

ABSTRACT
CBFβ was recently found to be a key regulator of the ability of human immunodeficiency virus type 1 (HIV-1) Vif to overcome host antiviral APOBEC3 proteins. However, the detailed molecular requirements for the Vif-CBFβ interaction are still not clear. Here, we mapped the minimum Vif domain required for CBFβ binding. In terms of CBFβ binding, the Vif N terminus was very sensitive to deletions. We determined that the Vif fragment from residues 5 to 126 was sufficient to form a stable complex with CBFβ in vitro. We also observed that ionic interactions were not the main contributor to the interaction between Vif and CBFβ. Instead, hydrophobic interactions were important for maintaining the Vif-CBFβ complex, since it could be disrupted by nonionic detergent. Site-directed mutagenesis of conserved hydrophobic amino acids revealed novel residues in Vif that were important for CBFβ binding and APOBEC3 inactivation. At least part of the well-characterized HCCH domain (residues 108 to 139) was required to form a stable Vif-CBFβ complex. Thus, the HCCH motif may have a dual role in binding both Cul5 and CBFβ. Considering the importance of Vif in HIV-1 infection, this unique Vif-CBFβ interaction represents an attractive pharmacological intervention target against HIV-1.
IMPORTANCE Vif-induced APOBEC3 protein degradation was the first host antiviral mechanism against HIV-1/simian immunodeficiency virus to be revealed, yet details regarding which proteins are degraded are not fully demonstrated. Recently, host cellular factor CBFβ was found to be essential for Vif to function and promote viral infectivity. In this study, we present more critical information on the Vif-CBFβ interaction by revealing that hydrophobicity contributes the most to the Vif-CBFβ interaction and locating several novel hydrophobic sites (tryptophans and phenylalanines) that are conserved among Vif proteins from different lentiviruses and essential for Vif binding to CBFβ. Mutations on these sites result in a reduced/abolished Vif-CBFβ interaction, leading to the attenuated potency of Vif on both inducing the degradation of antiviral factors like APOBEC3G and promoting HIV-1 infectivity. Therefore, information from this study will help people to further understand how Vif acts against host antiviral mechanism, which is important for novel anti-HIV-1 drug development.
INTRODUCTION
AIDS has been one of the world's challenges since the epidemic was first reported in 1981. Approximately 34 million people were living with human immunodeficiency virus type 1 (HIV-1) in 2011, according to UNAIDS (1). Every year, millions of new HIV-1 infections are reported worldwide. Despite the obvious benefits of anti-HIV drugs, the potential spread of resistant HIV strains is a major public health concern.
HIV-1 encodes a virion infectivity factor (Vif) that is a potential anti-HIV drug target (2, 3). Vif is needed by the virus to reduce the expression and function of the host restriction factor APOBEC3 (4–13) in HIV's natural target cells, such as CD4+ T cells and macrophages (14, 15). Recently, CBFβ has been shown to regulate HIV-1 Vif activity against APOBEC3 proteins (16–23). The requirement for CBFβ indicates a significant difference between HIV-1 Vif and cellular substrate receptor proteins, which do not require CBFβ to recruit the cellular Cullin5 (Cul5)-RING ubiquitin ligase complex (CRL5). This difference suggests that the Vif-CBFβ interaction constitutes a unique anti-HIV drug target. It is well established that the BC box in Vif binds to elongin B/C (EloB/C) (9, 10, 24–29), and the HCCH motif in Vif is required for Cul5 binding (9, 12, 27, 29–38). The N-terminal domain of Vif is involved in APOBEC3 binding (39–51). Previously, we have shown that Vif residues 1 to 140 are sufficient to directly bind CBFβ (16). In addition, Vif W21A and W38A mutants have a reduced ability to bind CBFβ (17). However, it is unclear whether additional Vif residues contribute to the interaction with CBFβ.
Here, we show that Vif residues 5 to 126 are required to form a stable complex with CBFβ. Furthermore, we demonstrate that nonionic detergent disrupts the Vif-CBFβ interaction, suggesting that hydrophobic interactions are important for this interaction. Mutation of clustered Trp and Phe residues to Ala in the N-terminal half of Vif revealed that many of these hydrophobic residues support the interaction with CBFβ. In addition, these CBFβ-binding-deficient mutants were also deficient in binding Cul5. Finally, all these mutants lost their ability to efficiently degrade A3G and restore HIV-1 infectivity in the presence of A3G.
MATERIALS AND METHODS
Plasmid construction.
For expression in Escherichia coli, full-length and truncated mutants of HIV-1 NL4-3 Vif were constructed in the pET21a vector (ampicillin resistant) without tags, and CBFβ (residues 1 to 140) was constructed in the pRSF-duet vector (kanamycin resistant) with a 6× His tag. For expression in 293T cells, Vif with a hemagglutinin (HA) tag was constructed in the VR1012 vector, CBFβ with a Myc tag was constructed in pcDNA3.1, and A3G with a V5 tag was constructed in the pcDNA vector.
Transfection, immunoprecipitation, and Western blot analysis.
Transfections were performed as previously described (17). Harvested cells were rinsed with phosphate-buffered saline (PBS) and lysed in radioimmunoprecipitation assay buffer (20 mM Tris-HCl, pH 7.5, with 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, and 1 mM Na3VO4). For immunoprecipitation assays, cell lysates were centrifuged at 18,000 × g for 10 min at 4°C. An anti-c-Myc agarose affinity gel produced in rabbits (catalog no. A7470; Sigma-Aldrich) was added to the supernatant, and the mixture was incubated at 4°C for 1 h. The beads were then collected by a quick spin and washed five times in PBS. The protein in the beads was then eluted with 50 μl of elution buffer (100 mM glycine, pH 2.5), and 0.5 μl of 5 M NaOH was added to adjust the pH. Then, 6× SDS-PAGE buffer was added, and the samples were heated at 95°C for 5 min for SDS-PAGE, followed by transfer to nitrocellulose membranes (Bio-Rad). After blocking with PBS–Tween 20 containing 5% bovine serum albumin for 1 h at room temperature, the blots were incubated with a specific antibody overnight at 4°C. After three washes, the blots were stained with an alkaline phosphatase-conjugated secondary antibody (Sigma) for 1 h at room temperature. After three washes with PBS–Tween 20, the blots were reacted with nitroblue tetrazolium (NBT) and 5-bromo-4-chloro-3′-indolylphosphate (BCIP) (Sigma). Protein band intensities were quantified using Image J software (http://rsbweb.nih.gov/ij/index.html) as previously described (52).
