

ABSTRACT
Natural killer (NK) cells and the complement system play critical roles in the first line of defense against pathogens. The synthesis of complement components C4 and C3 is transcriptionally downregulated by hepatitis C virus (HCV) core and NS5A proteins, and this negative regulation is apparent in chronically HCV-infected patients. In this study, we have examined the potential contribution of an NK cell line as a model in regulating complement synthesis. Coculture of NK cells (NK3.3) with human hepatoma cells (Huh7.5) expressing HCV core or NS5A protein led to a significant increase in C4 and C3 complement synthesis via enhanced specific transcription factors. Reestablishment of complement protein expression was found to be mediated by direct interaction between NKG2D on NK cells and the hepatocyte protein major histocompatibility complex class I-related chains A and B (MICA/B) and not to be associated with specific cytokine signaling events. On the other hand, C4 and C3 synthesis remained impaired in a coculture of NK cells and Huh7.5 cells infected with cell culture-grown HCV. The association between these two cell types through NKG2D and MICA/B was examined further, with MICA/B expression in HCV-infected hepatocytes found to remain inhibited during coculture. Further experiments revealed that the HCV NS2 and NS5B proteins are responsible for the HCV-associated decrease in MICA/B. These results suggest that HCV disables a key receptor ligand in infected hepatoma cells, thereby inhibiting the ability of infected cells to respond to stimuli from NK cells to positively regulate complement synthesis.
IMPORTANCE The complement system contributes to the protection of the host from virus infection. However, the involvement of complement in viral hepatitis has not been well documented. Whether NK cells affect complement component expression in HCV-infected hepatocytes remains unknown. Here, we have shown how HCV subverts the ability of NK cells to positively mediate complement protein expression.
INTRODUCTION
Natural killer (NK) cells represent a large proportion of the lymphocyte population in the liver and are involved in the early innate immune response to pathogen infection (1–3). During infection, there is a remarkable increase of hepatic NK cells, possibly due to the expansion of resident liver NK cells and/or recruitment of NK cells from the blood. The liver maintains intrahepatic NK cells in a functionally hyporesponsive state compared to splenic NK cells. NK cells in the liver display a reduced gamma interferon (IFN-γ) response to interleukin-12 (IL-12)/IL-18 stimulation (3). The liver contains a large population of functionally hyporesponsive NK cells that express high levels of the inhibitory receptor NKG2A and lack expression of major histocompatibility complex (MHC) class I-binding Ly49 receptors (4). NK cells from hepatitis C virus (HCV)-infected patients overexpress inhibitory receptors and produce cytokines, such as transforming growth factor β (TGF-β) and IL-10, and attenuate the adaptive immune response (5).
HCV affects NK cell activity through direct cell-to-cell interaction via CD81 or NK cell receptors or in an indirect manner via cytokine or TRAIL release (6–9). HCV E2 glycoprotein is suggested to inhibit NK cells directly by cross-linking CD81 (6, 10). However, E2 does not efficiently cross-link CD81 on NK cells when it is part of infectious virions, and NK cell function remains intact after in vitro exposure to cell culture-grown HCV (11). NK cells interact with hepatocytes through the interaction between NKG2D from NK cells and NKG2D ligands from hepatocytes. Major histocompatibility complex class I-related chains A and B (MICA/B) constitute one of the NKG2D ligands, which are expressed in human hepatocellular carcinoma (HCC) tissues and hepatoma cell lines (12). Although the expression of NKG2D ligands on HCV- or HBV-infected hepatocytes in humans has not yet been explored, it is expected to be elevated because in several murine models of liver injury, upregulated ligands have been detected on stressed hepatocytes (13, 14). In this study, we also examined the regulation of MICA/B in HCV-infected or uninfected hepatoma cells.
Activation of the complement system triggers a wide range of cellular responses, ranging from apoptosis to opsonization. Complement activation indirectly activates dendritic cell-mediated NK cell activation by inducing TGF-β1 (15). Although the complement system contributes to the protection of the host from virus infection, the involvement of complement in viral hepatitis has not been well documented. The complement system may inactivate NK cell function through C3 and TGF-β1 induction (15, 16), but whether NK cells affect complement component expression in HCV-infected hepatocytes remains unknown. In this study, we have examined the regulation of complement components by an established NK cell line (NK3.3) as a model (17) in the presence of HCV. Our results suggest that repression of C4 and C3 by Huh7.5 cells expressing HCV core or NS5A can be relieved by coculture with NK cells. However, NK cells exposed to cell culture-grown HCV-infected hepatocytes were unable to increase complement synthesis due to inhibition of MICA/B protein expression, thereby maintaining a potential lesion in the innate immune response via a decrease in the ability of the infected cell to respond to mitigating cellular factors.
MATERIALS AND METHODS
Cells, transfections, and NK cell stimulation.
Plasmid DNA from a mammalian expression vector (pcDNA3) carrying the HCV genotype 1a specific genomic region under the control of a cytomegalovirus promoter was transfected into Huh7.5 cells using Lipofectamine 2000 (Life Technologies, Inc., MD). Stable cell colonies were selected using neomycin and were pooled to avoid artifactual results from clonal variation. Stable transfectants of Huh7.5 cells were maintained in Dulbecco's modified Eagle medium containing 10% fetal calf serum and selection antibiotic. The expression of HCV proteins (core or NS5A) was verified by immunofluorescence or Western blot analysis (18, 19). Immortalized human hepatocytes (IHH) were generated and maintained as previously described (20). The NK3.3 cell line (17), kindly provided by Jackie Kornbluth (Saint Louis University, MO), was maintained in RPMI 1640 supplemented with 15% fetal bovine serum (FBS), 1% glutamine, 1% penicillin-streptomycin, and 200 IU/ml recombinant IL-2 (rIL-2) (R & D Systems, Inc., MN). Huh7.5 control cells or HCV core- or NS5A-expressing Huh7.5 cells were seeded on 35-mm dishes. NK cells stimulated with or without IL-2 were likewise seeded, and both cells were grown overnight. NK cells were transferred for coculture with the hepatoma cell line or to Transwells chamber (0.45-μm pore size, 24-mm diameter; Corning Costar Corporation, MA) for subsequent experiments. NK cells were cocultured with hepatoma cells (1:1 ratio). NK cells were exposed to cell culture-grown HCV at a multiplicity of infection (MOI) of ∼1 or to conditioned medium (CM) from infected hepatoma cells as a control for 24 h. After washing nonadherent HCV with PBS, NK cells were cocultured with hepatoma cells and subjected to flow cytometry.
