

Abstract
The isogenic host attachment spike protein recombinant demyelinating strain of mouse hepatitis virus (MHV) (RSA59) and the nondemyelinating strain (RSMHV2) differ in their abilities to infect distinct types of neural cells, spread from cell to cell, and induce subsequent demyelination and axonal loss. The differential demyelination properties of RSA59 and RSMHV2 may be a function of spike protein-mediated neuronal transport. Disruption of microtubules with colchicine and vinblastine significantly blocks neuronal transport and reduces the replication of RSA59, whereas RSMHV2 remains unaffected.
TEXT
RSA59 (demyelinating [DM]) and RSMHV2 (nondemyelinating [NDM]), enhanced green fluorescent protein (EGFP)-expressing recombinant strains of mouse hepatitis virus (MHV) are isogenic except for the spike gene, which encodes a glycoprotein expressed on the virion envelope that mediates many biological properties of MHV (1–5). Histopathological studies demonstrated that brain pathology from infection with either strain consists of encephalitis, characterized by parenchymal lymphocytic infiltrates and microglial activation with associated lymphocytic meningitis, but the strains differ in their abilities to induce demyelination and axonal loss. Evaluation of axonal loss and demyelination in spinal cord, where there is a clear separation of gray matter and large white-matter tracts, showed that RSA59 infection begins in the neuronal cell body, propagates centripetally to the axon, and subsequently induces axonal degeneration and demyelination (1, 3). RSMHV2 infection is mainly restricted to gray matter, is unable to spread from gray to white matter, and as a result cannot induce demyelination. In optic nerve (another white-matter tract), RSA59 induces inflammation and subsequent demyelination and axonal loss following intracranial inoculation, whereas RSMHV2 does not (3, 4, 6). RSMHV2 shows impaired spread in spinal cord and optic nerve compared to RSA59, highlighting the important role of spike-mediated transneuronal spread in axonal damage and demyelination. Differential spreading in the CNS could be due to interactions between the spike protein and host neuronal cytoskeleton proteins involved in anterograde and retrograde movement along the microtubule tracks, as seen in some human neurotropic viruses (7–12). Thus, the failure of NDM strains to trigger myelin damage could be entirely a function of failure of microtubule-mediated transport to the white matter. Indeed, evidence shows that the JHM strain of MHV spreads transneuronally, with MHV protein trafficking dependent on the presence of microtubules (13). Therefore, possible involvement of microtubule-mediated axonal transport of RSA59 in comparison to RSMHV2 was studied in colchicine- or vinblastine-treated Neuro2a cells to determine whether spike protein mediates microtubule-mediated neuronal transport. Colchicine and vinblastine are known microtubule-depolymerizing agents that have been shown to be effective blockers of the axonal transport system both in vivo and in vitro (14–16).
Neuro2a cells were phenotypically characterized by immunofluorescence analysis performed with primary antibodies directed against axonal and dendritic microtubules TAU (Sigma) and microtubule-associated protein-2 (MAP2) (2A + 2B) (Sigma) in combination with tetramethyl rhodamine isothiocyanate (TRITC)-conjugated secondary goat anti-mouse IgG to distinguish between axonal and somatodendritic compartments, respectively. Immunostained cells were then mounted in Vectashield with DAPI (4′,6-diamidino-2-phenylindole; Vector Laboratories). Cell organelle-specific TAU and MAP2 staining demonstrated that under our culture conditions, Neuro2a cells differentiated into a neuronal phenotype with dendritic processes (Fig. 1A) and axonal projections (Fig. 1B).
FIG 1.

