

Abstract
All currently identified primary receptors of adeno-associated virus (AAV) are glycans. Depending on the AAV serotype, these carbohydrates range from heparan sulfate proteoglycans (HSPG), through glycans with terminal α2-3 or α2-6 sialic acids, to terminal galactose moieties. Receptor identification has largely relied on binding to natural compounds, defined glycan-presenting cell lines, or enzyme-mediated glycan modifications. Here, we describe a comparative binding analysis of highly purified, fluorescent-dye-labeled AAV vectors of various serotypes on arrays displaying over 600 different glycans and on a specialized array with natural and synthetic heparins. Few glycans bind AAV specifically in a serotype-dependent manner. Differential glycan binding was detected for the described sialic acid-binding AAV serotypes 1, 6, 5, and 4. The natural heparin binding serotypes AAV2, -3, -6, and -13 displayed differential binding to selected synthetic heparins. AAV7, -8, -rh.10, and -12 did not bind to any of the glycans present on the arrays. For discrimination of AAV serotypes 1 to 6 and 13, minimal binding moieties are identified. This is the first study to differentiate the natural mixed heparin binding AAV serotypes 2, 3, 6, and 13 by differential binding to specific synthetic heparins. Also, sialic acid binding AAVs display differential glycan binding specificities. The findings are relevant for further dissection of AAV host cell interaction. Moreover, the definition of single AAV-discriminating glycan binders opens the possibility for glycan microarray-based discrimination of AAV serotypes in gene therapy.
INTRODUCTION
Adeno-associated viruses (AAVs) represent a family of helper-dependent parvoviruses composed of single-stranded DNA genomes packaged into icosahedral capsids. AAV capsids directly interact with specific host cell receptors. Various AAV serotypes of human and primate origin (AAV1 to AAV13) that differ in the structures of their capsids and display variable cell or tissue tropism have been defined (1). AAV-derived vectors are increasingly used in gene therapy. The differential tropism of various AAV serotypes is ideal for directing the vector to a certain cell type or tissue for gene therapy.
For many AAV serotypes, it has been shown that binding to cell surface glycans is required for infection (2–7). Cell surface glycans are commonly attached to proteins (glycoproteins and proteoglycans) or lipids (sphingolipids) of the cell membrane. These carbohydrates are structurally the most complex building blocks of life. The diversity of mammalian glycan biopolymers is achieved by alternate sequences of 10 different building blocks of monosaccharides, variable glycosidic linkages, and branching and modifications of saccharides. In addition, attachment to different proteins is achieved via asparagine (N-linked glycans) or serine or threonine (O-linked glycans) residues or via lipids (8). Virtually all membrane proteins are glycosylated, but their glycosylation patterns differ in different tissues (9). The biological roles of glycans are diverse and not yet fully understood. They have been associated with a variety of cellular processes, including protein folding (10) and signal transduction (11). In addition, surface-exposed glycans are recognized by various virus families that exploit them as host cell receptors (12).
The first identified receptor for AAV was heparin sulfate proteoglycan (HSPG), described as a primary receptor for prototype AAV2. Competition experiments with soluble heparin and pretreatment of cells with heparinase helped to identify its cell receptor (2). HSPG and the closely related heparin represent mixtures of naturally occurring, polydisperse linear polysaccharides that are composed of alternate units of glucosamine (GlcN) and uronic acid, either a glucuronic acid (GlcA) or an iduronic acid (IdoA). The alternate units are joined by 1 to 4 glycosidic linkages and can be variably modified by sulfates or acetyl groups. Heparin displays a higher degree of sulfation than HSPGs. Heparins are stored in vesicles of mast cells and are released upon stimulation. In contrast, HSPGs are ubiquitously found on cell surfaces, covalently linked to proteoglycan core proteins (13).
HSPG was subsequently confirmed as the primary receptor for AAV3 and AAV13 (3, 7). Furthermore, AAV6 was shown to bind to heparin in vitro. However, in contrast to the results with AAV2, AAV3, or AAV13, AAV6 infection in vivo was resistant to inhibition by soluble HSPG (14). Other AAV serotypes are heparin insensitive (15). Instead, cell pretreatment with neuraminidases revealed that sialic acids were required for infection with AAV serotype 1, 4, 5, or 6 (4, 5, 16). Sialic acids represent N- or O-substituted derivatives of neuraminic acid, a 9-carbon monosaccharide. The most common sialic acid, N-acetyl-neuraminic acid (Neu5Ac), is typically found at terminating branches of N-glycans, O-glycans, or glycosphingolipids (17). AAV4 was reported to require O-linked α2-3 sialic acids for infection, whereas AAV1, AAV5, and AAV6 bound to N-linked α2-3 or α2-6 sialic acids (4, 5, 18).
In the present study, we set out to directly compare the glycan binding specificities of AAV serotypes 1 to 8, 10, 12, and 13 using highly purified fluorescently labeled AAV vectors. Their binding profiles on arrays of synthetic glycans and heparins identified AAV serotype-specific capsid-carbohydrate interactions, allowing AAV capsid differentiation by glycan/heparin binding patterns.
MATERIALS AND METHODS
Cloning and mutagenesis.
For the introduction of mutations into the capsid genes of AAV2 and AAV3, the respective cap genes were subcloned from pDG or pDP3rs (19) into pBluescript II SK(+). The capsid gene of AAV13 was mutated directly in the pAAV-13-cap plasmid. Using the QuikChange site-directed mutagenesis protocol, the double-exchange mutation R585A/R588A was introduced into the cap gene of AAV2, the point mutation R594A into the cap gene of AAV3, and the point mutation K528E into the cap gene of AAV13. The DNA sequence-verified cap genes were cloned back into pDG or pDP3rs, respectively.
Cell culture.
HEK 293- or HeLa-derived C12 cells were cultivated as adherent monolayers at 37°C and 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) (Gibco) supplemented with 2.5 g/liter glucose, 100 μg/ml streptomycin, 100 U/ml penicillin (PAA), and 10% (vol/vol) fetal calf serum (FCS) (Gibco). Sf9 cell lines for recombinant AAV (rAAV) production expressing Rep and Cap of various serotypes were described previously (20). Sf9 cells were maintained in suspension culture under constant agitation with serum-free Spodopan medium (Pan-Biotech) supplemented with 200 μg/ml streptomycin, 200 U/ml penicillin, and 250 ng/ml amphotericin B (Invitrogen) at 27°C.
rAAV production in 293 cells.
HEK 293 cells were seeded at 25 to 33% confluence. The cells were transfected 24 h later with plasmids for AAV rep, cap, and adenovirus type 5 (Ad5) helper genes and an additional plasmid for the rAAV cassette expressing green fluorescent protein (GFP) under the control of the cytomegalovirus (CMV) promoter (pTR-UF5) using the calcium phosphate cotransfection method as described previously (21). rAAV1, rAAV2, and rAAV3 subtype vectors and mutants derived from them were produced by the two-plasmid transfection method (19) using pDP1, pDG, pDG-R585A-R588A, pDP3, or pDP3-R594A as a helper plasmid. Generation of rAAV13 or the mutant derived from it required triple-plasmid transfections of pTR-UF5, pDGdeltaVP, and pAAV13-cap or pAAV13-cap-K528E, respectively. For the production of rAAV12 vectors, transfection of four plasmids, pTR-UF5, pAAV12-Rep, pAAV12-Cap, and pHelper, was required (15). After 12 h, the culture medium was replaced by medium with 2% FCS. Cells and medium were harvested 72 h after transfection and lysed by three freeze-thaw cycles. The crude lysates were treated with 250 U benzonase (Merck) per ml of lysate at 37°C for 1 h to degrade input and unpackaged AAV DNA before centrifugation at 8,000 × g for 30 min to pellet cell debris.
rAAV production in Sf9 cells.
