

ABSTRACT
Glycoprotein B (gB), the fusogen of herpes simplex virus (HSV), is a class III fusion protein with a trimeric ectodomain of known structure for the postfusion state. Seen by negative-staining electron microscopy, it presents as a rod with three lobes (base, middle, and crown). gB has four functional regions (FR), defined by the physical location of epitopes recognized by anti-gB neutralizing monoclonal antibodies (MAbs). Located in the base, FR1 contains two internal fusion loops (FLs) and is the site of gB-lipid interaction (the fusion domain). Many of the MAbs to FR1 are neutralizing, block cell-cell fusion, and prevent the association of gB with lipid, suggesting that these MAbs affect FL function. Here we characterize FR1 epitopes by using electron microscopy to visualize purified Fab-gB ectodomain complexes, thus confirming the locations of several epitopes and localizing those of MAbs DL16 and SS63. We also generated MAb-resistant viruses in order to localize the SS55 epitope precisely. Because none of the epitopes of our anti-FR1 MAbs mapped to the FLs, we hyperimmunized rabbits with FL1 or FL2 peptides to generate polyclonal antibodies (PAbs). While the anti-FL1 PAb failed to bind gB, the anti-FL2 PAb had neutralizing activity, implying that the FLs become exposed during virus entry. Unexpectedly, the anti-FL2 PAb (and the anti-FR1 MAbs) bound to liposome-associated gB, suggesting that their epitopes are accessible even when the FLs engage lipid. These studies provide possible mechanisms of action for HSV neutralization and insight into how gB FR1 contributes to viral fusion.
IMPORTANCE For herpesviruses, such as HSV, entry into a target cell involves transfer of the capsid-encased genome of the virus to the target cell after fusion of the lipid envelope of the virus with a lipid membrane of the host. Virus-encoded glycoproteins in the envelope are responsible for fusion. Antibodies to these glycoproteins are important biological tools, providing a way of examining how fusion works. Here we used electron microscopy and other techniques to study a panel of anti-gB antibodies. Some, with virus-neutralizing activity, impair gB-lipid association. We also generated a peptide antibody against one of the gB fusion loops; its properties provide insight into the way the fusion loops function as gB transits from its prefusion form to an active fusogen.
INTRODUCTION
Herpes simplex virus (HSV) has four envelope glycoproteins that are essential for virus entry into cells: glycoproteins gD, gH, gL, and gB. All herpesviruses use a combination of gB and the heterodimer gH/gL to carry out virus-cell fusion, with current evidence indicating that gB is the fusion protein (1–4). Like most alphaherpesviruses, HSV also requires the receptor-binding protein gD to carry out fusion. Our current model of HSV fusion starts with the binding of gD to one of its receptors (nectin-1, herpesvirus entry mediator [HVEM], or 3-O-sulfotransferase [3-OST] heparan sulfate) (5), transmitting a physical signal to gH/gL, which, in turn, acts upon gB to trigger fusion (2).
HSV-1 gB (gB1) is a 904-amino-acid (aa) type I membrane glycoprotein whose crystal structure identifies it as a class III fusion protein (1, 6). The gB1 ectodomain comprises residues 31 to 773, and several crystal structures have been determined for residues 103 to 725, all of them in the postfusion form. These structures show a homotrimer with a long central coiled-coil and internal bipartite fusion loops (fusion loops 1 and 2 [FL1 and FL2]) (Fig. 1A). A similar crystal structure has been observed for gB from the betaherpesvirus Epstein-Barr virus (EBV) (7). In the absence of sequence similarity, EBV gB and HSV-1 gB share a high degree of structural similarity with other class III fusion proteins, including vesicular stomatitis virus (VSV) glycoprotein G (8) and baculovirus gp64 (9).
FIG 1.

(A) Surface representation of the crystal structure of the HSV-1 gB trimer. One monomer is colored according to the structural domain designations (I to V) given by Heldwein et al. (1), while the other two are in gray. An enlarged view of the gB base, highlighting the three pairs of FLs (colored pink for FL1 and cyan for FL2), is also shown. (B) Ribbon representation of one gB monomer labeled with the locations of relevant FR and domains, colored as in panel A. (C) HSV gB MAb “tree” showing the relationships between the different antibodies. The anti-gB MAbs are arranged by FR. Group names are given according to structural domain (from Bender et al. [14]) and are colored as shown in panel B. Arrows between groups denote binding competition among MAbs. Amino acid residues for known epitopes (determined through peptide analysis, truncation mutants, or mar mutant viruses) are shown. The antibodies studied in this paper are listed in boldface. MAb SS63, which was originally assigned to group 5b (FR1) (14), has been reclassified to group 4c (FR3) on the basis of the findings presented in this report.
The atomic structure of postfusion gB shows a subunit with five structural domains (domains I to V), four of which are similar to those of other class III fusion proteins (1, 7–9). Visualized at a lower resolution by electron microscopy (EM), the trimer appears as a rod with three distinguishable lobes (6, 10–13), to which we refer as the base, middle, and crown. Additionally, we mapped the epitopes of a panel of neutralizing monoclonal antibodies (MAbs) to distinct regions of the gB structure, thereby defining four functional regions (FR) (14). According to this mapping, FR1 includes structural domains I and V and forms the base of the gB trimer (Fig. 1B). Structural domain I contains the fusion loops (FLs) and is referred to as the fusion domain. Mutations within the FLs block virus entry and egress, as well as cell-cell fusion and virus spread (15–18). Certain MAbs with epitopes in FR1 also block gB-cell binding (19), gB-liposome binding (16), and gB-gH/gL association (20, 21). Importantly, cryo-EM studies show that gB binds to liposomes via the fusion loops of FR1 (13). Taken together, existing data suggest that FR1 is directly involved in gB-lipid association during virus-cell and cell-cell fusion.
FR2 maps to structural domain II and is located in the middle lobe of gB (Fig. 1B). Because MAbs to this FR block gB-gH/gL association (20), we hypothesize that FR2 is the site of at least one gB-gH/gL interaction. FR3 overlaps structural domain IV and is located at the crown of gB. Certain MAbs that map to this FR block gB-cell binding (19), suggesting that a potential gB receptor binds to this region (16). Mutations that affect the rate of fusion also map to FR3 (17). Finally, FR4 (residues 31 to 86) corresponds to the N terminus of gB (22–25), for which the crystal structure has not yet been solved (1, 6). MAbs within each FR have been further subdivided into groups based on peptide and gB truncation mapping and their abilities to compete with other MAbs for binding to gB (Fig. 1C) (14).
Our data show that the two FLs within FR1 constitute a “subdomain” wherein key hydrophobic amino acids form a ridge that is supported on both sides by charged residues. It is hypothesized that the hydrophobic residues interact with target membranes (13, 15, 16), helping gB to form the fusion pore. Soluble gB proteins, which are in a postfusion form, have an intrinsic ability to associate with target membranes via their fusion domains; amino acid substitutions in either FL abrogate this association (16). In the prefusion forms of most class I and class II viral fusion proteins, the fusion peptides/loops are buried (26). Although the prefusion form of gB has not been solved, we postulate that the hydrophobic membrane-proximal region (MPR) (residues 731 to 773) masks the FLs, since the presence of the MPR reduces the capacity of the gB ectodomain trimer to bind liposomes (27).
As yet, we do not know how FR1 works to direct the FLs and, consequently, to promote the process of membrane fusion. Antibodies (Abs) provide an indirect way of examining this region. How do MAbs with epitopes in FR1 neutralize the virus and impair cell-cell fusion? Furthermore, what does information of this kind say about how gB works to achieve fusion? In order to answer these questions, we need to first define the epitopes of these MAbs. From our panel of anti-gB FR1 MAbs (14), we selected SS55, which mapped to domains I and II, and SS144 and SS63, which mapped to domain V, for further study.
We examined Fab-gB complexes by EM, choosing them from each FR in addition to SS55, SS144, and SS63. We also included MAb DL16, which does not neutralize virus but is trimer specific (14). These data show that SS55 binds to the base of gB (FR1), a finding that, combined with previous results, assigns its epitope to domain I. This epitope was further narrowed by generating and sequencing MAb-resistant (mar) mutants. The EM data also confirmed the positioning of SS144 at the base of gB (14). Interestingly, the Fabs for both SS55 and SS144 neutralized virus and impaired liposome binding in vitro, suggesting that these activities are intrinsic to the MAbs as they engage their epitopes and are not due to IgG cross-linking. Finally, EM results positioned the epitope of DL16 within the base of gB and that of SS63 within the crown (FR3).
Because none of the anti-FR1 MAbs mapped directly to the FLs, we next generated polyclonal antibodies (PAbs) against peptides spanning either FL1 or FL2. While the anti-FL1 PAb failed to bind gB, the anti-FL2 PAb bound gB and neutralized virus, indicating that portions of the FLs are accessible to antibody either in prefusion gB or as an intermediate that forms as gB moves from its prefusion to its postfusion conformation. Presently, all of our structural data are derived from postfusion gB; anti-FR1 antibodies, such as anti-FL2, may help us dissect the dynamics of the fusion process at earlier time points.
MATERIALS AND METHODS
Cells and soluble proteins.
African green monkey kidney (Vero) cells were grown in Dulbecco's modified Eagle medium (DMEM) with 5% fetal bovine serum (FBS). 293T cells were grown in DMEM containing 10% FBS. Soluble gB1(730t) (gB1 truncated at aa 730), gB2(727t), and gB1(773t) were purified from baculovirus-infected insect (Sf9) cells by use of a DL16 immunosorbent column as described previously (27–29).
Antibodies.
Rabbit PAbs R238 and R239 were generated against a 20-amino-acid gB peptide (residues 167 to 186) corresponding to FL1 and its surrounding amino acids. Likewise, PAbs R240 and R241 were generated against a gB peptide encompassing FL2 (residues 252 to 271). Since antibodies from each pair of rabbits behaved identically, data from only one rabbit are shown in the results; these PAbs are referred to as anti-FL1 or anti-FL2 for ease of presentation. Rabbit PAb R242 was generated against a protein corresponding to the cytoplasmic tail of gB (residues 801 to 904) (30), a gift from E. E. Heldwein. Anti-peptide PAbs were made as described previously (31). gB-specific MAbs C226, DL16, SS10, SS55, SS63, and SS144 have been characterized previously (14, 19). To prepare Fabs for EM, IgGs were digested by immobilized papain (Thermo Scientific) overnight at 37°C. Papain-agarose beads were removed by centrifugation, and the supernatant was passed over a protein A column (GE Healthcare). The protein A flowthrough (containing Fab fragments) was collected and dialyzed against phosphate-buffered saline (PBS), and SDS-PAGE was run, followed by Coomassie staining, for verification of Fab isolation.
Transmission electron microscopy and image analysis.
