

ABSTRACT
Hepatitis B virus (HBV) DNA replication occurs within the HBV icosahedral core particles. HBV core protein (HBc) contains an arginine-rich domain (ARD) at its carboxyl terminus. This ARD domain of HBc 149-183 is known to be important for viral replication but not known to have a structure. Recently, nucleocapsid proteins of several viruses have been shown to contain nucleic acid chaperone activity, which can facilitate structural rearrangement of viral genome. Major features of nucleic acid chaperones include highly basic amino acid residues and flexible protein structure. To test the nucleic acid chaperone hypothesis for HBc ARD, we first used the disassembled full-length HBc from Escherichia coli to analyze the nucleic acid annealing and strand displacement activities. To exclude the potential contamination of chaperones from E. coli, we designed synthetic HBc ARD peptides with different lengths and serine phosphorylations. We demonstrated that HBc ARD peptide can behave like a bona fide nucleic acid chaperone and that the chaperone activity depends on basic residues of the ARD domain. The loss of chaperone activity by arginine-to-alanine substitutions in the ARD can be rescued by restoring basic residues in the ARD. Furthermore, the chaperone activity is subject to regulation by phosphorylation and dephosphorylation at the HBc ARD. Interestingly, the HBc ARD can enhance in vitro cleavage activity of RNA substrate by a hammerhead ribozyme. We discuss here the potential significance of the HBc ARD chaperone activity in the context of viral DNA replication, in particular, at the steps of primer translocations and circularization of linear replicative intermediates.
IMPORTANCE Hepatitis B virus is a major human pathogen. At present, no effective treatment can completely eradicate the virus from patients with chronic hepatitis B. We report here a novel chaperone activity associated with the viral core protein. Our discovery could lead to a new drug design for more effective treatment against hepatitis B virus in the future.
INTRODUCTION
Hepatitis B virus (HBV) is a human pathogen that chronically infects about 350 million people worldwide. Chronic HBV carriers have an increased risk of developing cirrhosis and hepatocellular carcinoma (1–4). As an enveloped DNA virus, HBV reverse transcribes an encapsidated pregenomic RNA (pgRNA) to generate a double-strand DNA genome with a relaxed circular (RC) conformation (rcDNA).
The full-length HBV core protein (HBc) consists of 183 to 185 amino acid residues. It contains two distinct domains connected by a hinge region. The N terminus is an assembly domain of HBc 1-140, and the C terminus is the arginine-rich domain (ARD) of HBc 150-183 (5, 6) (see Fig. 2A). The ARD is dispensable for capsid assembly in Escherichia coli but is required for pgRNA packaging (5, 7, 8). The ARD can be phosphorylated predominantly at serines 155, 162, and 170. HBV RNA encapsidation, DNA synthesis, and virion secretion are known to be regulated by serine phosphorylation at the ARD domain (9–13).
FIG 2.
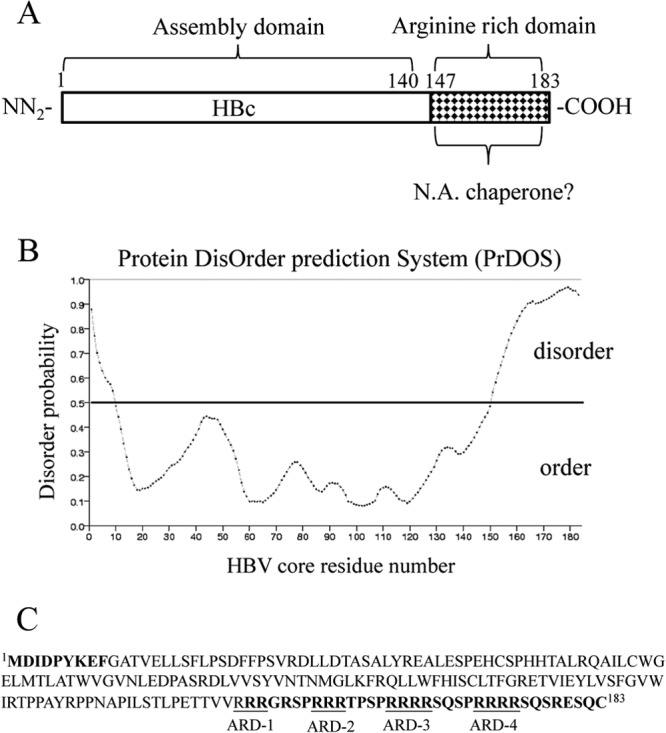
Prediction of disorder in HBV core protein (HBc). (A) Full-length HBc contains a capsid assembly domain and an arginine-rich domain (ARD). Here we asked whether the nucleic acid chaperone activity of HBc could reside in the ARD domain. (B) Disordered regions in HBV core protein (accession number J02203) were predicted using the Protein DisOrder prediction system (http://prdos.hgc.jp/cgi-bin/top.cgi). If the amino acid score is over 0.5, this amino acid is considered to reside in a disordered environment. If the amino acid score is less than 0.5, this amino acid is within an ordered region. (C) The disordered residues are displayed in boldface, and four arginine-rich subdomains (ARD I to IV) are included in this region.
Proteins with nucleic acid chaperone activity can either prevent RNAs from misfolding or help to resolve misfolded RNA without ATP (14–16). These chaperones generally bind RNA only weakly and with low specificity, indicating that the interaction between the RNA and nucleic acid chaperone is transient and predominantly electrostatic. The nucleic acid chaperone activity of proteins can be determined by strand annealing or strand displacement assays. A more complex assay is its ability to rescue the formation of misfolded complex structures of a catalytic RNA, such as the hammerhead ribozymes (HHRs) (17–19). Nucleic acid chaperone activity has been identified in nucleocapsid proteins of retroviruses, coronaviruses, hepatitis delta virus, hantavirus, and some Flaviviridae (20–24). Relative to the capsid proteins of many other viruses, HBc contains a very high helical content (25, 26). To date, it remains unclear whether such a highly structured HBc protein could still contain any nucleic acid chaperone activity.
In this study, we examined the possibility that HBc could display any nucleic acid chaperone activity. We demonstrated here that HBc can facilitate DNA annealing and unwinding, as well as hammerhead ribozyme cleavage in vitro. These chaperone activities can be regulated by phosphorylation and dephosphorylation of HBc ARD peptides. Loss of chaperone activity in vitro by mutations in the ARD is associated with loss of viral DNA replication in cell culture. Conversely, significant chaperone activity in vitro is associated with significant viral DNA replication in cell culture. We discuss the potential significance of HBc chaperone activity in viral DNA synthesis in hepatocytes, particularly at the steps of primer translocation and circularization from linear to relaxed circular forms. Our study provides the first experimental demonstration that the nucleic acid chaperone activity of a viral nucleocapsid protein could be regulated by serine phosphorylation and dephosphorylation.
MATERIALS AND METHODS
Plasmid DNAs.
DNA fragments Hammerhead F′ (5′-GCGATGACCTGATGAGGCCGAAAGGCCGAAACGTTCCC-3′) and Hammerhead R′ (5′-GGGAACGTTTCGGCCTTTCGGCCTCATCAGGTCATCGC-3′) were used to generate recombinant plasmid pT-HH in the T-A cloning site of a T&A cloning vector (RBC Bioscience). The plasmid pET15bs was described previously (24). HBc R-to-A mutants ARD-I (R151A and R152A, created with oligonucleotide 5′-CTACTGTTGTTAGAGCCGCCGGCAGGTCCCC-3′), HBc ARD-II (R157A and R158A, created with oligonucleotide 5′-GGCAGGTCCCCTGCCGCCAGAACTCCCTCGCCTCGC-3′), ARD-III (R165A and R166A, created with oligonucleotide 5′-CTCCCTCGCCTCGCGCCGCCAGGTCTCAATCGCCGCG-3′), and ARD-IV (R173A and R174A, created with oligonucleotide 5′-GGTCTCAATCGCCGCGT GCCGCCAGATCTCAATCTCGGGAATCTC-3′) were generated with a site-directed mutagenesis kit (Stratagene, USA) using wild-type (WT) HBV replicon pCH-9/3091 as a template (27). Underlined sequences represent mutation sites. HBc R-to-K substitution mutants ARD-III (R165K and R166K, created with oligonucleotide 5′-CTCCCTCGCCTCGC AAGAAGAGGTCTCAATCGCCG-3′) and ARD-IV (R173K and R174K, created with oligonucleotide 5′-GGTCTCAATCGCCGCGTAAGAAGAGATCTCAATCTC GGGAATC-3′) were also generated with the site-directed mutagenesis kit (Stratagene, USA). All mutants were confirmed by DNA sequencing. For HBV DNA replication analysis, the WT or HBc mutants were cotransfected with HBV tandem dimer 1903, which is a core-deficient construct, as described previously (28).