Generation of cell culture-grown HCV.
IHH were used to grow HCV genotype 1a (clone H77) or 2a (clone JFH1), as previously described (21). Cell culture supernatant was filtered through a 0.45-μm cellulose acetate membrane (Nalgene, NY), aliquoted as virus stocks, and stored at −70°C for single use. HCV RNA was quantified (IU/ml) by real-time PCR (Prism 7500 real-time thermocycler; ABI, CA) with the use of analyte-specific reagents (ASR, Abbott, IL) in the Pathology Clinical Laboratory at Saint Louis University. The infectious HCV titer from cell culture supernatant was also measured by fluorescence-focus forming units (FFU) using an NS5A-specific monoclonal antibody (9E10) kindly provided by Charlie Rice (Rockefeller University, NY). Expression of HCV core and NS5A was verified by immunofluorescence as previously described (22, 23).
Western blots analysis.
Proteins in cell lysates were resolved by SDS-PAGE, transferred onto nitrocellulose membranes, and blotted with a rabbit anti-C4 primary antibody. Positive signals were detected using a peroxidase-conjugated secondary antibody and an enhanced chemiluminescence Super Signal West Pico detection kit (Thermo Chemical Company, IL). Cellular actin was detected similarly for comparison of the protein load in each lane. Experiments were done at least three times and exhibited consistent results. Representative data are shown in Results.
Flow cytometry.
NK cells incubated with cell culture-grown HCV or mock treated were analyzed for surface expression of NKG2D by flow cytometry. NK cells were also cocultured with HCV-infected Huh7.5 cells for 24 h for comparison. For fluorochrome staining, cells were incubated with an antibody to NKG2D (Santa Cruz, CA) in phosphate-buffered saline (PBS) containing 0.5% bovine serum albumin (BSA) for 30 min at room temperature. After washing, cells were treated with Alexa Fluor 647-conjugated secondary antibody for 30 min and resuspended in PBS. NKG2D-stained cells were gated according to their size (forward light scatter) and granularity (side light scatter) using a flow cytometer (Becton Dickinson, CA). Surface marker expression on gated cells was analyzed using CellQuest software (BD Immunocytometry Systems).
Antibodies.
Western blot analyses were performed using antibodies to C4, C3, IRF-1, USF-1, C/EBP-β, phospho-C/EBP-β, or NKG2D (Santa Cruz Biotechnology, CA) or MICA/B (R & D Systems, MN). Matched control unrelated antibodies, goat antiserum to thrombin R, and mouse monoclonal antibody (MAb) IgG2a to HNF3β (Santa Cruz) were also used.
Statistical analysis.
Results were expressed as the mean ± standard deviation (SD), and statistical analyses were performed using the 2-tailed unpaired Student t test in GraphPad Prism 5 (GraphPad, La Jolla, CA). A P value of <0.05 was considered statistically significant.
RESULTS
Interactions between NK cells and hepatoma cells expressing HCV protein induce an increase in C4 synthesis.
Hepatocytes in the liver are the primary host for HCV replication and are the main source for complement synthesis. We have previously identified C4 downregulation in hepatoma cells expressing HCV core or NS5A protein (24). NK cells play an important role in controlling viral hepatitis, liver fibrosis, and liver tumorigenesis and also contribute to the pathogenesis of liver injury and inflammation (25). Thus, we examined the effect of activated NK cells on C4 recovery. IL-2, a cytokine with pleotropic effects, is required for proliferation and activation of many cell types, including T and NK lymphocytes. IL-2 drives the secretion of IFN-γ and other cytokines and the expression of the activation markers CD25 and CD69 on the NK cell surface. Since IL-2 increases activation of NK cells (26), we used rIL-2 (200 IU/ml) in our experiment. IL-2-treated NK cell activation induced C4 recovery in Huh7.5 cells expressing HCV core or NS5A, while untreated NK cells did not (Fig. 1A). Prior to lysis of Huh7.5 cells, NK cells were removed and the adherent cell layer was rinsed. Thus, the observations suggested that coculture with NK cells leads to recovery of C4 expression and that cell activation is important for C4 recovery in HCV core- or NS5A-expressing cells.
FIG 1.

Association between activated NK cells and hepatoma cells and role of cytokines in augmenting C4 expression by HCV proteins. (A) NK3.3 cells were preincubated with or without 200 IU/ml rIL-2 for 24 h and cocultured for 24 h with Huh7.5 cells stably transfected with HCV core or NS5A. (B) Huh7.5 cells stably transfected with HCV core or NS5A were incubated with NK3.3 conditioned medium (NKCM) for 24 h. NKCM containing rIL-2 was collected after 24 h of culture. Cell lysates were analyzed for C4 protein by Western blotting. (C) Huh7.5 cells stably transfected with HCV core or NS5A were cocultured separately with rIL-2-activated NK3.3 cells in a transwell chamber for 24 h. Hepatoma cells were lysed and C4 expression status analyzed by Western blotting.