Antibodies.
The antibodies used in this study were specific for Cul5 (catalog no. sc-13014; Santa Cruz), Vif (catalog no. 2221; contributed by D. Gabuzda, AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, National Institutes of Health), CBFβ (catalog no. ab11921; Abcam), elongin B (catalog no. sc-11447; Santa Cruz), elongin C (catalog no. 610760; BD Transduction Lab), β-actin (catalog no. A3853; Sigma), HA (catalog no. MMS-101R-1000; Covance), Myc (catalog no. 05-724; Upstate), and V5 (catalog no. 46-0705; Invitrogen).
Gel filtration chromatography.
Vif and CBFβ were coexpressed in E. coli BL21 cells and purified as previously described (16). Purified proteins from a pull-down via the 6× His tag in CBFβ were loaded onto a Superdex 200 10/300 GL column (GE Healthcare) with a 500-μl loop and run at a flow rate of 0.5 ml/min. The collected peak fractions were subjected to SDS-PAGE, followed by Coomassie staining or Western blotting with the indicated specific antibodies. The gel filtration column was calibrated using vitamin B12 (1,370 Da), myoglobin (17,000 Da), ovalbumin (44,000 Da), gamma globulin (158,000 Da), and thyroglobulin (670,000 Da) as standards.
Recombinant full-length HIV-1 Vif was coexpressed with CBFβ140-His in E. coli at 16°C for 18 h by induction with 0.1 mM isopropyl-d-thiogalactopyranoside (IPTG). Harvested cells were lysed in 20 mM Tris-HCl, pH 8.0, with 150 mM NaCl and clarified by sonication and centrifugation at 18,000 × g for 30 min. For nickel affinity pull-down, the supernatant was transferred to Ni-nitrilotriacetic acid (NTA) beads, and the flowthrough was loaded onto Ni-NTA beads for two more passages. After washing with 20 mM Tris-HCl, pH 8.0, with 150 mM NaCl and 40 mM imidazole, the nickel beads were divided into multitubes, and each tube was treated by the use of one of the different conditions (various pHs, salt concentrations, or detergent concentrations) at 4°C for 30 min with shaking. The nickel beads completed the treatment, and samples were eluted with 20 mM Tris-HCl, pH 8.0, with 150 mM NaCl and 200 mM imidazole. Sample buffer (6×) was added to the eluted samples. After boiling at 95°C for 5 min, the samples were analyzed by SDS-PAGE and visualized with Coomassie staining.
Viral infection and MAGI cell assays.
HEK 293T cells cultured in Dulbecco modified Eagle medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) were seeded in a 12-well plate. After the cells were cultured overnight, the medium was removed. The 293T cells were cotransfected with the HIV-1 NL4-3/deltaVif plasmid and the A3G and Vif mutant plasmids. Viruses were collected after 48 h. MAGI–CCR-5 cells for infection were prepared in 12-well plates in D-10 medium (DMEM with 10% wt/vol FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, 0.25 μg/ml fungizone, and 300 μg/ml glutamine) 1 day before infection, and cells were at 30 to 40% confluence at the time of infection. MAGI cells were infected by removing the medium from each well and adding dilutions of virus in a total volume of 500 μl of complete DMEM with a final concentration of 20 μg/ml of DEAE dextran. After a 2-h incubation at 37°C in a 5% CO2 incubator, 2 ml of complete DMEM was added to each well. After culture for 48 h, the medium was removed; the cells were fixed with 1% formaldehyde in PBS for 40 min and then washed three times with PBS. They were then stained with staining solution (0.4 mM potassium ferricyanide, 0.4 mM potassium ferrocyanide, 2 mM MgCl2, 40 μg/ml X-Gal [5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside]) for 50 min at 37°C. HIV-1-infected MAGI cells containing β-galactosidase were stained blue. After the staining solution was removed, the cells were washed three times with PBS. Positive blue dots were counted with an inverted light microscope. Viral infectivity was determined after normalizing the amount of virus input by the results for the p24 antigen.
RESULTS
Vif residues 5 to 126 are required to form a stable complex with CBFβ.
Previously, we have demonstrated that Vif residues 1 to 140 are sufficient to bind CBFβ directly (16). We were curious whether further Vif truncations would support the interaction with CBFβ. Thus, we created additional truncated constructs of Vif including residues 1 to 110, 1 to 126, 5 to 192, 6 to 192, and 8 to 192 to map the minimum CBFβ-binding region (Fig. 1A). Next, the Vif constructs were coexpressed with His-tagged CBFβ residues 1 to 140 in E. coli BL21 (Fig. 1B). CBFβ and the interacting Vif proteins were pulled down from soluble lysates using Ni-NTA beads. Eluted proteins were separated by SDS-PAGE and visualized by Coomassie stain and Western blotting. As shown in Fig. 1B, truncated Vif protein was expressed at a level comparable to that of full-length Vif (residues 1 to 192) when expressed alone (lanes 2 to 7) or when coexpressed with CBFβ (lanes 8 to 13). The expression of Vif and CBFβ was further confirmed by Western blotting using specific antibodies (Fig. 1B, bottom). Vif residues 1 to 192 and Vif residues 5 to 192 were efficiently pulled down by CBFβ (Fig. 1C, lanes 8 and 9). However, Vif from residues 6 to 192 was much less efficient at binding CBFβ (lane 10), and the interaction between Vif residues 8 to 192 and CBFβ was nearly abolished (lane 11). The pulldown results were further confirmed by Western blotting (Fig. 1C, bottom). These results suggested that N-terminal residues from Vif Trp5 are required for the CBFβ-Vif interaction. In addition, Vif residues 1 to 126 could be pulled down efficiently by CBFβ (Fig. 1C, lane 13); Vif residues 1 to 110 were pulled down less efficiently by CBFβ (Fig. 1C, lane 12, and D). As expected, untagged full-length Vif and Vif truncation mutants were not pulled down by nickel beads in the absence of CBFβ (Fig. 1C, lanes 2 to 7).
