ribose 2′-O methylations by nonstructural protein 5. J. Virol. 80:8362–8370. 10.1128/JVI.00814-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dong HP, Ren SP, Zhang B, Zhou YS, Puig-Basagoiti F, Li HM, Shi PY. 2008. West Nile virus methyltransferase catalyzes two methylations of the viral RNA cap through a substrate-repositioning mechanism. J. Virol. 82:4295–4307. 10.1128/JVI.02202-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lyles DS, Rupprecht CE. 2007. Rhabdoviridae, p 1363–1408 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed, vol 1 Lippincott Williams & Wilkins Co., Philadelphia, PA [Google Scholar]
- 51.Qanungo KR, Shaji D, Mathur M, Banerjee AK. 2004. Two RNA polymerase complexes from vesicular stomatitis virus-infected cells that carry out transcription and replication of genome RNA. Proc. Natl. Acad. Sci. U. S. A. 101:5952–5957. 10.1073/pnas.0401449101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Publicover J, Ramsburg E, Rose JK. 2004. Characterization of nonpathogenic, live, viral vaccine vectors inducing potent cellular immune responses. J. Virol. 78:9317–9324. 10.1128/JVI.78.17.9317-9324.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Flanagan EB, Schoeb TR, Wertz GW. 2003. Vesicular stomatitis viruses with rearranged genomes have altered invasiveness and neuropathogenesis in mice. J. Virol. 77:5740–5748. 10.1128/JVI.77.10.5740-5748.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bukreyev A, Skiadopoulos MH, Murphy BR, Collins PL. 2006. Nonsegmented negative-strand viruses as vaccine vectors. J. Virol. 80:10293–10306. 10.1128/JVI.00919-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ahmed M, McKenzie MO, Puckett S, Hojnacki M, Poliquin L, Lyles DS. 2003. Ability of the matrix protein of vesicular stomatitis virus to suppress beta interferon gene expression is genetically correlated with the inhibition of host RNA and protein synthesis. J. Virol. 77:4646–4657. 10.1128/JVI.77.8.4646-4657.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van Bleek GM, Osterhaus AD, de Swart RL. 2011. RSV 2010: recent advances in research on respiratory syncytial virus and other pneumoviruses. Vaccine 29:7285–7291. 10.1016/j.vaccine.2011.07.114 [DOI] [PubMed] [Google Scholar]
- 57.Zust R, Cervantes-Barragan L, Habjan M, Maier R, Neuman BW, Ziebuhr J, Szretter KJ, Baker SC, Barchet W, Diamond MS, Siddell SG, Ludewig B, Thiel V. 2011. Ribose 2 ′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol. 12:137–146. 10.1038/ni.1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li SH, Dong H, Li XF, Xie X, Zhao H, Deng YQ, Wang XY, Ye Q, Zhu SY, Wang HJ, Zhang B, Leng QB, Zuest R, Qin ED, Qin CF, Shi PY. 2013. Rational design of a flavivirus vaccine by abolishing viral RNA 2′-O methylation. J. Virol. 87:5812–5819. 10.1128/JVI.02806-12 [DOI] [PMC free article] [PubMed] [Google Scholar]