

ABSTRACT
Cells that undergo apoptosis in response to chemical or physical stimuli repress inflammatory reactions, but cells that undergo nonapoptotic death in response to such stimuli lack this activity. Whether cells dying from viral infection exhibit a cell death-type modulatory effect on inflammatory reactions is unknown. We compared the effects on macrophage inflammatory responses of cells dying an apoptotic or a nonapoptotic death as a result of adenoviral infection. The results were exactly opposite to the predictions from the conventional paradigm. Cells dying by apoptosis induced by infection with an adenovirus type 5 (Ad5) E1B 19-kilodalton (E1B 19K) gene deletion mutant did not repress macrophage NF-κB activation or cytokine responses to proinflammatory stimuli, whereas cells dying a nonapoptotic death from infection with E1B 19K-competent, wild-type Ad5 repressed these macrophage inflammatory responses as well as cells undergoing classical apoptosis in response to chemical injury. The immunorepressive, E1B 19K-related cell death activity depended upon direct contact of the virally infected corpses with responder macrophages. Replacement of the viral E1B 19K gene with the mammalian Bcl-2 gene in cis restored the nonapoptotic, immunorepressive cell death activity of virally infected cells. These results define a novel function of the antiapoptotic, adenoviral E1B 19K protein that may limit local host innate immune inflammation during accumulation of virally infected cells at sites of infection and suggest that E1B 19K-deleted, replicating adenoviral vectors might induce greater inflammatory responses to virally infected cells than E1B 19K-positive vectors, because of the net effect of their loss-of-function mutation.
IMPORTANCE We observed that cells dying a nonapoptotic cell death induced by adenovirus infection repressed macrophage proinflammatory responses while cells dying by apoptosis induced by infection with an E1B 19K deletion mutant virus did not repress macrophage proinflammatory responses and enhanced some cytokine responses. Our results define a new function of the antiapoptotic, adenoviral protein E1B 19K, which we have termed “apoptotic mimicry.” Our studies suggest the possibility that the presence or absence of this E1B 19K function could alter the immunological outcome of both natural and therapeutic adenoviral infections. For example, emerging, highly immunopathogenic adenovirus serotypes might induce increased host inflammatory responses as a result of altered E1B 19K function or expression. It is also possible that engineered variations in E1B 19K expression/function could be created during adenovirus vector design that would increase the therapeutic efficacy of replicating adenovirus vectors for vaccines or oncolytic viral targeting of neoplastic cells.
INTRODUCTION
Eukaryotic cells undergo different types of cell death responses. Apoptosis, or physiological cell death, is an active process in which cells proceed through an ordered pathway of destruction of many intracellular components, in most instances requiring the activity of cellular caspases, a family of cysteine proteases. Apoptosis is characterized by nuclear condensation prior to the loss of cell membrane integrity. Discrimination by macrophages of cells dying by apoptosis or nonapoptotic mechanisms affects the level of macrophage-mediated amplification of the host inflammatory response that occurs during phagocytic cell interactions with dying cells (1, 2). To date, all stimuli that induce apoptosis have been reported to generate dying cells that repress macrophage-induced inflammatory responses (3, 4). This has been proposed as a homeostatic mechanism that prevents autoimmunity during clearance of the large numbers of cells that die during normal, “physiological” cell turnover (5, 6). Conversely, the failure of cells dying by pathogen-induced nonapoptotic death to repress macrophage-mediated inflammatory responses may be essential for enhancement of local, anti-infective inflammation.
The morphological appearance of mammalian cells dying from viral infection in vitro has been termed cytopathic effect (CPE). CPE induced by viral infection can be categorized further by the cell death phenotype of the infected cells. For example, CPE induced by wild-type (wt) adenovirus (Ad) infection is distinctly nonapoptotic in nature, because of the blockade of apoptosis by the viral E1B 19-kilodalton protein (E1B 19K) (7–10). E1B 19K shares functional activity with the product of the antiapoptotic mammalian gene, Bcl-2, and is considered to be a Bcl-2 family member (8). E1B 19K gene deletion from adenovirus converts the death of cells undergoing Ad-induced CPE to a clearly apoptotic phenotype (9). These differences in the cell death phenotypes of cells dying as a result of infection with either wt Ad5 or E1B 19K-deleted Ad5 provided a congenic comparative system with which we could test the hypothesis that virally infected cells undergoing apoptosis are predictably immunorepressive for responder macrophages whereas virally infected cells undergoing nonapoptotic cell death are not.
The surprising result of these studies was that the immunomodulatory effects of Ad-induced CPE cells were exactly opposite to what was predicted from data with apoptotic and nonapoptotic cells dying after exposure to noninfectious injuries, where apoptotic cells are highly immunorepressive and nonapoptotic cells are not. Specifically, CPE corpses dying from infection with E1B 19K-negative mutant adenovirus underwent classical apoptosis but failed to repress macrophage responses and could even enhance those responses. Conversely, expression of the Bcl-2-like activity of E1B 19K protein during wt Ad5-induced CPE simultaneously blocked apoptosis and conveyed to the virally infected CPE corpses a trait that caused high-level, cell contact-dependent repression of macrophage inflammatory responses. Therefore, the data indicate that E1B 19K expression causes nonapoptotic, Ad-infected cells to mimic the macrophage repressive effect of cells undergoing apoptosis in response to chemical stimuli. The possible implications of these data are discussed, as related to the local innate immune responses to Ad-infected human cells.
MATERIALS AND METHODS
Cell lines.
Human (A549, HeLa, and MRC-5) and mouse (MH-S and RAW 264.7) cells were obtained from ATCC. Human AD293 cells were obtained from Clontech (Mountain View, CA). Stable κB-luciferase reporter 293 cells have been described (4). MRC-5 cells stably transfected with the Ad5 E1B 19K gene were provided by Keith Peden. A549, HeLa, and AD293 cells were grown in Dulbecco's modified Eagle medium (DMEM) supplemented with 5% bovine calf serum (BCS). MRC-5, MH-S, and RAW cells were grown in RPMI medium supplemented with 10% fetal bovine serum (FBS). All cells were cultured at 37°C in 5% CO2.
Viruses.
Wild-type adenovirus type 5 (Ad5; VR-5) was obtained from ATCC. Adenovirus mutants containing deletions of E1B 19K- or 55K-encoding regions (H5dl337 and H5dl338, respectively) and the parental E1B gene-expressing virus from which these viral mutants were derived, H5dl309, were provided by Tom Shenk. The E1B-19K-deleted virus containing inserted Bcl-2 expressed under the control of the cytomegalovirus (CMV) promoter was provided by George Chinnadurai. Virus stocks were grown in either A549 cells or AD293 cells (for H5dl337 and H5dl338). Adenovirus titers were determined by plaque-forming assays in A549 or AD293 cells and expressed as PFU/ml. Where indicated, Ad5 virus inocula were neutralized by using Ad5-specific, rabbit neutralizing antibody as reported earlier (11). Influenza H1N1 virus was obtained from the Clinical Microbiology Laboratory at the University of Illinois at Chicago as a seed stock grown on MDCK/A549 cells and was amplified, and plaque titers in A549 cells were determined. Influenza A/WSN/33 (H1N1) and A/Wyoming/3/2003 (H3N2) viruses were provided by Zhiping Ye as titered stocks grown in eggs to use as controls for group and subtype determination. The group and subtype of the influenza A H1N1virus isolate were confirmed using PCR (12).
Viral infection of cells and collection of CPE corpses.
Cells were infected for 1 h at 37°C in suspension with virus at a multiplicity of infection (MOI) of 10:1 PFU/cell, unless otherwise indicated, and then were plated in 100-mm dishes and allowed to proceed to complete CPE (approximately 96 h for Ad5 and 24 h for influenza virus). Floating cells were collected, and any remaining adherent cells were collected by incubation with EDTA at 37°C for 3 min. Cell corpses were washed repeatedly in phosphate-buffered saline (PBS) and resuspended in complete medium for immediate analysis or were fixed in 125 mM formaldehyde followed by repeated washes with complete medium and stored at 4°C for later analysis (4).
Noninfectious generation of apoptotic and necrotic corpses.
To induce apoptosis by chemical injury, cells were treated overnight with 1 μM staurosporine at 37°C and then washed repeatedly. Heat-killed necrotic cells were used to test an alternative type of cell death control to compare with apoptotic and virally infected cells, since such necrotic cells are not immunorepressive (13). Necrosis was induced by incubating cells at 56°C for 20 min.
Flow cytometry.
Unfixed corpses were collected and prepared for flow cytometry by washing in PBS + 3% FBS and staining with Hoechst dye and propidium iodide (PI) (14). Externalization of phosphatidylserine residues on the outer plasma membranes of apoptotic cells was assayed by staining CPE corpses with anti-annexin V antibody and 7-amino actinomycin D (7-AAD) according to the manufacturer's recommendations (BD Pharmingen). Apoptosis was inhibited by incubating cells with the pancaspase inhibitor zVAD (Calbiochem) at 50 μM during the infection and then adding zVAD to the cultures every 24 h at 50 μM until CPE corpses were harvested. Three cycles of centrifugation washing were sufficient to remove most debris from the CPE corpse preparations, as evidenced by flow analysis.
Assays of κB-luciferase-dependent transcription.
Studies of the effects of cellular corpses on NF-κB-dependent transcription were done with the nonmacrophage cell line 293, stably transfected with the κB luciferase reporter, as described previously (4). Plated 293-κB-luciferase cells were stimulated for 18 h with 2 nM phorbol myristate acetate (PMA; Sigma) in the absence or presence of cell corpses and were then washed and lysed for analysis of luciferase response. A stable RAW 264.7 κB-luciferase reporter macrophage cell line was created using the same method as for the 293 reporter cells. Luciferase expression was determined by using the Luciferase Assay kit (Promega, Madison, WI) and expressed as fold induction of stimulated cells versus unstimulated control cells.
Proinflammatory mediator measurement.