FIG 1.

HIV-1 Vif truncations competent for CBFβ binding. (A) Vif truncated mutants were created to map the CBFβ-binding region. PPLP, conserved Pro-Pro-Leu-Pro sequence in Vif residues 161 to 164. (B) Vif truncated mutants (with or without CBFβ residues 1 to 140; CBFβ residues 1 to 140 are fully functional in forming a Vif-CBFβ-EloB/C-Cul5 complex) were expressed in E. coli. The cell lysates were checked by SDS-PAGE, followed by Coomassie brilliant blue (CBB) staining or Western blotting (WB). (C) Expressed Vif mutants from the experiment whose results are shown in panel B were pulled down via the 6× His tag in CBFβ by nickel beads. CBFβ alone was used as a control. Vif mutants without coexpressed CBFβ were used as controls. (D) Percent pull-down by CBFβ in Vif truncated mutants.
In repeated experiments, we have observed that Vif residues 1 to 110 run slower than Vif residues 1 to 126 in SDS-polyacrylamide gels. The Vif fragment from residues 1 to 110 had a relative high pI of 10.13, whereas the pI was 9.6 for Vif residues 1 to 126. In particular, Vif residues 111 to 126 had a very low pI (4.18). Since protein electrophoresis is dependent on protein charges, it is possible that these two Vif fragments have different mobilities in SDS-polyacrylamide gels because of their differences in charged residues.
The Vif residue 1 to 110–CBFβ and Vif residue 1 to 126–CBFβ complexes were further characterized by gel filtration following nickel affinity purification. Interestingly, although we could detect both Vif residues 1 to 110 and CBFβ after nickel purification (Fig. 2A, right, lane Input), we were unable to detect the Vif-CBFβ complex after gel filtration, even though CBFβ eluted from the column (Fig. 2A). It is possible that this complex was retained in the column. Alternatively, it is possible that the complex was disrupted during the gel filtration process and only Vif residues 1 to 110 were retained in the column.
FIG 2.

The Vif C terminus up to residue 126 is required to form a stable complex with CBFβ. Vif residues 1 to 110 or Vif residues 1 to 126 and CBFβ residues 1 to 140 were purified by nickel pull-down and loaded onto Superdex 200 for gel filtration (left). The fractions were collected and subjected to SDS-PAGE, followed by Coomassie staining (right). (A) Gel filtration of Vif residues 1 to 110 and CBFβ. Vif did not elute with CBFβ. (B) Gel filtration of Vif residues 1 to 126 and CBFβ. Vif and CBFβ eluted in the same peak. (C) Gel filtration of CBFβ alone. mAu, milliabsorbance units. (D) Region important for the interaction between Vif and CBFβ.
In contrast, the Vif residue 1 to 126–CBFβ complex could be eluted from the size exclusion column (Fig. 2B), suggesting that the two molecules formed a relatively stable complex. Vif from residues 1 to 126 was pulled down more efficiently by CBFβ than Vif from residues 1 to 110 (Fig. 1C, lanes 12 and 13) when both proteins were present at comparable concentrations in the lysates (Fig. 1B, lanes 12 and 13). Thus, some residues between residues 111 and 126 may contribute to the association between Vif and CBFβ. Taken together, these data suggest that Vif residues 5 to 126 are required to form a stable complex with CBFβ (Fig. 2D).
Hydrophobic interactions are important for Vif-CBFβ complex formation.
Protein-protein interactions include hydrophilic interactions, hydrophobic interactions, ionic attractions, and hydrogen bonds. Vif is rich in basic residues and is known to have a high pI (∼10.5 for HIV-1 NL4-3 Vif). CBFβ, however, is a relatively acidic protein (pI 5.1). Thus, we first asked whether ionic attractions constituted the major type of interaction between Vif and CBFβ. To answer this question, we incubated the Vif-CBFβ complex with nickel beads and washed the beads using a variety of buffer conditions (various pHs, salt concentrations, and nonionic detergent concentrations). The complex was eluted from the nickel beads using imidazole for protein analysis (Fig. 3A). To our surprise, neither pH (6.0 to 9.0) (Fig. 3B) nor salt concentration (0.15 M to 2 M) (Fig. 3C) significantly affected the Vif-CBFβ interaction. This result suggested that ionic attractions were most likely not the main contributors to the Vif-CBFβ interaction.
FIG 3.

Hydrophobic interactions are important for the Vif-CBFβ interaction. (A) Strategy used to test Vif-CBFβ complex stability under various conditions. Vif residues 1 to 192 and CBFβ140-His were expressed in E. coli and purified by nickel beads via a 6× His tag in CBFβ. (B to D) Vif-CBFβ complex stability under conditions with various pHs (B), salt (NaCl) concentrations (C), and detergent (Triton X-100) concentrations (D).
We then asked whether hydrophobic interactions played a role in the Vif-CBFβ interaction. Nonionic detergents such as Triton X-100 disrupt hydrophobic interactions between proteins. We treated the Vif-CBFβ complex on nickel beads with different concentrations of Triton X-100. As shown in Fig. 3D, the amount of Vif protein decreased with increasing Triton X-100 concentrations, suggesting that hydrophobic interactions contribute to the Vif-CBFβ interaction.
Vif residues W5, W11, W21, W89, and F115 contribute to hydrophobic interactions with CBFβ.
Sequence analysis of HIV-1 Vif suggested that several hydrophobic residues are clustered in Vif's N-terminal domain, especially in residues 5 to 115. This region falls within our mapped CBFβ-binding region (Fig. 2C). These hydrophobic residues included W5, W11, W21, W38, F39, W70, W79, W89, F112, and F115 and accounted for 9% of all the residues in this region. Since we showed (above) that hydrophobic interactions may play an important role in mediating the interaction between Vif and CBFβ, we hypothesized that this cluster of Trp and Phe residues may be critical for the Vif-CBFβ interaction. Of note, 8 of 10 such residues are conserved in HIV-1 and simian immunodeficiency virus (SIV), which both require CBFβ for Vif function. This sequence similarity is consistent with our truncation studies suggesting that Vif residues 5 to 126 are required for a stable Vif-CBFβ complex.