Peptides.
HBc peptides were purchased from Yao-Hang Co., Taiwan. These peptides were characterized by high-performance liquid chromatography (HPLC) and mass spectrometry (MS). Crude peptides were dissolved in a stock buffer (20 mM Tris-HCl [pH 7.5], 500 mM NaCl, 1 mM EDTA, and 20% glycerol) and stored at −80°C.
Expression and purification of HBV capsids in E. coli.
Expression, purification, and characterization of the E. coli-produced HBV capsids have been described previously (5, 29).
Transfection, core particle isolation, and core-associated HBV DNA extraction.
Wild-type and mutant cores in pCH-9/3091 plasmids were cotransfected with plasmid 1903 (28) into Huh7 cells using transfection reagent PolyJet (SignaGen Laboratories) according to the manufacturer's protocol. Intracellular core particles and core-associated HBV DNAs were purified as previously described.
Antibodies.
The antibodies used in this study included rabbit polyclonal anti-HBc antibodies (Dako) and mouse monoclonal anti-alpha-tubulin antibody (ICON-GeneTex, Taiwan). The anti-HBc antibody can recognize both full-length HBc 183 and HBc capsid. Secondary antibodies for Western blotting included polyclonal goat anti-mouse (Santa Cruz) and goat anti-rabbit–horseradish peroxidase (HRP) (Santa Cruz).
TAR DNA.
The following 56-mer oligonucleotides used for DNA annealing and duplex conversion are derived from the HIV-1 TAR sequences (MAL strain) in both sense and antisense orientations: Tar(+) (sense), 5′-GGTCTCTCTTGTTAGACCAGGTCGAGCCCGGGAGCTCTCTGGCTAGCAAGGAACCC-3′; Tar(−) (antisense), 5′-GGGTTCCTTGCTAGCCAGAGAGCTCCCGGGCTCGACCTGGTCTAACAAGAGAGACC-3′; and Tar(−)m5 (with 5 mismatch mutations), 5′-GGGTTCCTTGCTAGCCAGAGAGCTCCCGGGCTCGACCTGGTCTAACTTGTGTGTCC-3′. Tar(+) was 32P labeled with [32P]ATP using T4 polynucleotide kinase (New England BioLabs), and labeled Tar(+) was purified by 10% PAGE with 7 M urea in TBE buffer (90 mM Tris-HCl, 90 mM boric acid, and 1 mM EDTA). 32P-Tar(+) was recovered, ethanol precipitated, and dissolved in TE buffer (10 mM Tris-HCl [pH 8.0] and 1 mM EDTA) before use. The concentrations of labeled Tar(+) were calculated from its specific radioactivity.
In vitro RNA synthesis.
Hammerhead ribozyme (HHR) and 15bs RNAs were synthesized by using T7 RNA polymerase (Promega) with linearized plasmid DNAs as templates. Plasmids pT-HH and pET22b-15bs were digested with BamHI and XbaI, respectively. These RNAs were labeled with [α-32P]UTP in an in vitro transcription reaction and purified from 10% polyacrylamide–7 M urea gels. The concentration of labeled RNA was calculated from its specific radioactivity.
DNA annealing and strand displacement.
A 1 nM concentration of 32P-labeled Tar(+) and equal amounts of unlabeled Tar(−) were incubated with HBc peptides in 10 μl reaction buffer (40 mM Tris-HCl [pH 7.5], 50 mM NaCl, 0.01 mM EDTA, and 2% glycerol). Annealing reactions were performed at 37°C for 5 min, followed by treatment with 0.2 mg/ml proteinase K at 37°C for 30 min. A positive control for the annealing reaction mixture was incubated at 62°C for 30 min. The reactions were stopped with 3 μl of stop solution (50 mM EDTA [pH 8.0], 37% glycerol, 0.01% xylene cyanol, and 0.01% bromophenol blue). Duplex DNA and single-stranded oligonucleotide were resolved on 8% acrylamide gels in TBE buffer with 0.1% SDS at 4°C. Gels were dried and autoradiographed.
The preannealed DNA duplex was made by mixing 0.5 μM 32P-labeled Tar(+) with 0.5 μM complementary Tar(−)m5 in annealing buffer (40 mM Tris-HCl [pH 7.5] and 50 mM NaCl). The DNA mixture was heated at 90°C for 5 min and then allowed to cool slowly to room temperature (for at least 2 h) and stored at −20°C before use. The 32P-labeled preannealed DNA duplex was diluted in TE buffer and mixed with a competitor Tar(−) DNA in the presence or absence of HBc peptides in 10 μl reaction buffer at 37°C for 5 min. Electrophoresis of DNA samples and data analysis were performed as described for the annealing assay.
HHR cleavage.
Hammerhead ribozyme (HHR) and 15bs substrate RNAs were independently heated for 5 min at 90°C in a dry bath, and heated RNAs were slowly cooled to room temperature for at least 30 min. The two RNAs (2 nM HHR and 1 nM 15bs) were mixed and preincubated with HBc ARD peptides in a 10-μl reaction buffer at 37°C for 30 min. The cleavage reaction mixture, with reaction initiated by the addition of MgCl2 to a final concentration of 12 mM, was allowed to incubate at 37°C for 30 min, followed by treatment with 0.2 to 0.7 mg/ml proteinase K at 37°C for 30 min. RNA samples were then added to 10 μl formamide loading buffer (80% formamide, 10 mM EDTA [pH 8.0], 1 mg/ml xylene cyanol and 1 mg/ml bromophenol blue) before heating for 2 to 5 min. Heated RNA samples were chilled on ice and analyzed on a 12% denaturing polyacrylamide gel containing 7 M urea in TBE buffer. The radioactivity of each ribozyme cleavage product was visualized by autoradiogram, followed by quantification using a PhosphorImager (ImageQuant; Amersham Biosciences).
Dephosphorylation of HBc ARD peptides by lambda phosphatase treatment.
Wild-type and phosphorylated HBc ARD peptides (5 μM) were treated with 20 U lambda protein phosphatase (Millipore) in 50 μl phosphatase buffer (50 mM HEPES [pH 7.5], 0.1 mM EDTA, 2 mM MnCl2, and 5 mM dithiothreitol [DTT]) for 2 h at 37°C. Phosphatase-treated HBc peptides were serially diluted in dilution buffer (20 mM Tris-HCl [pH 7.5], 500 mM NaCl, 1 mM EDTA, and 20% glycerol) before assays for DNA annealing and strand displacement activities.
Micrococcal nuclease treatment of E. coli-derived HBV capsids.
Disassembly of HBV capsids was as described previously (29). In brief, purified HBV capsids in Tris-buffered saline (0.1 M NaCl, 2 mM KCl, 25 mM Tris, pH 7.4) were diluted 20-fold into distilled water before incubation overnight at 37°C with an excess amount of micrococcal nuclease S7 in the presence of 8 mM CaCl2 and 25 mM DTT. The effect of micrococcal nuclease S7 digestion was evaluated on 1% agarose gels sequentially stained with SYBR green II and SYPRO Ruby.
Nucleic acid chaperone activities of disassembled HBc 1-183 particles.
S7-induced capsid disassembly was terminated by adding EDTA to a final concentration of approximately 25 mM. After incubation at room temperature for 10 min, full-length HBc in disassembled capsid particles was analyzed by DNA annealing and strand displacement assays. An aliquot of 2 to 5 μl S7-induced and uninduced samples of HBV capsids were incubated with DNA annealing or strand displacement reaction buffer in 40 mM Tris-HCl (pH 7.4) and 50 mM NaCl. The reaction conditions, gel electrophoresis, and data analysis were as described above.
RESULTS
Full-length disassembled HBV core exhibits nucleic acid chaperone activity.