Sf9 cell-derived AAV rep-cap-expressing cell lines (20, 22) were continuously held in suspension culture within the logarithmic growth phase prior to infection with the recombinant baculovirus Bac-rAAV-GFP (multiplicity of infection [MOI] = 5). The infected cells were incubated for 72 h at 27°C under constant agitation. The cells were pelleted at 2,000 × g for 5 min. The cell pellets were resuspended in lysis buffer containing 10 mM Tris-HCl (pH 8.5), 150 mM NaCl, 1 mM MgCl2, and 1% (vol/vol) Triton X-100. Crude lysates from 1-liter cell suspensions were treated with 3,000 U of benzonase (Merck) at 37°C for 1 h to degrade unpackaged AAV DNA and centrifuged at 8,000 × g for 30 min.
rAAV purification.
rAAV vectors were further purified from the benzonase-treated, cleared freeze-thaw supernatants by one-step AVB Sepharose affinity chromatography using 1-ml prepacked HiTrap columns on an ÄKTA purifier (GE Healthcare) as follows. Freeze-thaw supernatants were diluted 1:1 in 1× phosphate-buffered saline (PBS) supplemented with 1 mM MgCl2 and 2.5 mM KCl (PBS-MK) prior to loading on the column. The loading rate of the sample was set to 0.5 ml/min. Washing of the column was performed with 20 ml of 1× PBS-MK at a flow rate of 1 ml/min. AAV vectors were eluted with 0.1 M sodium acetate, 0.5 M NaCl, pH 2.5, at a flow rate of 1 ml per min and neutralized immediately with 1/10 volume of 1 M Tris-HCl, pH 10. The peak fractions of purified rAAV preparations were dialyzed against 1× PBS-MK using Slide-A-Lyzer dialysis cassettes (molecular weight cutoff [MWCO], 10,000; Thermo Scientific).
Quantification of rAAV vector preparations.
Highly purified rAAV vector preparations or rAAV-containing freeze-thaw supernatants were digested with proteinase K (Roth, Germany) to release the vector genomes from the capsids. Aliquots of the vector preparation were incubated in buffer containing 25 mM Tris-HCl (pH 8.5), 10 mM EDTA (pH 8.0), 1% (wt/vol) N-lauroyl sarcosinate, 40 μg proteinase K, and 1 μg pBluescript carrier plasmid for 2 h at 56°C. DNA was purified by extraction with phenol-chloroform-isoamyl alcohol (25:24:1) and precipitated with ethanol. The DNAs were analyzed by quantitative Light-Cycler PCR with a Fast Start DNA Master SYBR green kit (Roche) with dilutions depending on the subtype. Primers specific for the bovine growth hormone-derived poly(A) site of the vector backbone were used (forward primer, 5′-CTAGAGCTCGCTGATCAGCC-3′, and reverse primer, 5′-TGTCTTCCCAATCCTCCCCC-3′).
Fluorescence labeling of rAAV vectors.
Highly purified rAAV vectors with a titer of at least 1 × 1013 genomic particles per ml were fluorescently labeled using the DyLight488 Labeling Kit (Pierce) following the manufacturer's protocol. Labeled rAAV vector preparations were dialyzed twice against 1× PBS-MK using Slide-A-Lyzer dialysis cassettes (MWCO, 10,000; Thermo Scientific) to remove unbound dye from the vector preparation. To verify a successful labeling reaction, 10-μl aliquots of the vector preparations were lysed in 2× SDS protein sample buffer for 5 min at 95°C and analyzed on SDS-polyacrylamide gels. The DyLight-labeled AAV capsid bands could be visualized under UV light (data not shown).
Glycan array screening.
Fluorophore-labeled rAAV vectors were sent to the Consortium for Functional Glycomics (CFG) for glycan binding analysis on microarrays. The procedure is described in detail on the CFG webpage (http://www.functionalglycomics.org). In brief, up to 611 (version 5.0) or 610 (version 5.1) different glycan structures are printed on microscope glass slides, each available in replicates of six. PBS-MK was used as a binding and wash buffer during this assay. Dried slides are analyzed at the fluorophore-emitting wavelength in a PerkinElmer ScanArray scanner. The spots on the slide are aligned with the help of a grid and biotin control spots using Imagene software. Once aligned, the amount of fluorophore binding to each spot is quantified. The data sets were analyzed by averaging the six replicates after elimination of the two spots with the highest and lowest intensity, respectively. Full data sets are available at the website of the CFG (http://www.functionalglycomics.org/), and the addresses of individual AAV screens are provided in Table 1.
TABLE 1.
Web addresses of full data sets of primary AAV screens in the CFG database
Glycan array binding of fluorophore-labeled AAV serotypes 1 to 12 was detected directly. Unlabeled AAV2* was detected by MAb A20.
Heparin array screening.
Fluorophore-labeled rAAV vectors were analyzed for binding to chemically defined heparins on a glycan array. N-Hydroxysuccinimide (NHS)-activated CodeLink slides (SurModics, Inc., Eden Prairie, MN, USA) containing synthetic heparan sulfate (HS)/heparin oligosaccharides and 5-kDa natural heparin were prepared as described previously (23, 24). Briefly, heparin oligosaccharides bearing an aminopentyl linker at the reducing end were procured through a modular chemical synthesis approach. The oligosaccharides were immobilized on NHS-activated slides via their terminal amine groups using a piezoelectric spotting device (S3; Scienion, Berlin, Germany). Heparin oligosaccharides were spotted at concentrations of 1, 0.25, 0.063, and 0.016 mM dissolved in 50 mM phosphate buffer, pH 8.5 (10 spots for each concentration). The microarray slides were incubated in a humid chamber for 24 h to complete the reaction; quenched with 50 mM aminoethanol solution, pH 9, for 1 h at 50°C; washed three times with deionized water; and stored desiccated until use. For binding analyses, the slides were blocked at room temperature for 1 h with 1% (wt/vol) bovine serum albumin (BSA) in PBS-MK, washed three times with PBS-MK, and dried by centrifugation (5 min; 300 × g). The slides were incubated at 4°C overnight with purified and fluorescence-labeled rAAV vectors at a concentration of 100 μg/ml in PBS-MK with 0.01% (vol/vol) Tween 20 in a humid chamber. Then, the slides were washed three times with PBS-MK containing 0.1% (vol/vol) Tween 20, rinsed once with water, and dried by centrifugation. The slides were scanned with a GenePix 4300A microarray scanner (Molecular Devices). Fluorescence was excited at 488 nm, and fluorescence intensities were determined with GenePix Pro 7 software (Molecular Devices). The photomultiplier tube (PMT) voltage was adjusted so that scans were free of saturation signals. The mean fluorescence intensity (MFI) values were exported to Microsoft Excel for further analysis.
Heparin competition assay.
HeLa C12 cells were seeded in 24-well plates at 50% confluence and infected 24 h later with adenovirus type 2 for 1 h (MOI = 10). AAV-GFP vectors were incubated with serial dilutions of heparins in a total volume of 50 μl of DMEM at 37°C for 1 h. After aspiration of the adenovirus-containing cell medium, the rAAV vector-heparin mixture was added to the cells and incubated for 1 hour with occasional tilting. To remove nontransduced AAV vectors, the cells were washed twice with PBS. The cells were cultivated for 40 h in medium with 2% FCS, and the proportion of GFP-positive cells was determined by fluorescence-activated cell sorter (FACS) analysis (FACSCalibur; Becton Dickinson) according to the manufacturer's protocol. For each sample, 100,000 cells were counted. Cells that exceeded the cutoff of 40 in the FL1-H channel were regarded as GFP positive.
RESULTS
Generation of fluorescently labeled vectors of various AAV serotypes.
