

ABSTRACT
Herpes simplex virus type 1 immediate-early protein ICP0 is an E3 ubiquitin ligase of the RING finger class that degrades several cellular proteins during infection. This activity is essential for its functions in stimulating efficient lytic infection and productive reactivation from latency. ICP0 targets a number of proteins that are modified by the small ubiquitin-like SUMO family of proteins, and it includes a number of short sequences that are related to SUMO interaction motifs (SIMs). Therefore, ICP0 has characteristics that are related to those of cellular SUMO-targeted ubiquitin ligase enzymes. Here, we analyze the impact of mutation of a number of SIM-like sequences (SLSs) within ICP0 on HSV-1 replication and gene expression and their requirement for ICP0-mediated degradation of both sumoylated and unmodified promyelocytic leukemia (PML) and other sumoylated cellular proteins. One SLS in the central portion of the ICP0 sequence (SLS4) was found to be absolutely required for targeting cellular sumoylated species in general and sumoylated forms of PML other than those of PML isoform I. Mutation of a group of SLSs in the C-terminal quarter of ICP0 also reduced ICP0-mediated degradation of sumoylated PML in a cooperative manner. Although mutation of individual SLSs caused only modest decreases in viral replication, combined mutation of SLS4 with SLS sequences in the C-terminal quarter of the protein reduced plaque formation efficiency by up to two orders of magnitude. These results provide further evidence that the biological activities of ICP0 are connected with host cell sumoylation events.
IMPORTANCE Herpes simplex virus type 1 protein ICP0 plays important roles in regulating the initial stages of lytic infection and productive reactivation from latency. ICP0 mediates its effects through inducing the degradation of cellular proteins that have repressive effects on viral gene expression. An increasing number of cellular proteins are known to be sensitive to ICP0-mediated degradation; therefore, it is important to understand how ICP0 selects its substrates for degradation. This study identifies sequence motifs within ICP0 that are involved in targeting cellular proteins that are modified by the SUMO family of ubiquitin-like proteins and describes how mutation of combinations of these motifs causes a 100-fold defect in viral infectivity.
INTRODUCTION
Herpes simplex virus type 1 (HSV-1) immediate-early protein ICP0, a member of the RING finger class of E3 ubiquitin ligases, is required for efficient lytic infection and productive reactivation from latency (1–3). These biological functions of ICP0 require its biochemical ubiquitin ligase activity that results in the degradation of several cellular proteins. While these basic aspects of ICP0 have been firmly established, several questions remain concerning the mechanisms by which ICP0 selects its substrates for degradation and the significance of individual substrates for the outcome of infection. It has long been established that ICP0 initially localizes to cellular nuclear substructures known as ND10 or promyelocytic leukemia nuclear bodies (PML NBs) during the early stages of infection, where it brings about the proteasome-dependent degradation of the promyelocytic leukemia protein PML and the dispersal of other PML NB constituent proteins (4, 5). PML is heavily modified by the ubiquitin-like SUMO family of proteins, and both PML and its sumoylation are required for the proper assembly of PML NBs (6). ICP0 targets PML for degradation through two mechanisms, one being the direct recognition of the most abundant PML isoform, PML.I, in a SUMO-independent manner (7) and the other resulting in the loss of the sumoylated forms of all PML isoforms (4, 8). ICP0 also reduces the level of sumoylated protein species in the cell in general, especially at later times of infection (4, 8), indicating that substrate selection by ICP0 is in part determined by sumoylation.
The biological significance of these findings are illustrated not only by the role of PML in intrinsic resistance to HSV-1 and several other viruses (9–11) but also by indications that sumoylation is involved in several mechanisms that regulate the replication of HSV-1 and a number of other viruses (7, 8, 12, 13). The experiments reported in this paper were initiated on the basis of our previous work that identified a number of potential SUMO interaction motifs (SIMs) within the ICP0 sequence (here termed SIM-like sequences [SLSs]) (8). We presented evidence that some of these sequences could interact with SUMO family members in yeast 2-hybrid or glutathione S-transferase (GST) pulldown assays, and using cell lines that express ICP0 in an inducible manner, we found that these sequences are involved in the efficiency of degradation of SUMO-modified PML and other sumoylated proteins (8). Here, we extend these analyses by constructing HSV-1 mutants that express ICP0 proteins with lesions in one or more SLSs. Mutation of one particular SLS (SLS4), which was previously shown to impart interaction of ICP0 with SUMO2 (8), did not affect the activity of ICP0 on PML.I but eliminated the ability of ICP0 to degrade sumoylated PML isoform II and sumoylated species in general. This result identifies a short specific region of ICP0 that is required for targeting sumoylated proteins. Surprisingly, mutation of SLS4 by itself caused only a modest defect in viral replication, but a combination of this mutation with SLS mutations in the C-terminal quarter of the protein reduced the plaque-forming efficiency of HSV-1 by up to two orders of magnitude. These results strengthen the hypothesis that the biological activities of ICP0 are linked to SUMO-related pathways.
MATERIALS AND METHODS
Cells.
Human diploid fibroblasts (HFs) and U2OS cells were grown in Dulbecco's modified Eagles' medium (DMEM) supplemented with 10% fetal calf serum (FCS). Baby hamster kidney (BHK) cells were grown in Glasgow modified Eagles' medium (GMEM) supplemented with 10% newborn calf serum and 10% tryptose phosphate broth. HepaRG cells (14) were grown in William's medium E supplemented with 10% fetal bovine serum gold (PAA Laboratories Ltd.), 2 mM glutamine, 5 μg/ml insulin, and 0.5 μM hydrocortisone. Derivatives of HepaRG cells depleted of endogenous PML and expressing enhanced yellow fluorescent protein (EYFP)-linked PML isoform I or II at close to endogenous levels have been described previously (15). All cell growth media were supplemented with 100 U/ml penicillin and 100 μg/ml streptomycin. Lentivirus-transduced cells were maintained with continuous antibiotic selection as appropriate.
Plasmids.
Plasmids with ICP0 cDNAs in an inducible expression vector with mutations in the SIM-like sequences SLS4 (mSLS4), SLS5 and SLS7 (mSLS5/7), and SLS4, SLS5, and SLS7 (mSLS4/5/7) in combination have been described previously (8). Analogous plasmids with mutations in SLS6 (mSLS6; L667A/I669A/L670A), the two residues mutated in the mutant known as R8507 (D671A/E673A) (16), and SLS5, SLS6, and SLS7 (mSLS5/6/7) in combination were constructed by PCR splicing using mutagenic oligonucleotides. The mutations described above were transferred to plasmids containing a genomic version of the ICP0 gene with the IE1 promoter and 3′ flanking sequences to enable construction of mutant viruses.
Propagation and maintenance of virus stocks.
All viruses were grown in BHK cells and titrated in U2OS cells using human serum in the overlay. ICP0 is not required for HSV-1 plaque formation in U2OS cells (17), allowing a true comparison of the titers of wt and mutant virus stocks. HSV-1 wild-type (wt) strain 17 and the ICP0 null mutant dl1403 (18) and HSV-1 mutants with specific deletion mutations in ICP0 (D12, D13, and E52X) have been described previously (19, 20).