Escherichia coli-expressed HBc can spontaneously self-assemble into 28-nm capsid particles, and such capsid particles have been shown to package RNAs transcribed in E. coli (5, 7, 8, 30, 31). In a previous study (29), Newman et al. tested the “electrostatic interaction hypothesis” (called the “charge balance hypothesis” in reference 32) of HBV capsid stability and assembly and established an in vitro capsid disassembly/reassembly system using bacterially expressed HBV capsids. Taking advantage of this system, we disassembled HBc particles by removing the packaged RNA via micrococcal nuclease S7 treatment (Fig. 1A). The digestion of encapsidated RNAs was monitored by SYBR green II staining of encapsidated RNA on a native agarose gel (Fig. 1B, lanes 1 and 2). To monitor whether the capsids were indeed disassembled by the micrococcal nuclease treatment, the same gel was later restained with SYPRO Ruby for protein (Fig. 1B, lanes 3 and 4). As an internal control, HBc monomer from micrococcal nuclease-treated and untreated samples was visualized by denaturing SDS-PAGE and Western blotting using anti-HBc antibody (Fig. 1B, lower panel). Using this full-length HBc protein prepared from disassembled HBc capsid particles, we examined the nucleic acid chaperone activity of full-length HBc by performing both DNA annealing (Fig. 1C, D, and E) and strand exchange (displacement) (Fig. 1F and G) assays. As mentioned in the introduction, a nucleic acid chaperone could resolve misfolded structure and promote the formation of the most stable nucleic acid conformation. This process includes duplex dissociation and strand annealing, which could be analyzed by the strand annealing and exchange assays.
FIG 1.

Full-length HBc 1-183 of disassembled HBc particles can promote DNA annealing and strand displacement activities. (A) Schematic presentation of induction of capsid disassembly by treating HBc particles with nuclease S7 and low salt (29). According to the charge balance hypothesis (29, 32, 62), capsid instability and disassembly can be induced by the loss of encapsidated nucleic acid macromolecules upon nuclease S7 digestion. Capsid particle assembly and disassembly can be visualized in native agarose gel electrophoresis by SYPRO Ruby staining for protein or by SYBR green II staining for encapsidated RNA (29). Disassembled capsid protein monomers can be monitored in denaturing SDS-PAGE by Western blotting. (B) Encapsidated RNA of HBV capsids on a native agarose gel was stained with SYBR green II, and the same gel was later stained with SYPRO Ruby for HBc protein. No RNA or HBc protein signals were detected after treatment with micrococcal nuclease S7. Lower panel, full-length HBc protein monomer was detected by SDS-PAGE and Western blot analysis, indicating that HBc protein remains intact and full length after S7 treatment. (C) Cartoon illustration of the nucleic acid annealing assay. Radioactively labeled Tar(+) DNA and the complementary Tar(−) DNA oligonucleotides were incubated in the presence of HBc particles with or without capsid disassembly pretreatment. Efficient formation of the Tar(+)/Tar(−) duplex was detected only in the presence of disassembled particles. The oligonucleotides used in the experiment are indicated. (D) The kinetics of DNA annealing activity in the presence of assembled versus disassembled HBc particles were measured. A time course from 15 s to 5 min was conducted by using the Tar(−) and Tar(+) DNA annealing assay as detailed for panel C. (E) Graphic comparison of DNA annealing activity between assembled and disassembled HBc particles based on the data in panel D. The annealing activity (%) was calculated as the ratio of the banding intensities between duplex molecules and total intensities: [32P-labeled Tar(+)/Tar(−)]/{[32P-labeled Tar(+)/Tar(−)] + 32P-labeled Tar(+)}. (F) Schematic presentation of the DNA strand exchange assay. Radioactively labeled Tar(+) DNA was preannealed to the complementary Tar(−)m5 DNA, which contained a 5-nucleotide mismatch. Perfectly matched complementary Tar(−) DNA was added in the absence or presence of HBc ARD peptides. The oligonucleotides used in the experiment are indicated. (G) The kinetics of strand exchange (displacement) activity in the presence of assembled versus disassembled HBc particles were measured. A time course from 15 s to 5 min was conducted by using the Tar(−) and Tar(+) strand exchange assay as detailed for panel F.
As shown in a time course experiment (Fig. 1D), disassembled HBc particles (lanes 7 to 11) could facilitate duplex formation of Tar(+)/Tar(−) more efficiently than untreated assembled HBc particles (lanes 2 to 6) in a time-dependent manner (15 s to 5 min). Figure 1E shows the kinetic difference in DNA annealing activity between disassembled and assembled HBc particles (Fig. 1D) in a graphic format. For the strand exchange assay (Fig. 1F), an imperfect DNA duplex Tar(+)/Tar(−)m5 was formed by heating and annealing, followed by adding HBc particles (disassembled versus assembled) and another DNA oligonucleotide, Tar(−), which has the potential to form a perfect duplex with Tar(+). After incubation, HBc particles were removed by proteinase K digestion, and the ratio between the perfect and imperfect DNA duplexes was measured by polyacrylamide gel electrophoresis. Migrations of the two DNA duplexes and single-stranded Tar(+) were resolved and visualized. As shown in a time course experiment (Fig. 1G), disassembled particles (lanes 6 to 10) can kinetically more efficiently facilitate the strand exchange between perfectly matched Tar(−) and mismatched Tar(−)m5 than assembled HBc particles (lanes 1 to 5).
It cannot be rigorously excluded that the chaperone activity of disassembled HBc particles shown in Fig. 1 could originate from contamination of HBc-unrelated chaperone proteins from E. coli. Another issue is where on the full-length HBc 1-183 such a chaperone activity is located. We attempted to address these two issues by using synthetic HBc peptides which are thought to be free from any E. coli chaperone contamination.
One hallmark of nucleic acid chaperones is the lack of highly ordered structure (6, 25). To assess the intrinsic disorder in the core protein of the HBV ayw (J02203) strain, we used the Protein DisOrder prediction program, developed by Ishida and Kinoshita (33) (http://prdos.hgc.jp/cgi-bin/top.cgi) (Fig. 2B). The prediction identified two highly disordered regions at HBc 1-9 and HBc 151-183 (Fig. 2C). This prediction is consistent with the lack of structure at the C terminus of HBc in the context of an icosahedral particle as determined by cryo-electron microscopy (cryo-EM) (25, 34–37) and X-ray crystallography (38).
To assess the putative nucleic acid chaperone activity, we asked whether ARD peptide HBc 147-183 could enhance the annealing of two complementary Tar DNA oligonucleotides (Fig. 3A). Spontaneous annealing between the 56-mer Tar(+) and Tar(−) DNA oligonucleotides can occur at 37°C (39). Strand annealing can be facilitated at high temperatures or in the presence of a protein with nucleic acid chaperone activity (40). Tar(−) and 32P-labeled Tar(+) were incubated with increasing amounts of HBc 147-183 peptides at 37°C for 5 min (Fig. 3A, lanes 2 to 11). Formation of a Tar(+)/Tar(−) hybrid was analyzed by polyacrylamide gel electrophoresis after removing the peptides by protease K digestion. In the absence of ARD peptides, no significant annealing was detected at 37°C (Fig. 3A, lane 2). The annealing activity of ARD peptide HBc 147-183 was dose dependent within a narrow range of peptide concentration, and the annealing reaction appeared to be cooperative (Fig. 3A and B). In contrast, we detected no DNA annealing activity using peptide HBc 1-15 (data not shown).
FIG 3.
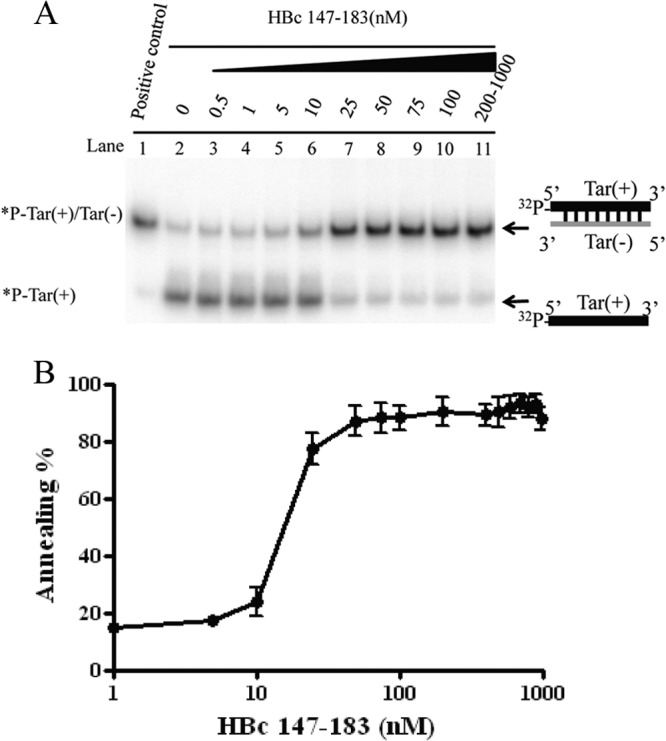
The arginine-rich domain (ARD) of HBc 147-183 exhibits DNA annealing activity. (A) DNA annealing activity is dependent on HBc ARD peptides in a dose-dependent manner. Annealing of Tar(+)/Tar(−) was promoted by HBc ARD peptides. 32P-labeled Tar(+) DNA (1 nM) was incubated with unlabeled Tar(−) DNA (1 nM) in the presence of increasing amounts of HBV core peptides HBc 147-183 (0.5 to 1000 nM) at 37°C for 5 min. (B) The annealing reaction is plotted against HBc ARD peptide concentrations based on the data in panel A. The reaction kinetics appeared to be cooperative after 10 nM before reaching a plateau. The annealing activity (percent) was calculated as the ratio of the banding intensities between duplex molecules and total intensities: [32P-labeled Tar(+)/Tar(−)]/{[32P-labeled Tar(+)/Tar(−)] + 32P-labeled Tar(+)}.