To prepare gB-Fab complexes, 300 μg SS10 Fab was incubated with 100 μg gB1(730t) in a 250-μl total volume (PBS) at room temperature (RT) for 1 h. The complex was then filtered through a 0.22-μm Spin-X filter (Corning) and was loaded onto a Superdex 200 column (Äkta purifier system) at 0.2 ml/min. The gB-Fab complex eluted from the column at fractions 13 to 15 (0.5 ml per fraction). Purified gB-Fab samples (15 μg/ml) were adsorbed onto grids bearing a carbon-coated support film, negatively stained with 2% uranyl acetate, and imaged with a CM120 transmission electron microscope (FEI, Hillsboro, OR) using a 1,024- by 1,024-pixel charge-coupled device (CCD) camera (Gatan, Warrendale, PA). The nominal magnification was ×60,000 (0.33 nm/pixel). EMAN2 (32) was then used to align, classify, and average the gB or gB/Fab particles using an iterative multivariate statistical analysis reference-free classification algorithm as implemented in e2refine2d. The number of particles contributing to each class average ranged from 25 to 60. To confirm that the crown and base domains could be reliably distinguished, the resulting 2-dimensional (2D) class averages were compared with model projections calculated from the crystal structure (Protein Data Bank [PDB] code 3NWF [6]) and band-limited to 25 Å resolution, using Bsoft (33).
Selection of SS55 mar viruses.
Monoclonal antibody-resistant (mar) viruses were generated according to the protocol outlined by Cairns et al. (34). HSV-2 strain 333 was added to confluent Vero cell monolayers in 6-cm tissue culture dishes (approximately 500 PFU/plate). After 1 h at 37°C, the virus inoculum was removed, and the plates were overlaid with 5 ml DMEM, 5% heat-inactivated FBS, 1× penicillin-streptomycin, 0.5% carboxymethyl cellulose, and 100 μg/ml SS55 IgG. Plates were incubated at 37°C for 3 to 4 days. Plaques were picked and amplified on Vero cells and were then subjected to a second round of MAb selection followed by plaque purification. The final gradient-purified stocks of SS55 mar viruses were able to infect cells in the presence or absence of SS55.
Plasmid DNAs.
Plasmids pPEP98 (wild-type [WT] gB) and pCAGGS/MCS were gifts from P. Spear (35, 36). Plasmids pCW1029 (R335Q) and pCW1028 (A203T) were generated by PCR amplification of the gB open reading frame (ORF) from SS55-resistant HSV-1. To prepare the total-cell infected DNA for cloning, Vero cells (2 × 105) were seeded into individual wells of a 24-well plate. The following day, cells were infected at a multiplicity of infection (MOI) of 1. Cells were recovered after 24 h, pelleted for 30 s at 13,000 × g in a microcentrifuge, and resuspended in 10 mM Tris (pH 7.5). Samples were adjusted to 200 mM Tris (pH 8), 50 mM EDTA, 0.5% SDS, and 100 μg/ml proteinase K and were incubated for 1 h at 65°C. Finally, samples were extracted twice with phenol-chloroform (24:1), ethanol precipitated, and dissolved in sterile water. This material was used as the template for PCR (Pfu Ultra) amplification of gB sequences using the following primers: 5′-GCGGTACCCGCCATGCACCAGGGCGCCCCCTCGT (KpnI site underlined; start codon in boldface) and 5′-CGCTCGAGTGCGCATGCGGTTTAACACCCGTGGTT (XhoI site underlined). Finally, the PCR-amplified gB ORFs were cloned into vector pCAGGS/MCS, which had been digested with KpnI and XhoI. Plasmid pBH805 (D199A) was created using QuikChange XL site-directed mutagenesis (Stratagene Cloning Systems) (37) with pPEP98 as the template. The gB genes in each plasmid were sequenced (University of Pennsylvania Cell Center DNA Sequencing Core Facility) to screen out PCR errors.
Virus neutralization assays.
Serial dilutions of antibody (IgG or Fab) were mixed with HSV-1 (KOS) or HSV-2 (333), and the mixture was incubated at 37°C for 1 h. Monolayers of Vero cells grown in 48-well plates were then incubated with the Ab-virus mixture for 24 h. Cells were fixed with methanol-acetone (2:1), and plaques were visualized by the black plaque assay (38).
Purification of gB-specific IgGs from total rabbit IgGs. (i) gB immunosorbent column.
Briefly, 1.1 g activated CNBr Sepharose 4B (Pharmacia) was swollen in 0.001 N HCl at RT for 1 h. The gel was washed with 50 to 100 ml of 0.001 N HCl on a glass filter. Soluble, purified gB2(727t) (10 mg) (29) in 2.8 ml PBS plus 2.8 ml of freshly made 0.1 M carbonate buffer (0.2 M sodium bicarbonate, 1 M NaCl [pH 8.3]) was mixed with the gel overnight at 4°C. Next, the mixture was washed with 1 M ethanolamine (pH 8.0), then with 0.1 M sodium acetate–1 M NaCl (pH 4.0), and finally with 0.1 M sodium borate–1 M NaCl (pH 8.0), all at RT. The protein-gel mixture was then equilibrated with TS washing buffer (10 mM Tris-HCl [pH 7.2], 0.5 M NaCl) at 4°C and was loaded onto a column.
(ii) Selection of gB-specific IgGs through the gB2(727t) column.
The gB2(727t) column was washed extensively with TS buffer before being loaded with total anti-FL IgGs. The flowthrough was collected and was recycled through the column 5 times, followed by washing with TS buffer. The gB-specific IgGs were eluted with 3 ml of 0.1 M ethanolamine (pH 11.5). The eluted sample was further dialyzed with PBS and was concentrated with centrifugal filter units (Millipore).
ELISA. (i) Capture ELISA.
Prior to the enzyme-linked immunosorbent assay (ELISA), 12-well plates seeded with 293T cells were transfected with plasmids expressing mutant or WT gB via the GenePORTER protocol (Gene Therapy Systems, Inc.). Two days posttransfection, cells were lysed using 5% NP-40–PBS and were immediately used for the capture ELISA approach (39). Each well of a 96-well plate was washed 3 times with PBS and was then coated with 50 μl of PAb R242 (10 μg/ml) for 2 h. The plate was next washed 3 times with blocking buffer (PBS, 0.05% Tween 20, 0.5% bovine serum albumin [BSA]), after which the cell lysates were added for 1 h. The plate was then blocked with 5% milk-PBS-0.05% Tween (T) for 30 min, followed by addition of the MAbs to be tested (10 μg/ml in 5% milk-PBS-T) for 1 h. gB-MAb binding was detected by incubation with a horseradish peroxidase (HRP)-conjugated goat anti-mouse antibody for 30 min at a dilution of 1:200 in 5% milk-PBS-T, followed by the addition of ABTS [2,2′-azinobis(3-ethylbenzthiazolinesulfonic acid)] substrate, and the signal was read at 405 nm. For each antibody, the signal generated from a control well containing no lysate was subtracted from the final reading.
(ii) Peptide ELISA.
After gB column purification, PAbs R240 and R241 (anti-FL2p) were tested for binding to a series of overlapping peptides corresponding to the gB ectodomain (14). Fifty microliters of each peptide at 1 μM (in PBS) was placed in each well of a 96-well plate. The wells were washed and were then incubated in blocking buffer. After 30 min at 37°C, 5-fold dilutions of IgG or preimmune sera were added, and the plates were incubated for an additional 1 h at 37°C. Antibodies were detected using an HRP-conjugated goat anti-mouse antibody as described above.
Biosensor/surface plasmon resonance (SPR) experiments.
Experiments were performed using a Biacore 3000 or Biacore X100 optical biosensor (GE Healthcare, Biacore Life Sciences) at 25°C. Filtered and degassed HBS-N buffer (10 mM HEPES [pH 7.4], 150 mM NaCl) was used in all liposome association experiments. We used an L1 sensor chip (Biacore) for binding liposomes and followed our previous protocol (27). Liposomes were purchased from Encapsula NanoSciences (Nashville, TN) at a size of 400 nm, containing a 1.7:1 molar ratio of soy-phosphatidylcholine to cholesterol. The chip surface was prepared for liposome binding by sequential washes with 20 μl of 1% octyl-β-d-glucopyranoside, 20 μl of 0.5% SDS, 10 μl of 1% octyl-β-d-glucopyranoside, and 10 μl of 30% ethanol. Liposomes (1 mM, diluted in HBS-N) were injected until the chip was saturated, giving a signal of approximately 8,000 response units (RU). Once bound, the liposomes remained on the chip, and there was no appreciable dissociation (no measurable off-rate). Purified soluble gB(730t) (10 μg/ml) was then injected for 240 s, followed by injection of an antibody (100 μg/ml for 240 s). Alternatively, 1 μg of soluble gB was incubated with 50 μg of antibody in 50 μl total of PBS for 1 h, and the solution was then brought to 150 μl with HBS-N buffer; this gB-Ab mixture was then flowed across a liposome-covered chip for 240 s. After each experiment, the surface preparation protocol was performed to remove protein and liposomes from the chip, regenerating the surface to the RU baseline. All injections were performed at a flow rate of 5 μl/min.
RESULTS
FR1, which is located at the base of postfusion gB, is composed of two structural domains (domains I and V, colored blue and red, respectively, in Fig. 1A and B) and contains two fusion loops (termed FL1 and FL2) per protomer. In our panel of anti-gB virus-neutralizing MAbs, many of those with epitopes that map within this broad region block cell-cell fusion and prevent gB from associating with liposomes; however, the mechanism of action of MAbs in each of these events is unknown (2, 14, 16, 20). To study FR1 in more detail, we first needed to define more precisely the epitopes that mapped to this region (14). Previously, we and others identified >15 anti-gB MAbs that mapped to FR1, including many with virus-neutralizing activity (14, 23–25, 40–42) (data not shown). Among them, we chose four to study by EM: SS144 and SS63, previously assigned to FR1; SS55, whose epitope was localized within a large region corresponding to FR1 and FR2 (residues 98 to 472) via deletion mutants; and DL16, recently suggested to be in FR1 (10). We also included MAbs C226 and SS10 as controls and representatives for FR2 (located in the middle lobe of gB) and FR3 (located in the crown), respectively. In each case, we purified the Fab fragments (Fig. 2A), combined them with soluble gB(730t), and purified the Fab-gB complex by gel filtration on a Superdex 200 column. Figure 2B shows the separation of SS55 Fab-gB from free Fab. Fractions representing the complex were pooled, concentrated, and used for EM studies.
FIG 2.
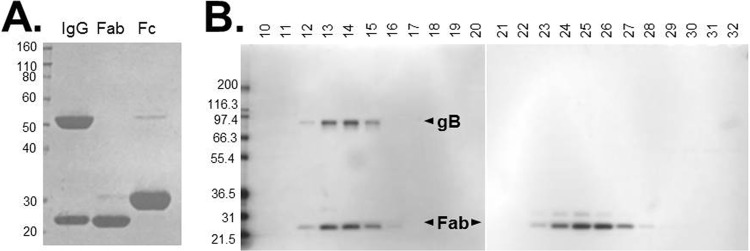
(A) Coomassie stain of purified Fab from MAb SS55. (B) Fractionation of the gB1(730t)-SS55 Fab complex via size exclusion chromatography. Fractions were analyzed on a denaturing SDS-PAGE gel and were visualized by silver staining. Fraction numbers are shown at the top; molecular size standards (in kilodaltons) are indicated on the left.