Cytokine and chemokine (here referred to as proinflammatory mediators) production by MH-S mouse alveolar macrophages was determined using the Bio-Plex Suspension Array system (Bio-Rad, Hercules, CA). Adherent MH-S cells (5 × 105 cells/well; 24-well plates) were stimulated for 18 h with lipopolysaccharide (LPS) (1 μg/ml) and gamma interferon (IFN-γ) (50 ng/ml) in the absence or presence of cell corpses at the indicated ratios. Media were collected, cleared of cells and debris by centrifugation, and frozen for later proinflammatory mediator analysis.
Statistical analysis.
The significance of differences in comparative data was estimated using Student's t test with SigmaPlot software.
RESULTS
Cell death phenotypes induced by human lung cell infection with wt Ad5 or Ad5 E1B deletion mutants.
The cell death phenotypes of cells exhibiting CPE in response to infection with wt Ad5 (nonapoptotic) and E1B 19K-deleted Ad (apoptotic) have been reported for different cell types (7–9, 15). To confirm these reported cell death phenotypes for the human lung cells (A549) used in these experiments, A549 cells were analyzed 48 h after infection by flow cytometry for DNA condensation and cell membrane integrity, using Hoechst and PI staining, respectively (Fig. 1A). Staining patterns were compared with those of control (untreated), necrotic (heat-treated), and apoptotic (staurosporine-treated) A549 cells. Heat-treated, necrotic cells stained positively for both Hoechst and PI (Hoechst+/PI+), as evidenced by a shift to the right upper quadrant of the flow cytogram (Fig. 1A, upper center). Staurosporine-treated cells showed the characteristic apoptotic phenotype of increased Hoechst staining intensity prior to increased PI staining (Hoechst+/PI−), as evidenced by a shift to the right lower quadrant of the flow cytogram (Fig. 1A, upper right). The cell death phenotype was assessed for A549 cells infected with wt Ad5 and four different Ad5 mutant viruses. A549 CPE cells infected with wt Ad5 (middle left cytogram) exhibited a relatively pure pattern of double-positive Hoechst+/PI+ staining, with minimal evidence of Hoechst+/PI− staining (i.e., little or no “apoptosis signal”). An identical nonapoptotic pattern of staining was observed with cells infected with the parental control virus H5dl309 (lower left cytogram), from which the two E1B-deleted mutant viruses tested were derived. In contrast, A549 infection with H5dl337 (middle center cytogram), an Ad5 mutant that does not express the antiapoptotic viral E1B 19K protein, induced an apoptotic cell death phenotype, similar to that observed with staurosporine-treated cells (16, 17). Staurosporine is a potent, prototypic apoptosis-inducing chemical agent (18, 19), which might explain the greater proportion of treated cells exhibiting the Hoechst+/PI− apoptotic pattern of staining than was observed with CPE cells infected with the E1B 19K-deleted virus, H5dl337. As another control for comparison, using viral infection instead of chemical treatment, A549 cells were infected with influenza virus, a well-known inducer of apoptotic death (20). The general pattern of Hoechst/PI staining and the proportion of virally infected cells exhibiting the Hoecsht+/PI− phenotype were similar for cells infected with E1B 19K-deleted H5dl337 and influenza virus (Fig. 1A, middle center versus middle right cytograms, respectively). In contrast, A549 infection with H5dl338, an Ad mutant that expresses E1B 19K normally but does not express E1B 55K (a unique E1B protein encoded from an alternative reading frame), resulted in a nonapoptotic phenotype identical to that of cells infected with wt Ad5 and the parental, E1B-competent virus, H5dl309 (Fig. 1A, lower center versus lower left cytograms, respectively). Since E1B 19K is a Bcl-2 family member, it was postulated that the apoptotic death phenotype of H5dl337-infected CPE cells resulted from the absence of E1B 19K antiapoptotic activity during viral infection. This was tested by analyzing CPE cells infected with an Ad5 virus, Ad-Bcl-2, in which E1B 19K was deleted and replaced by insertion of the mammalian Bcl-2 gene (7). Ad-Bcl-2 CPE cells exhibited a nonapoptotic pattern of staining identical to that of wt Ad5 (Fig. 1A, lower right compared with middle left cytograms, respectively).
FIG 1.

Nonapoptotic cell death induced by Ad5 infection is dependent on the expression of the adenoviral E1B 19K protein. (A) Changes in Hoechst/PI staining of A549 cells following either 48-h infection with different Ads or 24-h infection with influenza A virus. Viable (Control), necrotic (heat), and apoptotic (stauro) A549 cells were used as controls. Infection of A549 cells with wt Ad5, H5dl309, H5dl338, or Ad-Bcl-2 resulted in nonapoptotic cell death as evidenced by Hoechst/PI staining that was primarily Hoechst+/PI+, a pattern similar to that seen with necrotic (heat) A549 cells. In contrast, H5dl337 and influenza A virus infection resulted in apoptotic cell death, as evidenced by accumulation of Hoechst+/PI− cells (lower right quadrant of the cytograms), prior to loss of cell membrane integrity when cells were permeable to PI (Hoechst+/PI+ staining), a pattern similar to that observed with apoptotic (stauro) cells. (B) Externalization of phosphatidylserine residues as an indicator of apoptotic cell death. Forty-eight hours postinfection, cells were harvested and stained with annexin V and 7-AAD to assess cell surface phosphatidylserine exposure and cell membrane integrity, respectively. Infection of A549 cells with H5dl337 resulted in apoptotic cell death, as evidenced by the high percentage of cells showing phosphatidylserine residue externalization, prior to loss of cell membrane integrity (annexin V+/7-AAD− cells). In contrast, infection of A549 cells with E1B 19K-competent wt Ad5 resulted in repression of phosphatidylserine residue externalization prior to loss of membrane integrity, compared with cells infected with E1B 19K-negative H5dl337 (i.e., 24% versus 69% annexin V+/7-AAD− cells in right lower quadrants, respectively). (C) Caspase dependence of H5dl337-induced apoptotic cell death. A549 cells were infected with H5dl337 in the presence of zVAD (50 μM). Forty-eight hours postinfection, cells were harvested and stained with either annexin V/7-AAD or Hoechst/PI, to examine the caspase dependence of phosphatidylserine residue externalization or DNA condensation prior to loss of cell membrane integrity, respectively. Infection with H5dl337 in the presence of zVAD blocked the apoptotic phenotype and resulted in a reduction in the annexin V+/7-AAD− and Hoechst+/PI phenotypes of infected cells, resulting in a staining pattern that was identical to that seen with wt Ad5 infection (compare panel C results with those for wt Ad5 in panels A and B).
To confirm the apoptotic nature of the cell death observed with H5dl337-infected cells, we examined the externalization of phosphatidylserine residues to the cell surface and dependence of the cell death phenotype on caspase activity, two well-established characteristics of apoptotic cell death (Fig. 1B and C). Cells were costained with annexin V and 7-amino actinomycin D (7-AAD), a membrane-impermeable dye that, like PI, is excluded from cells with intact cell membranes. Cells infected with H5dl337 exhibited a predominant annexin V-positive, 7-AAD-negative phenotype, indicating externalization of phosphatidylserine residues prior to the loss of cell membrane integrity (Fig. 1B, middle cytogram), a phenotype that is typical for cells undergoing apoptotic death. In contrast, cells infected with E1B-expressing, wt Ad5 were less likely to exhibit single-positive annexin V staining than H5dl337-infected cells and were more likely to exhibit double-positive staining (Fig. 1B, right cytogram), indicating that annexin V staining occurred after loss of cell membrane integrity. This pattern of E1B 19K expression-related apoptotic cell death repression was identical to comparative cytogram patterns observed using Hoechst/PI staining of infected cells, in which single-positive Hoechst staining (nuclear condensation, before membrane permeabilization) was observed with H5dl337-infected cells but repressed in cells infected with wt Ad5 (Fig. 1A, middle center versus middle left cytograms, respectively). As reported for apoptotic cell death induced by noninfectious cellular stimuli (15), the apoptotic phenotype of H5dl337-infected cells was blocked by treatment of infected cells with the pancaspase inhibitor, zVAD, as evidenced by testing with both the annexin V/7-AAD and the Hoechst/PI staining assays (Fig. 1C). Specifically, single-positive annexin V and single-positive Hoechst staining were both repressed by zVAD treatment. Taken together, these flow cytometry data show that Ad infection of A549 cells in the absence of E1B 19K expression (H5dl337) induces classical, caspase-mediated apoptotic cell death that is repressed by E1B 19K expression during A549 infection with wt Ad5.
Ad5-infected cells repress NF-κB-dependent proinflammatory macrophage responses, as a result of expression of the antiapoptotic adenoviral protein, E1B 19K.
Chemically induced apoptotic corpses repress macrophage production of proinflammatory mediators, in contrast to heat-killed nonapoptotic cells, which lack this activity (13, 21). Cvetanovic et al. reported that chemically induced apoptotic corpses cause contact-dependent repression of LPS-induced, NF-κB-dependent macrophage cytokine and chemokine responses (4, 13). We used this cell system and our data on the cell death phenotypes of Ad-infected A549 cells (Fig. 1) to determine whether virally infected cells followed the model of apoptosis-related immunorepression that was developed from studies of noninfectious cellular injuries. Macrophage stimulation in the absence of cell corpses resulted in increased production of interleukin-6 (IL-6), granulocyte colony-stimulating factor (G-CSF), monocyte chemoattractant protein 1 (MCP-1), and IL-1β, compared with control, unstimulated cells (Fig. 2, Stim versus C, respectively). Macrophages cocultured with heat-killed, nonapoptotic corpses (Fig. 2, N) either enhanced (G-CSF and MCP-1) or failed to repress (IL-6 and IL-1β) mediator production. Addition of staurosporine-killed apoptotic corpses (A) repressed induction of all 4 proinflammatory mediators to levels similar to those observed with unstimulated control responder cells. Surprisingly, however, apoptotic CPE corpses infected with the E1B 19K-deleted adenovirus, H5dl337, lacked any significant immunorepressive activity, similar to necrotic control cells. In contrast, nonapoptotic CPE corpses infected with E1B 19K-competent wt Ad5 were as immunorepressive as staurosporine-treated apoptotic cells (Fig. 2, wt Ad5 versus A, respectively). Therefore, these data on the effects of Ad-infected CPE cells on macrophage cytokine and chemokine responses were completely opposite to what would be predicted by the conventional apoptosis-related immunorepression paradigm.