For the analysis of AAV capsid-glycan interactions, AAV vectors of serotypes 1 to 8, rh.10, 12, and 13 were produced either in Sf9 cells or in 293 cells. Virus binding to glycan microarrays is typically detected by specific antibodies visualized by fluorophore-labeled secondary antibodies. Direct fluorophore labeling of AAV capsids was expected to yield less nonspecific background signals. The required high AAV titers and extensive virus purification were achieved by AVB-Sepharose affinity chromatography, as described in Materials and Methods. Concentrated AAV preparations exhibiting 1 × 1013 genomic particles (gp) per ml were analyzed by electron microscopy (EM) analysis and on silver gels to confirm AAV purity (data not shown). Unfortunately, AAV9 and AAV11 were refractory to sufficient purification. Therefore, these serotypes were omitted from the analysis. The AAVs were labeled with the fluorescent dye DyLight488, which is conjugated to surface-exposed lysines on the capsids. Their numbers vary among different serotypes. AAV2 capsids expose significantly fewer lysines than capsids from other AAV subtypes. This led to variable labeling efficiencies of individual AAV serotypes. The number of dye molecules per capsid was calculated to range from approximately 10 in the case of AAV2 to up to 30 for other AAV serotypes. Only a small fraction of surface-exposed lysines are tagged; many more unmodified lysines are available for receptor attachment. Side-by-side transduction analysis of labeled and unlabeled AAV vectors showed comparable transduction efficiencies (data not shown).
CFG glycan array screening of various AAV serotypes.
The panel of fluorescently labeled AAV vectors was analyzed for binding to the glycan array offered by the CFG. The array is composed of 610 (v5.1) or 611 (v5.0) different natural or synthetic glycan structures, each in replicates of six. The original data sets, including full statistical analysis of all glycan screens, are available online (Table 1) (www.functionalglycomics.org/). Conclusive data for the interaction of AAV capsids with specific glycans on the array were observed for AAV1, AAV4, AAV5, and AAV6 (Fig. 1A).
FIG 1.

CFG glycan array screening of AAV serotypes 1 to 6. (A) Binding efficiencies of fluorescently labeled AAV1 to -6 vectors on CFG glycan arrays displaying 611 different glycan structures, each in replicates of six. Glycans with significantly higher binding efficiencies are highlighted with the corresponding CFG number. Due to different versions of the CFG glycan screen, the glycan numbers of the AAV6 screen glycan were adapted to the numbering of the newer version (5.1) to facilitate direct comparison to the AAV1 binding profile. Note that the binding signals for glycans 81, 265, and 518 were removed from the AAV2 and -3 screens based on a statement of the CFG that these glycans repeatedly produced nonspecific signals in unrelated glycan arrays performed with the same array batch. (B to D) Glycans that were identified in panel A to interact with the capsids of a particular serotype (42). Shown is a comparison of AAV serotype-dependent binding to families of closely related glycan structures present on the CFG array. Relative binding efficiencies are represented as percentages of that of the most efficiently binding glycan for the depicted AAV serotype. Error bars represent standard deviations.
The capsids of the two closely related AAV serotypes 1 and 6 exhibited binding to the same predominant glycan (number 237) shown in Fig. 1A and B. This glycan displays a terminal α2-3-linked sialic acid (Neu5Ac), which has been described as a receptor for either serotype (5). Our data show that the α2-3-linked sialic acid is not sufficient for AAV1 or -6 binding (Fig. 1B). Of over 70 alternative glycans with terminal α2-3 sialic acids present on the array, only glycan 237 showed significant AAV1/6 binding. The combination of a linked N-acetylgalactosamine and an additional β1-4-linked N-acetylglucosamine appears to be required. Minor changes of the glycan structure, as displayed in Fig. 1B, led to a reduction in binding efficiency in the range of 1 to 8% of that observed for glycan 237. On the AAV6 screen, glycan 237 was the only glycan binding reproducibly (1,153 ± 40 relative fluorescence units [RFU]). AAV6 showed multiple additional binding signals in the range of 40% of that of glycan 237 (Fig. 1A). They were disregarded, either because of large standard deviations or because of binding to a single monosaccharide (mannose). The relevance of mono- or disaccharide binding is unclear, since CFG binding data have previously shown nonreproducible results with alternative AAV serotypes.
AAV5 has been reported to require either terminal α2-3 or α2-6 sialic acids (4). Here, we show specific binding to glycan 46 and 230, both of which display terminal α2-3 sialic acids (Fig. 1A). AAV5 did not bind to any of over 70 alternative glycans with terminal α2-3 sialic acids or to any of 52 alternative glycans with terminal α2-6 sialic acids. In addition, a terminal α2-3 sialic acid was obviously not sufficient for binding. Linkage to a sulfated galactose in the second position was required, and the galactose moiety could not be replaced by acetylglucosamine (Fig. 1C). Fucosylation of the N-acetylglucosamine in the third position was not required but moderately improved AAV5 binding. Furthermore, certain biantennary glycans, with 483 as a prototype, showed interaction with AAV5, though with reduced binding efficiencies (Fig. 2B). Although not sulfated at the galactose in the second position, these glycans confirmed the requirement for terminal α2-3 sialic acids. It appears that glycans with terminal α2-3 sialic acids are bound, but only when attached to the mannose α1-3 arm of the biantennary glycan (Fig. 2B). However, an even more complex glycan structure (e.g., glycan 462 of the CFG array) (Fig. 2A) possessed an identical arm with the terminal α2-3 sialic acid but was not bound by AAV5. In summary, the minimal glycan binding structure for AAV5 appears to be composed of a terminal α2-3 sialic acid linked to sulfated galactose, followed by a β1-4-linked N-acetylglucosamine.
FIG 2.

Differential binding of branched glycans with the capsids of AAV4 or AAV5. (A) Branched glycan 463 binding to AAV4 as shown in Fig. 1A compared to closely related glycan structures with reduced binding efficiencies, shown as percentages of that of glycan 348. (B) Branched glycan 483 binding to AAV5 as shown in Fig. 1A compared to closely related glycan structures with reduced binding efficiencies, shown as percentages of that of glycan 230.
The glycan array for AAV4 displays the highest and most specific glycan binding pattern of all AAVs analyzed. The predominant binders, glycans 348 and 351, share a backbone of six sugar molecules extended by a terminal α2-6 sialic acid in the case of glycan 348 (Fig. 1A and D). Since the AAV4 binding efficiencies differ only marginally, the presence of the terminal sialic acid appears not to be required. On the other hand, the α1-6 linkage between two internal mannose moieties appears to be essential. Identical glycans with an α1-3 linkage between the two mannose moieties lose AAV4 binding. Two α1-6-linked mannoses alone, however, were not sufficient to bind AAV4. Branched glycan 463 displaying a binding efficiency about one-third that of glycan 348 carries terminal α2-3 sialic acids. Their replacement by α2-6 sialic acids leads to a dramatic drop in AAV4 binding, even below background levels (Fig. 2A). In view of the very clear AAV4 binding profile, it is not obvious how the findings can be reconciled with O-linked terminal α2-3 sialic acid being described as a primary receptor for AAV4 (4).
Screening the CFG glycan array using fluorescently labeled AAV vectors of serotypes 2 and 3 (Fig. 1A), 7, 8, rh.10, or 12 (Fig. 3) did not lead to any significant interaction with a specific glycan. In the case of AAV2, a second screen was performed with unlabeled virus, which was detected by AAV2 capsid-specific monoclonal antibody (MAb) A20 (25). As mentioned above, this procedure markedly increased the background. Nevertheless, the negative results with the directly labeled viral particles were confirmed (Table 1). The lack of specific glycan interaction of the heparin binders AAV2 and AAV3 was expected, since heparins were not present on the CFG array.
FIG 3.