Electron microscopy localizes important gB epitopes.
As in earlier studies (6, 10–13), when soluble gB is prepared for EM by negative staining, the protein tends to orient preferentially with its long axis lying in the plane of the support film (Fig. 3A and B). In these side views, it appears as a three-lobed rod, ∼18 nm long and ∼7 nm wide, whose ends are readily distinguishable, one having the concave crown domain and the other occupied by the oval base domain, which often has a patch of negative stain at its center (Fig. 4A to C).
FIG 3.
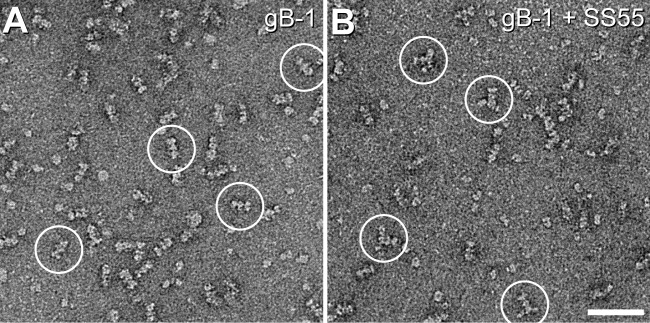
Negative-stain electron microscopy of gB. Shown are negatively stained low-magnification fields of gB1(730t) (A) and gB1(730t)-SS55 Fab (B). Some representative particles of each sample are circled. Bar, 50 nm.
FIG 4.
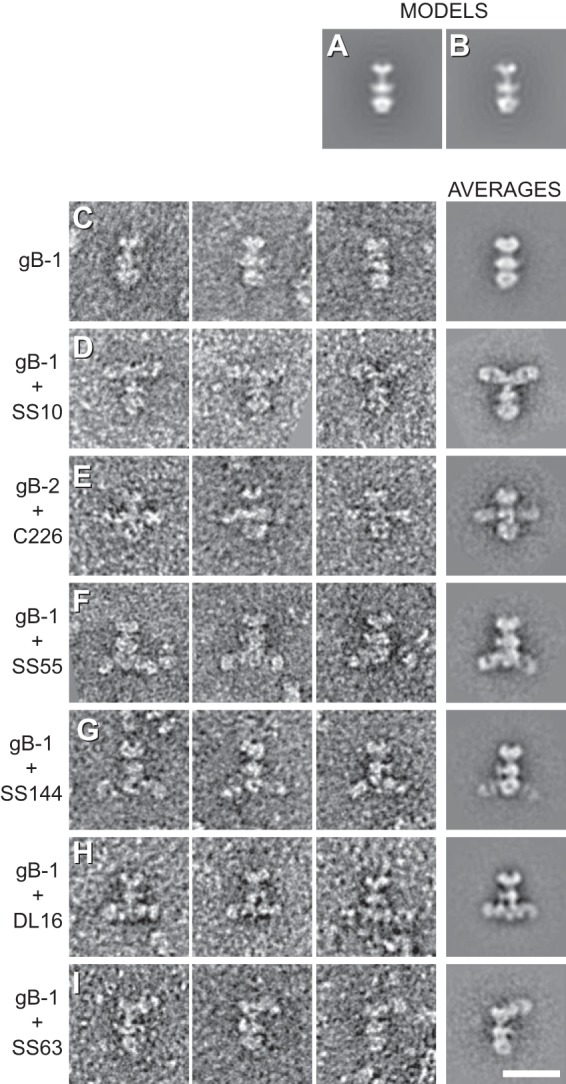
Two-dimensional class averaging of gB and Fab-bound gB. (A) Projection of a rotationally symmetrized gB model (PDB code 3NWF). (B) Projection of the gB model without imposed symmetry. (C) gB only. (D to I) Fab-bound gB. The figure shows three representatives (left panels) and the 2D class averages (rightmost panels) of each sample imaged. Bar, 20 nm.
When Fabs are complexed with gB, one or two extra densities are seen emanating laterally from gB (Fig. 3B and 4D to I); these densities represent bound Fabs. The location of the SS10 epitope (Fig. 4D) within the crown (FR3) agrees with the results of mapping done with gB truncation constructs, which localized the epitope of MAb SS10 to residues 640 to 670 (14). The binding of MAb C226 was previously localized via a series of deletion mutants to a region spanning aa 234 to 472 (14), which included portions of both FR1 and FR2. However, a C226 mar mutation at HSV-2 gB (gB2) residue 414 suggested that the C226 epitope was closer to FR2 (the middle lobe of gB) than to FR1 (14). Indeed, the EM data confirmed that C226 binds to the lateral knob-like structure in the central region of gB (Fig. 4E). Note that because C226 binds preferentially to gB2, we complexed its Fab with gB2. However, no difference in structure was seen between gB1(730t) and gB2(724t) (compare Fig. 4C and E). The epitope of MAb SS55 was also localized by use of deletion mutants, to a large region spanning residues 98 to 472 (14). However, in this case, the EM data prove that the epitope is within the base of gB (i.e., FR1) (Fig. 4F). Thus, the EM data clearly show that C226 and SS55 bind to different regions of gB. We previously mapped MAb SS144 within two overlapping synthetic peptides (residues 697 to 725). Because the N-terminal portion of this range is buried in gB(730t) (1), we further narrowed the SS144 epitope to surface-exposed residues 715 to 725 (14). These data agree with the EM results that locate its binding site near the base and to the side of gB, within FR1 (Fig. 4G).
MAb DL16 was of particular interest to us, because although it is nonneutralizing, it is trimer specific (14). Most other conformation-dependent MAbs to gB recognize both the trimer and a faster-migrating form (by SDS-PAGE) of the protein that corresponds in apparent size to a monomer (14). The EM data clearly show that DL16 binds to the base of gB within FR1 (Fig. 4H), verifying the recent mapping data for this MAb using C-terminal gB truncations (10). This observation was very interesting, since DL16 competes with MAb H1817, which binds within the first 43 amino acids of the gB N terminus (FR4) (14) (Fig. 1C). FR4 was not part of the gB ectodomain that was crystallized, and its location within the postfusion trimer is unknown (Fig. 1B). FR4 residues may stretch downward (with respect to the solved structure), thereby accounting for competition with DL16 (which was also performed using a postfusion, soluble form of gB). Regardless, the EM data clearly show that DL16 binds to the gB base, and therefore, its epitope is in FR1.
Finally, the SS63 epitope had been assigned to FR1 previously (14). However, EM analysis of SS63 Fab-gB definitively places the epitope for this MAb in the crown of gB (FR3) (Fig. 4I). We have accordingly reassigned SS63 to MAb group 4c (Fig. 1C). The original mapping of the epitope of MAb SS63 relied solely on peptide binding data and did not take into account the antibody competition data. That said, there is at least one stretch of amino acids within the crown (aa 98 to 115) that has a fair amount of sequence identity with the peptide to which SS63 bound.
In summary, the EM analyses of Fab-gB complexes provided compelling grounds for assigning MAb epitopes to FR of gB. For the remainder of this study, we concentrate on the following anti-FR1 antibodies with epitopes in domains I and V: the neutralizers SS55 and SS144 and the nonneutralizer DL16.
mar mutants further map the SS55 epitope.
We had proposed that the neutralizing mechanisms of SS55 and SS144 are based on obstructing the FL-lipid interaction (16). Yet the EM data show that SS55 and SS144 Fabs bind on the “side” of the base (Fig. 4F and G) and not at its apex, where the FLs are located (Fig. 1A). SS144 was previously mapped via overlapping peptides and surface availability to a linear stretch of residues in FR1 (residues 715 to 725) (14). To further define the discontinuous SS55 epitope, we followed up on the EM data by using the MAb to select for mar mutant viruses (34). Two separate mar viruses were obtained, both of which infected Vero cells in the presence or absence of SS55 (data not shown). Sequencing of the gB open reading frames from these mutants revealed two different amino acid changes. In one mutant, there was an A-to-T change at residue 203; in the other, an R-to-Q change at residue 335. Although quite far apart in the linear protein sequence, these two amino acids lie close to each other in domain I of the postfusion structure (Fig. 5A) and fit well with the position of SS55 Fab binding to gB (Fig. 4F).
FIG 5.
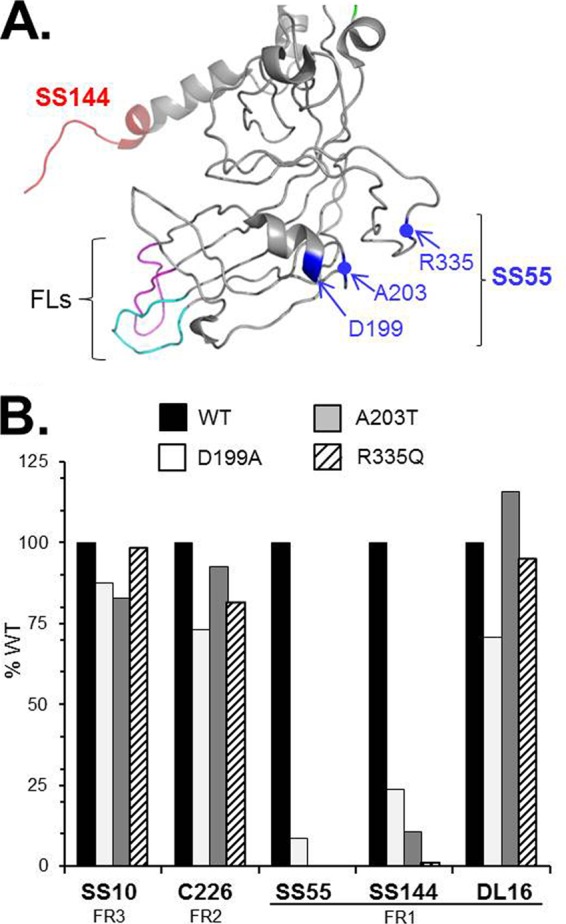
Identification of SS55 mar mutants. (A) Structure of FR1 from a gB monomer, based on the work of Stampfer et al. (6), with resolution of the loop between A328 and A338 at a low pH. The epitope of MAb SS144 is shown in red, and the fusion loops are shown in pink (FL1) and cyan (FL2). SS55 mar residues A203, R335, and D199 are highlighted in blue. Blue dots indicate the positions of A203 and R335 on the ribbon. (B) Full-length gB from transfected cell extracts was captured via PAb R242 (against the gB cytoplasmic tail) and was probed with anti-gB MAbs in an ELISA. Results of a representative experiment are shown. The percentage of WT absorbance was calculated as follows: (absorbance of test sample at 405 nm/absorbance of WT sample) × 100.
