

ABSTRACT
Atypical porcine reproductive and respiratory syndrome (PRRS) caused by highly pathogenic porcine reproductive and respiratory syndrome virus (HP-PRRSV) is characterized by high fever and high mortality. However, the mechanism underlying the fever induction is still unknown. Prostaglandin E2 (PGE2), synthesized by cyclooxygenase type 1/2 (COX-1/2) enzymes, is essential for inducing fever. In this study, we found that PGE2, together with COX-1, was significantly elevated by HP-PRRSV. We subsequently demonstrated that extracellular signal-regulated kinase 1/2 (ERK1/2) and phosphorylated ERK (p-ERK) were the key nodes to trigger COX-1 expression after HP-PRRSV infection. Furthermore, we proved the direct binding of p-C/EBP-β to the COX-1 promoter by luciferase reporter and chromatin immunoprecipitation assays. In addition, silencing of C/EBP-β remarkably impaired the enhancement of COX-1 production induced by HP-PRRSV infection. Taken together, our results indicate that HP-PPRSV elicits the expression of COX-1 through the ERK1/2-p-C/EBP-β signaling pathway, resulting in the increase of PGE2, which might be the cause of high fever in infected pigs. Our findings might provide new insights into the molecular mechanisms underlying the pathogenesis of HP-PRRSV infection.
IMPORTANCE The atypical PRRS caused by HP-PRRSV was characterized by high fever, high morbidity, and high mortality in pigs of all ages, yet how HP-PRRSV induces high fever in pigs remains unknown. In the present study, we found out that HP-PRRSV infection could increase PGE2 production by upregulation of COX-1, and we subsequently characterized the underlying mechanisms about how HP-PRRSV enhances COX-1 production. PGE2 plays a critical role in inducing high temperature in hosts during pathogen infections. Thus, our findings here could help us have a better understanding of HP-PRRSV pathogenesis.
INTRODUCTION
Porcine reproductive and respiratory syndrome (PRRS) is an important infectious disease worldwide in the swine industry and is characterized by respiratory disorders in piglets and reproductive failure in sows (1). The causative agent, porcine reproductive and respiratory syndrome virus (PRRSV), is an enveloped, single-strand positive RNA virus which is a member of the genus Arterivirus, family Arteriviridae, order Nidovirales (2). In 2006, a highly pathogenic strain of PRRSV (HP-PRRSV) with a discontinuous 30-amino-acid depletion in the Nsp2 protein was identified in China (3, 4). The atypical PRRS caused by HP-PRRSV was characterized by high fever (>41°C for at least 4 days), high morbidity, and high mortality in pigs of all ages. HP-PRRSV has spread rapidly (5–7), caused serious economic losses, and became the most epidemic strain of PRRSV in China, yet how HP-PRRSV induces high fever in pigs remains unknown.
Fever is the host's initial acute response to pathogen infection and plays a critical role in antiviral responses against viral infections, such as influenza virus, chickenpox virus, and respiratory syncytial virus infections (8–12). Prostaglandin E2 (PGE2) is an autocrine factor derived from arachidonic acid (AA) through the activation of cyclooxygenase (13–15). Interestingly, PGE2 synthase plays an important role in fever induction (11, 16, 17). PGE2 acts on neurons in the preoptic area (POA) through the EP3 receptor and sends the signal to the hypothalamus, leading to the activation of a sympathetic output system to cause thermogenesis (18). Neutralization of PGE2 with anti-PGE2 antibody delayed and abated lipopolysaccharide (LPS)-induced fever (19).
Cyclooxygenase, known as prostaglandin G/H synthase, is a key enzyme in the synthesis from arachidonic acid into PGE2, which plays a pivotal role in inflammation and respiratory tract hyperreactivity. There are two isoforms of cyclooxygenase (COX): COX type 1 (COX-1) and COX-2. COX-1 is constitutively expressed in most tissues, whereas COX-2 is an inducible isoform (20, 21). In LPS-induced PGE2 production in astrocytes, COX-2 was induced but COX-1 was downregulated through a MyD88-dependent pathway (22, 23). However, tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) induced the expression of COX-1 in a myeloid cell lineage, such as HL-60 cells, without affecting COX-2 expression and resulted in a significant increase of PGE2 production and release (24). The contribution of these enzymes in PGE2 formation depends on both stimulation and cell type.