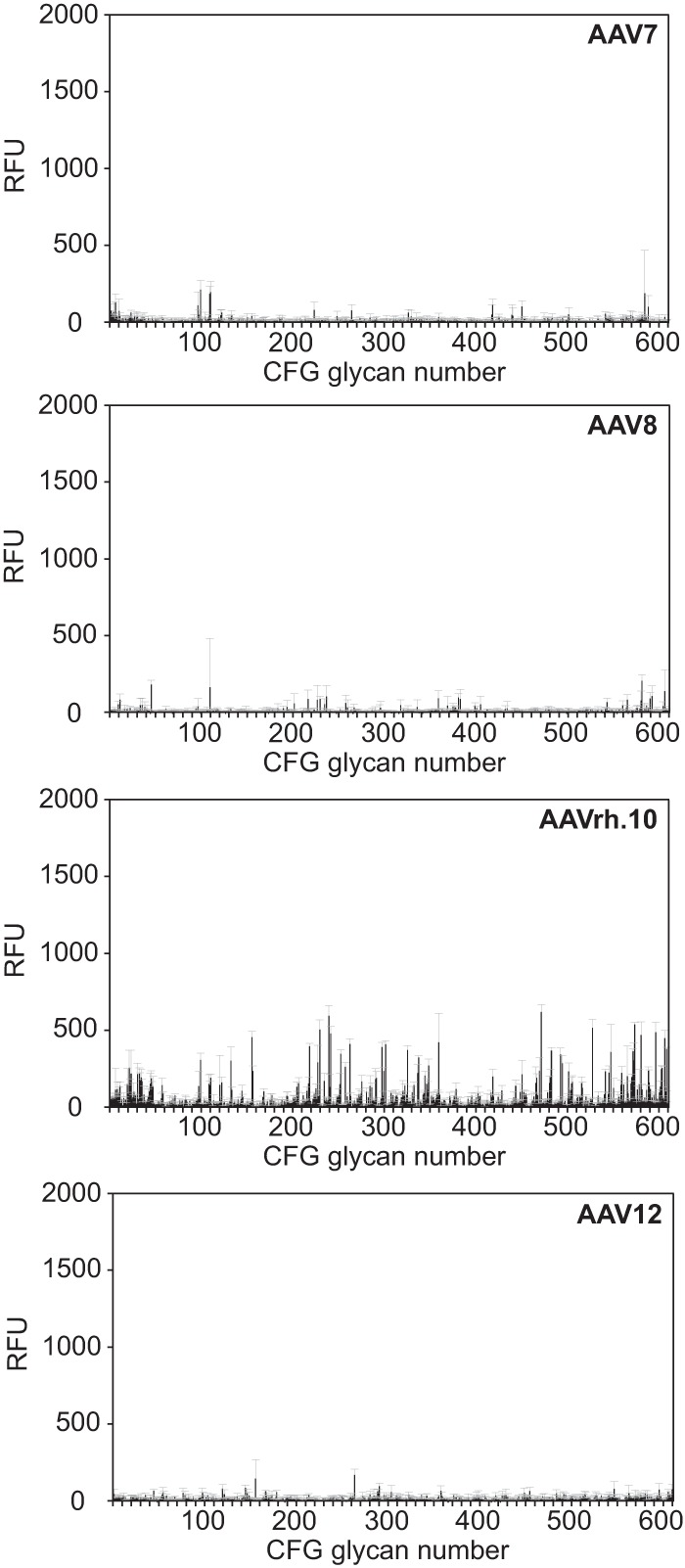
CFG glycan array screens of AAV7, -8, -rh.10, and -12. Shown are binding efficiencies of fluorescently labeled AAV7, -8, -rh.10, and -12 vectors on CFG glycan arrays displaying 611 different glycan structures, each in replicates of six. Note that binding signals for glycans 81, 265, and 518 were removed from the AAV7 and -rh.10 screens, respectively, based on a statement of the CFG that these glycans repeatedly produced nonspecific signals in unrelated glycan arrays performed with the same array batch. Error bars represent standard deviations.
Heparin array screening of various AAV serotypes.
AAV binding to defined heparin variants was analyzed on a specialized microarray with 13 synthetic single heparins and mixed natural low-molecular-weight heparin (26). All previous AAV heparin binding studies had relied on natural heparins as competitors. Natural heparins represent a mixture of heterogeneous biomolecules. Therefore, the exact composition and structure is unpredictable and varies from batch to batch. Single synthetic heparins offer the opportunity to differentiate the heparin binding specificities of different AAV serotypes, which has not been possible so far. The panel of fluorophore-labeled AAV preparations showed strong and specific heparin binding signals, but only in the cases of AAV2 and AAV3 (Fig. 4). The 2-fold-higher overall binding efficiency of AAV3 compared to AAV2 likely reflects the higher fluorophore-labeling efficiency. AAV6 and AAV13, which had been reported to require HSPG for infection (7), showed weaker binding. No binding to any of the heparins was detected for the other AAV serotypes (Fig. 4A and 5). These findings reflect previous observations that the majority of AAV serotypes were unable to bind to natural heparin.
FIG 4.

Heparin array screening of AAV serotypes 1 to 6 and 13. (A) Binding efficiencies of fluorescently labeled AAV1 to -6 and -13 vectors on heparin arrays displaying 13 different synthetic heparin structures and low-molecular-weight natural heparin 14, each in replicates of 10. Note that the binding signals of AAV13 were obtained from arrays displaying lower concentrations (0.25 mM) of the various heparin variants in contrast to all other AAV serotypes (1 mM). The error bars represent standard deviations. (B) Heparins that were identified in panel A to interact with the capsids of a particular serotype (42). Shown is a comparison of AAV serotype-dependent binding to related glycan structures present on the heparin array. Relative binding efficiencies are represented as percentages of that of the most efficiently binding glycan for the depicted AAV serotype.
FIG 5.
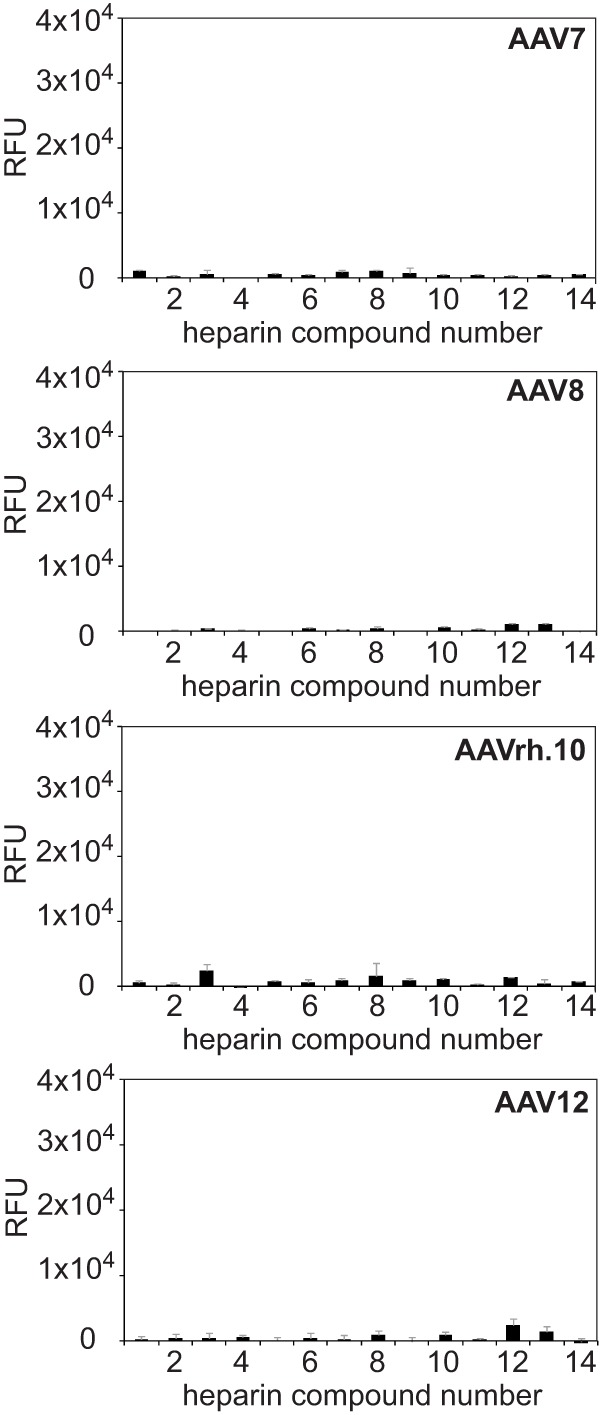
Heparin array screens of AAV7, -8, -rh.10, and -12. Shown are binding efficiencies of fluorescently labeled AAV7, -8, -rh.10, and -12 vectors on heparin arrays displaying 13 different synthetic heparin structures and low-molecular-weight natural heparin 14, each in replicates of 10. The error bars represent standard deviations.