We cloned both of these gB mutants into mammalian expression plasmids. Additionally, since residue A203 is located next to a two-turn α-helix (Fig. 5A), we wondered if the neighboring turn of the helix would also be within the SS55 epitope. Therefore, we mutated Asp199 to Ala in a cell extract containing full-length gB (which likely comprises both pre- and postfusion forms). We used ELISA to test MAb binding by each mutated form of gB by first capturing the proteins with an antibody to the gB cytoplasmic tail and then determining how well each MAb bound to gB-A203T, gB-R335Q, and gB-D199A, after normalizing the data to that for binding to WT gB (Fig. 5B). As expected, these three mutants bound to SS10 and C226; however, none of them reacted with SS55. We conclude that residues D199, A203, and R335 are all within the SS55 epitope in structural domain I. Remarkably, SS144 failed to bind to any of the SS55 mar proteins. Although the two epitopes are in different structural domains (Fig. 1A and 5A), changes to the SS55 epitope clearly affect the SS144 epitope. Interestingly, anti-FR1 MAb DL16 bound to all three mar proteins (Fig. 5B), indicating that not all MAbs that bind to the gB base are affected by the SS55 mar mutants.
Anti-FR1 IgGs and Fabs inhibit gB-liposome association.
To gain insight into how anti-FR1 antibodies affect gB function, we next tested their abilities to block the binding of gB(730t) to lipid by using a biosensor-based liposome binding assay (27). This postfusion form of gB associates with liposomes in vitro via the FLs (13), and this association is altered by either (i) mutations to residues in the FLs (15, 16), (ii) the presence of the membrane-proximal region (MPR; residues 731 to 773) (27), or (iii) blocking by IgG from anti-FR1 MAbs, such as SS55 or SS144 (16). Another anti-FR1 MAb, 2c, has been shown to neutralize virus via IgG cross-linking, since Fabs generated from this MAb did not inhibit virus infection (43). We therefore asked how the individual Fabs for SS55 and SS144 would behave in liposome binding and virus neutralization assays.
Previously, we used a liposome flotation assay to demonstrate inhibition of the gB-liposome association by IgGs (16). We adapted our method of determining the binding of gB to liposomes via biosensor analysis (27) to now measure the association of gB with liposomes in the presence of anti-gB IgGs or Fabs. In this assay, liposomes were immobilized on an L1 biosensor chip surface, and gB(730t) was flowed across the chip. As expected, we observed an increase in response units (RU), indicative of the binding of protein to the liposomes (Fig. 6A, black lines). As a negative control for blocking, we preincubated gB with the anti-FR2 MAb C226. The presence of C226 IgG or Fabs did not block the binding of gB to liposomes (Fig. 6); in fact, the percentage of liposome-bound gB increased when C226 IgG was present. This observation is likely due to the increased mass of gB-IgG complexes. With the nonneutralizing MAb DL16, both the IgG and Fabs partially blocked the association of gB with liposomes (Fig. 6). This was understandable in light of the location where DL16 binds (Fig. 4H). In contrast, when gB was preincubated with either SS55 or SS144 IgGs, the complexed gB bound poorly to liposomes; gB-liposome association was reduced to a level 20% that of WT gB (Fig. 6B). When gB was complexed with either SS55 or SS144 Fabs, binding was also reduced to <25%. Since the EM data suggest that these MAbs do not bind to the FLs, blocking (especially by the Fabs) could be caused by an indirect effect, such as a structural alteration upon MAb binding that, in turn, affects FL position. Alternatively, the blocking could be due to steric hindrance, even by the smaller Fabs.
FIG 6.
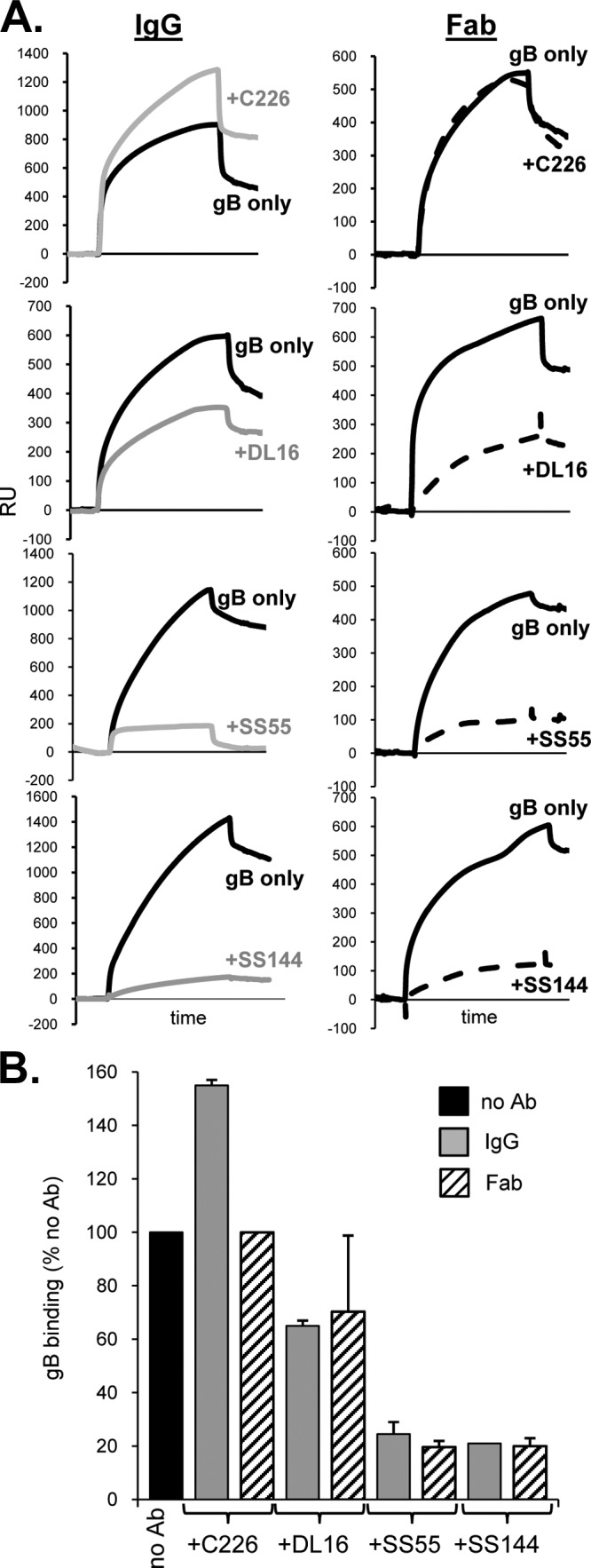
Antibodies against neutralizing epitopes in FR1 block gB-liposome binding. (A) Fifty micrograms of each antibody (IgG or Fab) was incubated with 1 μg of purified, soluble gB(730t) for 1 h, and the complex was then flowed across liposomes bound to an L1 biosensor chip. Solid black curves represent the RU of gB binding to liposomes; dashed curves represent those for gB plus Fab (right); and gray curves represent those for gB plus IgG (left). The response from Fab or IgG flowing across liposomes only (no gB) was subtracted from the curves. (B) Bar graph representing the percentage of gB bound to liposomes for each sample. The percentage of gB bound was calculated based on maximal binding after 240 s as follows: (RU of test sample [after injection of gB-Ab]/RU of WT) × 100. Error bars indicate standard errors for at least two experiments.
FR1 neutralizing epitopes remain accessible on lipid-bound gB.
Following up on these two possibilities, we asked if steric hindrance plays a role in the abilities of SS55 and SS144 to block the binding of gB to liposomes. If so, we reasoned that their epitopes should be inaccessible to antibody when gB was prebound to liposomes. To address this, we coated an L1 chip with liposomes and then injected gB(730t); gB binding to liposomes was seen as an increase in RU (Fig. 7). Next, IgGs were sequentially flowed across the chip surface. In the first experiment of this series (Fig. 7A), we injected first MAb SS55, then SS144, and finally SS10. SS55 was still able to bind, as evidenced by the increase in RU. SS144 gave no additional signal, since it was blocked from binding by SS55 (as seen in our previous competition studies using gB without liposomes [14]). As a control, the anti-FR3 MAb SS10, which does not compete with either SS55 or SS144, was injected last, and an increase in RU was seen, indicating that this epitope was still available.
FIG 7.
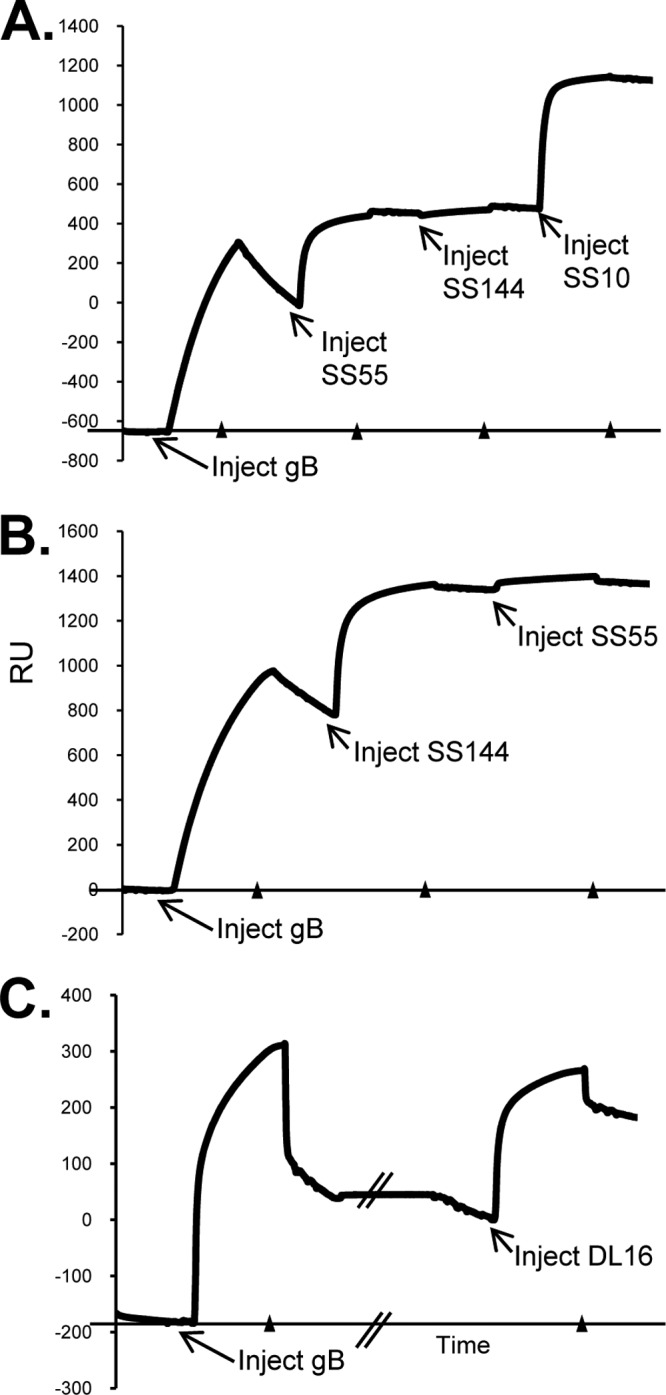
Liposome binding assay and biosensor analysis. Liposomes are injected into the L1 chip to saturation (∼8,000 RU) (not shown), and soluble gB(730t) is then injected at 5 μl/min for 240 s, followed by the indicated antibody (240 s); binding to liposomes is measured as an increase in RU. There is some day-to-day variability in the RU of gB captured on the liposomes (y axis values), because different batches of liposomes and different preparations of gB were used. However, the concentrations of liposomes and gB were always held constant, and the test samples were always compared to a control (IgG or Fab flowed across liposomes only) on the same day. The beginning of each injection is indicated with an arrow, and arrowheads along the x axis show where each injection was stopped. (A and B) Competition for binding to gB between MAbs SS55 and SS144. SS10, SS55, and SS144 were injected at a concentration of 75 μg/ml. (C) DL16 was injected at a concentration of 50 μg/ml. The double slash in the x axis indicates a pause before the injection of DL16.