FIG 2.
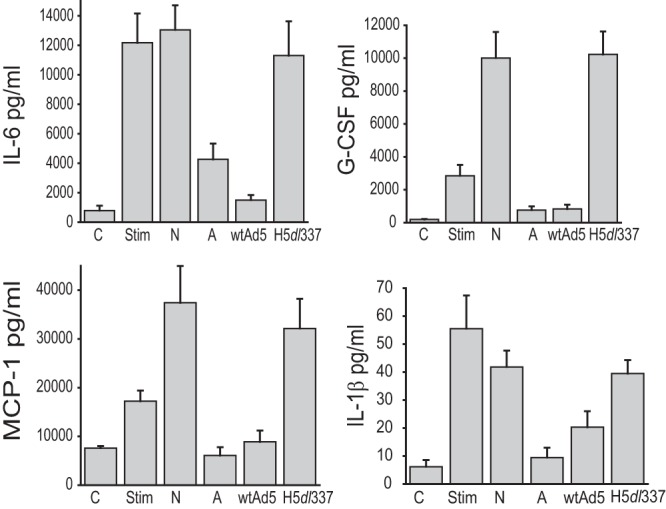
Adenovirus E1B 19K expression determines Ad CPE corpse immunomodulatory activity on proinflammatory cytokine/chemokine production by macrophages. A549 cells were infected with the indicated viruses, and the resulting corpses were fixed in formaldehyde. Macrophages were cultured under three conditions, (i) in the absence of LPS and IFN-γ stimulation or Ad CPE corpses (control [C]), (ii) with LPS and IFN-γ stimulation (Stim), or (iii) with LPS and IFN-γ stimulation, while being cocultured (10:1 corpse/macrophage ratio, 18 h) with heat-killed necrotic A549 corpses (N), staurosporine-killed apoptotic A549 corpses (A), or A549 corpses infected with either wild-type Ad5 (wtAd5) or the E1B 19K-deleted Ad5 mutant virus, H5dl337. E1B 19K deletion from the infecting virus resulted in a significant loss of Ad5 CPE corpse repression of mediator production by macrophages (wtAd5 versus H5dl337; values are means ± standard errors of the means [SEM], n = 3, P < 0.03).
All of these cytokines and chemokines, as well as most others involved in macrophage amplification of the host inflammatory response, are dependent upon NF-κB-dependent transcription for activation in response to proinflammatory stimuli. To test this cellular control mechanism in our system, two independent κB-luciferase reporter cell systems that were used by Cvetanovic et al. in their studies of apoptotic cell repression of NF-κB activation were used to measure the effects of Ad-infected CPE corpses on stimulus-induced, NF-κB-dependent transcription (Fig. 3) (4, 13). Viable (V) and heat-killed, necrotic (N) corpses had little effect on LPS-induced stimulation of κB-luciferase activity in mouse RAW 264.7 mouse macrophages that were transiently transfected with an NF-κB-luciferase reporter vector (Fig. 3A). In contrast, staurosporine-induced apoptotic corpses (A) repressed LPS stimulation of κB-luciferase activity to ∼10% of the levels of stimulated cells cultured without corpses (Fig. 3A, A versus None, respectively). CPE corpses infected with wt Ad5, in which apoptosis was repressed by E1B 19K expression (Fig. 1), repressed LPS stimulation of κB-luciferase activity in RAW 264.7 reporter cells to levels identical to those observed with chemically induced apoptotic corpses (Fig. 3A, Ad5 versus A, respectively).
FIG 3.
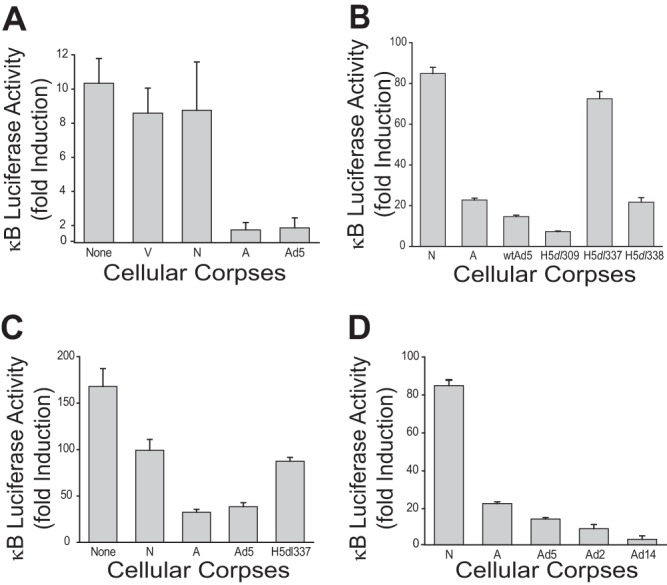
Ad5 CPE corpses repress NF-κB dependent transcription in mouse and human responder cells. (A) Luciferase activity from mouse RAW 264.7 macrophages transiently transfected with an NF-κB luciferase reporter. A549 cells were infected with Ad5. The resulting Ad CPE corpses and control cells were fixed and cocultured at a 10:1 ratio with transfected macrophages in the presence of 100 ng/ml LPS for 18 h. Apoptotic (staurosporine-treated [A]) and Ad5 CPE corpses significantly repressed LPS induction of NF-κB-dependent transcription in macrophages (means ± SEM, n = 3, P = 0.006), whereas viable (V) and heat-killed necrotic (N) cells failed to repress LPS induction of NF-κB-dependent transcription (means ± SEM, n = 3, no significant difference). (B) Luciferase activity from human 293 κB-luciferase epithelial reporter cells stimulated with 2 nM PMA for 18 h and cocultured at a 1:10 ratio with heat-killed necrotic corpses (N), staurosporine-treated apoptotic corpses (A), or cell corpses infected with the indicated viruses. A549 cells were infected with wild-type (wt) Ad5, H5dl309 (mutant control virus that expresses both E1B 19K and 55K proteins), or either of two Ad5 mutant viruses derived from H5dl309: E1B 19K-deleted H5dl337 or E1B 55K-deleted H5dl338 (both of which still express the alternative E1B protein). Only necrotic corpses (N) and corpses infected with E1B 19K-deleted virus (H5dl337) failed to significantly repress PMA-induced, NF-κB-dependent transcription (means ± SEM, n = 3, P < 0.001). (C) Luciferase activity from a stable human 293 NF-κB epithelial reporter cell line. HeLa cells were infected with either Ad5 or H5dl337. The resulting Ad CPE and control corpses were fixed and cocultured at a 10:1 ratio with reporter cells in the presence of 2 nM PMA for 18 h. Apoptotic (A) and Ad5 CPE corpses significantly repressed PMA induction of NF-κB-dependent transcription, compared with necrotic control cells (N) (means ± SEM, n = 3, P = 0.001). In contrast, there was no significant difference in the effects of apoptotic, H5dl337 CPE corpses and necrotic control cells. (D) Luciferase activity from a stable human 293 NF-κB epithelial reporter cell line cocultured with A549 cells infected with Ad5, Ad2, or Ad14 (a group B2 serotype Ad). The resulting Ad CPE and control corpses were fixed and cocultured at a 10:1 ratio with reporter cells in the presence of 2 nM PMA for 18 h. Ad CPE corpses infected with each of the three Ad serotypes significantly repressed PMA induction of NF-κB-dependent transcription in reporter cells (means ± SEM, n = 3, P < 0.001).
However, as observed in the studies of Ad5 CPE corpse effects on cytokine production by macrophages (Fig. 2), the NF-κB-repressive effect of Ad5 CPE corpses was strictly dependent upon E1B 19K expression by the infecting virus (Fig. 3B). Using a stable 293 κB-luciferase reporter system, Ad5 CPE repression of PMA-induced NF-κB activation was just as efficient as with wt Ad5-infected cells, when CPE cells were infected with the parental (and E1B competent) virus, H5dl309, from which the E1B mutant viruses were derived or with the E1B 55K-deleted Ad5 mutant, H5dl338. But there was no significant repression of stimulus-induced NF-κB activation in responder 293 cells when they were cocultured in contact with Ad CPE cells infected with the E1B 19K-deleted Ad5 mutant, H5dl337 (Fig. 3B). To determine if the observed immunomodulatory effects of Ad5 infection of A549 were restricted to just A549 cells, HeLa cells (another Ad-permissive cell line) were infected with either wt Ad5 or H5dl337. As shown in Fig. 3C, wt Ad5 CPE corpses repressed PMA-stimulated NF-κB activity of the 293 reporter cells as efficiently as apoptotic HeLa corpses (Ad5 versus A), whereas H5dl337 corpses behaved like necrotic HeLa corpses (H5dl337 versus N). Therefore, the NF-κB repressive effect of nonapoptotic Ad5 CPE corpses was not species specific or cell type restricted (i.e., the effect was observed with both mouse and human responder cells and with different types of Ad-infected stimulator cells).
The NF-κB repressive activity of Ad-infected CPE corpses was not unique to the wt Ad5 serotype, since the same effect was observed with A549 CPE cells infected with either wt Ad2 (a group C Ad serotype that is highly homologous with Ad5) or wt Ad14 (an Ad serotype from group B2 that has a lower level of sequence homology with Ad5) (Fig. 3D).
These results, combined with those in Fig. 2, indicated that repression of NF-κB-dependent transcription in responder cell and macrophage proinflammatory mediator production may be a general property of Ad-infected, nonapoptotic cells that is dependent on E1B 19K expression but independent of adenoviral serotype, infected cell type, or responder cell type. Furthermore, since both human and mouse responder cells exhibited the same responses to wt Ad5-infected, human Ad CPE corpses, the data indicated that there may be no species or genetic restriction of the signal(s) between the Ad-infected cells and the responder cells.
Adenovirus infection-induced nonapoptotic cell death mimics the immunorepressive effects of cells undergoing apoptosis in response to other noninfectious and infectious stimuli.