(A to J) Morphological characterization of microtubules in control Neuro2a cultures (A and B) in comparison to colchicine- and vinblastine-treated Neuro2a cells (C to J). (K and L) MTT assay results demonstrate cell viability after microtubule disruption. Neuro2a cells were characterized by immunofluorescence with dendritic marker anti-MAP2 (2A + 2B) and axonal microtubule marker anti-TAU antibodies. (A) The dendritic network was observed by MAP2 (2A + 2B) staining. Dendrites are clearly observed at high magnification (×60) as shown by arrows. (B) TAU antibody preferentially stained axonal projections and neuronal cell bodies (arrowheads mark axons). (C to J) Axonal and dendritic microtubules were disrupted using microtubule-depolymerizing agents colchicine and vinblastine at 5 different concentrations each (for colchicine, 125 nM, 1.25 μM, 12.5 μM, 125 μM, and 1.25 mM; for vinblastine, 30 nM, 300 nM, 3 μM, 30 μM, and 100 μM) for 24 h. Representative images from the lowest and highest concentrations of colchicine- and vinblastine-disrupted cells are shown. (Left panel) MAP2 (2A + 2B) staining in nontreated control Neuro2a cells (A) and in cells treated with 125 nM (lowest concentration) (C) and 1.25 mM (highest concentration) (G) colchicine and with 30 nM (lowest concentration) (E) and 100 μM (highest concentration) (I) vinblastine. (Right panel) Tau antibody staining in nontreated control Neuro2a cells (B) and in cells treated with 125 nM (lowest concentration) (D) and 1.25 mM (highest concentration) (H) colchicine and with 30 nM (lowest concentration) (F) and 100 μM (highest concentration) (J) vinblastine. The MTT assay was performed on Neuro2a cells with different concentrations of colchicine and vinblastine (n = 4) to assess the cell viability and repeated three times. Cell viability is plotted against the concentrations of colchicine (K) and vinblastine (L).
Confluent cultures of differentiated Neuro2a cells were treated with colchicine (12.5 pM to 12.5 mM) and vinblastine (30 nM to 3 mM) for 24 h. After 24 h of colchicine and vinblastine treatment, Neuro2a cells were processed for immunostaining with microtubule marker MAP2 and TAU to detect disruption of dendritic and axonal microtubules. Microtubule disruption was phenotypically obvious from microscopic observations in both colchicine- and vinblastine-treated cultures in a dose-dependent manner (Fig. 1C to J). Cytotoxicity of different concentrations of colchicine and vinblastine on Neuro2a cells was measured by MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide; Sigma catalog no. M5655] assays (Fig. 1K and L). Cell viability measured from three different sets of MTT assays revealed that 125 nM to 1.25 mM concentrations of colchicine and 30 nM to 100 μM concentrations of vinblastine were the most effective doses for axonal disruption studies.
Neuro2a cells cultured to 80% confluence were infected at a multiplicity of infection (MOI) of 2 (2 viral particles/cell) with RSA59 or RSMHV2 or were mock infected with phosphate-buffered saline (PBS) for in vitro time lapse video imaging in order to observe viral replication and spread over time. After viral adsorption for 1 h was allowed, cells were washed and placed in fresh media without virus. Viral replication and spread in real time in Neuro2a culture visualized as EGFP-positive (EGFP+) cells (RSA59-infected cells) were first observed 8 h postadsorption and continued until 36 h. RSA59-infected cells started to form syncytia as early as 9 h, consistent with prior studies of RSA59 or its MHV-A59 parental strain (17). The size of syncytia increased until 36 h before resolving. One such syncytium with viral spread is shown in Fig. 2. EGFP expression was evident in both dendrites (arrowheads) and axons (arrows) in newly infected neurons (as shown in Fig. 2C and D). At a later time point (4 h 15 min), viral antigen propagated to another neuron through axon terminals (Fig. 2E) and EGFP expression drastically decreased in the originally infected neuron (seen in Fig. 2C and D). The abilities of RSMHV2 to replicate and spread in Neuro2a cells were compared to those of RSA59. In RSMHV2-infected cultures (Fig. 2F to J), viral infection was restricted only to the initially infected neurons with little or no spread to adjacent cells observed until 36 h, when the infected cell typically lysed.
FIG 2.