Viruses expressing mSLS4, mSLS6, mSLS5/7, mSLS4/5/7, mSLS5/6/7, and R8507 versions of ICP0 were constructed by marker rescue of the ICP0 deletion mutant dl1403. Briefly, BHK cells were transfected with plasmid fragments including the complete ICP0 viral genomic coding and flanking regions and which contained the ICP0 mutations, and then they were infected with virus dl1403 the following day. Progeny virus was harvested, passaged in HFs three times to enrich for ICP0 activity greater than that of the parental null mutant, and then screened after plaque purification. Plaques with viruses expressing ICP0 protein were identified and then purified by repeated plaque isolation. Viral DNA was prepared from the final viral stocks and then analyzed by direct sequence analysis of PCR products to confirm introduction of the desired mutant ICP0 sequences.
Virus stocks for experimental analysis were prepared in batches by infection of 60-mm plates of BHK cells at low multiplicity (0.01 to 0.001, depending on the severity of the mutation), clarified supernatants were harvested 3 days later and stored at 4°C, and their titers were checked regularly. Comparisons were made using only batches of viruses prepared and titrated simultaneously. Infections to compare rates of viral protein expression were done at a multiplicity of infection (MOI) of 1, while infections to monitor rates of degradation of cellular proteins were performed at higher MOI to ensure that all of the cells in the population were expressing ICP0. This was checked by immunofluorescence analysis in parallel.
Infections, Western blotting, and antibodies.
Cells were seeded into 24-well dishes at 1 × 105 cells per well and then infected the following day. Cell monolayers were washed twice with PBS before harvesting in SDS-PAGE loading buffer. Proteins were resolved on 7.5% SDS gels and then transferred to nitrocellulose membranes by Western blotting. The following antibodies were used: anti-ICP0 mouse monoclonal antibody (MAb) 11060 (21); anti-actin MAb AC-40 (Sigma-Aldrich); anti-PML MAb 5E10 (22); anti-ICP4 MAb 58S (23); and anti-EGFP rabbit polyclonal antibody (rAb) ab290 (Abcam), anti-UL42 MAb Z1F11, SUMO1 with rAb ab32058 (Abcam), and anti-SUMO2/3 rAb ab3742-100 (Abcam). Anti-VP5 MAb DM165 was a gift from Frazer Rixon. Standard ECL reagents were used in most cases, with the exception of PML and SUMO2/3 blots, for which the enhanced ECL reagent (RPN2235; GE Healthcare) was used.
Immunofluorescence.
Cells on 13-mm glass coverslips were fixed with formaldehyde and prepared for immunofluorescence using standard methods. PML was detected with rAb ABD-030 (Jena Bioscience) or MAb 5E10, ICP4 with MAb 58S, ICP0 with MAb 11060, and SUMO2/3 with rAb ab3742-100 (Abcam). The secondary antibodies were fluorescein isothiocyanate (FITC)-conjugated sheep anti-mouse IgG, Alexa 555-conjugated goat anti-mouse IgG, and Alexa 633-conjugated goat anti-rabbit IgG (Invitrogen). The samples were examined using a Zeiss LSM 710 confocal microscope with 488-nm, 561-nm, and 633-nm laser lines, scanning each channel separately under image capture conditions that eliminated channel overlap. The images were exported as tif files, minimally adjusted using Photoshop, and then assembled into figures using Illustrator.
GST pull-down interaction assays.
The relevant coding regions of the various mutant versions of ICP0 were amplified from full-length ICP0 expression vectors by PCR using an N-terminal primer that added an EcoRI site at the 5′ end to allow precise ligation to and reconstruction of the fusion junction in a GST fusion protein, including ICP0 residues 594 to 775. A 3′ primer was used that enabled use of the SalI site near the C terminus of the ICP0 open reading frame as the 3′ ligation site. PCR products were cut with EcoRI and SalI and then inserted in place of the wt sequence. A GST-Ubc9 fusion protein (a kind gift from Ron Hay, University of Dundee) was used as a positive control. GST pulldown assays using the C-terminal quarter of ICP0 (residues 594 to 775) were performed as previously described, using bacterial extracts containing His-tagged SUMO1 and purified GST-ICP0 beads (8). Briefly, the beads were incubated in 500 μl buffer H (50 mM HEPES, pH 7.0, 150 mM NaCl, 5 mM β-mercaptoethanol, 0.1% NP-40) with 250 μl of 0.45-μm filtered bacterial supernatants containing His-tagged SUMO1. The samples were mixed end-over-end at 4°C overnight, and then the beads were washed three times in 1 ml buffer H and soluble complexes eluted in 90 μl of 1× SDS-PAGE loading buffer. Samples (30 μl) of eluent were resolved by SDS-PAGE and analyzed by Western blotting for SUMO1.
RESULTS
Mutations in the SIM-like sequences of ICP0 reduce HSV-1 plaque formation and gene expression.
Utilizing an inducible ICP0 expression system, we previously reported that mutation of SLSs within ICP0 reduced its ability to complement the plaque formation efficiency of an ICP0 null mutant HSV-1 and, to a lesser extent, to derepress quiescent HSV-1 genomes in cultured cells (8). The aims of the current study were to analyze these mutations in the context of the virus itself and to extend the work to include mutations in the SLS6 motif and neighboring residues that had previously been studied in a strain F mutant known as R8507 (16). For simplicity, we have retained this name for the D671A/E673A mutation, although in the work reported here the mutant was derived by homologous recombination into the strain 17 HSV-1 background rather than via a strain F bacmid. SLS4 confers the ability of ICP0 to bind to SUMO2/3 in a yeast 2-hybrid assay and lies in the central portion of the protein (8), while SLS5, SLS6, and SLS7 are situated close together within a region in the C-terminal quarter of ICP0 that had previously been identified as being required for efficient localization to PML NBs (24, 25) (Fig. 1A). After plaque purification, the identity of virus isolates was confirmed by direct DNA sequencing of viral DNA.
FIG 1.