HIV-1 NL4-3 Vif has 8 Trp residues and 3 Phe residues among its 192 amino residues. Ten of the 11 residues are located within the mapped CBFβ-binding region, while 1 (Trp174) lies outside. To determine which residues are important for CBFβ binding, we mutated all the Trp and Phe residues in the Vif sequence to Ala (i.e., W5A, W11A, W21A, W38A, W70A, W79A, W89A, W174A, F39A, F112A, or F115A; Fig. 4A) and examined their interaction with CBFβ and Cul5 by coimmunoprecipitation (co-IP) in 293T cells.
FIG 4.

Hydrophobic residues in HIV-1 Vif are important for CBFβ binding. (A) Vif mutants used in this study. (B and C) Coimmunoprecipitation with anti-Myc matrix to check for CBFβ-Vif interactions. WT and mutant Vif-HA expression vectors were cotransfected with the CBFβ-Myc expression vector into 293T cells; 36 h later, the cells were harvested and the cell lysates were analyzed by Western blotting (B) or coimmunoprecipitation analysis with an anti-Myc matrix, followed by Western blotting with the indicated antibodies (C). (D) Coimmunoprecipitation of EloB with WT and mutant Vif-HA. IP, immunoprecipitation.
For these experiments, Vif was HA tagged and CBFβ was Myc tagged. Vif-HA and full-length CBFβ vectors were cotransfected into 293T cells. Cell lysates were analyzed by Western blotting to check the expression of Vif-HA and CBFβ-Myc (Fig. 4B). Anti-Myc matrix beads were used to pull down CBFβ-Myc and interacting proteins. Wild-type (WT) Vif was pulled down, as expected (Fig. 4C, lane 3). However, Vif W5A, W21A, W38A, W89A, and F115A showed a reduced ability to interact with CBFβ (Fig. 4C). The Vif-CBFβ interaction was less affected in the case of F112A, yet this mutant lost the ability to interact with Cul5, suggesting that residue F112 is important for the Vif-Cul5 interaction.
Vif mutants (W5A, W21A, W38A, W89A, and F115A) that had an impaired binding ability were still capable of interacting with EloB (Fig. 4D). Moreover, these Vif mutants maintained the ability to interact with their substrate, A3G (Fig. 5). These data suggest that the overall structure of these Vif mutants was not destroyed by the mutations. The importance of these residues in mediating the Vif-CBFβ interaction was further evaluated using Vif mutants W5S, W21S, W38S, W89S, F112S, and F115S (Fig. 6A). Compared to WT Vif, all these mutants had a reduced ability to interact with CBFβ (Fig. 6B). On the other hand, the mutants could still interact with EloB (Fig. 6B) and A3G (Fig. 6C).
FIG 5.
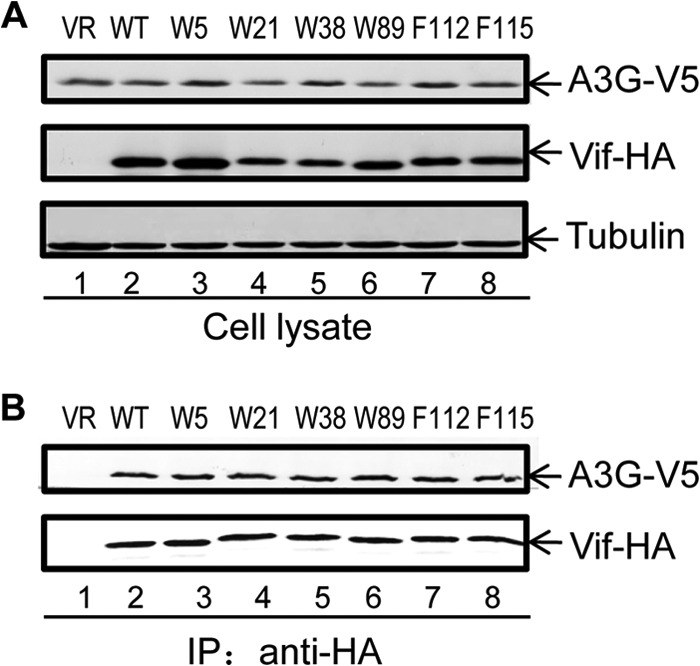
Interaction between HIV-1 Vif WT and mutant proteins and A3G. (A) 293T cells were cotransfected with a V5-A3G expression vector plus control vector (VR; lane 1), WT Vif-HA (lane 2), or one of the indicated Vif-HA mutants (lanes 3 to 8). Cell lysates were prepared and analyzed at 48 h after transfection (A) and immunoprecipitated with anti-HA affinity matrix (Roche) (B). The interaction of A3G with WT or mutated Vif-HA molecules was detected by Western blotting with anti-HA antibody to detect Vif-HA and anti-V5 antibody to detect V5-A3G.
FIG 6.

Mutation of hydrophobic residues in Vif to Ser reduced the interaction between mutant Vif and CBFβ. (A) Vif mutants (W5S, W21S, W38S, W89S, F112S, and F115S) used in this study. (B) Vif-HA WT or mutant expression vectors and the CBFβ-Myc expression vector were cotransfected into 293T cells; the cells were harvested after 36 h. Coimmunoprecipitation of CBFβ-Myc, Cul5, or EloB with Vif-HA was analyzed by Western blotting. (C) Vif-HA WT or mutant expression vectors and the A3G-V5 expression vector were cotransfected into 293T cells. Coimmunoprecipitation of A3G-V5 with Vif-HA was analyzed by Western blotting. Vif-HA mutant (or WT) plasmids and A3G-V5 plasmids were cotransfected into HEK 293T cells; the cells were harvested after 36 h. Coimmunoprecipitation with anti-HA matrix was used to detect the interaction between Vif mutants and A3G.
Vif mutants defective in CBFβ binding have a reduced ability to inactivate APOBEC3G and APOBEC3F.
We have shown previously that CBFβ is essential for Vif's ability to degrade APOBEC3 proteins and restore HIV-1 infectivity in the presence of APOBEC3 (17). We reasoned that if the motif that we identified as described above is important for the Vif-CBFβ interaction, mutation of this motif would reduce Vif's ability to destroy the APOBEC3 protein. To confirm our assumption, we cotransfected the Vif-HA expression vectors (WT or mutants) with an A3G (with a V5 tag) expression plasmid into 293T cells. After 36 h, the cells were harvested and analyzed for Vif and A3G expression by Western blotting.