Next, we reversed the order of addition of SS144 and SS55. In this case, MAb SS144 bound to gB-liposome complexes and consequently blocked the binding of SS55 (Fig. 7B). Finally, we performed the same experiment with DL16 and found that, like the other anti-FR1 MAbs, it also bound to liposome-associated gB (Fig. 7C). Thus, although SS55 and SS144 prevent soluble gB from interacting with lipids, they are still able to bind to gB when the protein is bound to lipid membranes. Notably, cryoelectron tomography shows that the interaction of the base of soluble gB with liposomes is limited to the outer leaflet of the lipid bilayer and that the FLs associate with liposomes but are not inserted (13), thus leaving space for each of these anti-FR1 MAbs to bind. Taking these results together with our previous data, we suggest that the FR1 neutralizing antibodies bind at sites distinct from the FLs to inhibit the association of gB with lipids, possibly through an alteration of FR1 structure and not through steric hindrance.
SS55 and SS144 Fabs neutralize better than their respective IgGs.
Our liposome experiments were done using gB(730t), a soluble postfusion form of gB. To understand how SS55 and SS144 function to block virus entry, we revisited virus neutralization. Our thinking was that if IgG cross-linking plays a role in the function of these antibodies, then neutralization by the full IgG would be better than that seen for the Fab. In order to easily compare IgG with Fab, the IgG concentrations in Fig. 8 were corrected for the amount of Fab by assuming that 1 μg of Fab equals 1.5 μg of IgG.
FIG 8.
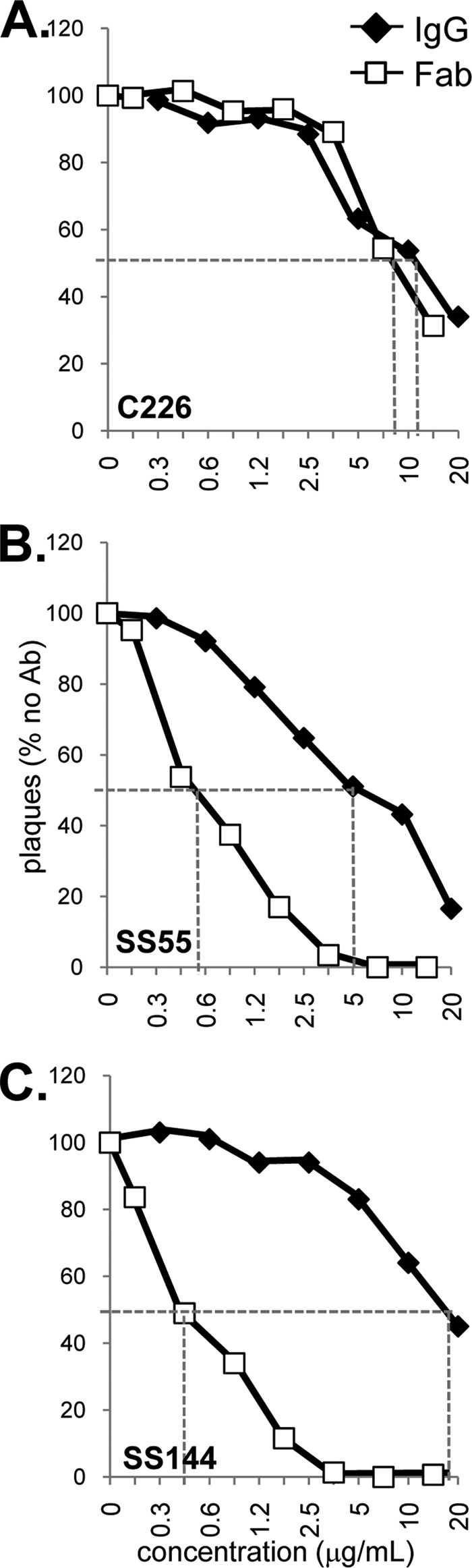
Virus neutralization. Serial dilutions of IgG or Fab of each MAb were mixed with HSV-1 KOS for 1 h at 37°C and were then added to Vero cells. Plaques were visualized 24 h later by the black plaque assay. Data are plotted as percentages of the result for the no-antibody sample. Dashed lines highlight the concentrations for 50% neutralization of virus. The IgG concentrations used were corrected for the amount of Fab by assuming that 1 μg of Fab equals 1.5 μg of IgG.
As a control, we tested both the IgG and the Fab of anti-FR2 MAb C226. In this case, both forms of the antibody neutralized virus infectivity with the same efficiency (Fig. 8A). As with C226, a number of other reported cases have shown that Fab activity is either the same as or less than that of its parental IgG (43–46). As expected from previous studies (14), the IgGs of MAbs SS55 and SS144 neutralized virus well; each inactivated the infectivity of 50% of the input virus at concentrations between 5 and 20 μg/ml (Fig. 8B and C, filled diamonds). In contrast, the Fabs for both SS55 and SS144 showed a marked improvement in neutralization activity. The 50% neutralization point for each Fab was ∼0.6 μg/ml, an 8- to 30-fold increase in potency over that of the parental IgGs (Fig. 8B and C, open squares). To our knowledge, this marked enhancement of neutralization by a Fab is unique. We believe that this improvement in neutralization reflects an improved ability of these two Fabs to reach their particular epitopes.
Generation of antibodies directed against the fusion loops.
Because none of the anti-FR1 MAbs in our collection bind directly to the FLs, we decided to prepare antibodies to the FLs themselves. To do this, we hyperimmunized rabbits with a peptide spanning FL1 (residues 167 to 186) or FL2 (residues 252 to 271) (Fig. 9A). Anti-FL1 failed to react with gB(730t) by Western blotting (data not shown) and was not studied further. In contrast, anti-FL2 bound denatured, but not native, gB (Fig. 9B, left). To enhance the reactivity of anti-FL2, we enriched for gB-specific IgGs by passing the total IgG for anti-FL2 over a gB2(724t) antigen-sorbent column (where soluble gB is covalently linked to Sepharose 4B). Column-purified antibody, termed anti-FL2p IgG (representing 0.5% of the total IgG), readily bound both native and denatured gB(730t) (Fig. 9B, right). Surprisingly, anti-FL2p also bound native gB(773t), a longer form that contains the structurally unresolved MPR but is likely also a postfusion form of gB (13, 27). The FLs were originally thought to be obscured (and perhaps inaccessible to antibody) in gB(773t), since this form exhibits reduced liposome binding (27).
FIG 9.
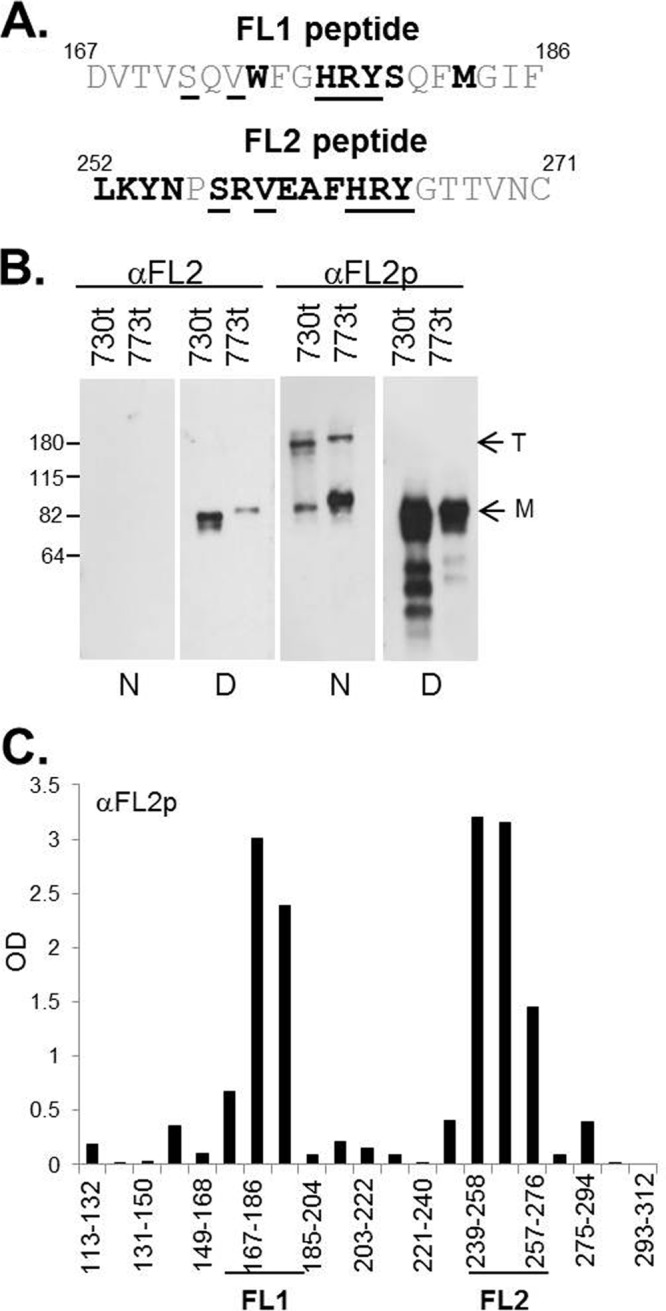
(A) gB peptides used to generate anti-FL1 and anti-FL2 PAbs. The sequence of each FL is identical for gB1 and gB2 (1). The numbers of the first and last amino acids of each peptide are shown. Residues that are accessible on the surface of the gB trimer are shown in boldface (1), and amino acids identical in FL1 and FL2 are underlined. (B) Western blots of purified, soluble gB probed with anti-FL2 or anti-FL2p. Molecular size standards (in kilodaltons) are shown on the left. T, trimer; M, monomer; N, native gel; D, denaturing gel. (C) ELISA using anti-FL2p against overlapping gB2 peptides that encompass the fusion domain. Each gB2 peptide (x axis) is 20 residues long and has a 10-residue overlap with peptides on either side. Peptides that contain FL1 and FL2 sequences are indicated.