To map the minimal essential region of HBc 147-183 required for DNA annealing activity, we designed serially deleted ARD peptides (Fig. 4A). HBc peptides containing four ARD subdomains enhanced the strongest duplex formation at 50 nM (Fig. 4B, lanes 3 and 6), relative to HBc peptides containing only three ARD subdomains (Fig. 4B, lanes 5, 7, and 8). HBc peptides containing only two ARD subdomains at 50 nM exhibited only borderline annealing activity (Fig. 4B, lanes 4 and 9). However, at higher peptide concentrations, moderate levels of annealing activity can be visualized (Fig. 4B, lanes 13 and 14). HBc 1-15 peptide did not promote duplex formation at either low (50 nM) or high (123 nM) peptide concentrations (Fig. 4B, lane 12). As shown in Fig. 4C, we compared the annealing activities of peptides containing various lengths and arginine contents. Longer peptides containing more arginine residues displayed stronger activity than shorter peptides containing a reduced number of arginine residues. Although the effects of length and arginine content were not thoroughly dissociated in this study, we detected no apparent difference in annealing between HBc 147-183 (Fig. 4B, lane 3) and HBc 147-175 (Fig. 4B, lane 6).
FIG 4.

Mapping the minimal essential region of HBc ARD peptides required for DNA annealing activity. (A) Amino acid sequences of HBc ARD peptides used in the deletion mapping experiment. (B) Labeled Tar(+) and cold Tar(−) were incubated with HBc ARD peptides with different lengths and arginine contents. The dotted line between lanes 6 and 7 indicates that lanes 1 to 6 and lanes 7 to 15 are spliced from the same gel. (C) Dose-response curves for DNA annealing activities were compared by using various HBc ARD peptides with different lengths and arginine contents. The calculation of the efficiency of DNA annealing activity was as described for Fig. 1. As shown in panel A, peptides HBc 1-15, 147-159, 147-167, and 147-183 contain 0, 2, 3, and 4 ARD subdomains, respectively.
There are three major serine phosphorylation sites at positions HBc 155, 162, and 170. Phosphorylation and dephosphorylation at these residues are known to be important for HBV replication (9–13). We asked whether serine phosphorylation at these sites could affect the DNA annealing activity of HBc ARD peptides. We designed ARD peptides containing phosphorylation at these three major sites, individually or in combination. These peptides were characterized by Tricine SDS-PAGE (Fig. 5A and B) before testing the DNA annealing activity. When HBc 147-183 peptides were phosphorylated, the DNA annealing activity was significantly reduced. The higher the serine phosphorylation of HBc peptides, the lower the DNA annealing activity and duplex formation (Fig. 5C). This result was confirmed by treatment with lambda phosphatase to remove the phosphorylated residues at HBc 155, 162, and 170. As expected, the DNA annealing activity of dephosphorylated ARD peptides could now be detected (Fig. 5D). In summary, the strand annealing activity of HBc 147-183 can be regulated by phosphorylation and dephosphorylation.
FIG 5.

Phosphorylation and dephosphorylation of HBc ARD peptides can influence DNA annealing activity. (A) Amino acid sequences of different HBc ARD peptides phosphorylated at different positions. (B) Characterization of HBc ARD peptides by Tricine SDS-PAGE. Peptides were stained with Green Angel. (C) Labeled Tar(+) and cold Tar(−) were incubated with 25 and 50 nM phosphorylated HBc ARD peptides at 37°C for 5 min. (D) Rescue of the reduced DNA annealing activity of HBc ARD peptides by dephosphorylation treatment with lambda phosphatase.
The HBc 147-183 peptide has DNA strand exchange activity.
A strand exchange assay was performed as described in Fig. 1E. As shown in Fig. 6A, ARD peptide HBc 147-183 facilitated the exchange of the Tar(−)m5 strand in the mismatched duplex for the perfect matched strand Tar(−) in a dose-dependent manner. In the deletion mapping experiment, no significant strand exchange (displacement) activity was detected by peptides HBc 1-15, HBc 147-159, and HBc 162-175 at 50 nM (Fig. 6B, lanes 2, 4, and 9) or higher concentration (Fig. 6B, lane 12). In general, at least three ARD subdomains are required to effect the strand exchange. Peptides containing only two ARD subdomains (HBc 147-159 and HBc 162-175) can also facilitate strand exchange, albeit to a measurable degree only at very high concentrations. Although arginine content per se is very critical for the strand exchange (Fig. 6B) or DNA annealing activities (Fig. 4B), it cannot be excluded that the effect of the total length of HBc peptides could be important as well. Next, we evaluated the effect of serine phosphorylation of peptide HBc 147-183 on the strand exchange activity (Fig. 6C). Similar to the results from the DNA annealing assay (Fig. 5), serine phosphorylation of HBc 147-183 reduced the formation of a perfect duplex. Again, the higher the serine phosphorylation of peptide HBc 147-183, the lower the formation of a perfect duplex. This result was confirmed by the increased DNA strand exchange activity in the phosphatase treatment experiment (Fig. 6D, lane 5 versus lane 6 and lane 11 versus lane 12). Altogether, these results demonstrated that peptide HBc 147-183 exhibited both DNA annealing and strand exchange activities, and these nucleic acid chaperone activities of HBV core protein can be regulated by serine phosphorylation and dephosphorylation.
FIG 6.

Strand exchange (displacement) activities of HBc ARD peptides. (A) A DNA duplex of 32P-Tar(+)/Tar(−)m5 (1 nM) was incubated with an excessive amount of Tar(−) DNA (2 nM) in the absence or presence of HBc ARD peptides at various concentrations at 37°C for 5 min. (B) Deletion mapping of the minimal essential region of HBc peptides important for strand displacement activity. (C) Serine phosphorylation can attenuate the strand displacement activity of HBc ARD peptides. (D) Dephosphorylation of HBc ARD peptides by lambda phosphatase treatment can restore the chaperone activity of HBc ARD peptides in the strand displacement assay.
Enhancement of HHR cleavage by HBc 147-183.
It has been reported in the literature that some nucleic acid chaperone proteins can stimulate hammerhead ribozyme (HHR) cleavage of an RNA substrate (Fig. 7A) (24, 41, 42). Ribozyme cleavage is possible only when the ribozyme and its substrate can form a very specific and precise complex conformation through inter- and intramolecular base pairing and RNA folding. In the absence of nucleic acid chaperones, hammerhead ribozyme-mediated cleavage is relatively slow, because the biologically active conformation and base pairing between the ribozyme and its RNA substrate cannot be formed efficiently. To investigate whether HBc ARD contains any chaperone-like activity for ribozyme cleavage, we established an in vitro assay using a hammerhead ribozyme reported previously in the literature (41, 42). Specifically, radiolabeled HHR (82 nucleotides [nt]) and 15bs substrate (57 nt) were boiled independently and cooled down to room temperature slowly. The ribozyme and substrate were first incubated at 37°C for 10 min, followed by the addition of ARD peptide HBc 147-183, and allowed to incubate for 30 min before the addition of MgCl2. HBc peptides were removed by protease K, and RNA products were analyzed by PAGE with 7 M urea.
FIG 7.

Enhancement of hammerhead ribozyme (HHR) cleavage by HBc ARD peptides. (A) Schematic diagrams of HHR and substrate 15bs RNAs. Only parts of the sequences of HHR and substrate 15bs are shown, and the arrow represents the cleavage point. (B) HBc ARD peptides promote hammerhead ribozyme cleavage. HHR RNA ribozyme and 15bs substrate RNA were incubated with various concentrations of HBc ARD peptides and analyzed as described in Materials and Methods. (C) Serine phosphorylation of HBc ARD peptides at lower concentrations attenuate the hammerhead RNA ribozyme cleavage activity yet increase the ribozyme activity at higher concentrations (above 120 nM). The cleavage activity of hammerhead ribozyme was calculated as the banding intensities of 44-nt and 13-nt cleaved substrates divided by the total intensities of 57-nt uncleaved substrates plus 44-nt and 13-nt cleaved substrates. (D) Hammerhead ribozyme cleavage activity using various phosphorylated ARD HBc 147-183 peptides.