HP-PRRSV infection causes high fever in pigs. Therefore, we asked whether HP-PRRSV increases PGE2 secretion and, if so, what the underlying mechanism involved in HP-PRRSV-induced PGE2 production is. In this study, we show that HP-PRRSV evokes a PGE2 increase in plasma and bronchoalveolar lavage fluid (BALF) in vivo. PGE2 secretion was induced by HP-PRRSV in porcine alveolar macrophages (PAMs) through the elevation of COX-1. In addition, activation of extracellular signal-regulated kinase 1/2 (ERK1/2)-phosphorylated C/EBP-β (p-C/EBP-β) was required for the enhancement of COX-1. Furthermore, we demonstrated that p-C/EBP-β moved into nuclei and interacted with the CCAAT/enhancer box motif on the COX-1 promoter to trigger the expression of COX-1, which resulted in the elevation of PGE2. Our findings may help us understand the molecular mechanisms underlying the high fever induced by HP-PRRSV infection.
MATERIALS AND METHODS
Cells and virus preparation.
PAMs were obtained by postmortem lung lavage of 8-week-old specific-pathogen-free (SPF) pigs and maintained in RPMI 1640 medium with 10% heat-inactivated fetal bovine serum (FBS) and penicillin-streptomycin. Cells of the Marc-145 cell line, a PRRSV-permissive monkey kidney cell line subcloned from MA-104 cells, were cultured in Dulbecco's minimum essential medium (DMEM) supplemented with 10% heat-inactivated FBS. All the cells were cultured and maintained at 37°C with 5% CO2.
Strain CH-1a (the first type 2 PRRSV strain isolated in China) was propagated in Marc-145 cells, and HP-PRRSV JXwn06 (a highly pathogenic PRRSV isolate) was propagated in PAMs. Virus preparations were titrated and then stored at −80°C. Inactivated HP-PRRSV JXwn06 was prepared by UV irradiation at 120 mJ cm−2 for 1 h using a Bio-Link cross-linker (VilberLourmat).
Virus preparations were titrated on PAMs. Briefly, a monolayer of PAMs was prepared in a 96-well plate and then infected with serially diluted PRRSV (10−1 to 10−10). PRRSV infection was determined at 72 h postinfection (hpi) using immunofluorescent staining for the PRRSV N protein. The viral titer was determined by the Reed-Muench method and expressed as the 50% tissue culture infective dose (TCID50).
Animal experiment.
Four-week-old SPF piglets were obtained from the Beijing Center for SPF Swine Breeding and Management and randomly divided into two groups (three piglets per group). All animal studies were performed according to protocols approved by the Animal Welfare Committee of China Agricultural University. Piglets were intranasally inoculated with 1 ml of PRRSV strain JXwn06 (105.2 TCID50s virus/ml). The rectal temperature of the piglets was monitored every day until the pigs died. Plasma was collected at 0, 3, and 7 days postinfection (dpi) (we added indomethacin to blood samples when we collected them), and BALF samples were collected from the necropsied dying pigs.
Reagents and antibodies.
An NF-κB inhibitor (BAY11-7082), a protein kinase C (PKC) inhibitor (GF-109203X), an MEK inhibitor (PD98059, U0126), a p38 inhibitor (SB203580), and a phosphatidylinositol 3-kinase (PI3K) inhibitor (LY294002) were purchased from Cell Signaling Technology, Inc. Poly(I·C) was obtained from Sigma-Aldrich. sc-560 (COX-1 inhibitor) and an enzyme-linked immunosorbent assay (ELISA) kit for porcine PGE2 were purchased from Cayman Systems. The COX-2 inhibitor celecoxib was from Sigma. All inhibitors were reconstituted in dimethyl sulfoxide (DMSO), and DMSO was used as the solvent control for all experiments involving treatment with inhibitors. The TRIzol reagent was from Invitrogen Life Technology. RNase-free DNase I and a Dual-Glo luciferase assay system were purchased from Promega.