The results to this point in the study showed that human cells dying a nonapoptotic death as a result of E1B 19K expression during wt Ad5 infection were comparable to staurosporine cells undergoing classical apoptosis when compared for repression of NF-κB-dependent cytokine and chemokine production by responder macrophages. But there were major differences in the infectious versus chemical nature of the cellular injuries of the dying cells used for these comparisons. We therefore took advantage of the characterization of the cell death phenotype of influenza virus-infected A549 cells that was done during the cytofluorometric analysis of the adenovirus-induced cell death response represented in Fig. 1. In those studies, influenza virus infection of A549 cells induced an apoptotic phenotype that was comparable to infection with the E1B 19K-deleted Ad5 mutant, H5dl337. But the influenza virus-induced apoptotic cell death response was distinctly different from the nonapoptotic cell death response induced by E1B 19K-competent wt Ad5 (Fig. 1). These observations were used for a direct comparison of the immunomodulatory effects of cells dying an apoptotic death from infection with influenza virus versus cells dying a nonapoptotic death from infection with wt Ad5. These experiments showed that wt Ad5 and influenza virus induced identical immunomodulatory phenotypes in infected A549 cells, despite their opposite cell death phenotypes. Both types of CPE corpses induced high-level repression of stimulus-induced macrophage production of proinflammatory mediators that was comparable to the immunorepressive effects of staurosporine-induced apoptotic cells (Fig. 4A). Furthermore, both types of virally infected, CPE corpses repressed stimulus-induced NF-κB activation in 293 reporter cells (Fig. 4B). These results supported the conclusion that cells dying as a result of wt Ad5 infection mimicked the immunorepressive effect of apoptotic cells induced by both noninfectious (e.g., staurosporine) and infectious (e.g., influenza virus) stimuli. We termed this immunorepressive effect of nonapoptotic, wt Ad5-infected cells “apoptotic mimicry.”
FIG 4.
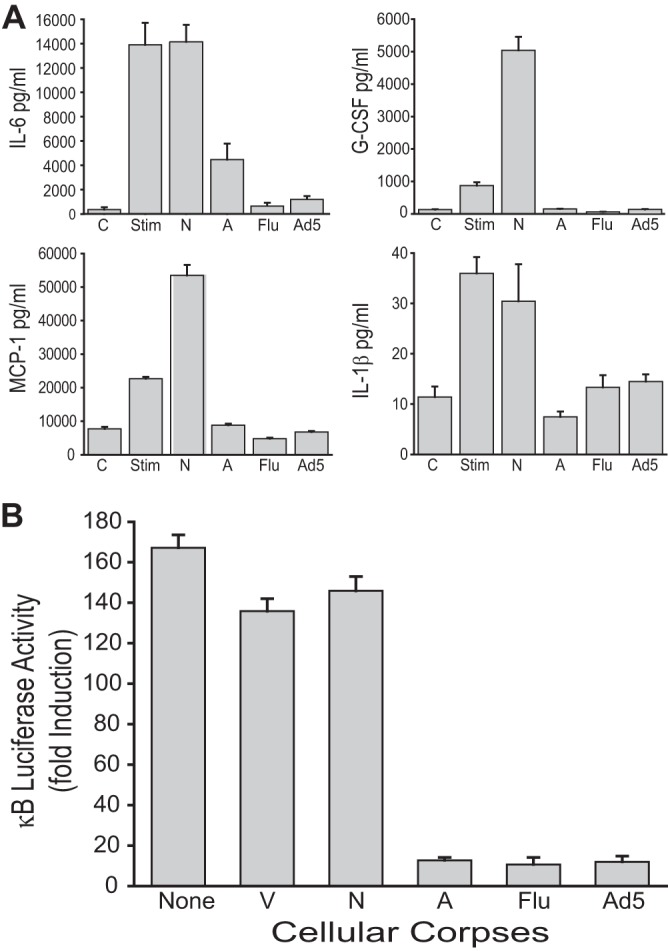
Nonapoptotic Ad5 CPE corpses mimic the immunosuppressive activities of apoptotic influenza A virus CPE corpses. (A) Cytokine and chemokine production by LPS+IFN-γ-stimulated mouse MH-S macrophages. Heat-killed necrotic (N) and staurosporine-killed apoptotic (A) A549 corpses served as controls. A549 infection with either influenza virus (Flu) or wild-type Ad5 produced corpses that significantly repressed macrophage production of all four proinflammatory mediators to an extent similar to that observed with staurosporine-induced apoptotic corpses (means ± SEM, n = 3, P < 0.01). (B) Luciferase activity from human 293 κB-luciferase epithelial reporter cells. A549 corpses were incubated with reporter cells at a 10:1 corpse/reporter cell ratio in the presence of 2 nM PMA for 18 h. Data are expressed as fold induction versus uninduced reporter cell activity. Viable cells (V) and heat-killed necrotic corpses (N) had no effect on NF-κB-dependent transcription, whereas staurosporine-induced apoptotic corpses (A), influenza A virus-infected corpses (Flu), and Ad5-infected corpses (Ad5) significantly repressed PMA-induced, NF-κB-dependent transcription to a similar extent (means ± SEM, n = 3, P < 0.001).
Expression of E1B 19K in trans restores apoptotic mimicry during infection with E1B 19K-deleted adenovirus.
The requirement for E1B 19K expression in Ad-infected cell corpses to repress NF-κB activation in stimulated responder cells was tested further by studying the effect of E1B 19K complementation in trans during viral infection. MRC-5 cells (nonimmortal, fetal human lung fibroblasts) stably transfected with the Ad5 E1B 19K gene were used to determine whether provision of E1B 19K in trans could complement the loss of apoptotic mimicry in cells infected with H5dl337. As observed with virally infected A549 cells, MRC-5 cells exhibited a nonapoptotic cell death phenotype (Hoechst+/PI+) when infected with wt Ad5 and an apoptotic (Hoechst+/PI−) cell death phenotype when infected with H5dl337 (Fig. 5A, upper middle cytogram versus upper right cytogram, respectively). Nonapoptotic MRC-5 cell corpses infected with wt Ad5 were highly repressive of PMA-induced NK-κB activation in 293 reporter cells, whereas MRC-5 cell corpses infected with H5dl337 were not (Fig. 5B, wtAd5 versus H5dl337, black bars). MRC-5 cells stably expressing E1B 19K (MRC-19K) were resistant to apoptotic cell death following infection with H5dl337 as evidenced by E1B 19K expression-related elimination of the Hoechst+/PI− cell population in flow cytometry studies of virally infected cells (Fig. 5A, lower right [19% single positive] versus upper right [63% single positive], respectively). These nonapoptotic H5dl337-infected, MRC-19K CPE corpses were as effective as wt Ad5-infected MRC corpses in repressing PMA-induced NF-κB activation in responder cells (Fig. 5B, H5dl337 versus wtAd5, gray bars). These transfected cell complementation studies showed that expression of E1B 19K was both necessary (Fig. 2 and 3) and sufficient (Fig. 5B) for Ad5-infected cells undergoing a nonapoptotic cell death to mimic the immunorepressive activity of cells undergoing classical, chemically induced apoptosis.
FIG 5.
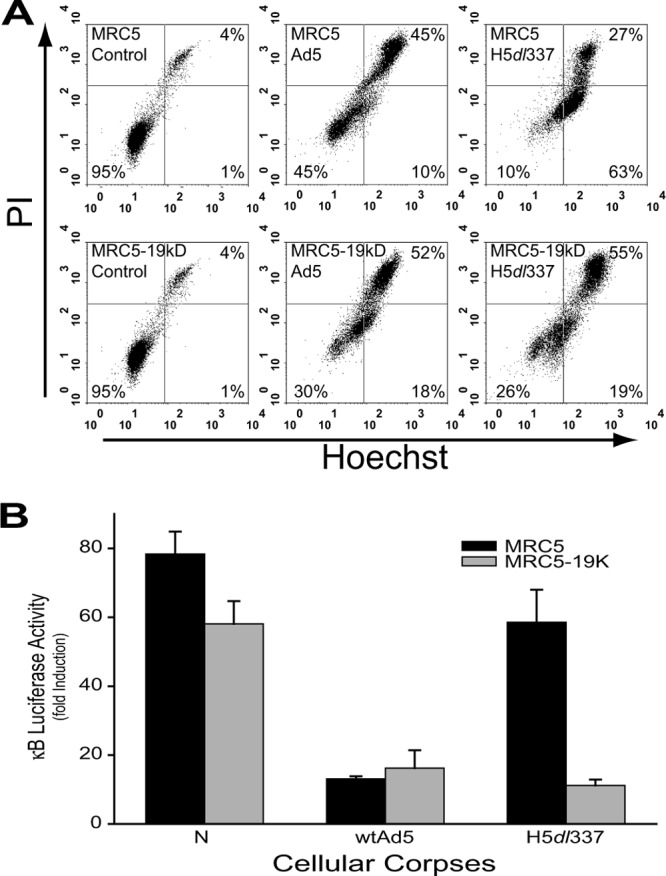
Restoration of Ad5 CPE corpse mediated immunosuppression through expression of E1B 19K in trans. (A) Expression of E1B 19K in trans inhibited the apoptosis induced by the H5dl337 infection. MRC5 cells or MRC5 cells stably expressing E1B 19K (MRC5-19kD) were infected with either wild-type Ad5 or H5dl337. Forty-eight hours postinfection, the cells were collected and stained with Hoechst/PI to assess the cell death response. MRC5 cells infected with wild-type Ad5 died a nonapoptotic death, as evidenced by the lack of accumulation of single-positive, Hoechst+/PI− cells (only 10% Hoechst+/PI− cells, upper center cytogram). In contrast, MRC5 cells infected with H5dl337 showed a marked accumulation of Hoechst+ cells prior to loss of membrane integrity (63% Hoechst+/PI−cells) indicative of apoptotic cell death (upper right cytogram). Ad5 infection of MRC5-19K cells exhibited the same pattern of nonapoptotic cell death as that seen with wild-type Ad5 infection of parental MRC5 cells (lower center cytogram). But H5dl337 infection of MRC5-19K cells did not induce the apoptotic cell death response (i.e., the majority Hoechst+/PI− phenotype) observed after infection of parental E1B 19K-negative cells with this mutant virus, indicating that expression of E1B 19K in trans restored the nonapoptotic cell death response to cells infected with H5dl337. (B) Luciferase activity of 293 κB-luciferase reporter cells stimulated with 2 nM PMA for 18 h and cocultured with either heat-killed necrotic corpses (N) or virally infected cell corpses, as indicated (10:1 corpse/reporter cell ratio). MRC5 or MRC5-19K (stably expressing E1B 19K) cells were infected with either wild-type (wt) Ad5 or the E1B 19K-deleted virus, H5dl337. Expression of E1B 19K in trans in MRC-19K cells significantly increased the repressive effect of H5dl337-infected cell corpses for NF-κB-dependent transcription in responder cells, compared with MRC5 parental cells (H5dl337 gray bar versus H5dl337 black bar, respectively; means ± SEM, n = 3, P < 0.008).