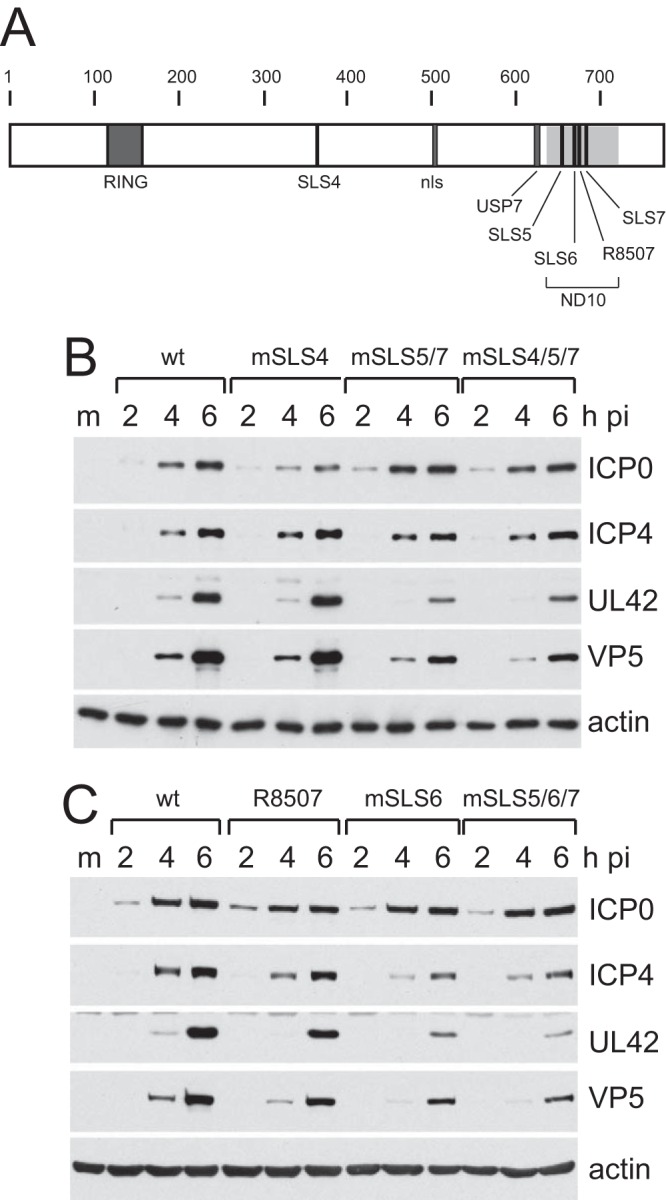
Locations of the mSLS mutations and their effects on viral gene expression. (A) A map of the coding sequence of ICP0, indicating the minimal RING domain from the first to last zinc-coordinating cysteine residues (residues 116 to 156); SIM-like sequence SLS4 (360 to 366); nuclear localization signal (nls; 501 to 510); core of the USP7 binding region (614 to 630); SIM-like sequences SLS5 (651 to 655), SLS6 (666 to 669), and SLS7 (681 to 684); and two point mutations adjacent to SLS6 previously defined by the mutation R8507 (670 and 672). The shaded area (633 to 720) indicates the region implicated in efficient localization of ICP0 to ND10 (PML NBs). (B and C) Comparison of the viral gene expression efficiencies of the mSLS viruses. HepaRG cells were infected at an MOI of 1 with the indicated viruses. Samples taken at the indicated time points were analyzed by Western blotting for ICP0, ICP4, UL42, and VP5 as representatives of the IE, early, and late classes of viral proteins. Actin provides the loading control. M, molecular size ladder.
Plaque assays indicated that mutations within SLS4 or SLS6 caused a modest 3-fold decrease in plaque formation in restrictive HepaRG cells compared to permissive U2OS cells, and a similar result was obtained with R8507 (Table 1). The latter result is consistent with an independent study (26). Combinations of double or triple mutations in the SLS5, SLS6, SLS7 region caused about a 10-fold defect, while the combination of the mSLS4 and mSLS5/7 mutations caused the most substantial defect in this study (60- to 70-fold in HepaRG cells) (Table 1). Similar results were observed in HFs, except that R8507 was not defective (confirmed by virus growth curves; data not shown), while the mSLS4/5/7 mutant had a 100-fold defect in plaque formation in this cell type (Table 1). These results are in line with the previous analysis of the SLS4, SLS5/7, and SLS4/5/7 mutants in the inducible cell line system (8) and of complementation analyses by expression of mutant proteins mSLS6, R8507, and mSLS5/6/7 in ICP0-inducible cells (data not shown). The data are consistent with the hypothesis that regions of the protein, including the SLS sequences, function in concert.
TABLE 1.
Plaque formation efficiencies of ICP0 mutant viruses
| Virus | Avg plaque formation efficiencya |
|
|---|---|---|
| HepaRG/U20S | HF/U2OS | |
| wt strain 17 | 1.0 | 1.0 |
| mSLS4 | 0.35 ± 0.19 | 0.48 ± 0.37 |
| mSLS6 | 0.34 ± 0.05 | 0.20 ± 0.07 |
| R8507 | 0.31 ± 0.09 | 1.28 ± 0.83 |
| mSLS5/7 | 0.073 ± 0.048 | 0.073 ± 0.029 |
| mSLS4/5/7 | 0.015 ± 0.009 | 0.010 ± 0.001 |
| mSLS5/6/7 | 0.10 ± 0.02 | 0.072 ± 0.043 |
Average plaque formation efficiencies of the mutant viruses normalized to those of the wt in the indicated cell type. The absolute efficiency of the wt in HepaRG versus U2OS was 3.8, and the average for HF versus U2OS was 3.0. The ICP0 RING finger deletion mutant virus gives a ratio of around 0.001 in both HFs and HepaRG cells.
Analysis of the rates of viral gene expression by this set of viruses indicated that they all expressed ICP0 at similar levels in HepaRG cells infected at an MOI of 1, and that the most defective viruses in terms of UL42 (early) and VP5 (late) gene expression were mSLS4/5/7, mSLS6, mSLS5/7, and mSLS5/6/7 (Fig. 1B and C). The differentials between the mutant and wt viruses are very sensitive to choice of input MOI, being reduced at higher MOI and augmented at lower MOI (data not shown); therefore, they are difficult to assess quantitatively. Qualitatively, however, the relative fitness of the viruses was reliable between different experiments and using different virus stocks. We conclude that point mutations in SIM-like sequences within ICP0 reduce the efficiency of viral gene expression.
Mutations in the SIM-like sequences of ICP0 affect its intracellular localization.
The mutant viruses enabled analysis of the intracellular localization of the mSLS mutant ICP0 proteins as infection progresses. During wt HSV-1 infection, ICP0 first colocalizes with PML and then rapidly induces degradation of PML and dispersal of PML NBs while becoming more diffusely spread through the nucleoplasm before beginning to accumulate in punctate foci in the cytoplasm (Fig. 2, upper 9 images) (5, 24, 25). The rates at which these changes occur vary with MOI and between individual cells; therefore, the assay is not easy to quantify. Nonetheless, we observed a number of reliable differences between the wt and mSLS mutant ICP0 proteins. Mutant mSLS4 behaved in a manner similar to that of the wt protein, except that it tended to be more diffuse within the nucleoplasm at 4 h and had migrated into the cytoplasm in a higher proportion of cells at 6 h (Fig. 2, lower left 9 images). Compared to the wt protein, mutant mSLS5/7 was clearly more diffuse at 2 h, it dispersed PML less efficiently at 4 h, and it remained entirely within the nucleoplasm at 6 h (Fig. 2, upper right 9 images). Mutant mSLS4/5/7 was again more diffuse than the wt in most cells and exhibited reduced colocalization with PML at 4 h, was less able to disperse PML even at 6 h, and again was retained in the nucleoplasm throughout this time course (Fig. 2, lower right 9 images). The most striking feature of mutants R8507 and mSLS6, particularly the latter, was the appearance of very bright, numerous, and distinct small nuclear foci at the 4-h time point, which were retained even at 6 h, while migration of both mutants into the cytoplasm was less marked than that with the wt protein (Fig. 3, lower left and upper right 9 images). PML dispersal with these mutants appeared slightly delayed compared to that of the wt (in the case of R8507, this finding is consistent with a previous report [16]). Mutant mSLS5/6/7 was similar to the double mutant mSLS5/7, in that it was more diffuse than the wt and was retained in the nucleoplasm throughout the time course (Fig. 3, lower right 9 images). Interestingly, mSLS5/6/7 accumulated in structures resembling viral replication compartments at the 6-h time point. This property could also be seen with mSLS5/7, particularly in heavily infected cells (data not shown).