In the absence of HBc peptides, ribozyme cleavage of the substrate occurred slowly (Fig. 7B, lane 2). In contrast, peptide HBc 147-183 stimulated ribozyme-directed cleavage of the 15bs RNA substrate (Fig. 7B, compare lane 1 and lanes 5 to 12). However, ribozyme cleavage activity became compromised when the concentration of peptide HBc 147-183 was too high (Fig. 7B, lanes 11 and 12) (17, 43). We also examined the effect of phosphorylation of HBc ARD peptide on the hammerhead ribozyme cleavage activity. As shown in Fig. 7D, serine phosphorylation of HBc ARD peptide at 25 and 50 nM reduced the ribozyme cleavage activity. However, at very high concentrations of wild-type HBc ARD peptide, ribozyme activity was reduced (Fig. 7C). Presumably, when the chaperone protein was present at too high a concentration, RNA molecules of both ribozyme and substrates were nonspecifically occupied, resulting in distortion of inter- and intramolecular RNA secondary structures (17).
We have demonstrated so far that HBc ARD peptides containing clustering arginines with positive charges could behave like nucleic acid chaperones in DNA annealing, strand displacement, and ribozyme cleavage (Fig. 3 to 7). Serine phosphorylation of HBc ARD peptides could dampen these activities, probably due to its negative charges of the phosphate groups. To directly demonstrate that positive charges from the arginine residues of HBc ARD could be important for the nucleic acid chaperone activity, we designed different arginine-to-alanine (R-to-A) substitutions in HBc ARD peptides (Fig. 8A) and assayed the nucleic acid chaperone activities of the mutant HBc ARD peptides. These mutant HBc peptides were first characterized by Tricine SDS-PAGE (Fig. 8A). R-to-A mutant HBc peptides, particularly ARD peptide III-IV-AA, exhibited the most significantly reduced DNA annealing activity at 25, 50, or 100 nM (Fig. 8B, lanes 2 to 5, 8 to 11, and 14 to 17). Such a reduction in DNA annealing activity was not observed with arginine-to-lysine (R-to-K) substitutions in ARD peptide III-IV-KK (Fig. 8B, lanes 6, 12, and 18). Similar to the results in Fig. 8B, both DNA strand exchange and hammerhead ribozyme cleavage activities were dependent on the positively charged amino acids, arginine or lysine (Fig. 8C and D).
FIG 8.

Positive charge content of HBc ARD is important for nucleic acid chaperone activity. (A) Amino acid sequences of mutant HBc ARD peptides containing various arginine-to-alanine (R-to-A) or arginine-to-lysine (R-to-K) substitutions and characterization of HBc ARD peptides by Tricine SDS-PAGE. These peptides were stained with Green Angel. (B) The DNA annealing activity of HBc ARD peptides was significantly reduced by R-to-A substitutions (lanes 3 to 5 and 9 to 11) but not by R-to-K substitutions (lanes 6, 12, and 18). (C) The strand displacement activity of HBc ARD peptides was significantly reduced by R-to-A substitutions (lanes 4 to 6 and 9 to 11) but not by R-to-K substitutions (lanes 7, 12, and 17). (D) The enhancement of hammerhead ribozyme cleavage activity by HBc ARD peptides was significantly reduced by R-to-A substitutions, but not by R-to-K substitutions, at 25 and 50 nM.
Role of HBc nucleic acid chaperone activity in HBV replication.
So far, our assays for the nucleic acid chaperone activities of HBc ARD peptides were performed in an in vitro cell-free setting. We attempted to extend the study to a more in vivo experimental setting in culture. The HBV (ayw) tandem dimer plasmid 1903 was replication defective due to the ablated AUG initiation codon of HBc at nucleotide 1903 (28). In a complementation assay, we cotransfected this core-deficient plasmid 1903 into Huh7 cells with the core-positive replicon plasmid pCH-9/3091 containing wild-type ARD or different HBc ARD mutations (Fig. 9A) (see Materials and Methods). As shown in Fig. 9B, core-associated HBV DNA synthesis was drastically decreased in R-to-A substitution mutants HBc ARD II-AA, ARD III-AA, and ARD IV-AA as determined by Southern blotting. In contrast, viral DNA synthesis was most resistant to HBc ARD I-AA mutation (Fig. 9B, lane 2), and R-to-K substitution mutants of HBc ARD III and IV were not replication defective (Fig. 9C). This result is consistent with our earlier report that SVC173RR and SVC173KK can replicate almost equally well, while mutant SVC173GG is replication defective (32). Internal controls of HBV capsid particles (native agarose gel), HBc core monomer (SDS-PAGE), and tubulin were assayed by Western blotting using anti-HBc antibody (Fig. 9C, lower panels). Together, the results show that positive charge content in the HBc ARD domain is important for both HBV DNA replication in vivo (Fig. 9) and nucleic acid chaperone activity in vitro (Fig. 8).
FIG 9.

Positive charge content of HBc ARD is important for HBV replication in Southern blot analysis. (A) Complementation analysis of HBV DNA replication was conducted by cotransfection of HBV replicon plasmid pCH-9/3091 containing R-to-A or R-to-K substitutions at the HBc ARD and core-deficient HBV tandem dimer plasmid p1903, which can provide functional polymerase. The amino acid sequences of HBc ARD mutations are designed to contain various positive charge contents by R-to-A or R-to-K substitutions. I to IV indicate the four different ARD subdomains. (B) HBV DNA replication of ARD mutants ARD-I to -IV in Huh7 cells was analyzed by Southern blotting assay. The most severe defect in replication was observed in HBc ARD-IIIAA and ARD-IVAA. The lower panel shows the control of intracellular HBV capsids from transfected cell lysates separated on 1% agarose gel and visualized by Western blotting using anti-HBc antibody. (C) However, the severe replication defect in mutant ARD-III AA and mutant ARD-IV AA was not observed for R-to-K substitutions in ARD-III and ARD-IV. Similarly, mutant ARD-III+IV AA, missing four arginines, is replication defective, yet no severe replication defect was observed for R-to-K substitutions in both ARD-III and ARD-IV. The lower panels (HBV capsids, HBV core protein, and tubulin) show controls for sample loading. This result is consistent with our previous studies of R-to-K substitutions in HBc ARD (32).
DISCUSSION
In this study, we demonstrated the nucleic acid chaperone activity of wild-type ARD peptide HBc 147-183 and disassembled HBc particles using assays for DNA annealing, DNA strand exchange, and hammerhead ribozyme cleavage. The chaperone activity of HBc ARD peptides is dependent on the content of positively charged amino acids, independent of ATP (data not shown), and is strongly influenced by serine phosphorylation and dephosphorylation. Mutations causing the loss of in vitro chaperone activity also caused the loss of viral replication in culture. Similarly, mutations which can rescue the in vitro chaperone activity also rescued the viral replication in culture. It is tempting to speculate here that the chaperone activity could contribute to viral DNA replication in natural infection (Fig. 10A).
FIG 10.