Since activated NK cells secret both proinflammatory and immunosuppressive cytokines in the culture medium (27), we examined the effect of NK cell conditioned medium (NKCM) on recovery of C4 from HCV protein-mediated suppression in Huh7.5 cells (Fig. 1B). C4 expression was inhibited by HCV core- or NS5A-expressing Huh7.5 cells compared to the parental control. NKCM did not promote C4 recovery, suggesting that soluble factors generated from NK cells may not have a role in upregulation of C4 synthesis. To further verify whether soluble factors affect C4 recovery in HCV core- or NS5A-expressing cells, NK cells were maintained in a transwell chamber (0.45-μm pore size) above cultured Huh7.5 cells for 24 h. As expected, C4 recovery was not observed during coculture in the transwell chamber (Fig. 1C), indicating that soluble cytokines or chemokines from NK cells did not promote C4 recovery under our experimental conditions. Thus, the results suggested that a contact between NK cells and hepatocytes is necessary to promote C4 expression.
NKG2D and MICA/B interaction promotes C4 synthesis.
NKG2D is one of the activation receptors expressed by NK cells, and it helps in associating with NKG2D ligands on hepatoma cells. To examine whether cell-to-cell interaction plays a role in the C4 recovery, we blocked the interaction between these two cell types using NKG2D-specific neutralizing antibody. The use of antibody led to a loss in C4 induction in HCV core- or NS5A-expressing cells in the coculture with NK cells (Fig. 2A). On the other hand, a significant loss of C4 induction was not observed when goat antiserum or a mouse monoclonal antibody (isotype 2a) to unrelated antigens was used as a negative control. Jinushi et al. (12) have shown that MICA/B is expressed in human HCC tissues and hepatoma cell lines. The expression of MICA/B on hepatocytes is involved in susceptibility to the effects of NKG2D by NK cells (28). We have observed that the use of a neutralizing MICA/B antibody blocks recovery of C4 in HCV core- or NS5A-expressing cells upon coculture with NK cells. Therefore, interactions between these two cell receptors appear to be important in modulating C4 expression.
FIG 2.
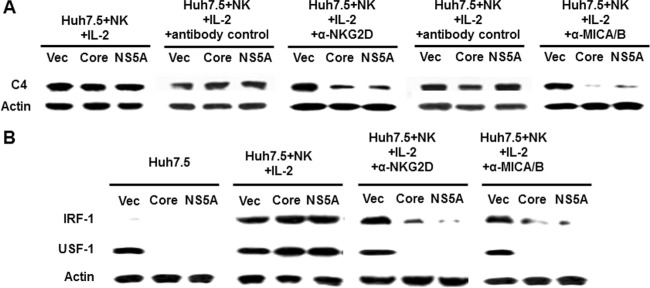
NKG2D and MICA/B interaction promotes C4 synthesis. rIL-2-activated NK3.3 and Huh7.5 cells stably transfected with HCV core or NS5A were preincubated for 30 min with anti-NKG2D, anti-MICA/B, or negative-control antibody and cocultured for 24 h. Nonadherent NK cells were removed, and the Huh7.5 cell monolayer was washed and analyzed for C4, IRF-1, USF-1, and actin expression by Western blotting. Cellular actin was used for comparison of protein load in each experiment.
We have previously observed that C4 synthesis is mediated by USF-1 and IRF-1 transcription factors (24), and thus we examined whether cell-to-cell interaction affects the status of these transcription factors for C4 synthesis. IRF-1 was expressed at a modest level in untreated cells, while a dramatic recovery of IRF-1 and USF-1 was observed after coculture of IL-2-activated NK cells and hepatoma cells (Fig. 2B). Recovery of the transcription factors did not occur when cells were treated with neutralizing antibody to NKG2D or MICA/B (Fig. 2A). Together, these results suggested that a cell-to-cell interaction between NK and hepatoma cells via NKG2D and MICA/B helps in the recovery of IRF-1 and USF-1 transcription factors for C4 synthesis.
NK cells fail to recover C4 expression in the presence of HCV.
C4 production from Huh7.5 cells was examined after exposure to HCV 2a and coculture with NK cells by Western blotting. NK cells in the absence of HCV increased C4 production in Huh7.5 cells, while HCV 2a exposure failed to increase C4 production (Fig. 3A). Densitometric scanning of the Western blot result displaying the relative induction of the C4 level is shown separately (Fig. 3B). Next, we examined the status of the IRF-1 and USF-1 transcription factors during coculture of Huh7.5 and NK cells in the presence of cell culture-grown HCV. The levels of IRF-1 in mock-infected Huh7.5 cells and HCV-infected cells were similar, and this could be due to a low basal expression in control cells. NK cell coculture with Huh7.5 cells increased both IRF-1 and USF-1 production. Interestingly, expression of these transcription factors was inhibited in the presence of HCV and NK cells (Fig. 3C). Coculture of NK cells and Huh7.5 cells increased IRF-1 and USF-1 expression, but it decreased in the presence of HCV 2a. Densitometric scanning of the Western blot result displayed relative expression of IRF-1 and USF-1 (Fig. 3D and E). Thus, the results suggest that HCV can impair NK cell function on C4 regulation via a mechanism that is independent of the effects of HCV core or NS5A, which occur prior to modulation of C4-specific transcription factors.
FIG 3.