FIG 2.

Immunofluorescence analysis of wt HSV-1 and mutant viruses mSLS4, mSLS5/7, and mSLS4/5/7. HFs on coverslips were infected at an MOI of 2, and then samples were analyzed at 2 h, 4 h, and 6 h after infection by immunofluorescence staining for ICP0 (green) and PML (red). Separated channels are shown in greyscale, with the color merged image to the right. The uppermost row shows PML and ICP0 staining in uninfected control cells.
FIG 3.

Immunofluorescence analysis of wt HSV-1 and mutant viruses R8507, mSLS6, and mSLS5/6/7. HFs on coverslips were infected at an MOI of 2, and then samples were analyzed at 2 h, 4 h, and 6 h after infection by immunofluorescence staining for ICP0 (green) and PML (red). Separated channels are shown in greyscale, with the color merged image to the right.
A clear general conclusion from these observations is that point mutations in the SIM-like sequences of ICP0 affect its intracellular localization and trafficking within the cell. The mSLS5/7 and mSLS5/6/7 mutations identify motifs that influence the initial efficiency of localization of ICP0 at PML NBs (or its retention within them [27]) and that are required for its later accumulation in the cytoplasm. Combining the mutations in SLS4, SLS5, and SLS7 caused a marked defect in PML NB localization and PML dispersal as well as eliminating later cytoplasmic accumulation.
The SIM-like sequences of ICP0 are involved in the degradation of PML.
Given that some of the mSLS mutants of ICP0 dispersed PML NBs less rapidly than the wt protein, we next analyzed the rate of degradation of endogenous PML induced by these viruses. HepaRG cells were infected at an MOI of 2, and then time course samples were analyzed by Western blotting for PML, ICP0, and actin. wt virus caused almost complete loss of PML by 4 h under these conditions (Fig. 4A). PML degradation by mSLS4 and mSLS5/7 was a little slower than that in the wt infection, but the mSLS4/5/7 mutant was substantially less active. The R8507, mSLS6, and mSLS5/6/7 mutants all induced degradation of PML at a reduced rate compared to wt HSV-1 (Fig. 4B), which may be related to their reduced abilities to disperse PML (described above; also see reference 16).
FIG 4.
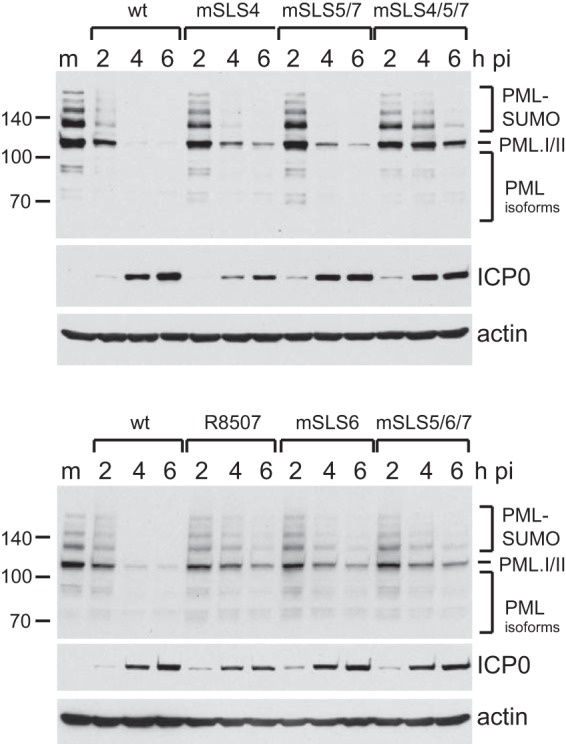
Degradation rates of endogenous PML by wt and mSLS mutant viruses. HepaRG cells were infected with the indicated viruses at an MOI of 2, and then samples harvested at 2 h, 4 h, and 6 h after infection were analyzed by Western blotting for endogenous PML (MAb 5E10), ICP0, and actin.
Although the effects of some of the mSLS mutations on PML stability have been reported previously in the inducible cell line system (8), the use of the viruses allows more controlled time course analysis. A significant issue is that, for reasons that remain unknown, mutants mSLS5, mSLS6, mSLS7, mSLS5/7, and mSLS5/6/7 all accumulate ICP0 to substantially greater levels than the wt in the inducible cell system (8 and data not shown), making it difficult to compare the efficiency of PML degradation at equalized levels of ICP0. This problem does not occur during virus infection. We conclude from these results that the combined mutation of mSLS4/5/7 causes a substantial decrease in the efficiency of degradation of endogenous PML, while all of the mutations in the SLS5/6/7 region reduce the rate of PML degradation compared to the wt.
Mutation of SLS4 within ICP0 compromises SUMO-dependent targeting of PML.
ICP0 induces degradation of PML through two mechanisms, one involving a SUMO-independent interaction with PML.I (the most abundant PML isoform) and the other resulting in the degradation of the sumoylated forms of all other PML isoforms (7, 8). We investigated the involvement of the SIM-like sequences of ICP0 in these two mechanisms by infecting HepaRG cells depleted of endogenous PML and expressing either EYFP-linked PML.I or PML.II (15). Consistent with the previous reports, wt HSV-1 degraded all forms of PML.I efficiently. The increased high-molecular-weight smear of PML.I in the 2-h infection sample most likely reveals directly ubiquitinated forms of the protein (Fig. 5A). The sumoylated forms of PML.II were rapidly lost during the infection, but there was an increase in the intensity and complexity of the unsumoylated forms, likely reflecting changes in other modifications, such as phosphorylation (Fig. 5A). All of the mutant viruses were analyzed in the same assay, comparing effects on PML.I and PML.II in parallel. The mSLS4 mutation had little effect on the rate of degradation of PML.I, but remarkably the mutant had no effect on sumoylated PML.II (Fig. 5B). This was also the case with mutant mSLS4/5/7, although this mutant also degraded PML.I at a reduced rate compared to the wt (Fig. 5C and H for a direct comparison of PML.I degradation by wt and mSLS4/5/7 at a reduced MOI). This striking result demonstrates that SLS4 is absolutely required for ICP0 to induce the degradation of the sumoylated forms of a PML isoform with which it does not directly interact (7). In contrast, mSLS5/7 retained the ability to degrade sumoylated PML.II (Fig. 5D). The mutants involving the SLS6/R8507 region had a more complex phenotype, with PML.I and sumoylated PML.II being more stable in all of these infections, especially in the case of R8507 (Fig. 5E to G). These results are broadly consistent with the analysis of endogenous PML degradation (Fig. 4). The efficient degradation of all forms of endogenous PML by mSLS4 (Fig. 4) may seem to be in conflict with its failure to degrade any of the modified forms of PML.II (Fig. 5B), but in the case of endogenous PML all isoforms interact with PML.I through their coiled-coil motifs (28). This may allow degradation of the more minor isoforms when in complex with PML.I.
FIG 5.