AAV2 efficiently bound to natural heparin 14 and two synthetic heparins, 1 and 7. These oligosaccharides have similar sequences but differ in length (Fig. 4B). Binding to these compounds is even more efficient than binding to mixed natural heparin. The same heparins are bound by AAV3, but in addition, AAV3 binds to heparin 5 (Fig. 4). Heparin 5 is a hexasaccharide that, in contrast to 1 and 7, is not 6-O-sulfated on glucosamine. The data suggest that AAV2 binds to heparins containing glucosamines that are both N- and 6-O-sulfated (Fig. 4B). Obviously, 6-O-sulfation of heparin is not necessary for AAV3 binding. Instead, heparins with N-sulfated glucosamines alternating with 2-O-sulfated iduronic acids are bound (Fig. 4B).
AAV6 is a special case, since it binds efficiently to α2-3-linked sialic acid containing glycan 237, as shown above. AAV6 was shown previously to weakly bind to natural heparin in vivo (14). On the heparin array, AAV6 did not bind to natural heparin 14 but showed weak binding signals to heparins 3 and 8 (Fig. 4A). The two heparins are related, as they are sulfated only on glucosamine but not on iduronic acid, and differ in chain length (Fig. 4B). More highly sulfated heparins, as well as singly sulfated heparins (number 6; 2-O-sulfation on iduronic acid), were not bound by AAV6.
AAV13, also called VR-942 (7), bound best to natural heparin 14 (Fig. 4A). The array contained heparins only up to a maximum concentration of 0.25 mM, whereas arrays with heparins up to 1 mM had been used for all other AAV serotypes. Comparing the binding intensities of all AAVs at 0.25 mM heparin showed that the efficiency of AAV13 binding to natural heparin 14 is in the range of that seen for AAV2 and AAV3 (data not shown). In addition, AAV13 showed enhanced binding to heparin 2. Heparin 2 is a hexasaccharide composed of acetylated, 6-O-sulfated glucosamines and 2-O-sulfated iduronic acids.
Binding efficiencies to variant heparins by wild-type or mutant capsids of AAV2 or AAV3.
To validate that the AAV2 or -3 capsid structures defined as binding sites for natural heparin also mediate binding to synthetic heparin variants, AAV2 (R585A/R588A) and AAV3 (R594A) capsid mutants that had been shown previously to be deficient for binding to natural heparin were generated (27, 28). As anticipated, fluorophore-labeled AAV2 or AAV3 capsid mutants were unable to bind to any of the specific heparins (Fig. 6A). Surprisingly, the AAV3 interaction with low-molecular-weight heparin 14 was not impaired. The interaction of AAV capsids and heparin was analyzed at increasing heparin concentrations, leading to saturation curves typical for receptor-ligand interactions (Fig. 6B). The apparently lower saturation levels of AAV2 and AAV3 binding to natural heparin 14 further emphasize the high-affinity binding of AAV2 and AAV3 to the synthetic heparins 1 and 7.
FIG 6.
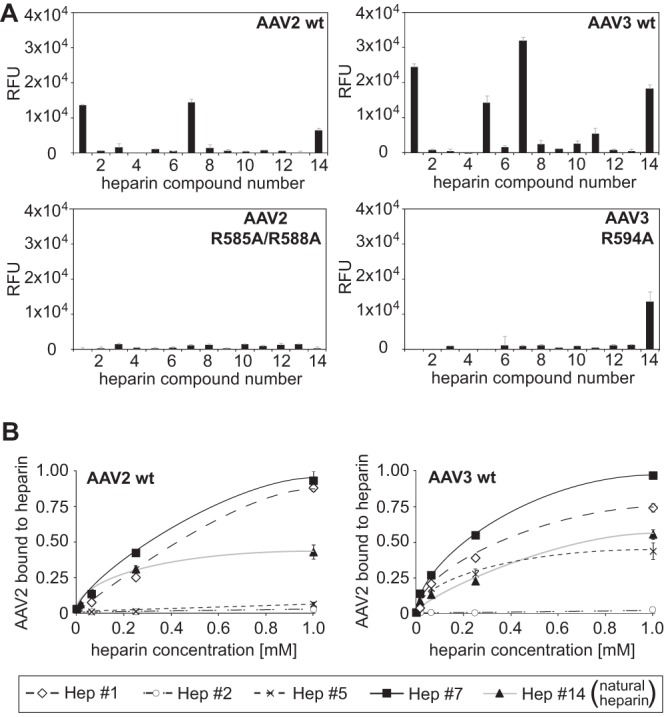
Specificities of AAV2 and -3 capsid binding to heparins. (A) Binding efficiencies of fluorescently labeled AAV2(R585A/R588A) and AAV3(R594A) vectors on heparin arrays displaying 13 different single synthetic heparin structures and low-molecular-weight natural heparin 14, each in replicates of 10. (B) Concentration-dependent binding of wild-type AAV2 and AAV3 to selected heparins. Heparins on arrays were present at increasing concentrations, 16 μM, 64 μM, 250 μM, and 1 mM, each in replicates of 10. The error bars represent standard deviations.
In vivo inhibition of AAV infection by natural and synthetic heparins.
To allow competition with heparin-like cell surface receptors on the target cell line, AAV infection neutralization assays were performed in the presence of soluble heparin. Increasing concentrations of heparin were preincubated with AAV before infection of C12 cells, as outlined in Materials and Methods. Heparin-dependent inhibition of AAV infection can be monitored by the expression of GFP as a transgene. The percentage of GFP-positive cells was determined by FACS analysis and served as a readout for AAV infection efficiency (Fig. 7A). The infection efficiency of wild-type AAV in the absence of heparin was arbitrarily set to 1.0. Infection by AAV2 and AAV13 was inhibited by more than 99% in the presence of increasing concentrations of heparin (Fig. 7B and C). The infectivity of the AAV2 and -13 heparin binding mutants was severely reduced and was unresponsive to increasing heparin concentrations (Fig. 7A, B, and C). AAV3 infection could not be fully blocked by heparin; approximately 20 to 30% of the initial infectivity remained at the highest heparin concentration of 1 mg/ml (P < 0.001) (Fig. 7D). The AAV3 heparin binding mutant was unresponsive to increasing heparin concentrations. Compared to wild-type (wt) AAV3, the mutant's infectivity was reduced to 50% in the absence of soluble heparin (Fig. 7D). The only partial neutralization of AAV3 by heparin may be taken as an indication of an alternative AAV3 entry mechanism, presumably by coreceptors, as described previously (29–31). As expected, AAV serotypes 1, 6, and rh.10 were resistant to increasing concentrations of natural heparin (Fig. 7E), as anticipated from the lack of binding to natural heparin on the array (Fig. 4A).
FIG 7.

In vivo AAV infection and competition by low-molecular-weight natural heparin. (A) GFP fluorescence of C12 cells infected with AAV2 wt or heparin binding site mutant AAV2(R585A/R588A) GFP vectors. AAV infection was performed in the presence of increasing concentrations of low-molecular-weight heparin as indicated. (B to E) Quantitative analysis (n = 3) of infection efficiencies of the indicated AAV serotypes or capsid variants derived from them in the presence of increasing concentrations of low-molecular-weight heparin. The error bars represent standard deviations.
The synthetic heparins were available only in limiting amounts, allowing competition assays up to a maximum concentration of 100 μg/ml. At this concentration, AAV2 had shown a clear inhibition by natural low-molecular-weight heparin 14 (Fig. 7B), and this subtype was therefore chosen to test neutralization by synthetic heparin 7, as described above. The results showed that synthetic heparin 7, in contrast to heparin 14, did not neutralize AAV infection (Fig. 8A). Since heparin 7 binding to AAV2 capsids on the array was more efficient than binding to natural heparin, we wondered whether components absent in synthetic heparin were needed for neutralization of AAV infection. To test this hypothesis, samples of natural heparin at concentrations of 1 μg/ml or 10 μg/ml were spiked with 10 μg/ml synthetic heparin 7 each during preincubation with AAV2 prior to infection. As shown in Fig. 8B, the addition of heparin 7 was unable to significantly enhance AAV neutralization by natural heparin. Heparin 7 is a rather short oligosaccharide. Although in vitro binding of pure heparin 7 to AAV2 is very efficient (Fig. 4B), a longer chain length may be required in vivo for AAV neutralization.