As expected, mutations of W5, W21, W38, W89, F112, and F115 indeed compromised HIV-1 Vif-mediated depletion of the A3G protein (Fig. 7A). The Vif W70A mutant, known to inefficiently bind A3G, was also unable to degrade A3G. Vif mutants W5A, W21A, W38A, W89A, F112A, and F115A also showed a reduced ability to induce the depletion of A3F (Fig. 7B). These results suggest that the Vif residues that are important for CBFβ binding are also important for Vif function.
FIG 7.
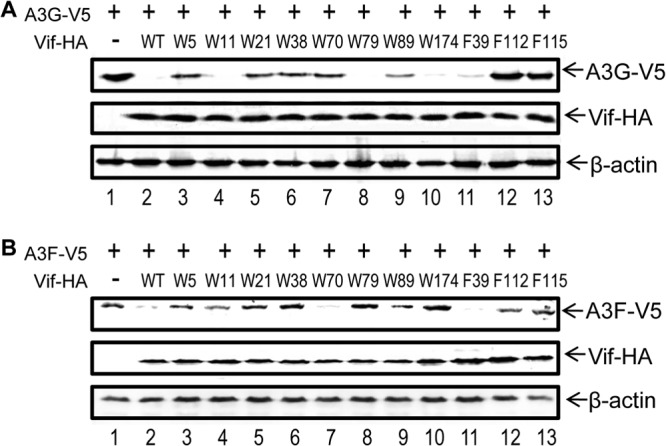
HIV-1 Vif mutants that have lost their ability to interact with CBFβ have a reduced ability to deplete APOBEC3 proteins. Expression vectors for Vif-HA (WT or mutants) were cotransfected with APOBEC3-V5 expression vectors into 293T cells. After 36 h, the cells were harvested, and the cell lysates were analyzed by Western blotting to check the expression of Vif-HA and A3G-V5 (A) or A3F-V5 (B). β-Actin was used as the loading control.
Next, we used the MAGI cell assay to investigate whether these Vif mutants were inefficient at restoring HIV-1 infectivity in the presence of A3G. Virus was prepared from 293T cells after transfection with Vif (WT or mutants), HIV-1 NL4-3ΔVif, and the A3G expression plasmids. MAGI cells were infected with the DEAE–HIV-1 mixture and incubated for 48 h. Next, cells were fixed and stained, and the infected cells were counted. The results for the WT Vif sample were set to 100%. As shown in Fig. 8A, Vif mutants W5A, W21A, W38A, W89A, F112A, and F115A had a reduced ability to promote HIV-1 infection in the presence of A3G compared to that of the WT Vif. Similar results were observed in the presence of A3F (Fig. 8B). These results further confirm the importance of these hydrophobic residues of Vif in mediating the interaction with CBFβ and inactivation of APOBEC3 proteins.
FIG 8.
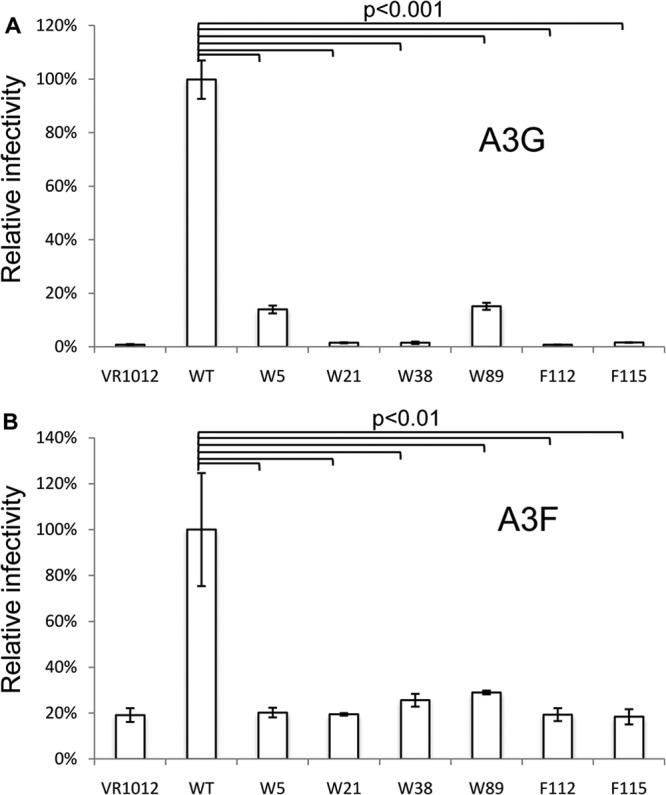
HIV-1 Vif mutants with reduced CBFβ binding also have an impaired ability to promote viral infectivity in the presence of APOBEC3 proteins. Vesicular stomatitis virus glycoprotein-pseudotyped HIV-1 ΔEnv ΔVif isolates labeled with enhanced green fluorescent protein were produced from 293T cells in the presence of A3G (A) or A3F (B) along with WT Vif or one of the Vif mutants. Viruses were then used to infect MAGI cells. Virus infectivity was determined 2 days after infection. Virus infectivity with WT Vif was set to 100%. All the data in this figure are representative of those from three independent experiments. The error bars indicate the SDs of three replicates within one experiment. Statistical analysis (Student's t test, two-tailed) was performed with Microsoft Excel software.
DISCUSSION
Most lentiviruses encode a Vif protein that is necessary for viral replication and survival in their natural hosts. Lentiviral Vif proteins have been shown to inactivate APOBEC3 antiviral factors from their respective hosts. Recently, CBFβ has been identified as a key regulator of HIV/SIV Vif function. CBFβ specifically interacts with HIV-1 Vif to enhance the solubility and stability of Vif as well as the assembly of the Vif-CRL5 E3 ubiquitin ligase. However, the determinant for Vif-mediated recruitment of CBFβ is largely unknown.