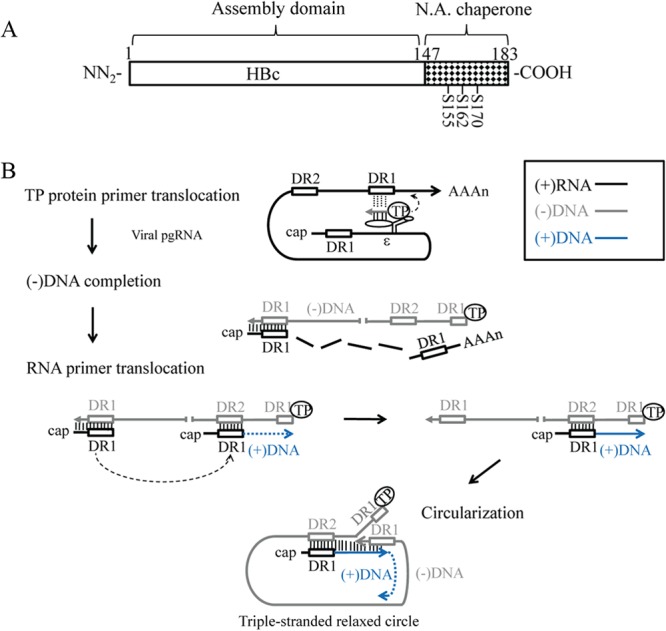
Nucleic acid chaperone hypothesis in HBV replication (A) Assignment of the nucleic acid (N.A.) chaperone activity to the ARD of HBc 147-183. Serines 155, 162, and 170 are subject to phosphorylation and dephosphorylation, which can modulate chaperone activity and viral replication. (B) A diagram of HBV DNA replication illustrates the specific steps where an HBc chaperone might play a role in nucleic acid structural rearrangement, such as annealing, unwinding, and strand displacement. Specifically, we hypothesize here that the HBc chaperone activity could facilitate TP protein primer translocation, which is important for HBV minus-strand DNA synthesis. In addition, it could facilitate RNA primer translocation, which is important for the initiation of plus-strand DNA synthesis. Finally, during the circularization of the linear minus-strand DNA, the 3′ DR1 could undergo a strand invasion and displacement of the 5′ DR1 sequences. Further details are as follows. The encapsidation signal ε is for TP protein binding. The two direct repeat sequences DR1 are located at the viral pgRNA 5′ and 3′-end regions, and the relative position of DR2 is also indicated. The viral pgRNA is associated with polymerase P protein and packaged into capsids during assembly. (i) For TP protein primer translocation, TP protein primes on the bulge of ε to initiate minus-strand synthesis. The nascent minus-strand DNA then translocates to the 3′ copy of DR1. (ii) The minus-strand DNA is elongated, and the pgRNA is degraded simultaneously by RNase H domain of polymerase. The 5′-end sequences of pgRNA, including the DR1, are somehow spared from RNase H degradation. (iii) For RNA primer translocation, this RNase H-spared 5′ pgRNA sequence could undergo a translocation from DR1 to DR2 and prime plus-strand DNA synthesis from DR2. (iv) For circularization, after the plus-strand DNA synthesis reaches the 5′ end of the minus-strand template, the 3′-end DR1 of the minus strand DNA could undergo a strand displacement of the 5′-end DR1 and form a relaxed-circle (RC) structure with a triple-strand region (53).
Implications in primer translocations and circularization.
The nucleocapsid protein (NCp7) of human immunodeficiency virus type 1 (HIV-1) was shown to contain nucleic acid chaperone activity and is likely to regulate RNA-RNA and RNA-DNA interactions and genomic RNA rearrangements in multiple steps of HIV replication (17, 43). In this study, on HBV capsid protein, it is natural to ask whether the in vitro nucleic acid chaperone activities also contribute to viral replication.
During HBV DNA replication (Fig. 10B), HBV pgRNA is packaged into capsid particles via an encapsidation signal, ε (44–46). After encapsidation, the bulge of ε on pgRNA is used as the template for reverse transcription (47, 48), and tyrosine 63 of the terminal protein (TP) domain of polymerase serves as the priming residue of minus-strand DNA synthesis (49, 50). The short, nascent minus-strand cDNA then translocates to a complementary DR1 sequence near the 3′ end of the pgRNA, which allows the minus-strand cDNA to continue its synthesis to completion (TP protein primer translocation) (47, 48, 51). During the minus-strand DNA synthesis, the majority of the pgRNA template is digested by the RNase H activity of the polymerase protein, except for the 5 to 17 nucleotides at the 5′ end of pgRNA (52). This undigested short RNA fragment could serve as an RNA primer for plus-strand DNA synthesis by undergoing a template switch from the 3′ end of the nascent minus-strand DNA to a cDNA sequence, termed DR2, near the 5′ end (RNA primer translocation) (52). When the elongating plus-strand DNA reaches the 5′ end of the minus-strand DNA template, another template translocation happens in which the 3′-end sequences of the minus-strand DNA could displace its own 5′-end sequences and establish base pairing with the complementary sequences of plus-strand DNA. As a consequence, a circular molecule containing a triple-strand discontinuity region can be generated, which then allows the elongating nascent plus-strand DNA to continue its DNA synthesis (circularization) (53, 54).
In summary, the in vivo replication defect of HBc mutant ARD-III+IV (Fig. 9) could in part result from the loss of the chaperone activity (Fig. 8). We hypothesize here that the chaperone activity of HBc ARD could contribute to the minus-strand DNA synthesis by affecting the DNA-RNA base pairing at the step of the TP-protein primer translocation to the 3′ DR1 on pgRNA (Fig. 10B). Indeed, it has been reported recently that a sequence element designated phi is complementary to the RNA packaging signal epsilon, and such complementarity is required for efficient minus-strand DNA synthesis, probably related to the TP-protein primer translocation from epsilon to DR1 (55). Similarly, the chaperone activity could contribute to the plus-strand DNA synthesis by affecting the DNA-RNA base pairing at the step of RNA primer translocation to DR2 (Fig. 10B), as well as DNA-DNA base pairing between cis-acting DNA sequences (h3E and hM) involved in template switching during relaxed-circle (RC) DNA synthesis (56). Furthermore, DNA-DNA base pairing and strand displacement at the step of the minus-strand DNA circularization are necessary for plus-strand DNA synthesis to continue and for formation of RC DNA (Fig. 10B). It is tempting to speculate whether the enhancement of ribozyme RNA cleavage activity by HBc ARD peptides (Fig. 7) could be related to the generation of the RNA primer for plus-strand DNA synthesis. The RNA primer can be spared from the progressive digestion of the RNA moiety of the pgRNA/(−)DNA hybrid by RNase H during reverse transcription.
Phosphorylation changes the nucleic acid chaperone activity of HBc.
It is known that phosphorylation and dephosphorylation of HBc in vivo play an important role in multiple steps in the HBV life cycle, including RNA encapsidation, capsid assembly, DNA replication, virion secretion, and nuclear import of HBc (9–13, 57–60). In the in vitro assays of DNA annealing, strand displacement, and hammerhead ribozyme cleavage, we observed that phosphorylation of HBc ARD peptides at either serine 155, 162, or 170, significantly attenuated nucleic acid chaperone activity. On the other hand, at higher HBc ARD peptide concentrations, phosphorylated HBc 147-183 peptides exhibited higher hammerhead cleavage activity (∼60%) than their unphosphorylated counterparts (∼40%) (Fig. 7E). By cryo-EM studies, phosphorylation of the HBc ARD domain appeared to affect the density and structural organization of encapsidated RNA (61). This phenomenon can be simply explained in the context of a previously proposed charge balance hypothesis (29, 32, 62). It would be interesting to demonstrate experimentally whether phosphorylation of HBc ARD could alter the RNA structure via a nucleic acid chaperone activity. In our current study, phosphorylation of HBc ARD peptides at either serine 155, 162, or 170 significantly attenuated DNA annealing and strand displacement activity in vitro (Fig. 5C and D and 6C and D), as well as ribozyme cleavage activity at lower peptide concentrations (Fig. 7C).
In theory, for each chaperone activity (annealing, strand displacement, or RNA cleavage), one can attempt to calculate the optimum stoichiometry between negative charges (from packaged nucleic acids and phosphorylated serine residues) and positive charges (from arginines at the HBc ARD) in the context of an icosahedral particle (T = 4). However, without specific information on the three-dimensional (3D) structure of the HBc ARD peptide and the detailed folding of the packaged pgRNA in the capsid interior, it remains uncertain whether such calculations could truly reflect the dynamic in vivo situation.
To our knowledge, this is the first direct experimental demonstration of nucleic acid chaperone activity associated with hepatitis B virus. Nucleic acid chaperone activity has been detected in the nucleocapsid proteins of several other viruses (20, 24, 27, 32, 55). These earlier studies with different viruses on the nucleic acid chaperone hypothesis in the literature can perhaps be extended to hepatitis B virus, as was discussed in the study of the arginine clusters of the carboxy-terminal domain of HBc (63). One major difficulty in testing this hypothesis in HBV by in vivo experiments is related to the multiple overlapping functions of the ARD domain in the life cycle of HBV, including capsid assembly/disassembly (29, 62), RNA encapsidation (10, 11, 30, 32), core protein trafficking (57, 58, 64–66), DNA synthesis (9–13, 32, 62, 63, 67), and antimicrobial activity (68). Given such an extreme degree of functional complexity associated with a very short HBc ARD domain (HBc 147-183), it is conceivable that attempts to dissociate nucleic acid chaperone activity of HBc ARD from its many other reported functions in vivo will be very challenging in the future. To the best of our knowledge, the current study provides the first experimental demonstration in vitro that the nucleic acid chaperone activity of viral nucleocapsids, such as in the case of HBV core protein, could be regulatable by serine phosphorylation and dephosphorylation.