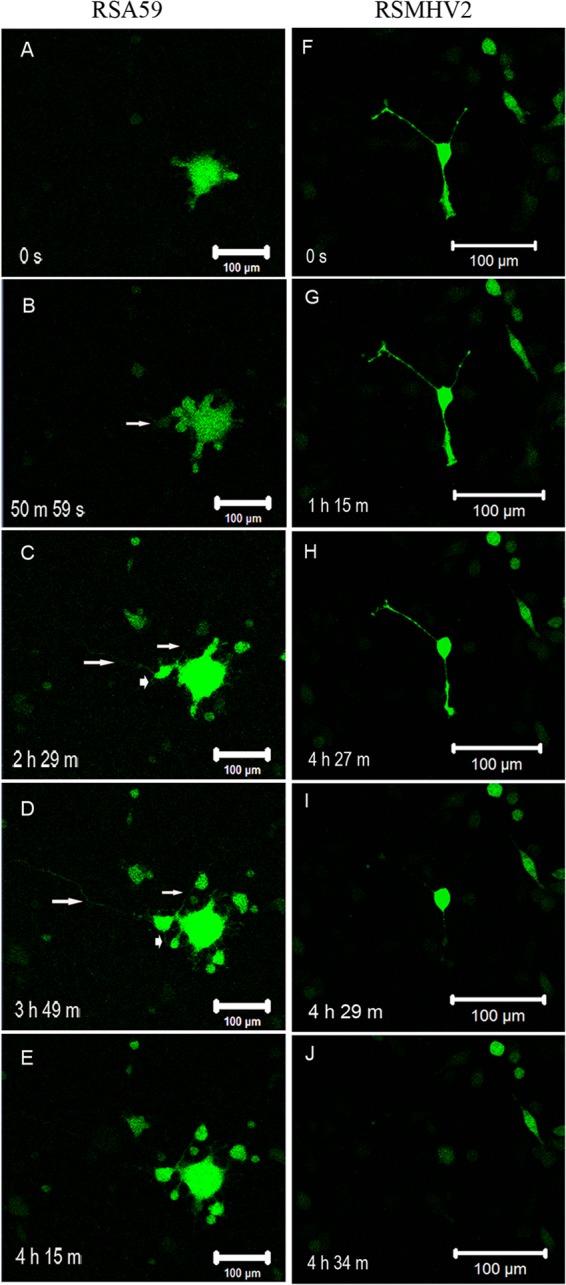
Still frames of video at different times p.i. demonstrate neuron-to-neuron spread of RSA59 and restriction of RSMHV2 to infected neurons only. (A to E) Selected still frames at different times p.i. from video of an RSA59-infected culture are shown to demonstrate the neuronal transport phenomenon of RSA59. (F to J) Sequential progress of RSMHV2 infection of a single neuron for 4 h is shown, demonstrating that RSMHV2 infects individual Neuro2a cells only without cell-to-cell spread (F). Though EGFP expression can be observed in axons of some cells as shown, the neurites contract over time and eventually dissolve with the cell body (G, H, and I) followed by a rapid lysis of the cell (J).
Parallel cultures of Neuro2a cells were treated with colchicine and vinblastine for 24 h and infected with RSA59 and RSMHV2 at an MOI of 0.5 (1 viral particle/2 cells) to measure viral replication. At 0, 6, 12, and 20 h postinfection (p.i.), cultures were monitored for replication and viral antigen (EGFP) spread by fluorescence microscopy. At 20 h p.i., culture supernatants were collected for viral titer estimation (viral replication) and images of infected cells were acquired and analyzed to assess viral antigen spread (by EGFP fluorescence). Infected cells were fixed using 4% paraformaldehyde (PFA), washed with PBS, and counterstained with DAPI for microscopic observations. The efficiency of RSA59 with respect to infecting cells and spreading from one neuron to another was reduced significantly with increasing doses of colchicine and vinblastine (Fig. 3). In control cultures, RSA59 viral antigen was initially observed in cell bodies with subsequent spread to neural processes followed by infection of the cells connected via the neural process (dendrites or axons). Viral antigen (EGFP) expression was significantly lower in the neuronal processes of cells in the colchicine- and vinblastine-treated cultures due to disruption of microtubules (Fig. 3). In contrast, in both colchicine- and vinblastine-treated and control cells, EGFP expression of RSMHV2-infected cells was mainly localized in cell bodies and axonal processes, demonstrating that axonal disruption did not significantly affect the spread of RSMHV2 from neuron to neuron (Fig. 3).