In the present study, we have identified a core region spanning amino acids 5 to 126 of HIV-1 Vif as a main binding domain for CBFβ. We have also demonstrated that hydrophobic interaction is a major contributor to the interaction between Vif and CBFβ. We further determined the critical hydrophobic residues in HIV-1 Vif that are important for the CBFβ interaction. In particular, W5, W21, W38, W89, and F115 were shown to be required for the binding of Vif to CBFβ. Kim et al. (22) have found that the hydrophobic residues L64 and I66 of HIV-1 Vif also contribute to the Vif-CBF interaction. Previous studies have also reported that hydrophobic residues in CBFβ, including F68, F69, and A71, may be important for the Vif-CBFβ interaction (16, 19, 20). All these results are consistent with a model in which hydrophobic interaction is a driving force for the binding of HIV-1 Vif to CBFβ.
We have also examined Vif W5S, W21S, W38S, W89S, F112S, and F115S for CBFβ binding. Compared to WT Vif, all these mutants had a reduced ability to interact with CBFβ (Fig. 6). Therefore, mutation of these aromatic hydrophobic residues in HIV-1 Vif to hydrophilic residues (Ser) or hydrophobic residues (Ala) with short side chains resulted in mutant proteins with impaired CBFβ binding. Determining whether these hydrophobic residues are directly involved in the interaction with CBFβ or are involved in maintaining certain Vif structures that are required for CBFβ binding requires more detailed structural information. It is worth pointing out that these mutants maintained the ability to interact with A3G and EloB, suggesting that the overall structure of these Vif mutants was not destroyed by the mutations.
Our identification of the importance of hydrophobic interaction also explains why the interaction with CBFβ improved the solubility of Vif (16, 19, 20). Since the hydrophobic residues are supposed to interact with other proteins, deletion of the partner would result in exposure of the hydrophobic residues, and their exposure would result in protein-protein interactions that could lead to oligomerization and aggregation.
When HIV-1 Vif was first identified, its unusually high content of W residues was recognized as a unique property (16, 19, 20). Interestingly, all the W residues in HIV-1 Vif are involved in A3 inactivation in one way or another. W11, W79 (16, 19, 20), and W174 are important for A3F binding and suppression (Fig. 7B). W70 appears to be uniquely required for suppression of A3G but not A3F (Fig. 7A). W5, W21, W38, and W89 participate in CBFβ binding and, thus, in Vif-CRL5 E3 ubiquitin ligase assembly. The requirement for CBFβ for the formation of the Vif-CRL5 E3 ubiquitin ligase is unique to HIV/SIV Vif, since cellular assembly of CRL5 E3 ubiquitin ligase does not involve CBFβ. This unique interface between Vif and CBFβ can potentially be explored to identify novel antiviral inhibitors.
ACKNOWLEDGMENTS
We are grateful to Egbert Hoiczyck and Kun Luo for advice and helpful discussions and to Nancy A. Speck and Alex Bullock for plasmids. We thank Deborah McClellan for editorial assistance.
This work was supported by grants from the Chinese Ministry of Science and Technology (no. 2012CB911100 and no. 2013ZX0001-005) and the Chinese Ministry of Education (IRT1016); the Key Laboratory of Molecular Virology, Jilin Province (20102209), China; and the NIH (2R56AI62644-6 and 5R21AI087192-02, F31 AI091326 [to S.L.E.], and 5R01DA026150-04).
Footnotes
Published ahead of print 18 December 2013
REFERENCES
- 1.UNAIDS. 2012. Global report: UNAIDS report on the global AIDS epidemic. UNAIDS, Geneva, Switzerland: http://www.unaids.org/en/media/unaids/contentassets/documents/epidemiology/2012/gr2012/20121120_UNAIDS_Global_Report_2012_en.pdf [Google Scholar]
- 2.Greene WC, Debyser Z, Ikeda Y, Freed EO, Stephens E, Yonemoto W, Buckheit RW, Esté JA, Cihlar T. 2008. Novel targets for HIV therapy. Antiviral Res. 80:251–265 [DOI] [PubMed] [Google Scholar]
- 3.Nathans R, Cao H, Sharova N, Ali A, Sharkey M, Stranska R, Stevenson M, Rana TM. 2008. Small-molecule inhibition of HIV-1 Vif. Nat. Biotechnol. 26:1187–1192. 10.1038/nbt.1496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Farrow MA, Sheehy AM. 2008. Vif and Apobec3G in the innate immune response to HIV: a tale of two proteins. Future Microbiol. 3:145–154. 10.2217/17460913.3.2.145 [DOI] [PubMed] [Google Scholar]
- 5.Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, Neuberger MS, Malim MH. 2003. DNA deamination mediates innate immunity to retroviral infection. Cell 113:803–809. 10.1016/S0092-8674(03)00423-9 [DOI] [PubMed] [Google Scholar]
- 6.Sheehy AM, Gaddis NC, Choi JD, Malim MH. 2002. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418:646–650. 10.1038/nature00939 [DOI] [PubMed] [Google Scholar]
- 7.Sheehy AM, Gaddis NC, Malim MH. 2003. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 9:1404–1407. 10.1038/nm945 [DOI] [PubMed] [Google Scholar]
- 8.Mariani R, Chen D, Schröfelbauer B, Navarro F, König R, Bollman B, Münk C, Nymark-McMahon H, Landau NR. 2003. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell 114:21–31. 10.1016/S0092-8674(03)00515-4 [DOI] [PubMed] [Google Scholar]
- 9.Yu Y, Xiao Z, Ehrlich ES, Yu X, Yu X-F. 2004. Selective assembly of HIV-1 Vif-Cul5-ElonginB-ElonginC E3 ubiquitin ligase complex through a novel SOCS box and upstream cysteines. Genes Dev. 18:2867–2872. 10.1101/gad.1250204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu XF. 2003. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 302:1056–1060. 10.1126/science.1089591 [DOI] [PubMed] [Google Scholar]