NK cells fail to promote C4 augmentation in the presence of HCV. (A) rIL-2-activated NK cells were cocultured with Huh7.5 cells infected with HCV genotype 2a for 24 h. Mock-infected cells were cocultured similarly to the control. After removal of nonadherent NK cells, Huh7.5 cells were lysed and subjected to Western blot analysis for C4 protein expression. (B) Densitometric scanning of the Western blot displaying relative C4 inhibition. The results are from 3 independent experiments, with standard deviations indicated by error bars. The P value for the Huh7.5 control versus HCV 2a-infected cells was 0.0011, that for the control versus coculture with NK cells was 0.0179, and that for the control versus HCV 2a-infected cells and coculture with NK cells was 0.0006. (C) The expression status of transcription factors IRF-1 and USF-1 from cell lysates was examined by Western blotting. Cellular actin was used for comparison of protein loads in each experiment. (D and E) Results are from 3 independent experiments, with standard deviations indicated by error bars. The difference in IRF-1 expression between Huh7.5 control and HCV 2a-infected cells was not statistically significant (P > 0.05). However, changes in IRF-1 or USF-1 expression were statistically significant in comparison among Huh7.5 control and all other experimental samples (P = 0.004 to 0.0001).
NK cells fail to recover C3 expression in the presence of HCV.
C3 is another important component of complement-mediated immunity. We previously reported that C3 synthesis in hepatocytes is inhibited by NS5A (29). C3 expression could be rescued in coculture of NS5A-expressing Huh7.5 cells with NK cells (Fig. 4A). The C3 promoter is regulated primarily via the activation of the transcription factor C/EBP-β. Thus, we examined whether cell-to-cell interaction affects C/EBP-β regulation. We examined the C/EBP-β activation status in NS5A-expressing Huh7.5 cells incubated alone or cocultured together with NK cells for 2 h. After removal of nonadherent NK cells, hepatoma cells were harvested and lysed for C/EBP-β expression and phosphorylation status from Western blot analysis (Fig. 4B). C/EBP-β production was inhibited in NS5A-expressing cells, while its expression was dramatically increased upon coculture with NK cells. Similarly, the presence of the phosphorylated form of C/EBP-β was inhibited by NS5A protein, while it was increased upon coculture with NK cells. The recovery of C3 in NS5A-expressing Huh7.5 cells after coculture with NK cells, however, did not occur in the presence of the neutralizing antibodies anti-NKG2D and anti-MICA/B (Fig. 4C), indicating that C3 induction in NS5A-expressing Huh7.5 cells is mediated by cell-to-cell interaction. Next, we examined the C3 level from HCV 2a-infected Huh7.5 cells after coculture with NK cells. Similar to the case for C4, C3 production was increased by NK cells in the absence of HCV, while it remained decreased in HCV 2a-infected Huh7.5 cells (Fig. 4D). We also examined the status of C/EBP-β during coculture of Huh7.5 and NK cells in the presence or absence of cell culture-grown HCV. Coculture of control NK cells with Huh7.5 cells increased both C/EBP-β and phospho-C/EBP-β production. However, activation of C/EBP-β was inhibited in the presence of HCV (Fig. 4E). Coculture of HCV-infected Huh7.5 cells with NK cells led to a significant decrease in C/EBP-β production as well as phosphorylation of C/EBP-β. Together, our results suggest that HCV impairs the ability of NK cells to positively regulate C3 production in hepatoma cells by the regulation of C/EBP-β.
FIG 4.
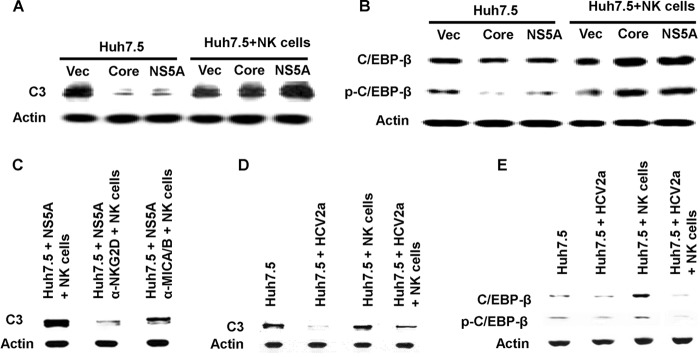
NK cells fail to promote C3 augmentation in the presence of HCV. (A) C3 expression in Huh7.5 cells stably transfected with NS5A following coculture with rIL-2-activated NK3.3 cells for 24 h was determined by Western blotting. (B) Huh7.5 cells stably transfected with HCV NS5A were cocultured with rIL-2-activated NK3.3 cells for 2 h, and cell lysates were analyzed for total and phospho-C/EBP-β expression by Western blotting. (C) rIL-2-activated NK3.3 and Huh7.5 cells stably transfected with HCV NS5A were preincubated for 30 min with anti-NKG2D and anti-MICA/B antibody and cocultured for 24 h. Nonadherent NK cells were removed, and the Huh7.5 cell monolayer was washed and analyzed for C3 expression by Western blotting. (D) rIL-2-activated NK cells were cocultured with Huh7.5 cells infected with HCV genotype 2a for 24 h. Mock-infected cells were cocultured similarly to the control. After removal of nonadherent NK cells, Huh7.5 cells were lysed and subjected to Western blot analysis for C3 protein expression. (E) The expression status of total C/EBP-β and phosphorylated C/EBP-β from a similar experiment was analyzed by Western blotting. Cellular actin was used for comparison of protein loads in each experiment.
HCV inhibits MICA/B expression in hepatoma cells but does not significantly inhibit NKG2D.
NKG2D appeared to play an important role in the recovery of C4 complement expression in a coculture of NK cells and Huh7.5 cells. We evaluated the expression of the NKG2D receptor following HCV exposure of NK cells or coculture with HCV-infected Huh7.5 cells. NKG2D expression on the NK cell surface was not significantly decreased (Fig. 5A) upon HCV exposure compared to that for mock-treated NK cells (71.2% for mock-infected versus 68.2% for HCV-exposed cells). NKG2D expression was also not significantly decreased in the coculture with HCV-infected Huh7.5 cells and NK cells (70.5% for NK plus Huh7.5 cells versus 63.8% for NK plus Huh7.5 cells plus HCV).