FIG 3.

Microtubule disruption altered the spread of RSA59 but not of RSMHV2. Cultures of Neuro2a cells were treated with colchicine and vinblastine for 24 h and infected with RSA59 and RSMHV2 at an MOI of 0.5. At 20 h p.i., infected cells were washed and counterstained with DAPI. EGFP expression denotes viral antigen-positive cells, whereas DAPI staining (blue) shows the nuclei of both infected and noninfected cells. Increasing levels of colchicine and vinblastine decreased the cell-to-cell spread of RSA59.
While colchicine and vinblastine in Neuro2a cells equally affected neuronal transport and subsequent spread of RSA59, neither colchicine nor vinblastine affected spread of RSMHV2. Similarly, neither microtubule-disrupting agent affected the fusion properties of RSA59, as fusion was evident in all colchicine- and vinblastine-treated cultures. The results are in agreement with previous findings that colchicine and cytochalasin do not affect the fusogenic property of MHV in fibroblast cultures (18).
For estimation of viral replication, culture supernatants from control mock-infected cultures, and from RSA59- and RSMHV2-infected cultures, were serially diluted and added to a confluent monolayer of L2 cells in 6-well plates for routine plaque assay (19). Interestingly, the viral titer showed a small decline in RSA59 numbers in microtubule-disrupted cultures, in a dose-dependent manner. As colchicine and vinblastine concentrations increased, the viral titer decreased accordingly (Fig. 4A). This indicates that viral replication and infection were dependent on microtubule-dependent axonal transport. In contrast, and consistent with microscopic observations shown in Fig. 4, no significant differences in titers were observed following RSMHV2 infection (Fig. 4B). This revealed that in RSMHV2 infection, replication and interneuronal spread were independent of axonal transport, in contrast to RSA59 infection.
FIG 4.
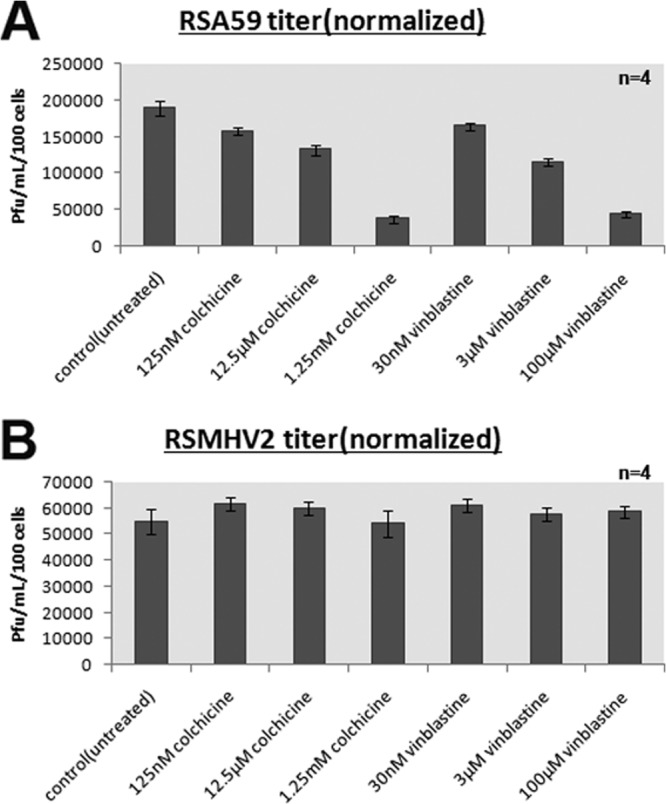
Colchicine and vinblastine treatment of Neuro2a cells reduces the growth of RSA59 but not RSMHV2. At 24 h posttreatment with colchicine and vinblastine, cells were infected with RSA59 or RSMHV2 at 0.5 MOI. Culture supernatant was collected for viral titer estimation, and viability of infected cells was measured by the MTT assay. Viral titers were normalized based on the live cell numbers measured by the MTT assay, and titers were plotted for RSA59 (A)- and RSMHV2 (B)-infected cultures. Mean and standard errors of the means (SEM) of titers from 4 wells in one representative experiment (of three) are shown.