- 11.Yu X-F. 2006. Innate cellular defenses of APOBEC3 cytidine deaminases and viral counter-defenses. Curr. Opin. HIV AIDS 1:187–193. 10.1097/01.COH.0000221590.03670.32 [DOI] [PubMed] [Google Scholar]
- 12.Liu B, Sarkis PTN, Luo K, Yu Y, Yu X-F. 2005. Regulation of Apobec3F and human immunodeficiency virus type 1 Vif by Vif-Cul5-ElonB/C E3 ubiquitin ligase. J. Virol. 79:9579–9587. 10.1128/JVI.79.15.9579-9587.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhen A, Wang T, Zhao K, Xiong Y, Yu X-F. 2010. A single amino acid difference in human APOBEC3H variants determines HIV-1 Vif sensitivity. J. Virol. 84:1902–1911. 10.1128/JVI.01509-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gabuzda DH, Lawrence K, Langhoff E, Terwilliger E, Dorfman T, Haseltine WA, Sodroski J. 1992. Role of vif in replication of human immunodeficiency virus type 1 in CD4+ T lymphocytes. J. Virol. 66:6489–6495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sakai H, Shibata R, Sakuragi J, Sakuragi S, Kawamura M, Adachi A. 1993. Cell-dependent requirement of human immunodeficiency virus type 1 Vif protein for maturation of virus particles. J. Virol. 67:1663–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou X, Evans SL, Han X, Liu Y, Yu X-F. 2012. Characterization of the interaction of full-length HIV-1 Vif protein with its key regulator CBFβ and CRL5 E3 ubiquitin ligase components. PLoS One 7:e33495. 10.1371/journal.pone.0033495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang W, Du J, Evans SL, Yu Y, Yu XF. 2012. T-cell differentiation factor CBF-beta regulates HIV-1 Vif-mediated evasion of host restriction. Nature 481:376–379. 10.1038/nature10718 [DOI] [PubMed] [Google Scholar]
- 18.Salter JD, Lippa GM, Belashov IA, Wedekind JE. 2012. Core-binding factor β increases the affinity between human Cullin 5 and HIV-1 Vif within an E3 ligase complex. Biochemistry 51:8702–8704. 10.1021/bi301244z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jager S, Kim DY, Hultquist JF, Shindo K, LaRue RS, Kwon E, Li M, Anderson BD, Yen L, Stanley D, Mahon C, Kane J, Franks-Skiba K, Cimermancic P, Burlingame A, Sali A, Craik CS, Harris RS, Gross JD, Krogan NJ. 2012. Vif hijacks CBF-beta to degrade APOBEC3G and promote HIV-1 infection. Nature 481:371–375. 10.1038/nature10693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hultquist JF, McDougle RM, Anderson BD, Harris RS. 2012. HIV type 1 viral infectivity factor and the RUNX transcription factors interact with core binding factor beta on genetically distinct surfaces. AIDS Res. Hum. Retroviruses 28:1543–1551. 10.1089/aid.2012.0142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hultquist JF, Binka M, LaRue RS, Simon V, Harris RS. 2012. Vif proteins of human and simian immunodeficiency viruses require cellular CBFβ to degrade APOBEC3 restriction factors. J. Virol. 86:2874–2877. 10.1128/JVI.06950-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim DY, Kwon E, Hartley PD, Crosby DC, Mann S, Krogan NJ, Gross JD. 2013. CBFbeta stabilizes HIV Vif to counteract APOBEC3 at the expense of RUNX1 target gene expression. Mol. Cell 49:632–644. 10.1016/j.molcel.2012.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Du J, Zhao K, Rui Y, Li P, Zhou X, Zhang W, Yu XF. 2013. Differential requirements for HIV-1 Vif-mediated APOBEC3G degradation and RUNX1-mediated transcription by core binding factor beta. J. Virol. 87:1906–1911. 10.1128/JVI.02199-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mehle A, Goncalves J, Santa-Marta M, McPike M, Gabuzda D. 2004. Phosphorylation of a novel SOCS-box regulates assembly of the HIV-1 Vif-Cul5 complex that promotes APOBEC3G degradation. Genes Dev. 18:2861–2866. 10.1101/gad.1249904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stanley BJ, Ehrlich ES, Short L, Yu Y, Xiao Z, Yu X-F, Xiong Y. 2008. Structural insight into the human immunodeficiency virus Vif SOCS box and its role in human E3 ubiquitin ligase assembly. J. Virol. 82:8656–8663. 10.1128/JVI.00767-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bergeron JRC, Huthoff H, Veselkov DA, Beavil RL, Simpson PJ, Matthews SJ, Malim MH, Sanderson MR. 2010. The SOCS-box of HIV-1 Vif interacts with ElonginBC by induced-folding to recruit its Cul5-containing ubiquitin ligase complex. PLoS Pathog. 6:e1000925. 10.1371/journal.ppat.1000925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shirakawa K, Takaori-Kondo A, Kobayashi M, Tomonaga M, Izumi T, Fukunaga K, Sasada A, Abudu A, Miyauchi Y, Akari H, Iwai K, Uchiyama T. 2006. Ubiquitination of APOBEC3 proteins by the Vif-Cullin5-ElonginB-ElonginC complex. Virology 344:263–266. 10.1016/j.virol.2005.10.028 [DOI] [PubMed] [Google Scholar]
- 28.Kobayashi M, Takaori-Kondo A, Miyauchi Y, Iwai K, Uchiyama T. 2005. Ubiquitination of APOBEC3G by an HIV-1 Vif-Cullin5-Elongin B-Elongin C complex is essential for Vif function. J. Biol. Chem. 280:18573–18578. 10.1074/jbc.C500082200 [DOI] [PubMed] [Google Scholar]
- 29.Wolfe LS, Stanley BJ, Liu C, Eliason WK, Xiong Y. 2010. Dissection of the HIV Vif interaction with human E3 ubiquitin ligase. J. Virol. 84:7135–7139. 10.1128/JVI.00031-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiao Z, Xiong Y, Zhang W, Tan L, Ehrlich E, Guo D, Yu X-F. 2007. Characterization of a novel Cullin5 binding domain in HIV-1 Vif. J. Mol. Biol. 373:541–550. 10.1016/j.jmb.2007.07.029 [DOI] [PubMed] [Google Scholar]
- 31.Xiao Z, Ehrlich E, Luo K, Xiong Y, Yu X-F. 2007. Zinc chelation inhibits HIV Vif activity and liberates antiviral function of the cytidine deaminase APOBEC3G. FASEB J. 21:217–222. 10.1096/fj.06-6773com [DOI] [PubMed] [Google Scholar]