FIG 8.
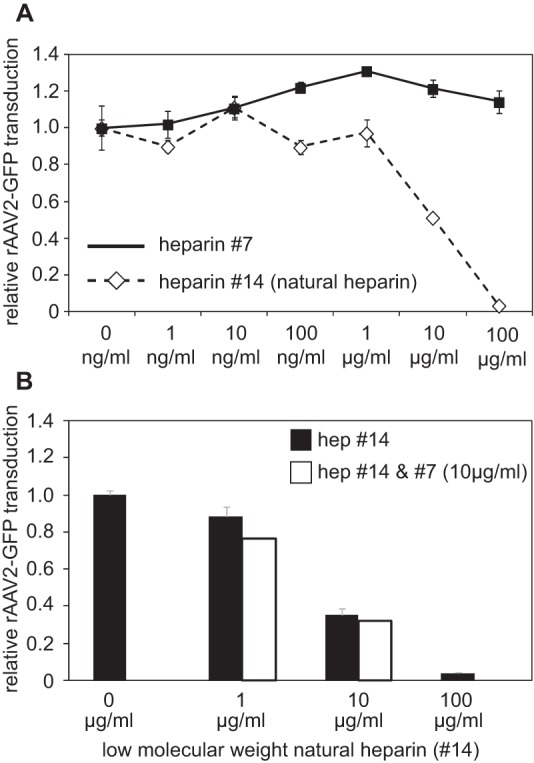
In vivo AAV infection and competition by synthetic heparin. (A) Quantitative analysis of AAV infection of AAV2 vectors in the presence of increasing concentrations of low-molecular-weight natural heparin 14 or synthetic heparin 7. The assay was performed as described in the legend to Fig. 4. (B) AAV2 infection in the presence of increasing concentrations of low-molecular-weight natural heparin 14, either alone or in the presence of 10 μg/ml synthetic heparin (hep) 7. The error bars represent standard deviations.
DISCUSSION
Various AAV serotypes differ in the structure of the exposed capsid surfaces (1) that represent the determinants for glycan binding and cell and tissue tropism (32). The primary receptors of eight AAV serotypes have been identified so far (2–7), and they can be assigned to three groups preferentially binding to (i) HSPG (AAV2, AAV3, AAV6, and AAV13), (ii) sialic acid (AAV1, AAV4, AAV5, and AAV6), and (iii) terminal galactose (AAV9). This is the first report to differentiate the HSPG-binding serotypes AAV2, AAV3, AAV6, and AAV13 by their affinities to specific synthetic heparin oligosaccharides. In addition, we further specify the exact nature of glycans bound by sialic acid binding AAVs. Thus, minimal structural glycan requirements could be defined for AAV1, AAV2, AAV3, AAV4, AAV5, AAV6, and AAV13, as summarized in Fig. 9.
FIG 9.

Discriminating minimal glycan structures bound by AAV serotypes 1 to 6 and 13. Definitions of the minimal glycan structure selected to allow discrimination of AAV serotypes 1, 2, 3, 4, 5, 6, and 13 are specified. Glycan structures are displayed in symbol nomenclature and in text nomenclature.
Glycan receptor specificities of AAV1 and AAV6.
AAV1 and AAV6 capsids differ by only 6 amino acids. Therefore, it is not surprising that the most prominent glycan recognized is the same. The minimal best binding structure, glycan 237, is composed of a terminal α2-3 sialic acid linked to N-acetylgalactosamine, followed by a β1-4-linked N-acetylglucosamine (Fig. 9). Binding to this glycan has been shown previously for AAV1 empty capsids using an earlier version of the CFG glycan array (5). This previous AAV1 screen was detected by antibodies, indicating that the fluorescent tag did not affect glycan binding specificity. Variants either displaying terminal α2-6 sialic acid or lacking a terminal sialic acid do not bind to either AAV1 or AAV6. Although we cannot exclude the possibility that particular glycans are missing on the array, we did not find evidence for α2-6 sialic acid-containing glycans as the AAV1/6 receptor. In addition, the minimal glycan structure we propose is more complex than previously postulated. Replacement of the middle N-acetylgalactosamine by galactose abolishes AAV binding (Fig. 1B). Although N-acetylglucosamine appears to be necessary (Fig. 1B), it cannot be determined whether alternative saccharides in this position would also allow binding to AAV1 or AAV6. The required glycan variants are not available on the array. Only AAV6 showed additional, weak interactions to specific heparin compounds, which were low sulfated, with a single N-sulfation (Fig. 4B). Natural heparins display a higher degree of sulfation (2.3 to 2.8 sulfates/disaccharide) than HSPG (0.6 to 1.5 sulfates/disaccharide) (13). This difference may explain why competition experiments using natural heparin and AAV6 were not successful in our study (Fig. 7E), as well as in a previous report (14).
Glycan receptor specificity of AAV5.
The current notion is that AAV5 capsids bind to both N-linked α2-3 and α2-6 sialic acids (4). Here, we confirm that AAV5 capsids recognize glycans with terminal α2-3 sialic acids, but only when they are linked to a sulfated galactose (Fig. 1C and 9). Although the structure is very similar to the one identified for AAV1 and AAV6, no cross-binding was detected. Multiple additional screens with AAV5 empty capsids deposited in the CFG database detected either by capsid-attached fluorophores or by AAV5 antibodies (ADK5) confirm the described binding pattern. All identified branched glycans share the common structure of N-glycans (33, 34). AAV5 did not bind to any of the available glycans that exclusively carry α2-6 sialic acid linkages. The lack of evidence for α2-6 sialic acid binding is difficult to reconcile with the description of the AAV5 receptor that has been identified by the use of α2-6-specific sialidases and glycan-expressing cell lines (4).
Glycan receptor specificity of AAV4.
The highest binding efficiency of AAV4, more than 100-fold above background, was observed for two linear glycans that share two internal mannose moieties linked by an α1-6 glycosidic bond. Variations of this connectivity were not tolerated (Fig. 1D). The two α1-6-linked mannose moieties alone, however, were not sufficient for AAV4 binding. Only a few more extended variations of the identified linear glycans are present on the CFG array, so the minimal binding structure could not be narrowed down any further. Therefore, glycan 351, which contains two mannose moieties but is devoid of sialic acids, emerged as the minimal best binder for AAV4 (Fig. 9). This finding is surprising in view of the O-linked terminal α2-3 sialic acids described previously as an AAV4 receptor (4). Although AAV4 binds to certain branched glycans displaying terminal α2-3 sialic acids, they more closely resemble N-glycans, rather than O-glycans. Recently, critical amino acids on the outer surface of the AAV4 capsid were characterized (35). Replacement of positively charged lysines by negatively charged glutamic acids dramatically reduced the in vivo AAV4 transduction efficiency of the heart and lung, but not of the liver (35). Sialic acids are negatively charged due to a carboxyl group that can interact with positively charged lysines, but not with negatively charged amino acids. Transductions with the described AAV4 mutants resulted in efficiencies comparable to those of AAV4 wild-type vectors after sialidase treatment of the target cells. This may be taken as evidence that the mutated amino acids are involved in sialic acid binding. Since infection of the liver remained largely unaffected, even in the presence of sialidases (35), an additional sialic acid-independent and as yet unidentified cell receptor appears to exist. The identified glycan 348-glycan 351 pair represents a good candidate for an alternative AAV4 cell receptor.
Glycan receptor specificities of AAV2, AAV3, and AAV13.