To confirm that the reduction of viral titer was due to reduced viral spread through neuron, and not due to cytotoxicity caused by colchicine and vinblastine, viability of infected cells was determined using the MTT assay at the time that culture supernatant was collected. Viral titers were normalized to the number of live cells determined by the MTT assay and plotted against concentrations of colchicine and vinblastine. Normalization data revealed that, irrespective of colchicine and vinblastine cytotoxicity, there was a reduction of viral replication in RSA59-infected but not in RSMHV2-infected cultures. Clear syncytium formation (observed in Fig. 3) at different concentrations demonstrates that the reduced titer was due to transport deficiency rather than altered fusion properties. Moreover, if colchicine and vinblastine affected cell-to-cell fusion in addition to neuronal spread, then drastic reduction of viral titer should have been observed. RSMHV2, irrespective of the presence or absence of colchicine or vinblastine treatment, was fusion defective.
It had been previously reported that microtubules play an important role in transneuronal spread of the JHM strain of MHV, but our studies reveal for the first time that microtubules also play a major role in transneuronal spread of isogenic strains of MHV, RSA59, and RSMHV2 that differ only in the spike gene. While some interaction of MHV nucleocapsid protein with the axonal transport machinery and accessory cargo proteins has been previously reported (13), the spike-mediated mechanisms of transport of MHV remain to be determined.
ACKNOWLEDGMENTS
This work was supported by a Research Grant from Department of Biotechnology (DBT) (BT/PR14260/MED/30/437/2010), India, Indian Institute of Science Education and Research-Kolkata (IISER-K), India start up Fund, and Research Grant RG3774A2/1 from the National Multiple Sclerosis Society to J.D.S. K.B. is supported by University Grant Commission (UGC), India.
We thank the Confocal Imaging Facility and Ritabrata Ghosh of IISER-K for confocal assistance. We thank Kenneth S. Shindler for critically editing the manuscript.
We declare that we have no financial conflicts of interest to disclose.
Footnotes
Published ahead of print 18 December 2013
REFERENCES
- 1.Das Sarma J. 2010. A mechanism of virus-induced demyelination. Interdiscip. Perspect. Infect. Dis. 2010:109239. 10.1155/2010/109239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Das Sarma J, Fu L, Tsai JC, Weiss SR, Lavi E. 2000. Demyelination determinants map to the spike glycoprotein gene of coronavirus mouse hepatitis virus. J. Virol. 74:9206–9213. 10.1128/JVI.74.19.9206-9213.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Das Sarma J, Kenyon LC, Hingley ST, Shindler KS. 2009. Mechanisms of primary axonal damage in a viral model of multiple sclerosis. J. Neurosci. 29:10272–10280. 10.1523/JNEUROSCI.1975-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Das Sarma J, Iacono K, Gard L, Marek R, Kenyon LC, Koval M, Weiss SR. 2008. Demyelinating and nondemyelinating strains of mouse hepatitis virus differ in their neural cell tropism. J. Virol. 82:5519–5526. 10.1128/JVI.01488-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Das Sarma J, Scheen E, Seo SH, Koval M, Weiss SR. 2002. Enhanced green fluorescent protein expression may be used to monitor murine coronavirus spread in vitro and in the mouse central nervous system. J. Neurovirol. 8:381–391. 