- 32.Xiao Z, Ehrlich E, Yu Y, Luo K, Wang T, Tian C, Yu X-F. 2006. Assembly of HIV-1 Vif-Cul5 E3 ubiquitin ligase through a novel zinc-binding domain-stabilized hydrophobic interface in Vif. Virology 349:290–299. 10.1016/j.virol.2006.02.002 [DOI] [PubMed] [Google Scholar]
- 33.Paul I, Cui J, Maynard EL. 2006. Zinc binding to the HCCH motif of HIV-1 virion infectivity factor induces a conformational change that mediates protein-protein interactions. Proc. Natl. Acad. Sci. U. S. A. 103:18475–18480. 10.1073/pnas.0604150103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reingewertz TH, Shalev DE, Friedler A. 2010. Structural disorder in the HIV-1 Vif protein and interaction-dependent gain of structure. Protein Pept. Lett. 17:988–998. 10.2174/092986610791498876 [DOI] [PubMed] [Google Scholar]
- 35.Giri K, Maynard EL. 2009. Conformational analysis of a peptide approximating the HCCH motif in HIV-1 Vif. Biopolymers 92:417–425. 10.1002/bip.21209 [DOI] [PubMed] [Google Scholar]
- 36.Giri K, Scott RA, Maynard EL. 2009. Molecular structure and biochemical properties of the HCCH-Zn2+ site in HIV-1 Vif. Biochemistry 48:7969–7978. 10.1021/bi900677w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mehle A, Thomas ER, Rajendran KS, Gabuzda D. 2006. A zinc-binding region in Vif binds Cul5 and determines cullin selection. J. Biol. Chem. 281:17259–17265. 10.1074/jbc.M602413200 [DOI] [PubMed] [Google Scholar]
- 38.Luo K, Xiao Z, Ehrlich E, Yu Y, Liu B, Zheng S, Yu X-F. 2005. Primate lentiviral virion infectivity factors are substrate receptors that assemble with cullin 5-E3 ligase through a HCCH motif to suppress APOBEC3G. Proc. Natl. Acad. Sci. U. S. A. 102:11444–11449. 10.1073/pnas.0502440102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen G, He Z, Wang T, Xu R, Yu X-F. 2009. A patch of positively charged amino acids surrounding the human immunodeficiency virus type 1 Vif SLVx4Yx9Y motif influences its interaction with APOBEC3G. J. Virol. 83:8674–8682. 10.1128/JVI.00653-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dang Y, Wang X, Zhou T, York IA, Zheng Y-H. 2009. Identification of a novel WxSLVK motif in the N terminus of human immunodeficiency virus and simian immunodeficiency virus Vif that is critical for APOBEC3G and APOBEC3F neutralization. J. Virol. 83:8544–8552. 10.1128/JVI.00651-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dang Y, Davis RW, York IA, Zheng Y-H. 2010. Identification of 81LGxGxxIxW89 and 171EDRW174 domains from human immunodeficiency virus type 1 Vif that regulate APOBEC3G and APOBEC3F neutralizing activity. J. Virol. 84:5741–5750. 10.1128/JVI.00079-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang W, Chen G, Niewiadomska AM, Xu R, Yu X-F. 2008. Distinct determinants in HIV-1 Vif and human APOBEC3 proteins are required for the suppression of diverse host anti-viral proteins. PLoS One 3:e3963. 10.1371/journal.pone.0003963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Russell RA, Smith J, Barr R, Bhattacharyya D, Pathak VK. 2009. Distinct domains within APOBEC3G and APOBEC3F interact with separate regions of human immunodeficiency virus type 1 Vif. J. Virol. 83:1992–2003. 10.1128/JVI.01621-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Russell RA, Pathak VK. 2007. Identification of two distinct human immunodeficiency virus type 1 Vif determinants critical for interactions with human APOBEC3G and APOBEC3F. J. Virol. 81:8201–8210. 10.1128/JVI.00395-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cadima-Couto I, Saraiva N, Santos ACC, Goncalves J. 2011. HIV-1 Vif interaction with APOBEC3 deaminases and its characterization by a new sensitive assay. J. Neuroimmune Pharmacol. 6:296–307. 10.1007/s11481-011-9258-7 [DOI] [PubMed] [Google Scholar]
- 46.Tian C, Yu X, Zhang W, Wang T, Xu R, Yu X-F. 2006. Differential requirement for conserved tryptophans in human immunodeficiency virus type 1 Vif for the selective suppression of APOBEC3G and APOBEC3F. J. Virol. 80:3112–3115. 10.1128/JVI.80.6.3112-3115.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schröfelbauer B, Senger T, Manning G, Landau NR. 2006. Mutational alteration of human immunodeficiency virus type 1 Vif allows for functional interaction with nonhuman primate APOBEC3G. J. Virol. 80:5984–5991. 10.1128/JVI.00388-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Simon V, Zennou V, Murray D, Huang Y, Ho DD, Bieniasz PD. 2005. Natural variation in Vif: differential impact on APOBEC3G/3F and a potential role in HIV-1 diversification. PLoS Pathog. 1:e6. 10.1371/journal.ppat.0010006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marin M, Rose KM, Kozak SL, Kabat D. 2003. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat. Med. 9:1398–1403. 10.1038/nm946 [DOI] [PubMed] [Google Scholar]
- 50.He Z, Zhang W, Chen G, Xu R, Yu X-F. 2008. Characterization of conserved motifs in HIV-1 Vif required for APOBEC3G and APOBEC3F interaction. J. Mol. Biol. 381:1000–1011. 10.1016/j.jmb.2008.06.061 [DOI] [PubMed] [Google Scholar]
- 51.Pery E, Rajendran KS, Brazier AJ, Gabuzda D. 2009. Regulation of APOBEC3 proteins by a novel YXXL motif in human immunodeficiency virus type 1 Vif and simian immunodeficiency virus SIVagm Vif. J. Virol. 83:2374–2381. 10.1128/JVI.01898-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wei W, Guo H, Han X, Liu X, Zhou X, Zhang W, Yu XF. 2012. A novel DCAF1-binding motif required for Vpx-mediated degradation of nuclear SAMHD1 and Vpr-induced G2 arrest. Cell. Microbiol. 14:1745–1756. 10.1111/j.1462-5822.2012.01835.x [DOI] [PubMed] [Google Scholar]