Specificity of targeting of PML.I, PML.II, and their SUMO-conjugated forms by wt and mSLS mutant viruses. (A to G) HepaRG cells expressing EYFP-PML.I (left) or EYFP-PML.II (right) at close to endogenous levels were infected with the indicated viruses at an MOI of 5, and then samples harvested at 2 h, 4 h, 6 h, and 8 h were analyzed by Western blotting for EYFP fusion proteins, ICP0, and actin. (H) Comparison of rates of degradation of EYFP-PML.I by wt and mSLS4/5/7 mutant viruses at an MOI of 2.
These experiments were performed because of a previous result indicating that the C-terminal quarter of ICP0 (residues 594 to 775) is required for efficient degradation of sumoylated PML.II under conditions in which PML.I is efficiently degraded (7). In this study, we confirmed this result and found that smaller deletions within this region (D12 [Δ594-632] and D13 [Δ633-680]) had a similar phenotype (data not shown). Taken together with the results for mutants mSLS5/6/7, mSLS6, and R8507, these results identify a second region of ICP0, in addition to that of SLS4, that is involved in the efficient degradation of sumoylated PML.II such that mutation of either is sufficient to inhibit this activity.
SUMO conjugate targeting by SLS mutants of ICP0.
ICP0 induces a substantial reduction in the overall level of high-molecular-weight SUMO conjugates during infection (4, 8). Given that mutation of SLS4 eliminated the ability of ICP0 to degrade sumoylated PML.II (Fig. 5B), we next investigated whether this defect extended to sumoylated species in general. This was indeed the case, with such species remaining stable in HepaRG cells infected with mSLS4 at an MOI of 5, while they were degraded in the parallel wt infection (Fig. 6A). As expected, mSLS4/5/7 gave a result similar to that of mSLS4. The sumoylated species were unstable in R8507 and mSLS6 infections but were significantly more stable in mSLS5/7 and mSLS5/6/7 infections than in the wt controls (Fig. 6A and B). These results are consistent with the analysis of Fig. 5, and they indicate that while SLS4 is essential for destabilizing a range of SUMO conjugates, the SLS5/6/7 region is also involved in this aspect of ICP0 activity and supports our previous conclusion that ICP0 has properties related to cellular STUbLs (8).
FIG 6.
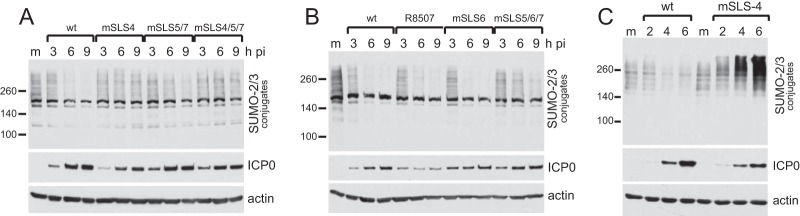
Degradation rates of high-molecular-weight SUMO2/3-conjugated proteins by wt and mSLS mutant viruses. HepaRG cells were infected with the indicated viruses at an MOI of 2, and then samples harvested at 2 h, 4 h, and 6 h after infection were analyzed by Western blotting for SUMO2/3, ICP0, and actin. (C) Infection of HFs by wt and mutant mSLS4 viruses at an MOI of 5, illustrating the substantial increase in high-molecular-weight SUMO conjugates that can occur at later times of mSLS4 infection.
High-MOI infection with ICP0 null mutant HSV-1 leads to an induction in the relative abundance of sumoylated proteins at later times of infection (8). The significance of this phenomenon remains unknown, but it also occurs during influenza virus infection (29). Remarkably, SUMO conjugate abundance also increased in HFs infected at high MOI with mSLS4, underscoring the significance of the SLS4 region in regulating sumoylated species during HSV-1 infection (Fig. 6C). A similar phenomenon could also be seen in HepaRG cells, but only at very high MOI (data not shown). We previously proposed that the general SUMO conjugate induction reflects an antiviral response of the cell (8), but this new observation necessitates reappraisal of this hypothesis, because mSLS4 has only a modest defect in plaque formation efficiency (Table 1). On the one hand, it could be argued that the increase in sumoylated species seen in mSLS4-infected cells cannot represent an antiviral response, because this virus does not have a large replication defect. On the other hand, it could be that despite being unable to impede the increase in sumoylated species, the mSLS4 protein remains able to counteract their antiviral effects. In favor of the latter argument are the observations that mSLS4 retains the ability to degrade sumoylated endogenous PML, and perhaps other specific sumoylated proteins among the general increase observed, and also that mSLS4 inhibits the recruitment of sumoylated proteins and the SIM-dependent recruitment (12) of hDaxx to viral genomes (see below).
The SIM-like sequences of ICP0 are involved in the inhibition of recruitment of PML and SUMO conjugates to HSV-1 genomes.
A prominent feature of ICP0 activity is its ability to inhibit the recruitment of PML, SUMO conjugates, and other PML NB proteins to sites that are closely associated with incoming HSV-1 genomes in cells at the edge of developing plaques during the earliest stages of infection (12, 30, 31). This property correlates well with the efficiency of ICP0-mediated stimulation of both lytic infection and reactivation of quiescent HSV-1 genomes (32). Therefore, we investigated if this hypothesis holds true for the mSLS set of viruses, with the prediction being that the most defective of the viruses, mSLS4/5/7 (Table 1), should be the least able to inhibit recruitment of PML- and SUMO-conjugated proteins to HSV-1 genomes. Because of variability between individual cells, this assay is difficult to quantify; therefore, the interpretation is limited to major differences. Recruitment of PML and SUMO2/3 is obvious in all cells infected with ICP0 null mutant HSV-1 and expressing ICP4 in characteristic asymmetric foci, but it is detectable in only a very small minority of wt-infected cells and only then at the very earliest stages of infection, when ICP4 (and ICP0) expression levels are very low. Consistent with the hypothesis posed above, recruitment of PML and SUMO2/3 was obvious in most mSLS4/5/7-infected cells but was rarely detectable or much less marked in cells infected with the other mutants (Fig. 7 and 8).
FIG 7.

Inhibition of recruitment of PML to viral genome foci by wt and mSLS mutant viruses. HFs on coverslips were infected at a low MOI, depending on the intrinsic plaque-forming efficiencies of the viruses, so that small virus plaques had been produced 24 h later. The cells were analyzed by immunofluorescence staining for PML (red) and ICP4 (green). Separated channels are shown in greyscale, with the color merged image to the right.
FIG 8.

Inhibition of recruitment of SUMO2/3-conjugated proteins to viral genome foci by wt and mSLS mutant viruses. HFs on coverslips were infected at a low MOI, depending on the intrinsic plaque-forming efficiencies of the viruses, so that small virus plaques had been produced 24 h later. The cells were analyzed by immunofluorescence staining for SUMO2/3 (red) and ICP4 (green). Separated channels are shown in greyscale, with the color merged image to the right.