To ensure that anti-FL2p IgG was specific for the FL region, we performed ELISA with overlapping 20-mer peptides that spanned the two FLs. Anti-FL2p IgG reacted with two peptides corresponding to FL2 (Fig. 9C) but also reacted with two peptides corresponding to FL1. The latter result is not surprising, since the two FLs share some sequence identity (Fig. 9A, identical residues underlined). Therefore, it is possible that some of the attributes of anti-FL2p are due to its ability to bind both FL1 and FL2, i.e., it binds a fusion region. Therefore, we will consider this antibody to react with both loops in the context of gB itself.
Anti-FL2p can bind lipid-bound gB and neutralize virus infectivity.
By the same method used for Fig. 7, we used anti-FL2p to test the accessibility of the antigenic sites on liposome-bound soluble gB (Fig. 10A). To our surprise, like the anti-FR1 MAbs, anti-FL2p readily bound to lipid-associated gB(730t). We conclude that a portion of FL2 and possibly the FL1 of gB(730t) is exposed even when the protein is bound to lipid.
FIG 10.
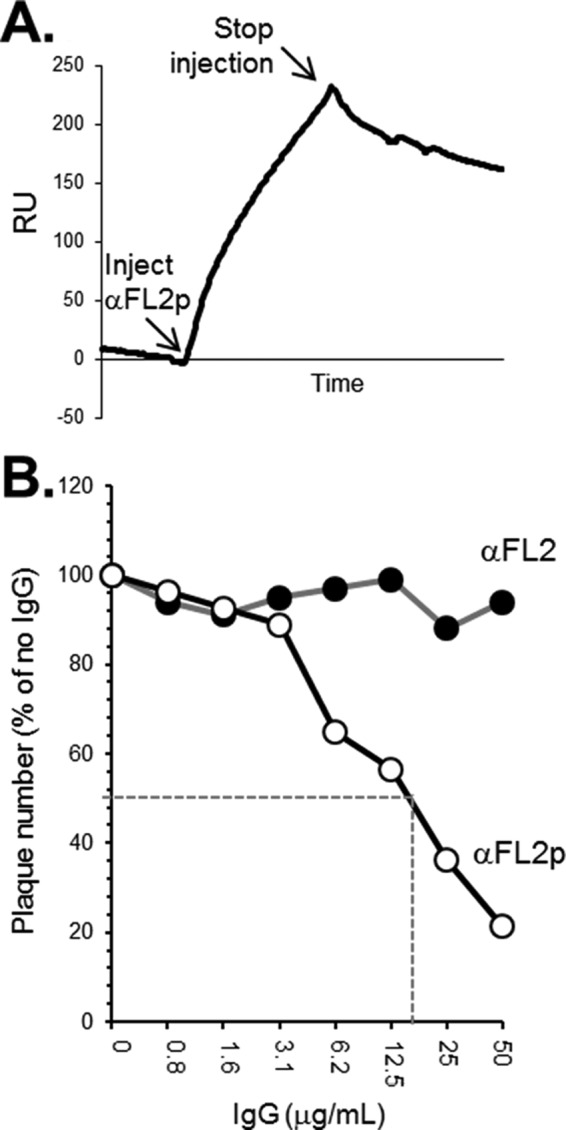
(A) Binding of anti-FL2p to liposome-associated gB was performed using biosensor analysis as described in the legend to Fig. 7. (B) Neutralization assays were performed as for Fig. 8, except that HSV-2 (333) was used. Filled circles, total IgG; open circles, gB-specific IgG. Dashed lines highlight the IgG concentrations for 50% neutralization of virus.
Since the fusion region is accessible when gB is associated with liposomes, we next asked if it would also be accessible on gB in the context of virus and, if so, whether anti-FL2p IgG could neutralize virus. One might expect that anti-FL2p would be either (i) nonneutralizing, because the FLs are inaccessible in prefusion gB on the virus, or (ii) neutralizing, because of direct interference with the gB-lipid binding step of fusion. To answer these questions, we tested anti-FL2p in our neutralization assay (Fig. 10B). Anti-FL2p IgG neutralized virus in a dose-dependent manner (50% neutralization at 40 μg/ml). In contrast, the total anti-FL2 IgG (not enriched for gB-specific IgGs) had no effect on virus infectivity. The fact that a PAb generated against the FL2 peptide neutralized virus suggests that the FLs are accessible to antibody on gB that is within the virus envelope at the time of infection.
Taken together, our data uncover how different antibodies with epitopes in FR1 neutralize virus infection. MAbs such as SS55 and SS144 do not bind the FLs directly but may instead alter them upon binding, thereby impairing gB-lipid association and membrane fusion. Lastly, a PAb generated against FL2 neutralizes virus, suggesting that the FLs are a viable target for antibody.
DISCUSSION
Our purpose here was to dissect the properties of gB functional region 1 (FR1), the portion of gB that interacts with lipid, houses the fusion domain, and contains the antigenic sites for numerous neutralizing antibodies.
Comments about the Fab/gB EM study.
We used EM to visualize the actual location of Fabs for antibodies binding to three of the four FR and to resolve ambiguities in our earlier mapping efforts (Fig. 11). These studies confirmed the locations of four antibodies chosen as representatives (SS10 in the crown of gB [FR3], C226 in the middle lobe [FR2], and SS55 and SS144 in the base [FR1]). Additionally, we determined the general locations of two other antibodies, SS63 and DL16. The SS63 epitope was originally assigned to FR1 (14), but EM clearly shows that it binds to the gB crown at the other end of the molecule (FR3). Because we use the nonneutralizing MAb DL16 extensively for gB purification, and because a number of other laboratories have taken advantage of its trimer-specific property (6, 10, 47–49), it was important to locate its epitope. While Vitu et al. (10) have suggested that DL16 binds to FR1 (residues 678 to 730 of structural domain V) based on its inability to bind C-terminal gB truncation constructs, our competition data showed that it competes with MAb H1817, known to bind within FR4 (residues 31 to 43) (Fig. 1C) (14). Since this region of gB is not resolved in the crystal structure, we originally thought that DL16 might bind near the N terminus, expected to be near the crown. Rather, our current EM data show DL16 binding to the base of gB (FR1). Taken together, the competition and EM data for DL16 suggest that perhaps FR4 stretches downward toward the gB base (FR1) when gB is in the postfusion form. In fact, when one compares the projections of the solved atomic structure (Fig. 4A and B) with the 2D class averages of negatively stained gB (Fig. 4C, right), it becomes apparent that the crown and the middle lobe are larger than the atomic model suggests. This indicates that the N terminus likely extends toward the base of the protein, interacting with domains IV, III, and II, and positioning FR4 (residues 31 to 86) more distant from the crown than originally expected, conclusions differing somewhat from those reached using cryo-EM (13). Thus, this approach has solidified and extended our immunological/biochemical analysis.
FIG 11.
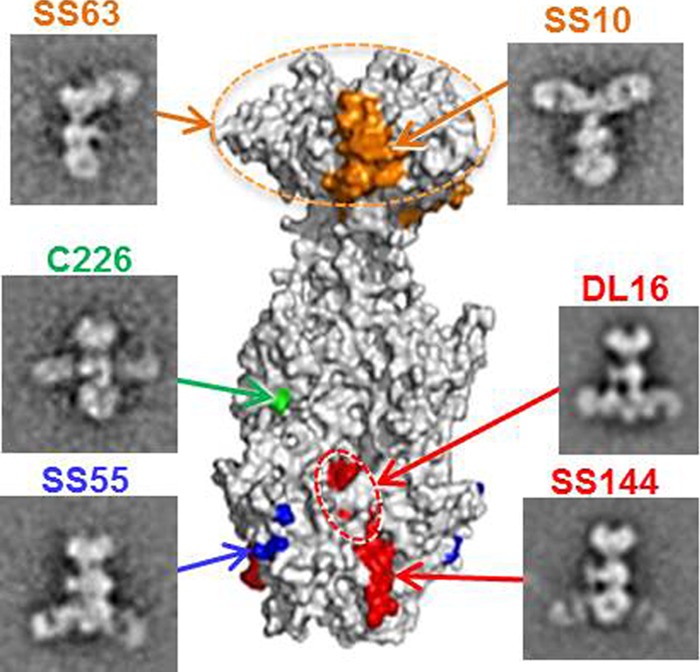
Epitopes of anti-gB MAbs displayed on a surface representation of the gB trimer. EMs showing gB-Fab complexes for each MAb are also shown, and arrows lead to their epitopes. Epitopes are colored according to the structural domain in which they are located, as in Fig. 1: SS10 (residues 640 to 670), orange; C226 (residue 419), green; SS55 (residues 199, 203, and 335), blue; DL16, red (circled); SS144 (residues 715 to 725), red. Epitope mapping data for MAbs SS10, C226, SS144, and DL16 have been published previously (10, 14). The epitope for MAb SS63 is localized to the crown via EM of gB-Fab complexes, and its residues are unknown (dashed orange circle).
gB(730t) is a trimer (1) and presumably presents three copies of each epitope at 120° intervals around its axis. However, we rarely saw clear-cut examples of decoration with three Fabs (Fig. 3B and 4). We attribute this to the fact that specimens tend to flatten onto the support film when air dried in a negative stain. As a result, the third Fab could become detached or perhaps protrude along the line of sight, out of the stain layer. It is also possible that the binding of two Fabs might preclude the binding of a third, due to an induced structural change. Notably, there is a precedent for nonstoichiometric Fab binding on trimeric proteins: the long tail-fibers of T-even bacteriophages, which, when decorated with specific Fabs and imaged by negative staining, show one Fab protruding on either side (50) despite the fact that they are trimeric, as established by scanning transmission EM (STEM) mass measurements (51) and crystal structure analysis (52).
How do SS55 and SS144 block gB binding to liposomes?
Originally, we mapped SS144 to structural domain V with synthetic peptides, since it binds a linear epitope, and this assignment is in agreement with the EM results presented here. On the other hand, SS55 binds to a conformation- and trimer-specific epitope that our EM data, in combination with the results from proteolytic fragments, localize to structural domain I. Yet these two antibodies compete for binding to the postfusion soluble form of gB [gB(730t)] (14). Since both MAbs bind to FR1 (albeit to different structural domains), one explanation for the observed competition is that the binding of one MAb blocks the other by steric hindrance. However, DL16 also binds within the base and does not hinder the binding of either SS55 or SS144 (14).
Both SS55 and SS144 (and their respective Fabs) block the binding of gB to liposomes, although they both still bind after gB is already liposome bound. We argue that these two MAbs influence conformational changes in gB that alter its ability to bind to liposomes. Much like the handles of scissors controlling the blades through a pivot point, the binding of SS55 or SS144 to its epitope could have an allosteric effect on the FLs. Such effects might also explain why SS55 mar mutants within domain I also fail to bind SS144 (Fig. 5B). The EM data show that when SS55 and SS144 bind to soluble gB(730t), the Fabs are angled slightly downward, toward the base (Fig. 4F and G). One could also speculate that the formation of this structure does not allow the FLs to get close enough to the lipid membrane to associate with it. It is also possible that liposome-bound gB binds SS55 and SS144 because only a subset of the FLs of the trimer are inserted into the liposome, while the remainder are exposed for antibody binding. This possibility is consistent with the EM data showing that many gB molecules have fewer than three Fabs binding to gB at once.