FIG 5.
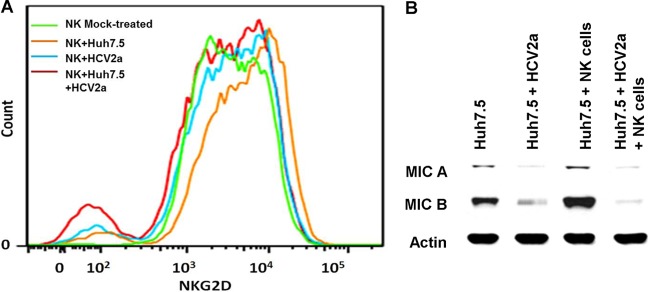
HCV-infected cells inhibit the expression of NKG2D or MICA/B. (A) Cell surface expression of NKG2D was analyzed in mock-treated NK cells, NK cells after coculture with Huh7.5, NK cells infected with HCV 2a, or NK cells incubated simultaneously with Huh7.5 cells and HCV genotype 2a. Representative results of fluorescence-activated cell sorter (FACS) analysis with anti-NKG2D antibody, followed by Alexa 647 Fluor-conjugated donkey anti-goat treatment, are shown. (B) Mock-treated or HCV genotype 2a-infected Huh7.5 cells were cocultured with NK cells. Hepatoma cells were lysed after removal of nonadherent NK cells and subjected to Western blot analysis for MICA/B. Cellular actin was used for comparison of protein loads in each well. In both the experiments, NK cells were activated with rIL-2 (200 IU/ml).
MICA/B is expressed in human HCC tissues and hepatoma cell lines (12). MICA/B expression was significantly decreased in HCV-infected Huh7.5 cells. NK cell coculture increased the expression of MICA/B in Huh7.5 cells to a level above that seen in the mock-treated control. Interestingly, MICA/B protein expression remained at a very low level in HCV-infected Huh7.5 cells despite the presence of NK cells (Fig. 5B), indicating that the presence of HCV strongly inhibits their expression in hepatocytes. Thus, the results suggest that the expression of MICA/B may be inhibited in hepatocytes of chronically HCV-infected patients, resulting in inefficient interaction with NK cells and failure to recover C4 synthesis.
HCV NS2 and NS5B inhibit MICA/B expression in Huh7.5 cells.
As prior analysis indicated that HCV NS5A or core was unable to inhibit NK cell-mediated enhancement of complement protein synthesis, we examined which HCV protein(s) may act to inhibit MICA/B expression in Huh7.5 cells. The MICA/B expression level was analyzed in cells transfected with empty vector, HCV core, E1/E2/p7, and an HCV subgenomic replicon encoding nonstructural proteins (BB7) after 24 h of coculture with NK cells. MICA/B expression in hepatoma cells expressing HCV structural proteins was increased by NK cell coculture compared to Huh7.5 cells alone, and a significant increase was seen in E1/E2/p7-expressing cells. MICA and B expression in BB7 cells remained inhibited after coculture with NK cells (Fig. 6A), indicating that one of the nonstructural proteins is involved in the inhibition of NK cell-mediated MICA/B expression in hepatoma cells. Further study revealed that among the nonstructural proteins, both HCV NS2 and NS5B may play a role in MICA/B inhibition during coculture of HCV-infected Huh7.5 cells with NK cells (Fig. 6B). As expected, no regulation of MICA/B was apparent in HCV core- or NS5A-expressing cells, allowing for the NK cell-mediated enhancement of C3 and C4 synthesis noted in these cell lines, which previously exhibited suppression of these proteins. Our results suggest that HCV NS2 and NS5B inhibit MICA/B expression in Huh7.5 cells, which might lead to limited interaction between HCV-infected Huh7.5 cells and NK cells, resulting in the impairment of NK cell-mediated C4 or C3 synthesis during HCV infection.
FIG 6.
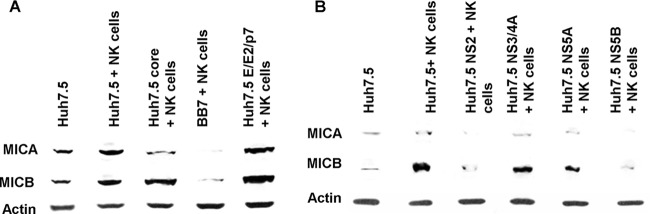
HCV NS2 and NS5B inhibit MICA/B expression in Huh7.5 cells in a coculture with NK cells. (A) MICA/B expression in HCV genomic region-transfected Huh7.5 cells was analyzed by Western blotting following coculture with rIL-2-activated NK cells. Stable transfectants with empty vector, core, nonstructural proteins (BB7), or E1/E2/p7 were used. (B) Huh7.5 cells transiently transfected with NS2, NS3/4A, NS5A, or NS5B were cocultured with rIL-2-activated NK cells, and MICA/B expression in Huh7.5 cells was analyzed by Western blotting. Cellular actin was used for comparison of protein loads in each experiment.