An important feature of these results is that despite its failure to degrade sumoylated proteins (Fig. 6), mSLS4 efficiently impeded recruitment of SUMO2/3-conjugated species (and hDaxx; data not shown) to sites of viral genomes (Fig. 8). Although these species would include sumoylated PML in the case of ICP0 null mutant HSV-1, recruitment of SUMO2/3 conjugates still occurs in PML-depleted cells (12); therefore, the lack of such recruitment in mSLS4 infections cannot be explained by degradation of PML (Fig. 4). Another feature of these results is that mutants R8507 and mSLS6 are less efficient than the wt in degrading endogenous PML (Fig. 4) and the various forms of introduced EYFP-PML.I and PML.II (Fig. 5), yet they efficiently inhibit recruitment of PML to the viral genomes (Fig. 7). This is reminiscent of the situation with the bovine herpesvirus 1, equine herpesvirus 1, and pseudorabies virus orthologues of ICP0, which have only small effects on the stability of PML yet inhibit its recruitment to HSV-1 genomes and stimulate ICP0-null mutant HSV-1 infection efficiently (33). Therefore, inhibition of recruitment of PML and other ND10 components serves as a good prediction factor for the ability of ICP0 proteins to stimulate lytic HSV-1 infection.
Influence of mutations in the C-terminal quarter of ICP0 on its interaction with SUMO1 in vitro.
We have reported previously that the C-terminal quarter of ICP0, residues 594 to 775, interacts with SUMO1 in GST pulldown assays in vitro (8). We extended this analysis by investigating the effects of all of the various mutations studied above on the efficiency of SUMO1 interaction (Fig. 9). Equalized amounts of GST fusion protein beads, including GST-Ubc9 and GST itself as positive and negative controls, were mixed with bacterial extracts containing His-tagged SUMO1. The bound proteins were purified and analyzed by Western blotting for SUMO1 and by Coomassie staining to confirm the qualities and quantities of the various fusion proteins. Compared to Ubc9, which is known to interact strongly with SUMO1 (34), the interaction of residues 594 to 775 of ICP0 was weak but greater than that of the GST negative control (Fig. 9). Consistent with our previous study (8), the SLS5 mutation decreased the interaction with SUMO1, but for unknown reasons the interaction of the mSLS5/7 mutant was similar to that of the wt. The SLS6 mutation had no effect on binding efficiency, but both R8507 and the triple mSLS5/6/7 mutants bound SUMO1 inefficiently. These data confirm that the C-terminal quarter of ICP0 interacts with SUMO1 in vitro, and they suggest that the region encompassing the SLS5, SLS6, SLS7, and R8507 motifs influences the efficiency of this interaction. We are unable to conclude, however, that any of the individual SLS motifs are capable of binding SUMO1 in a SIM-like-dependent manner. Furthermore, there is insufficient correlation between the effects of the mutations in this region on SUMO1 binding in vitro and phenotypes in our other assays to allow a direct connection to be made between SUMO1 binding and the role of this region in ICP0 biological activity during infection. For example, mutant mSLS5/7 retains SUMO1 binding but causes a clear defect in viral replication, whereas R8507 reduces SUMO1 binding but has a relatively modest effect in other assays. Therefore, the situation is complex, perhaps involving an overall structural fold encompassing the entire region and/or interactions in addition to those represented by monomeric interactions with SUMO1 in solution. In this regard, we note that this region is involved in the self-interaction of ICP0 (20, 35, 36), ICP0 intracellular localization during infection, and expression levels and/or stability of ICP0 in induced cells (8, 32).
FIG 9.
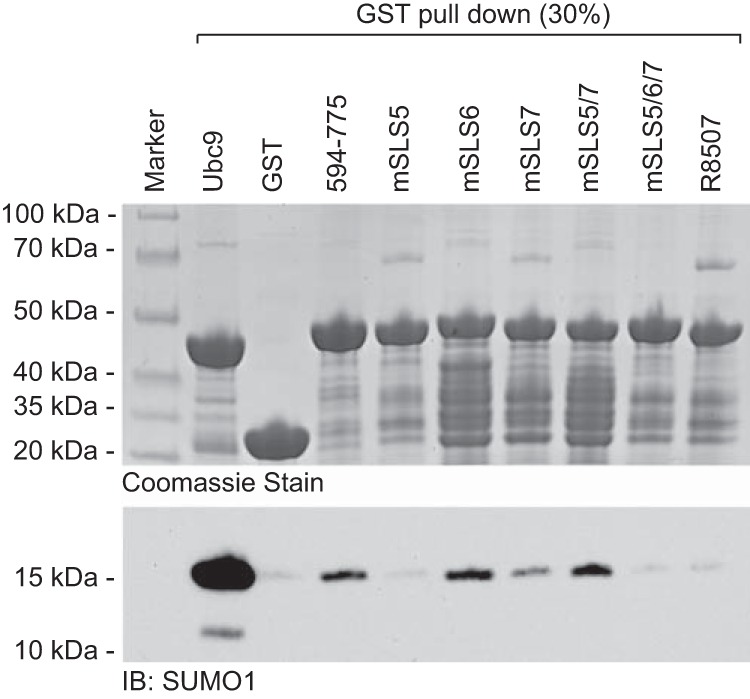
Influence of mutations in the C-terminal quarter of ICP0 on the interaction between ICP0 and SUMO1 in vitro. The C-terminal quarter of ICP0 (residues 594 to 775) and derivatives, including the indicated mutations, were expressed as GST fusion proteins in bacteria and then purified using glutathione beads. Samples of the beads were mixed with bacterial extracts containing His-tagged SUMO1, and the relative levels of bound SUMO1 were detected by Western blotting of purified washed beads (lower). The upper panel shows a Coomassie stain of the bead preparations, confirming equal loading. GST-Ubc9 and GST beads were used as positive and negative controls.
DISCUSSION
In this paper, we demonstrate that sequences related to SUMO interaction motifs within ICP0 contribute to its biological activities during HSV-1 infection and influence the substrate selection of its E3 ubiquitin ligase activity. Although overall sequence conservation among the alpha herpesvirus family of ICP0 proteins is limited outside the RING finger, many contain SIM-like sequences for which, in some cases, evidence exists for an interaction with SUMO (8, 37). Looking in more detail at the proteins most similar to ICP0 of HSV-1, namely, the related proteins of HSV-2 and herpesvirus B (HVB), the most highly conserved regions are the RING finger and sequences immediately downstream thereof; the region encompassing SLS4; the core USP7 binding site; the sequences including SLS5, SLS6, the R8507 mutations, and SLS7; and finally a block of sequence close to the C termini of the proteins (Fig. 10). The USP7 core binding site and C-terminal homology blocks are presented in Fig. 10 for reasons of completeness and will not be discussed further here. The RING finger itself has been extensively studied previously, while a recent paper implicates the sequences immediately downstream of the RING finger as also being involved in PML degradation (38). Outside all of these regions, the alignment between the ICP0 proteins of HSV-1, HSV-2, and HVB taken together shows considerably less sequence identity (Fig. 10A and data not shown), highlighting these conserved regions as being of potential importance among these members of the simplex virus genus.
FIG 10.

Comparison of the regions apart from the RING finger that are most highly conserved in the ICP0 proteins of HSV-1, HSV-2, and HVB. (A) A plot of sequence relatedness between the three proteins as compiled in Align-X (VectorNTI; Invitrogen). (B to E) Alignments and residue coordinates of the SLS4 (B), USP7 binding (C), SLS5/SLS6/SLS7/R8507 (HSV-1 ICP0 coordinates only) (D), and C-terminal regions (E) are shown, with completely conserved residues indicated by asterisks. Apart from the RING finger and its immediate downstream region (which has 66% identity between the three proteins over the 119-residue stretch corresponding to HSV-1 ICP0 coordinates 116 to 224; data not shown), there is only limited precise conservation in other regions of the proteins.