The EM data also suggest that although DL16 binds FR1, its binding differs from the binding of SS55 and SS144 (Fig. 4). When DL16 Fabs bind, they are positioned outward and away from the gB-lipid binding interface (Fig. 4H). Moreover, DL16 binds to both SS55 mar mutants, further evidence that it is binding to a different portion of FR1 than SS55 and SS144 (Fig. 5B). These data could explain why DL16, though binding within FR1, only partially blocks gB-liposome association.
In a previous study (13), we noted that when gB(730t) associates with liposomes, there are lateral projections of the base of the protein that form an extended surface, which we termed a “fusion patch.” We further suggested that the charged and hydrophobic residues of such a patch could anchor the protein on the membrane by interacting with the charged lipid head groups and hydrophilic carbon tails, respectively. We now propose that SS55 and SS144 prevent the formation of the fusion patch or the interaction of one or both FLs with the outer leaflet of a lipid bilayer.
Neutralizing properties of SS55, SS144, and C226.
It is intriguing that the Fabs for SS55 and SS144 neutralize virus significantly better than their parental IgGs, given that the majority of Fabs have equal or weaker neutralizing activity (43–46). A previous report regarding neutralizing anti-gB MAb 2c, whose epitope also maps to FR1 (43, 53), showed that this MAb required 94-fold more Fab than IgG to reduce virus infection to the same extent (43). The antiviral activity of MAb 2c is a consequence of gB cross-linking; the addition of secondary antibodies to the Fab restores its neutralization activity to near-IgG levels. We suggest that the epitopes of SS55 and SS144 are more accessible to Fabs than to full-sized IgG. This is not the case for C226 (FR2), whose Fab neutralizes virus to the same extent as its parental IgG; perhaps its epitope is equally accessible to both. It is important to remember that the gB crystal structure depicts the postfusion form of gB; these epitopes may be presented differently on prefusion gB (7, 17).
An antibody to FL2 provides further insight into the way FR1 functions.
In a variety of viruses, antibodies raised to fusion peptides and loops have neutralizing properties, and the prevailing thought is that these loops and peptides are hidden until the fusion protein goes from a pre- to a postfusion state (54–59). Yet an antibody against an FL2 peptide was neutralizing for both HSV-1 (data not shown) and HSV-2, implying that some portion of the FLs or the surrounding residues are exposed on full-length gB when it is in the viral membrane. These data suggest that HSV gB is akin to parainfluenza virus F and VSV G, which have partially exposed fusion peptides/loops in their prefusion forms (60, 61). Furthermore, anti-FL2p binds to gB(730t) when the latter is bound to liposomes, indicating that the extended surface in the fusion patch does not prevent the FLs from contacting antibodies. Additionally, FL2 is known to change conformation upon exposure to a low pH (and presumably during endosomal entry), adopting an “outward” conformation that moves it away from FL1 (6). It is possible that at a neutral pH, FL2 (and perhaps FL1) adopts a similar conformation upon exposure to lipid in order to achieve membrane association. In the future, we plan to test anti-FL2p against our panel of FL mutants to try to pinpoint its epitope further.
In summary, we have examined the interaction of a truncated postfusion form of gB with Fabs to localize epitopes and with artificial membranes (liposomes), and then we have tried to extrapolate those findings to a “real” situation, i.e., virus-cell fusion and virus neutralization. However, the nature of the prefusion form of gB remains elusive. Assuming that these in vitro events can be transposed onto actual fusion events, at which step(s) in the prefusion-to-postfusion structural cascade do FR1 neutralizing antibodies block the gB-lipid interaction? We envision that they should block just before or at the time when the FLs interact with a cell membrane to begin infection. We further propose that these antibodies alter the interaction of the fusion domain with the outer leaflet of the host membrane. Our data suggest that in virions, the FLs are likely exposed to the environment prior to the onset of fusion, i.e., when gB is in a prefusion or intermediate form. Clearly, more needs to be uncovered about the structure of the prefusion form(s) of gB and the positions of the FLs in that structure. In future studies, we plan to use anti-FR1 Fabs, including anti-FL2p or a monoclonal antibody to the FL2 peptide, to attempt to trap a prefusion form of gB, as has been done for respiratory syncytial virus F in complex with a Fab for a neutralizing MAb (59).
ACKNOWLEDGMENTS
This work was supported by the following grants from NIH: AI-076231 and AI-056045 (to R.J.E.) and AI-18289 (to G.H.C.). S.S.S. was funded in part by the VMD/Ph.D. program of the School of Veterinary Medicine at the University of Pennsylvania and by NIH training grant AI-07234. This research was also supported in part by the Intramural Research Program of the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health.
We thank S. Baxter (University of Pennsylvania Cell Center) for Fab fragmentation and B. P. Hannah for the construction of plasmid pBH805. We also acknowledge P. Spear and E. E. Heldwein for reagents. Special thanks to C. Krummenacher and L. King for critical readings of the manuscript.
Footnotes
Published ahead of print 18 December 2013
REFERENCES
- 1.Heldwein EE, Lou H, Bender FC, Cohen GH, Eisenberg RJ, Harrison SC. 2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313:217–220. 10.1126/science.1126548 [DOI] [PubMed] [Google Scholar]
- 2.Atanasiu D, Saw WT, Cohen GH, Eisenberg RJ. 2010. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J. Virol. 84:12292–12299. 10.1128/JVI.01700-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Atanasiu D, Cairns TM, Whitbeck JC, Saw WT, Rao S, Eisenberg RJ, Cohen GH. 2013. Regulation of herpes simplex virus gB-induced cell-cell fusion by mutant forms of gH/gL in the absence of gD and cellular receptors. mBio 4(2):e00046–13. 10.1128/mBio.00046-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chowdary TK, Cairns TM, Atanasiu D, Cohen GH, Eisenberg RJ, Heldwein EE. 2010. Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat. Struct. Mol. Biol. 17:882–888. 10.1038/nsmb.1837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spear PG. 2004. Herpes simplex virus: receptors and ligands for cell entry. Cell. Microbiol. 6:401–410. 10.1111/j.1462-5822.2004.00389.x [DOI] [PubMed] [Google Scholar]
- 6.Stampfer SD, Lou H, Cohen GH, Eisenberg RJ, Heldwein EE. 2010. Structural basis of local, pH-dependent conformational changes in glycoprotein B from herpes simplex virus type 1. J. Virol. 84:12924–12933. 10.1128/JVI.01750-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Backovic M, Longnecker R, Jardetzky TS. 2009. Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proc. Natl. Acad. Sci. U. S. A. 106:2880–2885. 10.1073/pnas.0810530106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roche S, Bressanelli S, Rey FA, Gaudin Y. 2006. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science 313:187–191. 10.1126/science.1127683 [DOI] [PubMed] [Google Scholar]
- 9.Kadlec J, Loureiro S, Abrescia NG, Stuart DI, Jones IM. 2008. The postfusion structure of baculovirus gp64 supports a unified view of viral fusion machines. Nat. Struct. Mol. Biol. 15:1024–1030. 10.1038/nsmb.1484 [DOI] [PubMed] [Google Scholar]
- 10.Vitu E, Sharma S, Stampfer SD, Heldwein EE. 2013. Extensive mutagenesis of the HSV-1 gB ectodomain reveals remarkable stability of its postfusion form. J. Mol. Biol. 425:2056–2071. 10.1016/j.jmb.2013.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma S, Wisner TW, Johnson DC, Heldwein EE. 2013. HCMV gB shares structural and functional properties with gB proteins from other herpesviruses. Virology 435:239–249. 10.1016/j.virol.2012.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Backovic M, Leser GP, Lamb RA, Longnecker R, Jardetzky TS. 2007. Characterization of EBV gB indicates properties of both class I and class II viral fusion proteins. Virology 368:102–113. 10.1016/j.virol.2007.06.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maurer UE, Zeev-Ben-Mordehai T, Pandurangan AP, Cairns TM, Hannah BP, Whitbeck JC, Eisenberg RJ, Cohen GH, Topf M, Huiskonen JT, Grunewald K. 2013. The structure of herpesvirus fusion glycoprotein B-bilayer complex reveals the protein-membrane and lateral protein-protein interaction. Structure 21:1396–1405. 10.1016/j.str.2013.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bender FC, Samanta M, Heldwein EE, de Leon MP, Bilman E, Lou H, Whitbeck JC, Eisenberg RJ, Cohen GH. 2007. Antigenic and mutational analyses of herpes simplex virus glycoprotein B reveal four functional regions. J. Virol. 81:3827–3841. 10.1128/JVI.02710-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hannah BP, Heldwein EE, Bender FC, Cohen GH, Eisenberg RJ. 2007. Mutational evidence of internal fusion loops in herpes simplex virus glycoprotein B. J. Virol. 81:4858–4865. 10.1128/JVI.02755-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hannah BP, Cairns TM, Bender FC, Whitbeck JC, Lou H, Eisenberg RJ, Cohen GH. 2009. Herpes simplex virus glycoprotein B associates with target membranes via its fusion loops. J. Virol. 83:6825–6836. 10.1128/JVI.00301-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Atanasiu D, Saw WT, Gallagher JR, Hannah BP, Matsuda Z, Whitbeck JC, Cohen GH, Eisenberg RJ. 2013. Dual split protein-based fusion assay reveals that mutations to herpes simplex virus (HSV) glycoprotein gB alter the kinetics of cell-cell fusion induced by HSV entry glycoproteins. J. Virol. 87:11332–11345. 10.1128/JVI.01700-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wright CC, Wisner TW, Hannah BP, Eisenberg RJ, Cohen GH, Johnson DC. 2009. Fusion between perinuclear virions and the outer nuclear membrane requires the fusogenic activity of herpes simplex virus gB. J. Virol. 83:11847–11856. 10.1128/JVI.01397-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bender FC, Whitbeck JC, Lou H, Cohen GH, Eisenberg RJ. 2005. Herpes simplex virus glycoprotein B binds to cell surfaces independently of heparan sulfate and blocks virus entry. J. Virol. 79:11588–11597. 10.1128/JVI.79.18.11588-11597.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Atanasiu D, Whitbeck JC, de Leon MP, Lou H, Hannah BP, Cohen GH, Eisenberg RJ. 2010. Bimolecular complementation defines functional regions of herpes simplex virus gB that are involved with gH/gL as a necessary step leading to cell fusion. J. Virol. 84:3825–3834. 10.1128/JVI.02687-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Atanasiu D, Whitbeck JC, Cairns TM, Reilly B, Cohen GH, Eisenberg RJ. 2007. Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion. Proc. Natl. Acad. Sci. U. S. A. 104:18718–18723. 10.1073/pnas.0707452104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pereira L, Ali M, Kousoulas K, Huo B, Banks T. 1989. Domain structure of herpes simplex virus 1 glycoprotein B: neutralizing epitopes map in regions of continuous and discontinuous residues. Virology 172:11–24. 10.1016/0042-6822(89)90102-5 [DOI] [PubMed] [Google Scholar]