ACKNOWLEDGMENTS
This work is supported by Academia Sinica, the National Health Research Institutes (NHRI), and the National Science Council, Taiwan.
Footnotes
Published ahead of print 18 December 2013
REFERENCES
- 1.Purcell RH. 1994. Hepatitis viruses: changing patterns of human disease. Proc. Natl. Acad. Sci. U. S. A. 91:2401–2406. 10.1073/pnas.91.7.2401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shih C. 2012. Chronic hepatitis B and C: basic science to clinical applications. World Scientific, Hackensack, NJ [Google Scholar]
- 3.Wieland SF, Chisari FV. 2005. Stealth and cunning: hepatitis B and hepatitis C viruses. J. Virol. 79:9369–9380. 10.1128/JVI.79.15.9369-9380.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seeger C, Mason WS. 2000. Hepatitis B virus biology. Microbiol. Mol. Biol. Rev. 64:51–68. 10.1128/MMBR.64.1.51-68.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Newman M, Suk FM, Cajimat M, Chua PK, Shih C. 2003. Stability and morphology comparisons of self-assembled virus-like particles from wild-type and mutant human hepatitis B virus capsid proteins. J. Virol. 77:12950–12960. 10.1128/JVI.77.24.12950-12960.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steven AC, Conway JF, Cheng N, Watts NR, Belnap DM, Harris A, Stahl SJ, Wingfield PT. 2005. Structure, assembly, and antigenicity of hepatitis B virus capsid proteins. Adv. Virus Res. 64:125–164. 10.1016/S0065-3527(05)64005-5 [DOI] [PubMed] [Google Scholar]
- 7.Birnbaum F, Nassal M. 1990. Hepatitis B virus nucleocapsid assembly: primary structure requirements in the core protein. J. Virol. 64:3319–3330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wingfield PT, Stahl SJ, Williams RW, Steven AC. 1995. Hepatitis core antigen produced in Escherichia coli: subunit composition, conformational analysis, and in vitro capsid assembly. Biochemistry 34:4919–4932. 10.1021/bi00015a003 [DOI] [PubMed] [Google Scholar]
- 9.Basagoudanavar SH, Perlman DH, Hu J. 2007. Regulation of hepadnavirus reverse transcription by dynamic nucleocapsid phosphorylation. J. Virol. 81:1641–1649. 10.1128/JVI.01671-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gazina EV, Fielding JE, Lin B, Anderson DA. 2000. Core protein phosphorylation modulates pregenomic RNA encapsidation to different extents in human and duck hepatitis B viruses. J. Virol. 74:4721–4728. 10.1128/JVI.74.10.4721-4728.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lan YT, Li J, Liao W, Ou J. 1999. Roles of the three major phosphorylation sites of hepatitis B virus core protein in viral replication. Virology 259:342–348. 10.1006/viro.1999.9798 [DOI] [PubMed] [Google Scholar]
- 12.Yu M, Summers J. 1994. Phosphorylation of the duck hepatitis B virus capsid protein associated with conformational changes in the C terminus. J. Virol. 68:2965–2969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu M, Summers J. 1994. Multiple functions of capsid protein phosphorylation in duck hepatitis B virus replication. J. Virol. 68:4341–4348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Semrad K. 2011. Proteins with RNA chaperone activity: a world of diverse proteins with a common task-impediment of RNA misfolding. Biochem. Res. Int. 2011:532908. 10.1155/2011/532908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rajkowitsch L, Chen D, Stampfl S, Semrad K, Waldsich C, Mayer O, Jantsch MF, Konrat R, Blasi U, Schroeder R. 2007. RNA chaperones, RNA annealers and RNA helicases. RNA Biol. 4:118–130. 10.4161/rna.4.3.5445 [DOI] [PubMed] [Google Scholar]
- 16.Doetsch M, Schroeder R, Furtig B. 2011. Transient RNA-protein interactions in RNA folding. FEBS J. 278:1634–1642. 10.1111/j.1742-4658.2011.08094.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cristofari G, Darlix JL. 2002. The ubiquitous nature of RNA chaperone proteins. Prog. Nucleic Acid Res. Mol. Biol. 72:223–268. 10.1016/S0079-6603(02)72071-0 [DOI] [PubMed] [Google Scholar]
- 18.Rajkowitsch L, Semrad K, Mayer O, Schroeder R. 2005. Assays for the RNA chaperone activity of proteins. Biochem. Soc. Trans. 33:450–456. 10.1042/BST0330450 [DOI] [PubMed] [Google Scholar]
- 19.Rajkowitsch L, Schroeder R. 2007. Dissecting RNA chaperone activity. RNA 13:2053–2060. 10.1261/rna.671807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zuniga S, Sola I, Cruz JL, Enjuanes L. 2009. Role of RNA chaperones in virus replication. Virus Res. 139:253–266. 10.1016/j.virusres.2008.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pong WL, Huang ZS, Teoh PG, Wang CC, Wu HN. 2011. RNA binding property and RNA chaperone activity of dengue virus core protein and other viral RNA-interacting proteins. FEBS Lett. 585:2575–2581. 10.1016/j.febslet.2011.06.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levin JG, Guo J, Rouzina I, Musier-Forsyth K. 2005. Nucleic acid chaperone activity of HIV-1 nucleocapsid protein: critical role in reverse transcription and molecular mechanism. Prog. Nucleic Acid Res. Mol. Biol. 80:217–286. 10.1016/S0079-6603(05)80006-6 [DOI] [PubMed] [Google Scholar]
- 23.Mir MA, Panganiban AT. 2006. Characterization of the RNA chaperone activity of hantavirus nucleocapsid protein. J. Virol. 80:6276–6285. 10.1128/JVI.00147-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang ZS, Wu HN. 1998. Identification and characterization of the RNA chaperone activity of hepatitis delta antigen peptides. J. Biol. Chem. 273:26455–26461. 10.1074/jbc.273.41.26455 [DOI] [PubMed] [Google Scholar]
- 25.Yu X, Jin L, Jih J, Shih C, Zhou ZH. 2013. 3.5A cryoEM structure of hepatitis B virus core assembled from full-length core protein. PLoS One 8:e69729. 10.1371/journal.pone.0069729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Conway JF, Watts NR, Belnap DM, Cheng N, Stahl SJ, Wingfield PT, Steven AC. 2003. Characterization of a conformational epitope on hepatitis B virus core antigen and quasiequivalent variations in antibody binding. J. Virol. 77:6466–6473. 10.1128/JVI.77.11.6466-6473.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Junker-Niepmann M, Bartenschlager R, Schaller H. 1990. A short cis-acting sequence is required for hepatitis B virus pregenome encapsidation and sufficient for packaging of foreign RNA. EMBO J. 9:3389–3396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yuan TT, Sahu GK, Whitehead WE, Greenberg R, Shih C. 1999. The mechanism of an immature secretion phenotype of a highly frequent naturally occurring missense mutation at codon 97 of human hepatitis B virus core antigen. J. Virol. 73:5731–5740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Newman M, Chua PK, Tang FM, Su PY, Shih C. 2009. Testing an electrostatic interaction hypothesis of hepatitis B virus capsid stability by using an in vitro capsid disassembly/reassembly system. J. Virol. 83:10616–10626. 10.1128/JVI.00749-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gallina A, Bonelli F, Zentilin L, Rindi G, Muttini M, Milanesi G. 1989. A recombinant hepatitis B core antigen polypeptide with the protamine-like domain deleted self-assembles into capsid particles but fails to bind nucleic acids. J. Virol. 63:4645–4652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hatton T, Zhou S, Standring DN. 1992. RNA- and DNA-binding activities in hepatitis B virus capsid protein: a model for their roles in viral replication. J. Virol. 66:5232–5241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Le Pogam S, Chua PK, Newman M, Shih C. 2005. Exposure of RNA templates and encapsidation of spliced viral RNA are influenced by the arginine-rich domain of human hepatitis B virus core antigen (HBcAg 165–173). J. Virol. 79:1871–1887. 10.1128/JVI.79.3.1871-1887.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ishida T, Kinoshia K. 2007. PrDOS: prediction of disordered protein regions from amino acid sequence. Nucleic Acids Res. 35(Suppl 2):W460–W464. 10.1093/nar/gkm363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crowther RA, Kiselev NA, Bottcher B, Berriman JA, Borisova GP, Ose V, Pumpens P. 1994. Three-dimensional structure of hepatitis B virus core particles determined by electron cryomicroscopy. Cell 77:943–950. 10.1016/0092-8674(94)90142-2 [DOI] [PubMed] [Google Scholar]