DISCUSSION
We have shown that HCV attenuates innate immune function in infected human hepatoma cells by reducing the ability of NK3.3 cells to stimulate C4/C3 complement synthesis. NK cells may directly target HCV-infected hepatocytes or indirectly influence immune cells, such as dendritic cells (DCs) or T cells for viral clearance. Perturbations in NK cell frequency, phenotype, and function have been shown in chronically HCV-infected patients (30). The peripheral blood NK cell number and percentage of total lymphocyte population are lower in HCV-infected individuals (8). Additionally, the proportion of NKp30- and NKp46-expressing cells is reduced in patients with chronic hepatitis C (31). NKG2D expression is lowered on circulating NK cells from patients with chronic hepatitis C (32). These features may lead to failure of C4/C3 recovery in chronically HCV-infected patients (8). Here, we have shown that IL-2-activated NK cells promote C4 recovery via the interaction between NKG2D and MICA/B, indicating that activation by cell-to-cell interaction may be important for complement regulation.
NK cells recover C4 production in core- or NS5A-expressing Huh7.5 cells through the augmentation of the IRF-1 and USF-1 transcription factors. However, NK cells fail to recover C4 augmentation in HCV-infected Huh7.5 cells. The fate of NK cells in HCV infection may be determined by the integration of signals generated by direct cell-to-cell interaction and cytokine stimulation. Engagement of CD81 to plate-bound anti-CD81 or recombinant HCV-E2 inhibits NK cell activation (6). Exposure to inhibitory cytokines, such as IL-10 and TGF-β, suppresses NK cell cytotoxicity, while chronic exposure to activating cytokine IFN-α contributes to the polarization of NK cells toward cytotoxicity (8). NK cells kill MHC class I-negative tumor targets (33). An interaction between MHC I (HLA-ABC) of the target cell and KIR of the NK cell surface inhibits NK cell activation. NK cells also seem to be inactivated by hepatocytes. Our cytokine array data have shown decreased expression of cytokines, including IFN-γ from NK cells in a coculture with hepatocytes (data not shown). Yoon et al. (9) have shown that NK cells increase NKG2D expression after coculture with Huh7.5 cells; however, NKG2D expression is inhibited following incubation with HCV-infected hepatocytes. On the other hand, coculture of Huh7.5 cells was shown to increase the expression of inhibitory receptors KIR2DL1 and KIR2DL2/DL3 in NK cells but not NKG2A. Thus, the interaction between NK cells and HCV-infected hepatocytes may result in inactivation of NK cells.
MICA/B plays an important role in the interaction between hepatocytes and NK cells. We have shown that MICA/B expression in hepatoma cells is downregulated upon HCV infection, and NKG2D expression from NK3.3 cells is also reduced by HCV. Therefore, cell-to-cell contact with HCV-infected cells negatively modulates the functional capacity of NK cells, and inhibition of NK cell function is associated with downregulation of activating receptors on NK cell surfaces. Physical interaction between NKG2D and MICA/B may increase cytokine and chemokine production from NK cells and hepatocytes, which may accelerate the synthesis of complement components in hepatocytes. However, C4/C3 production is not increased by NKCM or treatment with individual cytokines (TNF-α, IFN-γ, IL-2, or IL-6). NK cells express a low level of C3/C4 complement components (references 34 and 35 and our unpublished data). The variability in the levels of C3/C4 in our Western blots may result from contributions of NK cells. However, if NK cells have a significant influence, C3 and C4 would increase in the blocking experiments, instead of being decreased by anti-NKG2D or anti-MIC/B antibody in the coculture. Further studies are necessary to elucidate how the interaction between NK cells and hepatocytes may positively affect immune function against HCV infection.
IFN-γ is the major cytokine that NK cells secrete and is a critical factor for inhibition of viral replication (36). It is known that concurrent engagement of activating receptors and cytokine receptors on NK cells induces IFN-γ secretion by NK cells (8). Therefore, a decrease in activating receptor expression would likely be correlated with IFN-γ inhibition by CD56dim NK cells (1). In another study, a polarized NK cell phenotype was induced by chronic exposure to HCV-induced IFN-α. This phenotype may contribute to liver injury through TRAIL expression and cytotoxicity, whereas the lack of increase in IFN-γ production may facilitate the inability to clear HCV (37).
NK cells may directly target infected hepatocytes or may act indirectly by influencing other immune cells such as DCs or T cells. In the acute phase of HCV infection, expression of the activating receptor NKG2D is increased. Functional experiments also showed augmented IFN-γ production and cytotoxicity in these patients. Therefore, in the acute phase of HCV infection, there is activation of NK cells, indicating their role in the immune response, whereas chronically HCV-infected patients have shown perturbations in NK cell frequency, phenotype, and function.
Peripheral blood NK cell frequencies, both absolute numbers and percentages of the total lymphocyte population, are reduced in chronically HCV-infected patients compared to healthy individuals. The reduction of NK cell frequency and function may lead to failure of C4 recovery in chronically HCV-infected hepatocytes (8). Natural killer cells elaborate IFN-γ to mediate antiviral effects. However, HCV E2 protein binds the NK CD81 receptor, decreasing the release of IFN-γ and cytotoxic granules by NK cells (6, 10).
The expression of NKG2D ligands on HCV- or HBV-infected hepatocytes in humans has not yet been examined. However, NKG2D ligands are expected to be elevated, since several murine models of liver injury upregulated these ligands on stressed hepatocytes (38). Here, we examined the expression of one of NKG2D ligands, MICA/B. Interestingly, expression of MICA/B was observed in uninfected hepatoma cells but not in HCV-infected hepatoma cells following coculture with NK3.3 cells. In particular, MICA/B expression was downregulated by NS2 and NS5B protein expression, resulting in a loss of the C3/C4 recovery mechanism conveyed by NK cells in culture. We have also shown that the inhibition between MICA/B and NKG2D using neutralization antibodies results in an inhibition of C4/C3 production. NKG2D is a relatively conserved protein on NK cells, the function of which appears to be important for C3/C4 recovery as indicated by our observations. Its interaction with MICA/B is also well defined in the literature (30) and is the mechanism which we now believe to be relevant in C3/C4 synthesis and modulation in the face of added NK cell-mediated immune modulation.