10.1080/13550280260422686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shindler KS, Chatterjee D, Biswas K, Goyal A, Dutt M, Nassrallah M, Khan RS, Das Sarma J. 2011. Macrophage-mediated optic neuritis induced by retrograde axonal transport of spike gene recombinant mouse hepatitis virus. J. Neuropathol. Exp. Neurol. 70:470–480. 10.1097/NEN.0b013e31821da499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ye GJ, Vaughan KT, Vallee RB, Roizman B. 2000. The herpes simplex virus 1 U(L)34 protein interacts with a cytoplasmic dynein intermediate chain and targets nuclear membrane. J. Virol. 74:1355–1363. 10.1128/JVI.74.3.1355-1363.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim W, Tang Y, Okada Y, Torrey TA, Chattopadhyay SK, Pfleiderer M, Falkner FG, Dorner F, Choi W, Hirokawa N, Morse HC., III 1998. Binding of murine leukemia virus Gag polyproteins to KIF4, a microtubule-based motor protein. J. Virol. 72:6898–6901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jacob Y, Badrane H, Ceccaldi PE, Tordo N. 2000. Cytoplasmic dynein LC8 interacts with lyssavirus phosphoprotein. J. Virol. 74:10217–10222. 10.1128/JVI.74.21.10217-10222.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mueller S, Cao X, Welker R, Wimmer E. 2002. Interaction of the poliovirus receptor CD155 with the dynein light chain Tctex-1 and its implication for poliovirus pathogenesis. J. Biol. Chem. 277:7897–7904. 10.1074/jbc.M111937200 [DOI] [PubMed] [Google Scholar]
- 11.Raux H, Flamand A, Blondel D. 2000. Interaction of the rabies virus P protein with the LC8 dynein light chain. J. Virol. 74:10212–10216. 10.1128/JVI.74.21.10212-10216.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diefenbach RJ, Miranda-Saksena M, Diefenbach E, Holland DJ, Boadle RA, Armati PJ, Cunningham AL. 2002. Herpes simplex virus tegument protein US11 interacts with conventional kinesin heavy chain. J. Virol. 76:3282–3291. 10.1128/JVI.76.7.3282-3291.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kalicharran K, Dales S. 1995. Involvement of microtubules and the microtubule-associated protein tau in trafficking of JHM virus and components within neurons. Adv. Exp. Med. Biol. 380:57–61. 10.1007/978-1-4615-1899-0_8 [DOI] [PubMed] [Google Scholar]
- 14.Abbott CJ, Choe TE, Lusardi TA, Burgoyne CF, Wang L, Fortune B. 2013. Imaging axonal transport in the rat visual pathway. Biomed. Opt. Express. 4:364–386. 10.1364/BOE.4.000364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Altar CA, Cai N, Bliven T, Juhasz M, Conner JM, Acheson AL, Lindsay RM, Wiegand SJ. 1997. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature 389:856–860. 10.1038/39885 [DOI] [PubMed] [Google Scholar]
- 16.Takihara Y, Inatani M, Hayashi H, Adachi N, Iwao K, Inoue T, Iwao M, Tanihara H. 2011. Dynamic imaging of axonal transport in living retinal ganglion cells in vitro. Invest. Ophthalmol. Vis. Sci. 52:3039–3045. 10.1167/iovs.10-6435 [DOI] [PubMed] [Google Scholar]
- 17.Hingley ST, Leparc-Goffart I, Weiss SR. 1998. The spike protein of murine coronavirus mouse hepatitis virus strain A59 is not cleaved in primary glial cells and primary hepatocytes. J. Virol. 72:1606–1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Daya M, Cervin M, Anderson R. 1988. Cholesterol enhances mouse hepatitis virus-mediated cell fusion. Virology 163:276–283. 10.1016/0042-6822(88)90267-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lavi E, Gilden DH, Wroblewska Z, Rorke LB, Weiss SR. 1984. Experimental demyelination produced by the A59 strain of mouse hepatitis virus. Neurology 34:597–603. 10.1212/WNL.34.5.597 [DOI] [PubMed] [Google Scholar]