Bcl-2-like antiapoptotic activity of E1B 19K is required for Ad5-infected cell corpse repression of NF-κB activation in responder cells.
The E1B 19K gene shares a sequence motif and related antiapoptotic function with the human proto-oncogene Bcl-2 (8, 22). Bcl-2 can replace the E1B 19K function of deleted, mutant virus during infection, blocking apoptosis of virally infected cells and restoring viral titers to wild-type virus levels (8, 22). These observations suggested the possibility that the Bcl-2-like activity of E1B 19K is involved in the apoptotic mimicry of Ad5-infected cells. To test this hypothesis, similar studies were done using the Ad5-Bcl2 virus characterized in the flow cytometry studies of the cell death phenotypes of virally infected cells (Fig. 1). Ad5-Bcl-2 virus induced a nonapoptotic cell death phenotype similar to that of cells infected with wt Ad5, as predicted for Bcl-2 complementation of the loss of the antiapoptotic activity caused by the E1B 19K deletion. When Ad5-Bcl-2-infected CPE corpses were cocultured with 293 κB-luciferase reporter cells, PMA-induced NF-κB-dependent transcription was repressed as efficiently as with staurosporine-induced apoptotic corpses (A) or with wt Ad5-infected, nonapoptotic corpses (Ad5), and the Ad5-Bcl-2-infected corpses (Δ19kD+Bcl-2) were significantly more repressive than those infected with the E1B 19K-negative Ad5 mutant, H5dl337 (Fig. 6; P < 0.05). These results indicated that expression of the Bcl-2-like activity of E1B 19K is sufficient for the apoptotic mimicry observed with wild-type Ad5-infected cells.
FIG 6.
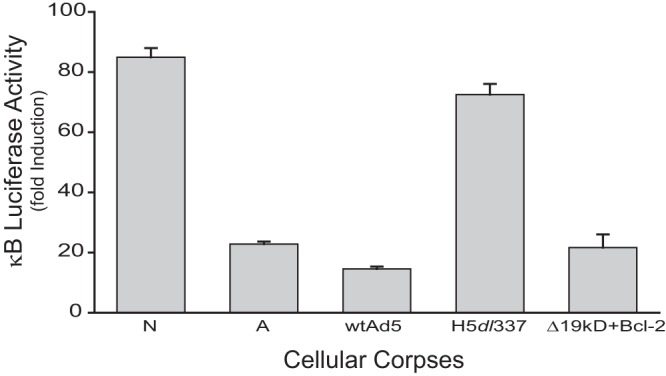
The role of the Bcl-2-like activity of E1B 19K in apoptotic mimicry. Luciferase activity of 293 κB-luciferase reporter cells stimulated with 2 nM PMA for 18 h and cocultured with heat-killed necrotic corpses (N), staurosporine-treated apoptotic corpses (A), or cell corpses infected with the indicated viruses (10:1 corpse/reporter cell ratio). A549 cells were infected with wtAd5, H5dl337, or an Ad5 mutant virus with E1B 19K deleted and replaced by CMV-Bcl-2 (Δ19kD+Bcl2). Expression of Bcl-2 in the recombinant virus complemented the loss of E1B 19K, resulting in a significant increase in infection-induced corpse repression of NF-κB-dependent transcription in responder cells, compared with corpses dying from infection with the E1B-deleted virus, H5dl337 (values are means ± SEM, n = 3, P < 0.001).
Ad5-infected CPE corpses exhibit E1B 19K-related, cell contact-dependent repression of virus-induced, NF-κB-dependent transcription activation responses of macrophages.
Adenovirus induces a proinflammatory response early after infection of the immunocompetent host (23, 24). At later times after infection, this response wanes, purportedly because the host immune response clears the viral infection and thus eliminates the proinflammatory stimulus. Based on the data to this point in the study, we postulated that modulation of the early, innate immune response by accumulation of virus-induced CPE corpses at the site of infection could be a previously unrecognized regulatory component of this adenovirus-host interaction. To test this hypothesis, we developed a culture system to analyze the relative roles of viral particles, CPE corpses dying from Ad infection, and E1B 19K expression in these corpses in determining the net effects on the macrophage-induced proinflammatory amplification response. We first tested the direct effect of Ad particles on NF-κB-dependent transcription responses of macrophages (Fig. 7A). We used RAW 264.7 macrophages stably expressing a κB-luciferase reporter and added increasing amounts of virus either directly to cells or in the upper chambers of a Transwell culture system, with macrophages in the bottom chamber. Addition of wt Ad5 at a multiplicity of infection of 10 PFU/cell induced approximately a 3-fold induction of macrophage κB-luciferase activity (Fig. 7A, closed circles). Progressively increasing the Ad5 dose up to 50-fold (500 PFU/cell) increased this transcription stimulation to approximately a 5-fold response. This level of virus-induced NF-κB activation in macrophages was unaffected by the Transwell insert (open circles), as would be expected for stimulating virus particles passing freely across the membrane. Virus-induced NF-κB activation in macrophages was eliminated by preincubation of the viral inoculum with Ad5-specific neutralizing antibody (Fig. 7A, closed triangles). These data indicated that the Transwell system could be used to explore the independent activities of Ad5 virions and Ad5 CPE corpses on macrophage inflammatory responses when both stimuli were present at the same time.
FIG 7.

Ad5 CPE corpses exhibit E1B 19K-dependent, cell-contact-dependent repression of Ad virion-induced proinflammatory responses by macrophages. (A) Luciferase activity of mouse RAW 264.7 κB-luciferase macrophages induced by viral particles. RAW reporter cells were stimulated with Ad5 virions added at ratios of 10, 100, 200, or 500 PFU per reporter cell. Virions added directly to reporter cell cultures (filled circles) or to a Transwell insert above reporter cell cultures (open circles) induced a 3- to 5-fold increase in NF-κB activation in reporter macrophages, unless the viral inoculum was treated with Ad5-specific neutralizing antibody (triangles), which significantly blocked viral particle stimulation of reporter cell transcription (means ± SEM, n = 3, P < 0.001). (B) Luciferase activity of mouse RAW 264.7 κB-luciferase macrophages cultured with virally infected cell corpses. RAW reporters were cocultured with unfixed A549 corpses infected with either wild-type (wt) Ad5 (circles) or H5dl337 (diamonds), added either directly to reporter cell cultures (filled symbols) or to Transwell inserts above reporter cell cultures (open symbols) at ratios of 1, 3, 5, or 10 corpses to reporter cells. Both wt Ad5 and H5dl337 CPE corpses stimulated NF-κB-dependent transcription when separated from reporter cells by Transwell inserts (open circles and diamonds, respectively), presumably through the stimulatory effects of Ad virions released from unfixed corpses. wt Ad5 CPE corpses significantly repressed this virion-induced NF-κB-dependent transcription through direct contact with reporter cells, when 3 or more corpses were added per reporter cell (filled circles) (values are means ± SEM, n = 3, P < 0.001 for corpse/responder cell ratios of 3:1, 5:1, and 10:1). In contrast, H5dl337 CPE corpses failed to repress this virion-induced NF-κB-dependent transcription when in direct contact with reporter cells (filled diamonds; nonsignificant). (C) Contact-dependent Ad-infected CPE corpse repression of virion-induced cytokine/chemokine expression. A549 cells were infected with wtAd5 or H5dl337. The resulting Ad corpses were added at a ratio of 10 corpses to 1 macrophage, either directly to MH-S macrophages (Transwell−) or in Transwell inserts above the macrophage cultures (Transwell+). wtAd5 virions (V) were added in the same manner at a ratio of 10 PFU per 1 MH-S cell, to stimulate responder macrophages. After 18 h of incubation, media were assayed for cytokine/chemokine levels from control, untreated macrophages (C), compared with all other conditions. Ad5 virions (V) stimulated macrophage mediator production when added directly to macrophages (Transwell−) or through the Transwell insert (Transwell+). Both wtAd5 and H5dl337 CPE corpses stimulated macrophage mediator production, when separated from macrophages by the insert (Transwell+). wt Ad5 CPE corpses significantly repressed this virion-induced macrophage mediator response when added directly to macrophage cultures (Transwell−; means ± SEM, n = 4, P < 0.01) but not when separated from macrophages by inserts (Transwell+; NS). In contrast, direct contact with H5dl337 CPE corpses (Transwell−) had no significant repressive effect on macrophage mediator responses (means ± SEM, n = 4, nonsignificant). Moreover, the levels of some macrophage mediator responses were significantly increased when the responder macrophages were in contact with H5dl337 CPE corpses (Transwell−) than when separated from the corpses by the membrane (H5dl337 Transwell− versus H5dl337 Transwell+; i.e., IL-6, G-CSF, and MCP-1; means ± SEM, n = 4, P < 0.001).