AAV2 and AAV3 have long been known to bind to HSPG, which serves as a primary host cell receptor. HSPGs are ubiquitous on mammalian cell membranes. However, there is considerable structural heterogeneity in terms of chain length, modifications, extent of sulfation, and epimerization within the heparan sulfate molecule (36). This is the first report to demonstrate binding of particular AAVs to chemically distinct synthetic heparins. The heparin binding array shows that both AAV2 and AAV3 bind to 6-O- and N-sulfated heparan sulfate, whereas AAV3 also binds to 2-O- and N-sulfated heparan sulfate. AAV13, although closely related to AAV3 (93% identity of the capsids) (7), differs in heparin binding properties. It binds to acetylated 6-O- and 2-O-sulfated heparins (Fig. 9). The variations in heparin binding specificities appear to reflect reported structural variations of AAV capsids and the relative positions of amino acids found to be critical for heparin binding (7, 37). A recent publication using chemically modified natural heparin further supports the notion that AAV2 binds to 6-O- and N-sulfated HSPGs (38), and the loss of function of the well-studied AAV2 heparin binding site mutant (R585A/R588A) shows that HSPG binding is critical for cell entry (28). In contrast to the situation with AAV2, transduction with AAV3 was only partially inhibited by soluble heparins. In addition, the heparin binding-deficient AAV3 capsid mutant R594 had retained some infectivity in the presence of heparin (Fig. 7D). Additional AAV3 capsid-exposed amino acids may mediate binding to alternative glycan variants present in mixed natural heparin 14, but not on the CFG glycan array or on the specialized heparin array.
The only synthetic glycan available in sufficient quantities for in vivo competition assays was synthetic heparin 7. Although synthetic heparin 7 was bound by AAV2 more efficiently than natural heparin 14, in vivo competition assays were unsuccessful. Heparin 7 is a tetrasaccharide of 1.3 kDa that is considerably shorter than natural low-molecular-mass heparin, with a mean mass of 5 kDa, translating into a 14-mer glycosaminoglycan. Previously published competition assays with short, size-fractionated heparins have shown only marginal inhibitory effects on AAV2 infection efficiencies (29). This is in line with the recent claim that a chain length of at least 13 would be needed to make full contact with the positively charged amino acids at the 3-fold symmetry axis on the capsid surface of AAV2 (38). The lack of AAV2 neutralization by heparin 7 could also be explained by lower affinity of AAV for pure synthetic heparins than for mixed natural heparin (Fig. 6B). In this case, AAV binding to heparin 7 might be outcompeted by components of cell culture medium and fetal calf serum or by higher-affinity cell surface-bound HSPG molecules.
Glycan receptor specificities of AAV serotypes 7 to 12.
For AAV serotypes 7 to 12, no primary receptor has been described, except for AAV9, where glycans with terminal galactoses were reported (6). Unfortunately, neither the CFG glycan array, with 611 different glycans, nor the heparin array revealed potential interaction partners for any of these serotypes. Since glycans can form an infinite variety of structures, the most likely explanation is that appropriate ligands were not present on the arrays. It will be interesting to see whether updated glycan arrays will detect receptor candidates for these AAV serotypes.
Implications and applications of the identified glycan receptor binding specificity.
HSPGs and sialic acids are present on the surface of almost every human cell, yet variant AAV serotypes display distinct cell- and tissue-specific targeting profiles. Their differentiation may have immediate implications for successful AAV infection. The currently best-examined viral-protein–glycan interaction is that of influenza virus hemagglutinin interacting with sialic acids on the cell surface. Human influenza virus strains bind to terminal α2-6 sialic acids linked to galactose, which primarily reside in the human upper respiratory tract, whereas avian influenza virus strains preferentially bind to α2-3 sialic acids linked to galactose. These types of glycans are prevalent in the intestinal mucosa of birds and the lower respiratory tract of humans (39). The variant cell receptor distribution determines the host and pathogenicity profiles of variant influenza virus strains. A comparable description of cell surface determinants would help to further define the targeting profiles of the increasing repertoire of AAV serotypes and their variants. This seems especially relevant in view of the increasing success of AAV vectors in human gene therapy. Research to characterize the glycomes of specific cell types and tissues (e.g., the CFG glycan-profiling program) is in progress. Most of the analyses were done by mass spectrometry, which cannot determine linkages in glycan structures. Our study demonstrates the importance of specific linkages between glycan moieties for binding to AAV capsids. Since these variations may determine if a cell can be infected, alternative methods, like nuclear magnetic resonance spectroscopy, are necessary for full characterization of the cell glycome (40). Further identification of the preferred binding structures for particular AAV variants may improve gene delivery, as shown recently for enhanced AAV9 cell transduction by prior modification of glycan target cell receptors (41).
ACKNOWLEDGMENTS
We thank John Chiorini (National Institutes of Health, Bethesda, MD, USA) for AAV plasmids and Stefan Weger and Catrin Stutika (Charité, Berlin, Germany) for critical readings of the manuscript.
Generous financial support was provided by the German academic exchange service DAAD (PROMOS) and the Max-Planck Society. A.R. thanks the Studienstiftung des Deutschen Volkes for a doctoral fellowship.
Footnotes
Published ahead of print 26 December 2013
REFERENCES
- 1.Agbandje-McKenna M, Kleinschmidt J. 2011. AAV capsid structure and cell interactions. Methods Mol. Biol. 807:47–92. 10.1007/978-1-61779-370-7_3 [DOI] [PubMed] [Google Scholar]
- 2.Summerford C, Samulski RJ. 1998. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J. Virol. 72:1438–1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Handa A, Muramatsu S, Qiu J, Mizukami H, Brown KE. 2000. Adeno-associated virus (AAV)-3-based vectors transduce haematopoietic cells not susceptible to transduction with AAV-2-based vectors. J. Gen. Virol. 81:2077–2084 http://vir.sgmjournals.org/content/81/8/2077.long. [DOI] [PubMed] [Google Scholar]
- 4.Kaludov N, Brown KE, Walters RW, Zabner J, Chiorini JA. 2001. Adeno-associated virus serotype 4 (AAV4) and AAV5 both require sialic acid binding for hemagglutination and efficient transduction but differ in sialic acid linkage specificity. J. Virol. 75:6884–6893. 10.1128/JVI.75.15.6884-6893.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu Z, Miller E, Agbandje-McKenna M, Samulski RJ. 2006. Alpha2,3 and alpha2,6 N-linked sialic acids facilitate efficient binding and transduction by adeno-associated virus types 1 and 6. J. Virol. 80:9093–9103. 10.1128/JVI.00895-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bell CL, Vandenberghe LH, Bell P, Limberis MP, Gao GP, Van Vliet K, Agbandje-McKenna M, Wilson JM. 2011. The AAV9 receptor and its modification to improve in vivo lung gene transfer in mice. J. Clin. Invest. 121:2427–2435. 10.1172/JCI57367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmidt M, Govindasamy L, Afione S, Kaludov N, Agbandje-McKenna M, Chiorini JA. 2008. Molecular characterization of the heparin-dependent transduction domain on the capsid of a novel adeno-associated virus isolate, AAV(VR-942). J. Virol. 82:8911–8916. 10.1128/JVI.00672-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bertozzi CR, Rabuka D. 2009. Structural basis of glycan diversity, p 23–36 In Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME. (ed), Essentials of glycobiology, 2nd ed. Cold Spring Harbor Press, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 9.Benallal M, Anner BM. 1994. Identification of organ-specific glycosylation of a membrane protein in two tissues using lectins. Experientia 50:664–668. 10.1007/BF01952869 [DOI] [PubMed] [Google Scholar]
- 10.Molinari M. 2007. N-glycan structure dictates extension of protein folding or onset of disposal. Nat. Chem. Biol. 3:313–320. 10.1038/nchembio880 [DOI] [PubMed] [Google Scholar]