Antibodies against C/EBP-β, p-C/EBP-β, and histone 3 were purchased from Santa Cruz Biotechnology. Anti-ERK1/2 and anti-phosphorylated ERK1/2 (anti-p-ERK1/2) antibodies were from Cell Signaling Technology, Inc. Antibodies against β-actin, α-tubulin, and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) were purchased from Sigma. Antibody against COX-2 was from Merck, and antibody against COX-1 was obtained from Abcam.
Real-time PCR.
Total RNA from cells was extracted using a total RNA extraction kit (Omega Scientific), and 1 μg of RNA was reverse transcribed to cDNA using Moloney murine leukemia virus (MMLV) reverse transcriptase (Promega). Real-time PCR was performed on an ABI 7500 real-time PCR system using a real-time SYBR master mix kit (TaKaRa). Gene-specific primers for porcine COX-1 were forward primer CGAGAAGTGCCTCCCAAACTCC and reverse primer AAGCCCATCTCGCCACCAAACG; primers for porcine COX-2 were forward primer GTGGGGCATGAGGTCTTTGG and reverse primer CAGCCTGCTCGTCTGGAACA, as described in a previous study (25). The gene expression was normalized to that for the gene for cyclophilin (as a housekeeping gene) (forward primer AATGGCACTGGTGGCAAGTC, reverse primer GATGCCAGGACCCGTATGC). The copy number of the ORF7 gene was calculated by using an ORF7-containing plasmid of known concentrations as a standard.
Inhibition of signal transduction pathways.
Cells were pretreated with DMSO, the PCK inhibitor GF-109203X (2 μM), the p38 mitogen-activated protein kinase (MAPK) inhibitor SB202190 (10 μM), the MEK inhibitor PD98059 (10 μM), the MEK inhibitor U0126 (5 μM), the NF-κB inhibitor BAY11-7082 (2 μM), or the PI3K inhibitor LY294002 (5 μM) for 1 h and then infected with PRRSV at a multiplicity of infection (MOI) of 2 in the presence of inhibitors. Twenty-four hours later, supernatants were harvested for the analysis of secreted PGE2 by ELISA, and cells were harvested for COX-1 mRNA analysis by real-time PCR and protein detection using Western blotting.
Western blotting.
Whole-cell extracts were prepared by lysing cells in radioimmunoprecipitation assay lysis buffer with 100 U proteinase inhibitor (Promega) and 20 μM NaF (Beyotime). Nuclei and cytoplasm protein samples were extracted with a nuclear and cytosol fractionation kit (BioVision Incorporated). The protein level in each sample was quantified with a bicinchoninic acid assay kit (Pierce Biotechnology, Inc.). A similar amount of protein from each sample was run on a 12% SDS-polyacrylamide gel and then transferred to polyvinylidene difluoride membranes (Millipore). After blocking with a 5% no-fat milk solution in Tris-buffered saline with 0.1% Tween 20, the membranes were incubated for 2 h at room temperature with the antibodies at a suitable dilution, as recommended (anti-C/EBP-β, p-C/EBP-β, ERK1/2, p-ERK1/2, COX-1, COX-2, and histone 3 at 1:1,000; anti-β-actin, α-tubulin, and GAPDH at 1:5,000). The membranes were then incubated with horseradish peroxidase-conjugated goat anti-mouse IgG or goat anti-rabbit IgG (Santa Cruz Biotechnology) as secondary antibodies for 1 h at a dilution of 1:5,000. The signals were recorded with enhanced chemiluminescence (ECL) reagents (Millipore ECL plus kit).
siRNA knockdown.
Cells were transfected with 200 pmol of small interfering RNA (siRNA) oligonucleotides specific for the C/EBP-β target gene, a nonspecific control (NC), or a 6-carboxyfluorescein-labeled control oligonucleotide (GenePharma Inc.) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. The efficiency of knockdown of protein expression was confirmed by immunoblotting. PRRSV infection (MOI = 2) was performed 12 h after transfection. At 24 h after PRRSV infection, cells were harvested for COX-1 analysis using Western blotting.
Construction of porcine COX-1 promoters.