In our previous experiments, we used fixed CPE corpses to examine the effect that the CPE corpses had on inflammatory responses independently of free virus. Using the Transwell system, it was possible to test the effects of both virions and CPE corpses together by using unfixed, virally infected CPE corpses. CPE corpses dying from infection with wt Ad5 or H5dl337 (without formaldehyde fixation) were then cocultured with responder macrophages either in the same culture chamber with the macrophages or separated from the macrophages by a Transwell membrane (Fig. 7B). When cocultured in the same chamber at a ratio of one Ad5-infected CPE corpse to one responder macrophage (1:1 corpse/responder cell ratio), there was a 3-fold induction of macrophage κB-luciferase activity (Fig. 7B, closed circles)—i.e., no difference compared with the Ad5 infection control, in the absence of cell corpses (Fig. 7A, closed circles). This similarity was explained by stimulation of macrophage responder cells by the infectious virus contaminating or produced by Ad5-infected CPE corpses, since addition of Ad5 neutralizing antibody suppressed Ad5-infected corpse induction of NF-κB activity (not shown). Addition of increasing numbers of Ad5-infected CPE corpses in the same culture chamber as the macrophage responder cells resulted in a corpse dose-related repression of the macrophage NF-κB activation response, with significant repression detected at a 3:1 corpse/macrophage ratio and to the point where the macrophage response was extinguished at a 10:1 corpse/macrophage ratio. These data showed that wt Ad5 CPE corpses can repress Ad5 virion-induced NF-κB-dependent transcription.
If the wt Ad5-infected CPE corpses were separated from responder macrophages by the Transwell membrane (Fig. 7B, open circles), the corpse-dose-related repressive effect on the NF-κB activation response of macrophages was lost, but the viral stimulatory effect persisted. It is noteworthy that preparations of unfixed Ad5-infected CPE corpses contained infectious virus (>10 PFU/ml [Fig. 7A]), despite repeated washing; therefore, infectious virus was present in both culture chambers and only the CPE corpses and macrophage responder cells were separated by the Transwell membrane. Like wt Ad5-infected CPE corpses, H5dl337-infected corpses (and the associated, contaminating virus) stimulated responder macrophage NF-κB activation at a 1:1 corpse/macrophage ratio (Fig. 7B, closed diamonds). The difference with H5dl337-infected CPE corpses (closed diamonds), compared with wt Ad5-infected corpses (closed circles), was the lack of repression of macrophage responses with increasing corpse/macrophage responder cell ratios with H5dl337-infected CPE corpses, whether the corpses were incubated in contact with macrophage responders or were separated by the Transwell membrane (Fig. 7B, closed versus open diamonds, respectively). In essence, the deletion of E1B 19K from the infecting virus eliminated the contact-dependent macrophage-repressive effect of the cocultured CPE cells. These data indicated that there was an E1B 19K-related, cell-contact-dependent macrophage-repressive effect of Ad-infected cells.
E1B 19K-dependent suppressive effect of Ad-infected CPE corpses for virus-induced, proinflammatory mediator responses of macrophages.
The same Transwell culture system was used to test the Ad CPE corpse/macrophage responder cell contact dependence of modulation of virus-induced macrophage inflammatory mediatory responses by Ad-infected cells dying a nonapoptotic (wt Ad5) or apoptotic (H5dl337) cell death. As a positive virus control for macrophage stimulation, wt Ad5 particles were added to macrophage cultures at a multiplicity of infection of 10 PFU/macrophage, in the absence of Ad CPE corpses (Fig. 7C, V-no Transwell [Transwell−] and V-Transwell [Transwell+]). This stimulation of macrophages by Ad5 virus particles induced IL-1β, IL-6, G-CSF, and MCP-1 production by macrophages (Fig. 7C, V, [Transwell−]). Interposition of the Transwell membrane between added virus particles (top chamber) and macrophage responder cells (bottom chamber) (Fig. 7C, V, [Transwell+]) had no significant effect on viral induction of these macrophage proinflammatory mediator responses. Cell corpses dying a nonapoptotic death from infection with wt Ad5 (E1B 19K positive) repressed macrophage cytokine/chemokine production (Fig. 7C, wtAd5, [Transwell−]), and the efficiency of macrophage repression was dependent on cell-to-cell contact, as evidenced by the reduced repression when wt Ad5 CPE corpses (top chamber) were separated from responder macrophages (bottom chamber) by Transwell membranes (Fig. 7C, wtAd5 [Transwell−] versus wtAd5 [Transwell+]). Unfixed CPE corpses dying an apoptotic death from infection with H5dl337 either failed to repress (IL-1β) or further enhanced (IL-6, G-CSF, and MCP-1) viral particle stimulation of macrophage cytokine/chemokine production. These data extend the implications of the data on the effects of virally infected cell corpses on macrophage NF-κB activation responses to similar and consistent effects of infected corpses on virus-induced macrophage production of proinflammatory mediators. Cells undergoing apoptotic death from infection with E1B 19K-negative H5dl337 not only failed to repress these virus-induced, NF-κB-dependent, proinflammatory responses but frequently increased the responses through a contact-dependent, macrophage activation mechanism. In contrast, cells dying from wt Ad5 infection, in which apoptosis was repressed by E1B 19K expression, were highly effective in repressing viral stimulation of these macrophage inflammatory responses, in a manner that is identical to that of immunorepressive cells undergoing classical apoptosis in response to chemical stimuli (e.g., Fig. 2, A versus Stim).
DISCUSSION
Observations from previous studies indicated that the host inflammatory response to dying cells is determined by the qualitative nature of cell death (25). These observations have indicated that if cells die by apoptosis, the cell corpses are cleared by phagocytic cells without eliciting an inflammatory response; furthermore, the apoptotic corpses actively repress inflammation (1, 3–6, 26). Conversely, it has been proposed that nonapoptotic cell death generates corpses that fail to repress inflammation and can even be proinflammatory. These differences in the inflammatory responses to apoptotic and nonapoptotic cell death have been studied using a variety of cell death stimuli but have not previously included studies of cells dying as a result of viral infection. The objective of this study was to determine whether the cell death phenotypes induced by adenoviral infections would predict the effects of dying cells on macrophage-mediated inflammatory responses and whether the response patterns would be similar to those observed with noninfectious cell death stimuli.
The results showed that the effects of adenoviral infection were discordant with the conventional model of apoptosis-mediated immunorepression. Apoptotic corpses infected with a mutant adenovirus lacking expression of the antiapoptotic viral protein E1B 19K were not immunorepressive and for some macrophage mediators were proinflammatory. In contrast, nonapoptotic corpses infected with E1B 19K-competent, wild-type adenovirus were highly immunorepressive. Adenoviral infection has long been known to induce nonapoptotic cell death, as a result of the Bcl-2-like function of Ad E1B 19K protein (10, 15). The data presented here show that this Ad-induced, E1B 19K-related nonapoptotic cell death causes an effect that mimics “classical apoptosis” by causing the same level of immunorepressive effect as that observed with cells dying from chemically induced apoptosis or apoptosis caused by influenza infection. We term this adenoviral activity “apoptotic mimicry.” This is the first report of direct, contact-dependent repression of a macrophage inflammatory response by virus-infected cell corpses that is independent of any macrophage-repressive effect of the infecting virus itself.
The general applicability of the results of these studies is suggested by the observations that Ad infection of three different types of human cells exhibited the same Ad-specific immunorepressive activities and that both mouse and human responder cells exhibited the same patterns of immunorepressive effect of Ad-infected cell corpses. Thus, Ad infection of two different human epithelial cell lines (A549 and HeLa) and nonimmortalized, diploid human fibroblasts (MRC-5) generated cell corpses with the same E1B 19K-induced immunorepressive activities (Fig. 2 to 7). The observation that Ad infection conveyed the same E1B 19K-specific immunomodulatory trait to nonimmortal human MRC-5 fibroblasts as that observed with the two virally infected human tumor cell lines, A549 and HeLa, indicated that the E1B 19K-related infected cell immunorepression trait was independent of tumor-related phenotypes. The diversity of the responder cells used in these assays supported the conclusion that the immunomodulatory effects of Ad-infected cell corpses may also be generally applicable to multiple responder cell types. Ad-CPE corpses repressed stimulus-induced, NF-κB-dependent transcription in both mouse macrophage (RAW 264.7) and human epithelial (293) responder cells (Fig. 3 to 7) and repressed stimulus-induced cytokine and chemokine responses in mouse macrophage cells (Fig. 2, 4, and 7). Studies are in progress to determine whether these same patterns of response are observed during interactions between Ad-infected human cells and primary human lymphoid responder cells, as a further step toward testing the in vivo translational relevance of these data.