This study identifies a clear function of SLS4, a region known to mediate SUMO2 binding: it is absolutely required for the ability of ICP0 to target the SUMO-modified forms of PML isoforms other than those of PML.I and also high-molecular-weight SUMO conjugates in general. Given that virus mSLS4 has only a small defect in plaque formation (Table 1), it appears that targeting SUMO conjugates in general may not of itself be of great importance in terms of ICP0 biological function, at least in these cultured cell assays. This does not, however, exclude the importance of the targeting of selected specific SUMO-conjugated proteins. For example, mutant mSLS4 efficiently degrades sumoylated PML.I and endogenous sumoylated forms of PML (Fig. 4 and 5), and it is possible or even likely that other specific sumoylated cellular proteins are also degraded by this mutant. The mSLS4 mutant also influences the behavior of sumoylated species in a manner that does not necessarily require degradation, as evidenced by its ability to inhibit the recruitment of sumoylated species to, and/or their induction at, sites associated with HSV-1 parental genomes and early replication compartments (Fig. 7 and 8).
The block of sequence including the SLS5, SLS6, SLS7, and R8507 mutations (residues 636 to 717) presents a substantial stretch of high homology among the proteins of HSV-1, HSV-2, and HVB (Fig. 10). This region overlaps extensively with that mapped as being required for self-multimerization of ICP0 (residues 633 to 771) (20, 35, 36), and deletion of either the N-terminal or C-terminal parts of this region results in failure to localize efficiently to PML NBs (or to be retained within them) (27). These deletions also cause retention of ICP0 in the nucleus as infection progresses (24, 25), a phenotype also observed with mSLS5/6/7. Based on published GST pull-down evidence (39), the C-terminal part of this region is included in the minimal region implicated in ICP0 interaction with CoREST (residues 693 to 716; note that this does not include SLS5/6/7), while the N-terminal part is not absolutely required for this activity. One other phenotype of note is that deletions in this region correlate with high levels of ICP0 accumulation in the inducible cell expression system (32), a phenotype also observed with mutants mSLS5/7 (8), mSLS5/6/7, and mSLS6 (but not R8507) (data not shown). It is possible that rather than defining individual entities, the SLS mutations in this region affect an overall structural fold. Consistent with this view, individual mSLS mutations may disturb this fold to only a relatively minor degree, while combinations of the mutations might cause more marked perturbations, resulting in greater defects in the various assays than any of the individual mutations by themselves. Whether or not SUMO1 binding is the primary function of this region remains an open question. Certainly at least some of the SLS mutations have an impact on the interaction in vitro between SUMO1 and this part of ICP0 (Fig. 9), and several of the mutations affect the efficiency with which sumoylated PML.II is degraded, especially when in combination. However, given the functional complexity of this region, it seems unlikely that interaction with SUMO1, independent of other factors, could explain all of the various associated phenotypes.
Finally, although the defect in HSV-1 replication associated with mSLS4 is only slight and that of mSLS5/6/7 is around 10-fold, combination of mutations in SLS4 and the SLS5, SLS6, SLS7 region causes a defect of up to two orders of magnitude. This correlates with a marked reduction in the rates of degradation of both sumoylated and unmodified PML (and probably of other sumoylated and unmodified ICP0 substrates as well) and a defect in the ability of ICP0 to inhibit recruitment of PML NB proteins and sumoylated species to sites of parental HSV-1 genomes. Therefore, these short sequence motifs clearly have a role in ICP0 activity in an apparently cooperative manner. It will be of interest to explore further the impact of these mutations on the targeting of specific substrates by ICP0.
ACKNOWLEDGMENTS
The work in the authors' laboratories is supported by the Medical Research Council. K.P. is supported by an MRC postgraduate studentship.
We thank Roel van Driel, Hans Will, Ron Hay, and Philippe Gripon for reagents.
Footnotes
Published ahead of print 18 December 2013
REFERENCES
- 1.Boutell C, Everett RD. 2013. Regulation of alphaherpesvirus infections by the ICP0 family of proteins. J. Gen. Virol. 94:465–481. 10.1099/vir.0.048900-0 [DOI] [PubMed] [Google Scholar]
- 2.Everett RD. 2011. The role of ICP0 in counteracting intrinsic cellular resistance to virus infection, p 39–50 In Weller SK. (ed), Alphaherpesviruses: molecular virology. Caister Academic Press, Norfolk, United Kingdom [Google Scholar]
- 3.Hagglund R, Roizman B. 2004. Role of ICP0 in the strategy of conquest of the host cell by herpes simplex virus 1. J. Virol. 78:2169–2178. 10.1128/JVI.78.5.2169-2178.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Everett RD, Freemont P, Saitoh H, Dasso M, Orr A, Kathoria M, Parkinson J. 1998. The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110- and proteasome-dependent loss of several PML isoforms. J. Virol. 72:6581–6591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maul GG, Guldner HH, Spivack JG. 1993. Modification of discrete nuclear domains induced by herpes simplex virus type 1 immediate early gene 1 product (ICP0). J. Gen. Virol. 74:2679–2690. 10.1099/0022-1317-74-12-2679 [DOI] [PubMed] [Google Scholar]
- 6.Shen TH, Lin HK, Scaglioni PP, Yung TM, Pandolfi PP. 2006. The mechanisms of PML-nuclear body formation. Mol. Cell 24:331–339. 10.1016/j.molcel.2006.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cuchet-Lourenco D, Vanni E, Glass M, Orr A, Everett RD. 2012. Herpes simplex virus 1 ubiquitin ligase ICP0 interacts with PML isoform I and induces its SUMO-independent degradation. J. Virol. 86:11209–11222. 10.1128/JVI.01145-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boutell C, Cuchet-Lourenco D, Vanni E, Orr A, Glass M, McFarlane S, Everett RD. 2011. A viral ubiquitin ligase has substrate preferential SUMO targeted ubiquitin ligase activity that counteracts intrinsic antiviral defence. PLoS Pathog. 7:e1002245. 10.1371/journal.ppat.1002245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Everett RD, Chelbi-Alix MK. 2007. PML and PML nuclear bodies: implications in antiviral defence. Biochimie 89:819–830. 10.1016/j.biochi.2007.01.004 [DOI] [PubMed] [Google Scholar]
- 10.Geoffroy MC, Chelbi-Alix MK. 2011. Role of promyelocytic leukemia protein in host antiviral defense. J. Interferon Cytokine Res. 31:145–158. 10.1089/jir.2010.0111 [DOI] [PubMed] [Google Scholar]
- 11.Tavalai N, Stamminger T. 2008. New insights into the role of the subnuclear structure ND10 for viral infection. Biochim. Biophys. Acta 1783:2207–2221. 10.1016/j.bbamcr.2008.08.004 [DOI] [PubMed] [Google Scholar]