- 11.Zhao YY, Takahashi M, Gu JG, Miyoshi E, Matsumoto A, Kitazume S, Taniguchi N. 2008. Functional roles of N-glycans in cell signaling and cell adhesion in cancer. Cancer Sci. 99:1304–1310. 10.1111/j.1349-7006.2008.00839.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nizet V, Esko JD. 2009. Bacterial and viral infections, p 537–552 In Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME. (ed), Essentials of glycobiology, 2nd ed. Cold Spring Harbor Press, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 13.Dreyfuss JL, Regatieri CV, Jarrouge TR, Cavalheiro RP, Sampaio LO, Nader HB. 2009. Heparan sulfate proteoglycans: structure, protein interactions and cell signaling. Ann. Acad. Bras. Cienc. 81:409–429. 10.1590/S0001-37652009000300007 [DOI] [PubMed] [Google Scholar]
- 14.Halbert CL, Allen JM, Miller AD. 2001. Adeno-associated virus type 6 (AAV6) vectors mediate efficient transduction of airway epithelial cells in mouse lungs compared to that of AAV2 vectors. J. Virol. 75:6615–6624. 10.1128/JVI.75.14.6615-6624.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmidt M, Voutetakis A, Afione S, Zheng C, Mandikian D, Chiorini JA. 2008. Adeno-associated virus type 12 (AAV12): a novel AAV serotype with sialic acid- and heparan sulfate proteoglycan-independent transduction activity. J. Virol. 82:1399–1406. 10.1128/JVI.02012-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walters RW, Yi SM, Keshavjee S, Brown KE, Welsh MJ, Chiorini JA, Zabner J. 2001. Binding of adeno-associated virus type 5 to 2,3-linked sialic acid is required for gene transfer. J. Biol. Chem. 276:20610–20616. 10.1074/jbc.M101559200 [DOI] [PubMed] [Google Scholar]
- 17.Varki A, Schauer R. 2009. Sialic acids, p 199–218 In Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME. (ed), Essentials of glycobiology, 2nd ed. Cold Spring Harbor Press, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 18.Walters RW, Pilewski JM, Chiorini JA, Zabner J. 2002. Secreted and transmembrane mucins inhibit gene transfer with AAV4 more efficiently than AAV5. J. Biol. Chem. 277:23709–23713. 10.1074/jbc.M200292200 [DOI] [PubMed] [Google Scholar]
- 19.Grimm D, Kay MA, Kleinschmidt JA. 2003. Helper virus-free, optically controllable, and two-plasmid-based production of adeno-associated virus vectors of serotypes 1 to 6. Mol. Ther. 7:839–850. 10.1016/S1525-0016(03)00095-9 [DOI] [PubMed] [Google Scholar]
- 20.Aslanidi G, Lamb K, Zolotukhin S. 2009. An inducible system for highly efficient production of recombinant adeno-associated virus (rAAV) vectors in insect Sf9 cells. Proc. Natl. Acad. Sci. U. S. A. 106:5059–5064. 10.1073/pnas.0810614106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Winter K, von Kietzell K, Heilbronn R, Pozzuto T, Fechner H, Weger S. 2012. Roles of E4orf6 and VA I RNA in adenovirus-mediated stimulation of human parvovirus B19 DNA replication and structural gene expression. J. Virol. 86:5099–5109. 10.1128/JVI.06991-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mietzsch M, Grasse S, Zurawski C, Weger S, Bennett A, Agbandje-McKenna M, Muzyczka N, Zolotukhin S, Heilbronn R. OneBac: platform for scalable and high-titer production of AAV serotype 1–12 vectors for gene therapy. Hum. Gene Ther. 10.1089/hum.2013.184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Noti C, de Paz JL, Polito L, Seeberger PH. 2006. Preparation and use of microarrays containing synthetic heparin oligosaccharides for the rapid analysis of heparin-protein interactions. Chemistry 12:8664–8686. 10.1002/chem.200601103 [DOI] [PubMed] [Google Scholar]
- 24.de Paz JL, Spillmann D, Seeberger PH. 2006. Microarrays of heparin oligosaccharides obtained by nitrous acid depolymerization of isolated heparin. Chem. Commun. 29:3116–3118. 10.1039/B605318A [DOI] [PubMed] [Google Scholar]
- 25.Wistuba A, Kern A, Weger S, Grimm D, Kleinschmidt JA. 1997. Subcellular compartmentalization of adeno-associated virus type 2 assembly. J. Virol. 71:1341–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yin J, Seeberger PH. 2010. Applications of heparin and heparan sulfate microarrays. Methods Enzymol. 478:197–218. 10.1016/S0076-6879(10)78009-5 [DOI] [PubMed] [Google Scholar]
- 27.Lerch TF, Chapman MS. 2012. Identification of the heparin binding site on adeno-associated virus serotype 3B (AAV-3B). Virology 423:6–13. 10.1016/j.virol.2011.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Opie SR, Warrington KH, Jr, Agbandje-McKenna M, Zolotukhin S, Muzyczka N. 2003. Identification of amino acid residues in the capsid proteins of adeno-associated virus type 2 that contribute to heparan sulfate proteoglycan binding. J. Virol. 77:6995–7006. 10.1128/JVI.77.12.6995-7006.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Messina EL, Nienaber J, Daneshmand M, Villamizar N, Samulski J, Milano C, Bowles DE. 2012. Adeno-associated viral vectors based on serotype 3b use components of the fibroblast growth factor receptor signaling complex for efficient transduction. Hum. Gene Ther. 23:1031–1042. 10.1089/hum.2012.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blackburn SD, Steadman RA, Johnson FB. 2006. Attachment of adeno-associated virus type 3H to fibroblast growth factor receptor 1. Arch. Virol. 151:617–623. 10.1007/s00705-005-0650-6 [DOI] [PubMed] [Google Scholar]
- 31.Ling C, Lu Y, Kalsi JK, Jayandharan GR, Li B, Ma W, Cheng B, Gee SW, McGoogan KE, Govindasamy L, Zhong L, Agbandje-McKenna M, Srivastava A. 2010. Human hepatocyte growth factor receptor is a cellular coreceptor for adeno-associated virus serotype 3. Hum. Gene Ther. 21:1741–1747. 10.1089/hum.2010.075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zincarelli C, Soltys S, Rengo G, Rabinowitz JE. 2008. Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. 16:1073–1080. 10.1038/mt.2008.76 [DOI] [PubMed] [Google Scholar]
- 33.Stanley P, Cummings RD. 2009. Structures common to different glycans, p 175–198 In Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME. (ed), Essentials of glycobiology, 2nd ed. Cold Spring Harbor Press, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 34.Stanley P, Schachter H, Taniguchi N. 2009. N-glycans, p 101–114 In Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME. (ed), Essentials of glycobiology, 2nd ed. Cold Spring Harbor Press, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 35.Shen S, Troupes AN, Pulicherla N, Asokan A. 2013. Multiple roles for sialylated glycans in determining the cardiopulmonary tropism of adeno-associated virus 4. J. Virol. 87:13206–13213. 10.1128/JVI.02109-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bishop JR, Schuksz M, Esko JD. 2007. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 446:1030–1037. 10.1038/nature05817 [DOI] [PubMed] [Google Scholar]
- 37.Lerch TF, Xie Q, Chapman MS. 2010. The structure of adeno-associated virus serotype 3B (AAV-3B): insights into receptor binding and immune evasion. Virology 403:26–36. 10.1016/j.virol.2010.03.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang F, Aguilera J, Beaudet JM, Xie Q, Lerch TF, Davulcu O, Colon W, Chapman MS, Linhardt RJ. 2013. Characterization of interactions between heparin/glycosaminoglycan and adeno-associated virus. Biochemistry 52:6275–6285. 10.1021/bi4008676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Esko JD, Sharon N. 2009. Microbial lectins: hemagglutinins, adhesins, and toxins, p 489–500 In Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME. (ed), Essentials of glycobiology, 2nd ed. Cold Spring Harbor Press, Cold Spring Harbor, NY: [PubMed] [Google Scholar]
- 40.Brisson JR, Vinogradov E, McNally DJ, Khieu NH, Schoenhofen IC, Logan SM, Jarrell H. 2010. The application of NMR spectroscopy to functional glycomics. Methods Mol. Biol. 600:155–173. 10.1007/978-1-60761-454-8_11 [DOI] [PubMed] [Google Scholar]
- 41.Shen S, Bryant KD, Brown SM, Randell SH, Asokan A. 2011. Terminal N-linked galactose is the primary receptor for adeno-associated virus 9. J. Biol. Chem. 286:13532–13540. 10.1074/jbc.M110.210922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harvey DJ, Merry AH, Royle L, Campbell MP, Dwek RA, Rudd PM. 2009. Proposal for a standard system for drawing structural diagrams of N- and O-linked carbohydrates and related compounds. Proteomics 9:3796–3801. 10.1002/pmic.200900096 [DOI] [PubMed] [Google Scholar]