The data presented here provide a list of basic observations regarding possible mechanism(s) through which E1B 19K expression enables nonapoptotic, Ad-infected cells to mimic the immunorepressive activity of classical apoptotic cells. This database of observations can be used for hypothesis building for studies to define the precise mechanism(s) of this E1B 19K-related immunomodulatory activity. This E1B 19K effect is not explained by an adventitious effect on macrophages of Ad particles contaminating or produced by infected cell corpses. Those viral particles stimulate responder macrophages, rather than suppressing their NF-κB-dependent activation (Fig. 7). This viral stimulation is probably mediated through interactions between viral structural proteins and RGD-responsive integrin receptors and coxsackievirus-adenovirus receptors (CAR) on macrophages (27–32). The E1B 19K-dependent macrophage-repressive activity of Ad-infected CPE cells is not explained by extracellular soluble mediators (e.g., cytokines) produced by virally infected cell corpses, since Transwell membrane separation of macrophages from the corpses eliminates immunorepressive activity and should not block passage of such cell-free mediators (Fig. 7B and C). The Transwell studies show that the Ad-infected CPE corpses must be in direct contact with the responder macrophages to repress their activation and that there is a dose-response relationship between the number of virally infected CPE corpses in contact with the macrophages and repression of NF-κB-dependent macrophage activation (Fig. 7B and C). Ratios of Ad CPE corpses to responder macrophages as low as 3:1 resulted in a significant repression of stimulus-induced NF-κB-dependent macrophage activation responses, and a further increase in the ratios of Ad CPE corpses to macrophages completely shut off Ad particle-induced NF-κB-dependent activation in responder cells (Fig. 7B). Testing of different types of infected cells and responder cells from different species revealed that the immunomodulatory cell membrane trait (or putative ligands) of Ad-infected cells and the putative receptors of responder cells are neither species specific nor genetically restricted. The observation that virally infected human cells repressed stimulus-induced activation of mouse macrophages excludes the possibility that the E1B 19K effect could be mediated by cellular interactions requiring identity between major histocompatibility complex (MHC) antigens on dying cells and macrophage receptors. The results indicate that the immunorepressive effect of E1B 19K expressing cells dying from Ad infection is not limited to a single responder cell pathway. For example, the same repressive effect was observed for stimulus-induced responder cell signaling through Toll-like receptor 4 (TLR4; LPS stimulation of macrophages), protein kinase C (PMA stimulation of 293 epithelial cells), and CAR/integrin (Ad particle stimulation of macrophages) activation pathways. This lack of responder cell pathway selectivity of repression by Ad-infected cells is probably explained by the centrality of NF-κB activation to the responder cell cytokine and chemokine responses measured and the Ad-infected cell-induced repression of NF-κB activation in all responder cell types tested. The data further suggest that the immunorepressive effect of Ad CPE corpses blocks viral induction of interferon-stimulated genes in responder cells. Ad particles are known to stimulate interferon-responsive genes in infected cells, and MCP-1 is a chemokine that is induced through cellular pathways triggered by type I and type II interferons (33, 34). The results of the studies represented in Fig. 7 showed that Ad5 viral stimulation of high-level macrophage expression of MCP-1 was blocked down to control levels by contact with CPE corpses dying from wild-type Ad5 infection (Fig. 7C, MCP-1, Ad5, −/+ Transwell insertion). In contrast, CPE corpses dying from infection with E1B 19K-negative H5dl337 had no repressive effect on virally induced MCP-1 expression (Fig. 7C, MCP-1, H5dl337, −/+ Transwell insertion). Finally, any molecular or cellular pathway mechanisms that are identified to explain the immunomodulatory effect of Ad-infected cells must account for the observation that this E1B 19K activity can be replaced in cis by Bcl-2 (Fig. 5 and 6). One hypothesis suggested by this observation is that E1B 19K expression might preserve pathways or cell surface molecules that are induced by viral infection and create the immunorepressive signal to responder cells but that, in the absence of E1B 19K, would be eliminated (or destroyed) by the cellular apoptotic environment. Collectively, these observations provide a set of operational rules against which candidate E1B 19K-related immunorepressive mechanisms can be tested.
The same requirement for cell-cell contact observed in our studies has been reported for immunorepression of macrophages by classical apoptotic cellular corpses created by noninfectious stimuli (4, 21). This suggests the possibilities that Ad infection and E1B 19K expression induce CPE cell surface expression of immunorepressive molecules or ligands that are also expressed on classical apoptotic cells or that there are different types of immunorepressive molecules expressed on Ad-infected cell surfaces compared with the surfaces of classical apoptotic cells, both of which have a similar negative effect on responder cell activation.
Ad-induced cell death was described initially as being either necrotic or necrotic-like but clearly not apoptotic, because of the Bcl-2-like antiapoptotic effect of Ad E1B 19K protein (8, 22). Recent studies indicate that cell death induced by wild-type Ad infection results from autophagy (35–38) and, furthermore, that Ad-induced autophagic cell death is caused, at least in part, by an inducing interaction of the Ad E1B 19K protein with the autophagy pathway member Beclin 1 (39). Coupled with the data in this report, these observations suggest the possibility that the more recently recognized, nonapoptotic cell death response, autophagy, may be the cellular trait through which Ad E1B 19K conveys macrophage-immunorepressive activity to dying cells.
Our data indicate that the Ad-infected CPE corpse signals that repress proinflammatory macrophage responses will depend on an E1B 19K activity that induces cells to express an “off” signal to virally stimulated macrophages in a way that mimics “physiological” apoptosis and that increases as the numbers of virally infected, dying cells increase in the microenvironment of the local, innate immune response to viral infection. We propose the following model as to how apoptotic mimicry induced by Ad infection regulates the local host inflammatory response to Ad infection (Fig. 8). Early during infection, there are large numbers of Ad virions produced in the local environment. The Ad virions interact with local macrophages and turn on proinflammatory responses through interaction with αVβ3,5 integrins and CARs on the surfaces of the macrophages. As infection progresses and more virally infected host cells are killed, Ad CPE corpses accumulate in the local environment and begin to repress the proinflammatory viral responses of tissue macrophages through E1B 19K-dependent apoptotic mimicry. Based upon this model, we speculate that this E1B 19K-dependent effect on Ad-infected cells could alter the immunological outcome of both natural and therapeutic Ad infections.
FIG 8.

Model of apoptotic mimicry as a regulator of the local inflammatory response during viral infection. Early in infection, Ad virions increase local inflammatory responses through their direct interactions with αVβ3 integrins and CARs on tissue macrophages. Later during infection, as virally infected, dying host cells accumulate, the virally infected CPE corpses repress the local Ad virion-induced, macrophage-mediated proinflammatory responses through E1B 19K-dependent apoptotic mimicry. Adenovirus vector image from Dvorak, Integration and Application Network, University of Maryland Center for Environmental Science (http://ian.umces.edu/imagelibrary/displayimage-4798.html).
It is interesting to consider how the antiapoptotic effect of E1B 19K and the immunomodulatory effect of cells dying from infection with E1B 19K-competent adenovirus might have evolved to affect viral replication, persistence, and transmission and how the collaborating effects of these two E1B 19K activities might influence the therapeutic outcomes of infections with conditionally replicating adenoviral vectors. Mammalian cells infected with adenovirus detach from substrates, exhibit a rounded morphology, and form grape-like clusters that typify the cytopathic effect of this infection, but these cells survive for several days after infection (40). This prolongation of Ad-infected cell survival is a trait mediated by E1B 19K expression (16). If E1B 19K is deleted, the viral DNA and the host cellular DNA are extensively degraded, and viral production is reduced 10-fold (16). It is therefore possible that E1B 19K expression was selected through evolution to prolong infected cell survival, increase the time for viral replication, protect viral and host cell DNA, and ultimately increase the yield of viral infection. Local, innate immune inflammatory responses at sites of initial adenoviral infection are another factor that could reduce the establishment of adenoviral infection, thereby providing another blockade to the success of infection, viral persistence, and infection transmission. It is therefore theoretically possible that evolutionary selection for E1B 19K expression could have involved the advantage of viral evasion and delay of the local, cytokine-mediated amplification of the host immune response until infection had proceeded beyond the level of immune-mediated elimination.
Both sides of this theoretical, evolutionary collaboration between E1B 19K-induced antiapoptotic and immunomodulatory effects might also affect the in vivo outcome of therapeutic infections with conditionally replicating adenovirus vectors. For example, E1B 19K expression of such vectors might prolong and protect cargo gene expression in infected cells and might cause those cells to modulate the local inflammatory response at sites of vector infection. Conversely, if the goal of adenoviral vector infection is targeted host cell destruction (such as in the case of vector-targeted tumor therapy), there could be an advantage to deletion of E1B 19K, which could result in increased infected-cell apoptosis and increased host innate immune-mediated inflammatory responses in the environment of vector-infected cells. These possibilities regarding the E1B 19K-related activities of Ad-infected cells, at the interface with the host innate immune response, might also have implications for the pathogenesis of emerging adenoviral variants. For example, emerging, highly pathogenic Ad serotypes could induce increased host inflammatory responses, caused by viruses that have altered E1B 19K function (41, 42). A better understanding of the mechanisms of E1B 19K-induced apoptotic mimicry might provide more clarity to such studies of Ad-related immunopathogenesis and therapeutic Ad vector strategies.
ACKNOWLEDGMENTS
This work was supported by Veterans Administration Career Development Award-2 to J.R.R., The James and Marion Grant Fellowship in Immunology and Infectious Diseases to J.R.R. and J.L.C., a laboratory development fund from Loyola University to J.L.C., and an NIH grant AG029633 to D.S.U.
We thank Keith Peden for the MRC5-E1B 19K cells, Govindaswamy Chinnadurai for the Ad-Bcl-2 virus, Thomas Shenk for the H5dl viruses, and Zhiping Ye for the H1N1 influenza virus strain.