- 35.Bottcher B, Wynne SA, Crowther RA. 1997. Determination of the fold of the core protein of hepatitis B virus by electron cryomicroscopy. Nature 386:88–91. 10.1038/386088a0 [DOI] [PubMed] [Google Scholar]
- 36.Conway JF, Cheng N, Zlotnick A, Wingfield PT, Stahl SJ, Steven AC. 1997. Visualization of a 4-helix bundle in the hepatitis B virus capsid by cryo-electron microscopy. Nature 386:91–94. 10.1038/386091a0 [DOI] [PubMed] [Google Scholar]
- 37.Roseman AM, Berriman JA, Wynne SA, Butler PJ, Crowther RA. 2005. A structural model for maturation of the hepatitis B virus core. Proc. Natl. Acad. Sci. U. S. A. 102:15821–15826. 10.1073/pnas.0504874102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wynne SA, Crowther RA, Leslie AG. 1999. The crystal structure of the human hepatitis B virus capsid. Mol. Cell 3:771–780. 10.1016/S1097-2765(01)80009-5 [DOI] [PubMed] [Google Scholar]
- 39.Cristofari G, Ivanyi-Nagy R, Gabus C, Boulant S, Lavergne JP, Penin F, Darlix JL. 2004. The hepatitis C virus Core protein is a potent nucleic acid chaperone that directs dimerization of the viral (+) strand RNA in vitro. Nucleic Acids Res. 32:2623–2631. 10.1093/nar/gkh579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang ZS, Chen AY, Wu HN. 2004. Characterization and application of the selective strand annealing activity of the N terminal domain of hepatitis delta antigen. FEBS Lett. 578:345–350. 10.1016/j.febslet.2004.11.043 [DOI] [PubMed] [Google Scholar]
- 41.Tsuchihashi Z, Khosla M, Herschlag D. 1993. Protein enhancement of hammerhead ribozyme catalysis. Science 262:99–102. 10.1126/science.7692597 [DOI] [PubMed] [Google Scholar]
- 42.Wang CC, Chang TC, Lin CW, Tsui HL, Chu PB, Chen BS, Huang ZS, Wu HN. 2003. Nucleic acid binding properties of the nucleic acid chaperone domain of hepatitis delta antigen. Nucleic Acids Res. 31:6481–6492. 10.1093/nar/gkg857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Darlix JL, Godet J, Ivanyi-Nagy R, Fosse P, Mauffret O, Mely Y. 2011. Flexible nature and specific functions of the HIV-1 nucleocapsid protein. J. Mol. Biol. 410:565–581. 10.1016/j.jmb.2011.03.037 [DOI] [PubMed] [Google Scholar]
- 44.Bartenschlager R, Junker-Niepmann M, Schaller H. 1990. The P gene product of hepatitis B virus is required as a structural component for genomic RNA encapsidation. J. Virol. 64:5324–5332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bartenschlager R, Schaller H. 1992. Hepadnaviral assembly is initiated by polymerase binding to the encapsidation signal in the viral RNA genome. EMBO J. 11:3413–3420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pollack JR, Ganem D. 1993. An RNA stem-loop structure directs hepatitis B virus genomic RNA encapsidation. J. Virol. 67:3254–3263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nassal M, Rieger A. 1996. A bulged region of the hepatitis B virus RNA encapsidation signal contains the replication origin for discontinuous first-strand DNA synthesis. J. Virol. 70:2764–2773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rieger A, Nassal M. 1996. Specific hepatitis B virus minus-strand DNA synthesis requires only the 5′ encapsidation signal and the 3′-proximal direct repeat DR1. J. Virol. 70:585–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang GH, Seeger C. 1992. The reverse transcriptase of hepatitis B virus acts as a protein primer for viral DNA synthesis. Cell 71:663–670. 10.1016/0092-8674(92)90599-8 [DOI] [PubMed] [Google Scholar]
- 50.Zoulim F, Seeger C. 1994. Reverse transcription in hepatitis B viruses is primed by a tyrosine residue of the polymerase. J. Virol. 68:6–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tavis JE, Perri S, Ganem D. 1994. Hepadnavirus reverse transcription initiates within the stem-loop of the RNA packaging signal and employs a novel strand transfer. J. Virol. 68:3536–3543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Will H, Reiser W, Weimer T, Pfaff E, Buscher M, Sprengel R, Cattaneo R, Schaller H. 1987. Replication strategy of human hepatitis B virus. J. Virol. 61:904–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shih C, Burke K, Chou MJ, Zeldis JB, Yang CS, Lee CS, Isselbacher KJ, Wands JR, Goodman HM. 1987. Tight clustering of human hepatitis B virus integration sites in hepatomas near a triple-stranded region. J. Virol. 61:3491–3498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Beck J, Nassal M. 2007. Hepatitis B virus replication. World J. Gastroenterol. 13:48–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Oropeza CE, McLachlan A. 2007. Complementarity between epsilon and phi sequences in pregenomic RNA influences hepatitis B virus replication efficiency. Virology 359:371–381. 10.1016/j.virol.2006.08.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lewellyn EB, Loeb DD. 2007. Base pairing between cis-acting sequences contributes to template switching during plus-strand DNA synthesis in human hepatitis B virus. J. Virol. 81:6207–6215. 10.1128/JVI.00210-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liao W, Ou JH. 1995. Phosphorylation and nuclear localization of the hepatitis B virus core protein: significance of serine in the three repeated SPRRR motifs. J. Virol. 69:1025–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kann M, Sodeik B, Vlachou A, Gerlich WH, Helenius A. 1999. Phosphorylation-dependent binding of hepatitis B virus core particles to the nuclear pore complex. J. Cell Biol. 145:45–55. 10.1083/jcb.145.1.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Perlman DH, Berg EA, O'Connor BP, Costello CE, Hu J. 2005. Reverse transcription-associated dephosphorylation of hepadnavirus nucleocapsids. Proc. Natl. Acad. Sci. U. S. A. 102:9020–9025. 10.1073/pnas.0502138102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pugh J, Zweidler A, Summers J. 1989. Characterization of the major duck hepatitis B virus core particle protein. J. Virol. 63:1371–1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang JC, Dhason MS, Zlotnick A. 2012. Structural organization of pregenomic RNA and the carboxy-terminal domain of the capsid protein of hepatitis B virus. PLoS Pathog. 8:e1002919. 10.1371/journal.ppat.1002919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chua PK, Tang FM, Huang JY, Suen CS, Shih C. 2010. Testing the balanced electrostatic interaction hypothesis of hepatitis B virus DNA synthesis by using an in vivo charge rebalance approach. J. Virol. 84:2340–2351. 10.1128/JVI.01666-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lewellyn EB, Loeb DD. 2011. The arginine clusters of the carboxy-terminal domain of the core protein of hepatitis B virus make pleiotropic contributions to genome replication. J. Virol. 85:1298–1309. 10.1128/JVI.01957-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eckhardt SG, Milich DR, McLachlan A. 1991. Hepatitis B virus core antigen has two nuclear localization sequences in the arginine-rich carboxyl terminus. J. Virol. 65:575–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li HC, Huang EY, Su PY, Wu SY, Yang CC, Lin YS, Chang WC, Shih C. 2010. Nuclear export and import of human hepatitis B virus capsid protein and particles. PLoS Pathog. 6:e1001162. 10.1371/journal.ppat.1001162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yeh CT, Liaw YF, Ou JH. 1990. The arginine-rich domain of hepatitis B virus precore and core proteins contains a signal for nuclear transport. J. Virol. 64:6141–6147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kock J, Nassal M, Deres K, Blum HE, von Weizsacker F. 2004. Hepatitis B virus nucleocapsids formed by carboxy-terminally mutated core proteins contain spliced viral genomes but lack full-size DNA. J. Virol. 78:13812–13818. 10.1128/JVI.78.24.13812-13818.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen HL, Su PY, Chang YS, Wu SY, Liao YD, Yu HM, Lauderdale TL, Chang K, Shih C. 2013. Identification of a novel antimicrobial peptide from human hepatitis B virus core protein arginine-rich domain (ARD). PLoS Pathog. 9:e1003425. 10.1371/journal.ppat.1003425 [DOI] [PMC free article] [PubMed] [Google Scholar]