In summary, our study suggests a multifaceted approach undertaken by HCV to impair normal immune function beyond the initial reduction of C3/C4 seen in HCV-infected patients. Further analysis details how HCV subverts the ability of NK3.3 cells to positively mediate complement protein expression via an additional inhibition of MICA/B signaling. This study highlights the cooperative approach of individual HCV proteins in the control of host immune function at multiple points to perpetuate virus fitness.
ACKNOWLEDGMENTS
We thank Lin Cowick for preparation of the manuscript.
This work was supported by research grant U54-AI057160 from the NIAID to the Midwest Regional Center of Excellence (MRCE) for Biodefense and Emerging Infectious Diseases Research, by DK080812 from the NIDDK, and by the Internal Blue Ribbon Fund of Saint Louis University.
Footnotes
Published ahead of print 18 December 2013
REFERENCES
- 1.Caligiuri MA. 2008. Human natural killer cells. Blood 112:461–469. 10.1182/blood-2007-09-077438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nellore A, Fishman JA. 2011. NK cells, innate immunity and hepatitis C infection after liver transplantation. Clin. Infect. Dis. 52:369–377. 10.1093/cid/ciq156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krueger PD, Lassen MG, Qiao H, Hahn YS. 2011. Regulation of NK cell repertoire and function in the liver. Crit. Rev. Immunol. 31:43–52. 10.1615/CritRevImmunol.v31.i1.40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lassen MG, Lukens JR, Dolina JS, Brown MG, Hahn YS. 2010. Intrahepatic IL-10 maintains NKG2A+Ly49− liver NK cells in a functionally hyporesponsive state. J. Immunol. 184:2693–2701. 10.4049/jimmunol.0901362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jinushi M, Takehara T, Tatsumi T, Kanto T, Miyagi T, Suzuki T, Kanazawa Y, Hiramatsu N, Hayashi N. 2004. Negative regulation of NK cell activities by inhibitory receptor CD94/NKG2A leads to altered NK cell-induced modulation of dendritic cell functions in chronic hepatitis C virus infection. J. Immunol. 173:6072–6081 [DOI] [PubMed] [Google Scholar]
- 6.Crotta S, Stilla A, Wack A, D'Andrea A, Nuti S, D'Oro U, Mosca M, Filliponi F, Brunetto RM, Bonino F, Abrignani S, Valiante NM. 2002. Inhibition of natural killer cells through engagement of CD81 by the major hepatitis C virus envelope protein. J. Exp. Med. 195:35–41. 10.1084/jem.20011124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Golden-Mason L, Rosen HR. 2006. Natural killer cells: Primary target for hepatitis C virus immune evasion strategies? Liver Transpl. 12:363–372. 10.1002/lt.20708 [DOI] [PubMed] [Google Scholar]
- 8.Cheent K, Khakoo SI. 2011. Natural killer cells and hepatitis C: action and reaction. Gut 60:268–278. 10.1136/gut.2010.212555 [DOI] [PubMed] [Google Scholar]
- 9.Yoon JC, Lim JB, Park JH, Lee JM. 2011. Cell-to-cell contact with hepatitis C virus-infected cells reduces functional capacity of natural killer cells. J. Virol. 85:12557–12569. 10.1128/JVI.00838-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tseng CT, Klimpel GR. 2002. Binding of the hepatitis C virus envelope protein E2 to CD81 inhibits natural killer cell functions. J. Exp. Med. 195:43–49. 10.1084/jem.20011145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoon JC, Shiina M, Ahlenstiel G, Rehermann B. 2009. Natural killer cell function is intact after direct exposure to infectious hepatitis C virions. Hepatology 49:12–21. 10.1002/hep.22624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jinushi M, Takehara T, Tatsumi T, Kanto T, Groh V, Spies T, Kimura R, Miyagi T, Mochizuki K, Sasaki Y, Hayashi N. 2003. Expression and role of MICA and MICB in human hepatocellular carcinomas and their regulation by retinoic acid. Int. J. Cancer 104:354–361. 10.1002/ijc.10966 [DOI] [PubMed] [Google Scholar]
- 13.Raulet DH. 2003. Roles of the NKG2D immunoreceptor and its ligands. Nat. Rev. Immunol. 3:781–790. 10.1038/nri1199 [DOI] [PubMed] [Google Scholar]
- 14.Kahraman A, Fingas CD, Syn WK, Gerken G, Canbay A. 2012. Role of stress-induced NKG2D ligands in liver diseases. Liver Int. 32:370–382. 10.1111/j.1478-3231.2011.02608.x [DOI] [PubMed] [Google Scholar]
- 15.Qing X, Koo GC, Salmon JE. 2012. Complement regulates conventional DC-mediated NK-cell activation by inducing TGF-β1 in Gr-1+ myeloid cells. Eur. J. Immunol. 42:1723–1734. 10.1002/eji.201142290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang SY, Veeramani S, Racila E, Cagley J, Fritzinger DC, Vogel CW, St John W, Weiner GJ. 2009. Depletion of the C3 component of complement enhances the ability of rituximab-coated target cells to activate human NK cells and improves the efficacy of monoclonal antibody therapy in an in vivo model. Blood 114:5322–5330. 10.1182/blood-2009-01-200469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kornbluth J, Flomenberg N, Dupont B. 1982. Cell surface phenotype of a cloned line of human natural killer cells. J. Immunol. 129:2831–2837 [PubMed] [Google Scholar]