- 12.Cuchet-Lourenco D, Boutell C, Lukashchuk V, Grant K, Sykes A, Murray J, Orr A, Everett RD. 2011. SUMO pathway dependent recruitment of cellular repressors to herpes simplex virus type 1 genomes. PLoS Pathog. 7:e1002123. 10.1371/journal.ppat.1002123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Everett RD, Boutell C, Hale BG. 2013. Interplay between viruses and host sumoylation pathways. Nat. Rev. Microbiol. 11:400–411. 10.1038/nrmicro3015 [DOI] [PubMed] [Google Scholar]
- 14.Gripon P, Rumin S, Urban S, Le Seyec J, Glaise D, Cannie I, Guyomard C, Lucas J, Trepo C, Guguen-Guillouzo C. 2002. Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. U. S. A. 99:15655–15660. 10.1073/pnas.232137699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cuchet D, Sykes A, Nicolas A, Orr A, Murray J, Sirma H, Heeren J, Bartelt A, Everett RD. 2011. PML isoforms I and II participate in PML-dependent restriction of HSV-1 replication. J. Cell Sci. 124:280–291. 10.1242/jcs.075390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gu H, Roizman B. 2009. The two functions of herpes simplex virus 1 ICP0, inhibition of silencing by the CoREST/REST/HDAC complex and degradation of PML, are executed in tandem. J. Virol. 83:181–187. 10.1128/JVI.01940-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yao F, Schaffer PA. 1995. An activity specified by the osteosarcoma line U2OS can substitute functionally for ICP0, a major regulatory protein of herpes simplex virus type 1. J. Virol. 69:6249–6258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stow ND, Stow EC. 1986. Isolation and characterization of a herpes simplex virus type 1 mutant containing a deletion within the gene encoding the immediate early polypeptide Vmw110. J. Gen. Virol. 67:2571–2585. 10.1099/0022-1317-67-12-2571 [DOI] [PubMed] [Google Scholar]
- 19.Everett RD. 1989. Construction and characterization of herpes simplex virus type 1 mutants with defined lesions in immediate early gene 1. J. Gen. Virol. 70(Part 5):1185–1202. 10.1099/0022-1317-70-5-1185 [DOI] [PubMed] [Google Scholar]
- 20.Meredith M, Orr A, Elliott M, Everett R. 1995. Separation of sequence requirements for HSV-1 Vmw110 multimerisation and interaction with a 135-kDa cellular protein. Virology 209:174–187. 10.1006/viro.1995.1241 [DOI] [PubMed] [Google Scholar]
- 21.Everett RD, Cross A, Orr A. 1993. A truncated form of herpes simplex virus type 1 immediate-early protein Vmw110 is expressed in a cell type dependent manner. Virology 197:751–756. 10.1006/viro.1993.1651 [DOI] [PubMed] [Google Scholar]
- 22.Stuurman N, de Graaf A, Floore A, Josso A, Humbel B, de Jong L, van Driel R. 1992. A monoclonal antibody recognizing nuclear matrix-associated nuclear bodies. J. Cell Sci. 101:773–784 [DOI] [PubMed] [Google Scholar]
- 23.Showalter SD, Zweig M, Hampar B. 1981. Monoclonal antibodies to herpes simplex virus type 1 proteins, including the immediate-early protein ICP 4. Infect. Immun. 34:684–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Everett RD, Maul GG. 1994. HSV-1 IE protein Vmw110 causes redistribution of PML. EMBO J. 13:5062–5069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maul GG, Everett RD. 1994. The nuclear location of PML, a cellular member of the C3HC4 zinc-binding domain protein family, is rearranged during herpes simplex virus infection by the C3HC4 viral protein ICP0. J. Gen. Virol. 75:1223–1233. 10.1099/0022-1317-75-6-1223 [DOI] [PubMed] [Google Scholar]
- 26.Ferenczy MW, Ranayhossaini DJ, Deluca NA. 2011. Activities of ICP0 involved in the reversal of silencing of quiescent herpes simplex virus 1. J. Virol. 85:4993–5002. 10.1128/JVI.02265-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gu H, Zheng Y, Roizman B. 2013. Interaction of herpes simplex virus ICP0 with ND10 bodies: a sequential process of adhesion, fusion, and retention. J. Virol. 87:10244–10254. 10.1128/JVI.01487-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jensen K, Shiels C, Freemont PS. 2001. PML protein isoforms and the RBCC/TRIM motif. Oncogene 20:7223–7233. 10.1038/sj.onc.1204765 [DOI] [PubMed] [Google Scholar]
- 29.Pal S, Santos A, Rosas JM, Ortiz-Guzman J, Rosas-Acosta G. 2011. Influenza A virus interacts extensively with the cellular SUMOylation system during infection. Virus Res. 158:12–27. 10.1016/j.virusres.2011.02.017 [DOI] [PubMed] [Google Scholar]
- 30.Everett RD, Murray J. 2005. ND10 components relocate to sites associated with herpes simplex virus type 1 nucleoprotein complexes during virus infection. J. Virol. 79:5078–5089. 10.1128/JVI.79.8.5078-5089.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Everett RD, Murray J, Orr A, Preston CM. 2007. Herpes simplex virus type 1 genomes are associated with ND10 nuclear substructures in quiescently infected human fibroblasts. J. Virol. 81:10991–11004. 10.1128/JVI.00705-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Everett RD, Parsy ML, Orr A. 2009. Analysis of the functions of herpes simplex virus type 1 regulatory protein ICP0 that are critical for lytic infection and derepression of quiescent viral genomes. J. Virol. 83:4963–4977. 10.1128/JVI.02593-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Everett RD, Boutell C, McNair C, Grant L, Orr A. 2010. Comparison of the biological and biochemical activities of several members of the alphaherpesvirus ICP0 family of proteins. J. Virol. 84:3476–3487. 10.1128/JVI.02544-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Capili AD, Lima CD. 2007. Structure and analysis of a complex between SUMO and Ubc9 illustrates features of a conserved E2-Ubl interaction. J. Mol. Biol. 369:608–618. 10.1016/j.jmb.2007.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ciufo DM, Mullen MA, Hayward GS. 1994. Identification of a dimerization domain in the C-terminal segment of the IE110 transactivator protein from herpes simplex virus. J. Virol. 68:3267–3282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mullen MA, Ciufo DM, Hayward GS. 1994. Mapping of intracellular localization domains and evidence for colocalization interactions between the IE110 and IE175 nuclear transactivator proteins of herpes simplex virus. J. Virol. 68:3250–3266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang L, Oliver SL, Sommer M, Rajamani J, Reichelt M, Arvin AM. 2011. Disruption of PML nuclear bodies is mediated by ORF61 SUMO-interacting motifs and required for varicella-zoster virus pathogenesis in skin. PLoS Pathog. 7:e1002157. 10.1371/journal.ppat.1002157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perusina Lanfranca M, Mostafa HH, Davido DJ. 2013. Two overlapping regions within the N-terminal half of the herpes simplex virus 1 E3 ubiquitin ligase ICP0 facilitate the degradation and dissociation of PML and dissociation of Sp100 from ND10. J. Virol. 87:13287–13296. 10.1128/JVI.02304-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gu H, Roizman B. 2007. Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the CoREST-REST complex. Proc. Natl. Acad. Sci. U. S. A. 104:17134–17139. 10.1073/pnas.0707266104 [DOI] [PMC free article] [PubMed] [Google Scholar]