Footnotes
Published ahead of print 18 December 2013
REFERENCES
- 1.Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. 1997. Immunosuppressive effects of apoptotic cells. Nature 390:350–351. 10.1038/37022 [DOI] [PubMed] [Google Scholar]
- 2.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. 1998. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Invest. 101:890–898. 10.1172/JCI1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patel VA, Longacre-Antoni A, Cvetanovic M, Lee DJ, Feng L, Fan H, Rauch J, Ucker DS, Levine JS. 2007. The affirmative response of the innate immune system to apoptotic cells. Autoimmunity 40:274–280. 10.1080/08916930701357463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cvetanovic M, Mitchell JE, Patel V, Avner BS, Su Y, van der Saag PT, Witte PL, Fiore S, Levine JS, Ucker DS. 2006. Specific recognition of apoptotic cells reveals a ubiquitous and unconventional innate immunity. J. Biol. Chem. 281:20055–20067. 10.1074/jbc.M603920200 [DOI] [PubMed] [Google Scholar]
- 5.Hanayama R, Tanaka M, Miyasaka K, Aozasa K, Koike M, Uchiyama Y, Nagata S. 2004. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science 304:1147–1150. 10.1126/science.1094359 [DOI] [PubMed] [Google Scholar]
- 6.Cook HT, Botto M. 2006. Mechanisms of disease: the complement system and the pathogenesis of systemic lupus erythematosus. Nat. Clin. Pract. Rheumatol. 2:330–337. 10.1038/ncprheum0191 [DOI] [PubMed] [Google Scholar]
- 7.Subramanian T, Tarodi B, Chinnadurai G. 1995. p53-independent apoptotic and necrotic cell deaths induced by adenovirus infection: suppression by E1B 19K and Bcl-2 proteins. Cell Growth Differ. 6:131–137 [PubMed] [Google Scholar]
- 8.Subramanian T, Tarodi B, Chinnadurai G. 1995. Functional similarity between adenovirus E1B 19-kDa protein and proteins encoded by Bcl-2 proto-oncogene and Epstein-Barr virus BHRF1 gene. Curr. Top. Microbiol. Immunol. 199(Part 1):153–161 [DOI] [PubMed] [Google Scholar]
- 9.White E, Grodzicker T, Stillman BW. 1984. Mutations in the gene encoding the adenovirus early region 1B 19,000-molecular-weight tumor antigen cause the degradation of chromosomal DNA. J. Virol. 52:410–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiou SK, Tseng CC, Rao L, White E. 1994. Functional complementation of the adenovirus E1B 19-kilodalton protein with Bcl-2 in the inhibition of apoptosis in infected cells. J. Virol. 68:6553–6566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cook JL, May DL, Lewis AM, Jr, Walker TA. 1987. Adenovirus E1A gene induction of susceptibility to lysis by natural killer cells and activated macrophages in infected rodent cells. J. Virol. 61:3510–3520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wright KE, Wilson GA, Novosad D, Dimock C, Tan D, Weber JM. 1995. Typing and subtyping of influenza viruses in clinical samples by PCR. J. Clin. Microbiol. 33:1180–1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cvetanovic M, Ucker DS. 2004. Innate immune discrimination of apoptotic cells: repression of proinflammatory macrophage transcription is coupled directly to specific recognition. J. Immunol. 172:880–889 http://www.jimmunol.org/content/172/2/880 [DOI] [PubMed] [Google Scholar]
- 14.Radke JR, Siddiqui ZK, Miura TA, Routes JM, Cook JL. 2008. E1A oncogene enhancement of caspase-2-mediated mitochondrial injury sensitizes cells to macrophage nitric oxide-induced apoptosis. J. Immunol. 180:8272–8279 http://www.jimmunol.org/content/180/12/8272 [DOI] [PubMed] [Google Scholar]
- 15.Chiou SK, White E. 1998. Inhibition of ICE-like proteases inhibits apoptosis and increases virus production during adenovirus infection. Virology 244:108–118. 10.1006/viro.1998.9077 [DOI] [PubMed] [Google Scholar]
- 16.Pilder S, Logan J, Shenk T. 1984. Deletion of the gene encoding the adenovirus 5 early region 1b 21,000-molecular-weight polypeptide leads to degradation of viral and host cell DNA. J. Virol. 52:664–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pilder S, Moore M, Logan J, Shenk T. 1986. The adenovirus E1B-55K transforming polypeptide modulates transport or cytoplasmic stabilization of viral and host cell mRNAs. Mol. Cell. Biol. 6:470–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Couldwell WT, Hinton DR, He S, Chen TC, Sebat I, Weiss MH, Law RE. 1994. Protein kinase C inhibitors induce apoptosis in human malignant glioma cell lines. FEBS Lett. 345:43–46. 10.1016/0014-5793(94)00415-3 [DOI] [PubMed] [Google Scholar]
- 19.Yue TL, Wang C, Romanic AM, Kikly K, Keller P, DeWolf WE, Jr, Hart TK, Thomas HC, Storer B, Gu JL, Wang X, Feuerstein GZ. 1998. Staurosporine-induced apoptosis in cardiomyocytes: a potential role of caspase-3. J. Mol. Cell. Cardiol. 30:495–507. 10.1006/jmcc.1997.0614 [DOI] [PubMed] [Google Scholar]
- 20.Takizawa T, Matsukawa S, Higuchi Y, Nakamura S, Nakanishi Y, Fukuda R. 1993. Induction of programmed cell death (apoptosis) by influenza virus infection in tissue culture cells. J. Gen. Virol. 74(Part 11):2347–2355. 10.1099/0022-1317-74-11-2347 [DOI] [PubMed] [Google Scholar]
- 21.Cocco RE, Ucker DS. 2001. Distinct modes of macrophage recognition for apoptotic and necrotic cells are not specified exclusively by phosphatidylserine exposure. Mol. Biol. Cell 12:919–930. 10.1091/mbc.12.4.919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Subramanian T, Boyd JM, Chinnadurai G. 1995. Functional substitution identifies a cell survival promoting domain common to adenovirus E1B 19 kDa and Bcl-2 proteins. Oncogene 11:2403–2409 [PubMed] [Google Scholar]
- 23.Moro MR, Bonville CA, Suryadevara M, Cummings E, Faddoul D, Kobayaa H, Branigan PJ, Domachowske JB. 2009. Clinical features, adenovirus types, and local production of inflammatory mediators in adenovirus infections. Pediatr. Infect. Dis. J. 28:376–380. 10.1097/INF.0b013e31819075a6 [DOI] [PubMed] [Google Scholar]
- 24.Nemerow GR. 2009. A new link between virus cell entry and inflammation: adenovirus interaction with integrins induces specific proinflammatory responses. Mol. Ther. 17:1490–1491. 10.1038/mt.2009.177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Birge RB, Ucker DS. 2008. Innate apoptotic immunity: the calming touch of death. Cell Death Differ. 15:1096–1102. 10.1038/cdd.2008.58 [DOI] [PubMed] [Google Scholar]
- 26.Fadok VA, Henson PM. 1998. Apoptosis: getting rid of the bodies. Curr. Biol. 8:R693–695. 10.1016/S0960-9822(98)70438-5 [DOI] [PubMed] [Google Scholar]
- 27.Liu Q, Muruve DA. 2003. Molecular basis of the inflammatory response to adenovirus vectors. Gene Ther. 10:935–940. 10.1038/sj.gt.3302036 [DOI] [PubMed] [Google Scholar]
- 28.Tamanini A, Nicolis E, Bonizzato A, Bezzerri V, Melotti P, Assael BM, Cabrini G. 2006. Interaction of adenovirus type 5 fiber with the coxsackievirus and adenovirus receptor activates inflammatory response in human respiratory cells. J. Virol. 80:11241–11254. 10.1128/JVI.00721-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muruve DA, Barnes MJ, Stillman IE, Libermann TA. 1999. Adenoviral gene therapy leads to rapid induction of multiple chemokines and acute neutrophil-dependent hepatic injury in vivo. Hum. Gene Ther. 10:965–976. 10.1089/10430349950018364 [DOI] [PubMed] [Google Scholar]
- 30.Cartmell T, Southgate T, Rees GS, Castro MG, Lowenstein PR, Luheshi GN. 1999. Interleukin-1 mediates a rapid inflammatory response after injection of adenoviral vectors into the brain. J. Neurosci. 19:1517–1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Otake K, Ennist DL, Harrod K, Trapnell BC. 1998. Nonspecific inflammation inhibits adenovirus-mediated pulmonary gene transfer and expression independent of specific acquired immune responses. Hum. Gene Ther. 9:2207–2222. 10.1089/hum.1998.9.15-2207 [DOI] [PubMed] [Google Scholar]
- 32.Higginbotham JN, Seth P, Blaese RM, Ramsey WJ. 2002. The release of inflammatory cytokines from human peripheral blood mononuclear cells in vitro following exposure to adenovirus variants and capsid. Hum. Gene Ther. 13:129–141. 10.1089/10430340152712683 [DOI] [PubMed] [Google Scholar]
- 33.Reich N, Pine R, Levy D, Darnell JE., Jr 1988. Transcription of interferon-stimulated genes is induced by adenovirus particles but is suppressed by E1A gene products. J. Virol. 62:114–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rusinova I, Forster S, Yu S, Kannan A, Masse M, Cumming H, Chapman R, Hertzog PJ. 2013. Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res. 41:D1040–D1046. 10.1093/nar/gks1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ito H, Aoki H, Kuhnel F, Kondo Y, Kubicka S, Wirth T, Iwado E, Iwamaru A, Fujiwara K, Hess KR, Lang FF, Sawaya R, Kondo S. 2006. Autophagic cell death of malignant glioma cells induced by a conditionally replicating adenovirus. J. Natl. Cancer Inst. 98:625–636. 10.1093/jnci/djj161 [DOI] [PubMed] [Google Scholar]
- 36.Jiang H, White EJ, Rios-Vicil CI, Xu J, Gomez-Manzano C, Fueyo J. 2011. Human adenovirus type 5 induces cell lysis through autophagy and autophagy-triggered caspase activity. J. Virol. 85:4720–4729. 10.1128/JVI.02032-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baird SK, Aerts JL, Eddaoudi A, Lockley M, Lemoine NR, McNeish IA. 2008. Oncolytic adenoviral mutants induce a novel mode of programmed cell death in ovarian cancer. Oncogene 27:3081–3090. 10.1038/sj.onc.1210977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abou El Hassan MA, Mastenbroek DC, Gerritsen WR, Giaccone G, Kruyt FA. 2004. Overexpression of Bcl2 abrogates chemo- and radiotherapy-induced sensitisation of NCI-H460 non-small-cell lung cancer cells to adenovirus-mediated expression of full-length TRAIL. Br. J. Cancer 91:171–177. 10.1038/sj.bjc.6601910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Piya S, White EJ, Klein SR, Jiang H, McDonnell TJ, Gomez-Manzano C, Fueyo J. 2011. The E1B19K oncoprotein complexes with Beclin 1 to regulate autophagy in adenovirus-infected cells. PLoS One 6:e29467. 10.1371/journal.pone.0029467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tollefson AE, Kuppuswamy M, Shashkova EV, Doronin K, Wold WS. 2007. Preparation and titration of CsCl-banded adenovirus stocks. Methods Mol. Med. 130:223–235. 10.1385/1-59745-166-5:223 [DOI] [PubMed] [Google Scholar]
- 41.Tate JE, Bunning ML, Lott L, Lu X, Su J, Metzgar D, Brosch L, Panozzo CA, Marconi VC, Faix DJ, Prill M, Johnson B, Erdman DD, Fonseca V, Anderson LJ, Widdowson MA. 2009. Outbreak of severe respiratory disease associated with emergent human adenovirus serotype 14 at a US air force training facility in 2007. J. Infect. Dis. 199:1419–1426. 10.1086/598520 [DOI] [PubMed] [Google Scholar]
- 42.Metzgar D, Osuna M, Kajon AE, Hawksworth AW, Irvine M, Russell KL. 2007. Abrupt emergence of diverse species B adenoviruses at US military recruit training centers. J. Infect. Dis. 196:1465–1473. 10.1086/522970 [DOI] [PubMed] [Google Scholar]