- 18.Banerjee A, Saito K, Meyer K, Banerjee S, Ait-Goughoulte M, Ray RB, Ray R. 2009. Hepatitis C virus core protein and cellular protein HAX-1 promotes 5-fluorouracil-mediated hepatocyte growth inhibition. J. Virol. 83:9663–9671. 10.1128/JVI.00872-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ait-Goughoulte M, Banerjee A, Meyer K, Mazumdar B, Saito K, Ray RB, Ray R. 2010. Hepatitis C virus core protein interacts with fibrinogen-β and attenuates cytokine stimulated acute phase response. Hepatology 51:1505–1513. 10.1002/hep.23502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ray RB, Meyer K, Ray R. 2000. Hepatitis C virus core protein promotes immortalization of primary human hepatocytes. Virology 271:197–204. 10.1006/viro.2000.0295 [DOI] [PubMed] [Google Scholar]
- 21.Kanda T, Basu A, Steele R, Wakita T, Ryerse JS, Ray R, Ray RB. 2006. Generation of infectious hepatitis C virus in immortalized human hepatocytes. J. Virol. 80:4633–4639. 10.1128/JVI.80.9.4633-4639.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Banerjee A, Meyer K, Mazumdar B, Ray RB, Ray R. 2010. Hepatitis C virus differentially modulates activation of forkhead transcription factors and insulin induced metabolic gene expression. J. Virol. 84:5936–5946. 10.1128/JVI.02344-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bose SK, Shrivastava S, Meyer K, Ray RB, Ray R. 2012. Hepatitis C virus activates mTOR/S6K1 signaling pathway in inhibiting IRS-1 function for insulin resistance. J. Virol. 86:6315–6322. 10.1128/JVI.00050-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Banerjee A, Mazumdar B, Meyer K, Di Bisceglie AM, Ray RB, Ray R. 2011. Transcriptional repression of C4 complement by hepatitis C virus proteins. J. Virol. 85:4157–4166. 10.1128/JVI.02449-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tian Z, Chen Y, Gao B. 2013. Natural killer cells in liver disease. Hepatology 57:1654–1662. 10.1002/hep.26115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lehmann C, Zeis M, Uharek L. 2001. Activation of natural killer cells with interleukin 2 (IL-2) and IL-12 increases perforin binding and subsequent lysis of tumour cells. Br. J. Haematol. 114:660–665. 10.1046/j.1365-2141.2001.02995.x [DOI] [PubMed] [Google Scholar]
- 27.Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Laneir LL, Yokoyama WM, Ugolini S. 2011. Innate or adaptive immunity? The example of natural killer cells. Science 331:44–49. 10.1126/science.1198687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bae DS, Hwang YK, Lee JK. 2012. Importance of NKG2D-NKG2D ligands interaction for cytolytic activity of natural killer cell. Cell Immunol. 276:122–127. 10.1016/j.cellimm.2012.04.011 [DOI] [PubMed] [Google Scholar]
- 29.Mazumdar B, Kim H, Meyer K, Bose SK, Di Bisceglie AM, Ray RB, Ray R. 2012. Hepatitis C virus proteins inhibit C3 complement production. J. Virol. 86:2221–2228. 10.1128/JVI.06577-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Varchetta S, Mele D, Mantovani S, Oliviero B, Cremonesi E, Ludovisi S, Michelone G, Alessiani M, Rosati R, Montorsi M, Mondelli MU. 2012. Impaired intrahepatic natural killer cell cytotoxic function in chronic hepatitis C virus infection. Hepatology 56:841–849. 10.1002/hep.25723 [DOI] [PubMed] [Google Scholar]
- 31.Nattermann J, Feldmann G, Ahlenstiel G, Langhans B, Sauerbruch T, Spengler U. 2006. Surface expression and cytolytic function of natural killer cell receptors is altered in chronic hepatitis C. Gut 55:869–877. 10.1136/gut.2005.076463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sène D, Levasseur F, Abel M, Lambert M, Camous X, Hernandez C, Pene V, Rosenberg AR, Jouvin-Marche E, Marche PN, Cacoub P, Caillat-Zucman S. 2010. Hepatitis C virus (HCV) evades NKG2D-dependent NK cell responses through NS5A-mediated imbalance of inflammatory cytokines. PLoS Pathog. 6:e1001184. 10.1371/journal.ppat.1001184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Elliott JM, Yokoyama WM. 2011. Unifying concepts of MHC-dependent natural killer cell education. Trends Immunol. 32:364–372. 10.1016/j.it.2011.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Finberg RW, White W, Nicholson-Weller A. 1992. Decay-accelerating factor expression on either effector or target cells inhibits cytotoxicity by human natural killer cells. J. Immunol. 149:2055–2060 [PubMed] [Google Scholar]
- 35.Morgan BP, Gasque P. 1997. Extrahepatic complement biosynthesis: where, when and why? Clin. Exp. Immunol. 107:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schoenborn JR, Wilson CB. 2007. Regulation of interferon-gamma during innate and adaptive immune responses. Adv. Immunol. 96:41–101. 10.1016/S0065-2776(07)96002-2 [DOI] [PubMed] [Google Scholar]
- 37.Ahlenstiel G, Titerence RH, Koh C, Edlich B, Feld JJ, Rotman Y, Ghany MG, Hoofnagle JH, Liang TJ, Heller T, Rehermann B. 2010. Natural killer cells are polarized toward cytotoxicity in chronic hepatitis C in an interferon-alfa-dependent manner. Gastroenterology 138:325–335. 10.1053/j.gastro.2009.08.066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raulet DH, Gasser S, Gowen BG, Deng W, Jung H. 2013. Regulation of ligands for the NKG2D activating receptor. Annu. Rev. Immunol. 31:413–441. 10.1146/annurev-immunol-032712-095951 [DOI] [PMC free article] [PubMed] [Google Scholar]