- 23.Navarro D, Paz P, Pereira L. 1992. Domains of herpes simplex virus I glycoprotein B that function in virus penetration, cell-to-cell spread, and cell fusion. Virology 186:99–112. 10.1016/0042-6822(92)90064-V [DOI] [PubMed] [Google Scholar]
- 24.Marlin SD, Highlander SL, Holland TC, Levine M, Glorioso JC. 1986. Antigenic variation (mar mutations) in herpes simplex virus glycoprotein B can induce temperature-dependent alterations in gB processing and virus production. J. Virol. 59:142–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Highlander SL, Dorney DJ, Gage PJ, Holland TC, Cai W, Person S, Levine M, Glorioso JC. 1989. Identification of mar mutations in herpes simplex virus type 1 glycoprotein B which alter antigenic structure and function in virus penetration. J. Virol. 63:730–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.White JM, Delos SE, Brecher M, Schornberg K. 2008. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol. 43:189–219. 10.1080/10409230802058320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shelly SS, Cairns TM, Whitbeck JC, Lou H, Krummenacher C, Cohen GH, Eisenberg RJ. 2012. The membrane-proximal region (MPR) of herpes simplex virus gB regulates association of the fusion loops with lipid membranes. mBio 3(6):e00429–12. 10.1128/mBio.00429-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bender FC, Whitbeck JC, Ponce de Leon M, Lou H, Eisenberg RJ, Cohen GH. 2003. Specific association of glycoprotein B with lipid rafts during herpes simplex virus entry. J. Virol. 77:9542–9552. 10.1128/JVI.77.17.9542-9552.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cairns TM, Whitbeck JC, Lou H, Heldwein EE, Chowdary TK, Eisenberg RJ, Cohen GH. 2011. Capturing the herpes simplex virus core fusion complex (gB-gH/gL) in an acidic environment. J. Virol. 85:6175–6184. 10.1128/JVI.00119-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Silverman JL, Greene NG, King DS, Heldwein EE. 2012. Membrane requirement for folding of the herpes simplex virus 1 gB cytodomain suggests a unique mechanism of fusion regulation. J. Virol. 86:8171–8184. 10.1128/JVI.00932-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aldaz-Carroll L, Whitbeck JC, Ponce de Leon M, Lou H, Hirao L, Isaacs SN, Moss B, Eisenberg RJ, Cohen GH. 2005. Epitope-mapping studies define two major neutralization sites on the vaccinia virus extracellular enveloped virus glycoprotein B5R. J. Virol. 79:6260–6271. 10.1128/JVI.79.10.6260-6271.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. 2007. EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 157:38–46. 10.1016/j.jsb.2006.05.009 [DOI] [PubMed] [Google Scholar]
- 33.Heymann JB, Belnap DM. 2007. Bsoft: image processing and molecular modeling for electron microscopy. J. Struct. Biol. 157:3–18. 10.1016/j.jsb.2006.06.006 [DOI] [PubMed] [Google Scholar]
- 34.Cairns TM, Shaner MS, Zuo Y, Ponce-de-Leon M, Baribaud I, Eisenberg RJ, Cohen GH, Whitbeck JC. 2006. Epitope mapping of herpes simplex virus type 2 gH/gL defines distinct antigenic sites, including some associated with biological function. J. Virol. 80:2596–2608. 10.1128/JVI.80.6.2596-2608.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Okuma K, Nakamura M, Nakano S, Niho Y, Matsuura Y. 1999. Host range of human T-cell leukemia virus type I analyzed by a cell fusion-dependent reporter gene activation assay. Virology 254:235–244. 10.1006/viro.1998.9530 [DOI] [PubMed] [Google Scholar]
- 36.Pertel PE, Fridberg A, Parish ML, Spear PG. 2001. Cell fusion induced by herpes simplex virus glycoproteins gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology 279:313–324. 10.1006/viro.2000.0713 [DOI] [PubMed] [Google Scholar]
- 37.Connolly SA, Landsburg DJ, Carfi A, Wiley DC, Eisenberg RJ, Cohen GH. 2002. Structure-based analysis of the herpes simplex virus glycoprotein D binding site present on herpesvirus entry mediator HveA (HVEM). J. Virol. 76:10894–10904. 10.1128/JVI.76.21.10894-10904.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tal-Singer R, Peng C, Ponce De Leon M, Abrams WR, Banfield BW, Tufaro F, Cohen GH, Eisenberg RJ. 1995. Interaction of herpes simplex virus glycoprotein gC with mammalian cell surface molecules. J. Virol. 69:4471–4483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Connolly SA, Landsburg DJ, Carfi A, Wiley DC, Cohen GH, Eisenberg RJ. 2003. Structure-based mutagenesis of herpes simplex virus glycoprotein D defines three critical regions at the gD-HveA/HVEM binding interface. J. Virol. 77:8127–8140. 10.1128/JVI.77.14.8127-8140.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kousoulas KG, Pellett PE, Pereira L, Roizman B. 1984. Mutations affecting conformation or sequence of neutralizing epitopes identified by reactivity of viable plaques segregate from syn and ts domains of HSV-1(F) gB gene. Virology 135:379–394. 10.1016/0042-6822(84)90194-6 [DOI] [PubMed] [Google Scholar]
- 41.Pellett PE, Kousoulas KG, Pereira L, Roizman B. 1985. Anatomy of the herpes simplex virus 1 strain F glycoprotein B gene: primary sequence and predicted protein structure of the wild type and of monoclonal antibody-resistant mutants. J. Virol. 53:243–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pereira L, Dondero D, Norrild B, Roizman B. 1981. Differential immunologic reactivity and processing of glycoproteins gA and gB of herpes simplex virus types 1 and 2 made in Vero and HEp-2 cells. Proc. Natl. Acad. Sci. U. S. A. 78:5202–5206. 10.1073/pnas.78.8.5202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krawczyk A, Krauss J, Eis-Hubinger AM, Daumer MP, Schwarzenbacher R, Dittmer U, Schneweis KE, Jager D, Roggendorf M, Arndt MA. 2011. Impact of valency of a glycoprotein B-specific monoclonal antibody on neutralization of herpes simplex virus. J. Virol. 85:1793–1803. 10.1128/JVI.01924-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Drew PD, Moss MT, Pasieka TJ, Grose C, Harris WJ, Porter AJ. 2001. Multimeric humanized varicella-zoster virus antibody fragments to gH neutralize virus while monomeric fragments do not. J. Gen. Virol. 82:1959–1963 http://vir.sgmjournals.org/content/82/8/1959.long [DOI] [PubMed] [Google Scholar]
- 45.Thullier P, Lafaye P, Megret F, Deubel V, Jouan A, Mazie JC. 1999. A recombinant Fab neutralizes dengue virus in vitro. J. Biotechnol. 69:183–190. 10.1016/S0168-1656(99)00037-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lamarre A, Talbot PJ. 1995. Protection from lethal coronavirus infection by immunoglobulin fragments. J. Immunol. 154:3975–3984 [PubMed] [Google Scholar]
- 47.Dollery SJ, Wright CC, Johnson DC, Nicola AV. 2011. Low-pH-dependent changes in the conformation and oligomeric state of the prefusion form of herpes simplex virus glycoprotein B are separable from fusion activity. J. Virol. 85:9964–9973. 10.1128/JVI.05291-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dollery SJ, Delboy MG, Nicola AV. 2010. Low pH-induced conformational change in herpes simplex virus glycoprotein B. J. Virol. 84:3759–3766. 10.1128/JVI.02573-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Siekavizza-Robles CR, Dollery SJ, Nicola AV. 2010. Reversible conformational change in herpes simplex virus glycoprotein B with fusion-from-without activity is triggered by mildly acidic pH. Virol. J. 7:352. 10.1186/1743-422X-7-352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Riede I, Schwarz H, Jahnig F. 1987. Predicted structure of tail-fiber proteins of T-even type phages. FEBS Lett. 215:145–150. 10.1016/0014-5793(87)80130-8 [DOI] [PubMed] [Google Scholar]
- 51.Cerritelli ME, Wall JS, Simon MN, Conway JF, Steven AC. 1996. Stoichiometry and domainal organization of the long tail-fiber of bacteriophage T4: a hinged viral adhesin. J. Mol. Biol. 260:767–780. 10.1006/jmbi.1996.0436 [DOI] [PubMed] [Google Scholar]
- 52.Bartual SG, Otero JM, Garcia-Doval C, Llamas-Saiz AL, Kahn R, Fox GC, van Raaij MJ. 2010. Structure of the bacteriophage T4 long tail fiber receptor-binding tip. Proc. Natl. Acad. Sci. U. S. A. 107:20287–20292. 10.1073/pnas.1011218107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Däumer MP, Schneider B, Giesen DM, Aziz S, Kaiser R, Kupfer B, Schneweis KE, Schneider-Mergener J, Reineke U, Matz B, Eis-Hubinger AM. 2011. Characterisation of the epitope for a herpes simplex virus glycoprotein B-specific monoclonal antibody with high protective capacity. Med. Microbiol. Immunol. 200:85–97. 10.1007/s00430-010-0174-x [DOI] [PubMed] [Google Scholar]
- 54.Prabhu N, Prabakaran M, Ho HT, Velumani S, Qiang J, Goutama M, Kwang J. 2009. Monoclonal antibodies against the fusion peptide of hemagglutinin protect mice from lethal influenza A virus H5N1 infection. J. Virol. 83:2553–2562. 10.1128/JVI.02165-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu JY, Gorny MK, Palker T, Karwowska S, Zolla-Pazner S. 1991. Epitope mapping of two immunodominant domains of gp41, the transmembrane protein of human immunodeficiency virus type 1, using ten human monoclonal antibodies. J. Virol. 65:4832–4838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Robinson WE, Jr, Gorny MK, Xu JY, Mitchell WM, Zolla-Pazner S. 1991. Two immunodominant domains of gp41 bind antibodies which enhance human immunodeficiency virus type 1 infection in vitro. J. Virol. 65:4169–4176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Earl PL, Broder CC, Doms RW, Moss B. 1997. Epitope map of human immunodeficiency virus type 1 gp41 derived from 47 monoclonal antibodies produced by immunization with oligomeric envelope protein. J. Virol. 71:2674–2684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zolla-Pazner S. 2004. Identifying epitopes of HIV-1 that induce protective antibodies. Nat. Rev. Immunol. 4:199–210. 10.1038/nri1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McLellan JS, Chen M, Leung S, Graepel KW, Du X, Yang Y, Zhou T, Baxa U, Yasuda E, Beaumont T, Kumar A, Modjarrad K, Zheng Z, Zhao M, Xia N, Kwong PD, Graham BS. 2013. Structure of RSV fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. Science 340:1113–1117. 10.1126/science.1234914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yin HS, Wen X, Paterson RG, Lamb RA, Jardetzky TS. 2006. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 439:38–44. 10.1038/nature04322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roche S, Rey FA, Gaudin Y, Bressanelli S. 2007. Structure of the prefusion form of the vesicular stomatitis virus glycoprotein G. Science 315:843–848. 10.1126/science.1135710 [DOI] [PubMed] [Google Scholar]