Genomic DNA was extracted from PAMs and purified by use of a DNA extraction kit (TaKaRa). The fragment of the porcine COX-1 gene promoter flanking the 5′ COX-1 gene (NC_010443.4) was cloned with specific primers. Gene-specific primers for the porcine COX-1 promoter were forward primer GGGGTACCGTTGGATCTGAGGGTAGTGG and reverse primer TCCCCCGGGGGTGCGGGATACAGACTGC. The obtained 476-bp Sus scrofa COX-1 promoter sequence (nucleotides −405 to +71) relative to the transcription initiation site (+1) was subcloned into the luciferase (luc) reporter vector pGL3-basic at the KpnI site (pGL3−405/+71-luc), and the nucleotide sequence was determined by DNA sequencing. The mutants with truncated mutations of the COX-1 promoter were then constructed and inserted into the luciferase reporter vector pGL3-basic (pGL3−215/+71-luc, pGL3−111/+71-luc, and pGL3−32/+71-luc). The truncated mutants of the COX-1 promoter were constructed using the primers listed in Table 1. Site-directed mutagenesis of the C/EBP-β site was performed by multiple rounds of PCR using pGL3−405/+71-luc as a template and specific primers with altered bases. The primers were reverse primer 5′-CAGAGATGATGcGCcAGTCACCCA-3′ and forward primer 5′-TGGGTGACTTGgACgTCATCTCT-3′, where the lowercase nucleotides indicate the mutant nucleotides. The mutated DNA was then subcloned into the pGL3-basic vector and verified by sequence analysis.
TABLE 1.
Primers for truncated sequence of COX-1 promotera
| Primer name | Sequence |
|---|---|
| +71-smaI | TCCCCCGGGGGTGCGGGATACAGACTGC |
| −215/71-luc | GGGGTACCTTTCCTATCTTCCTAATGGGC |
| −111/71-luc | GGGGTACCTAATTAATAAAATGTCTTGGCC |
| −37/71-luc | GGGGTACCGAGAGGTGGAGCCAGGGA |
Nucleotide +1 represents the site of transcription initiation of the COX-1 promoter, and a truncated COX-1 was cloned at the KpnI and SmaI sites in the pGL3-basic luciferase reporter vector. Underlining shows the KpnI enzyme site.
Luciferase reporter assays.
Marc-145 cells seeded on 24-well plates were transfected with the constructed plasmids (pGL3−405/+71-luc, pGL3−215/+71-luc, pGL3−111/+71-luc, pGL3−32/+71-luc, and pGL3−405/+71-C/EBP-β MUT-luc [where MUT-luc indicates mutant luciferase]; 2 μg for each plasmid) using the Lipofectamine LTX reagent. Twelve hours later, cells were infected with strain CH-1a at an MOI of 2. Cells were harvested at 36 hpi, and luciferase activity analysis was performed with extracts prepared using a dual-luciferase reporter assay system according to the manufacturer's instruction (Promega).
ELISA for PGE2.
The levels of PGE2 in BALF, plasma, and cell culture supernatants were measured using an ELISA kit (Cayman Systems) for porcine PGE2 according to the instruction manual. Cell culture supernatants and BALF were centrifuged at 720 × g for 5 min at 4°C and plasma was centrifuged at 1,000 × g for 15 min at 4°C before ELISA was performed.
ChIP assay.
To study the DNA-protein interaction of p-C/EBP-β and the COX-1 promoter, a chromatin immunoprecipitation (ChIP) assay was performed using a ChIP-IT Express enzymatic system (Active Motif) following the manufacturer's instructions. Briefly, PAMs (1.2 × 107 cells) were infected with or without HP-PRRSV JXwn06 at an MOI of 2. Six hours later, PAMs were fixed with 1% formaldehyde, which cross-links and preserves DNA-protein interactions, for 10 min at room temperature. After washing with ice-cold phosphate-buffered saline (PBS), fixation was stopped with glycine solution (0.1 M). Cells were then harvested with ice-cold PBS containing phenylmethylsulfonyl fluoride (PMSF; 0.5 mM). After centrifugation, cells were resuspended in 1 ml lysis buffer supplemented with PMSF and protease inhibitors and lysed using an ice-cold Dounce homogenizer. The nuclear pellet was collected and digested in digesting buffer. Cross-linked materials from the nuclei were digested into chromatin fragments with an average length of 200 to 800 bp using an enzymatic shearing cocktail in 50% glycerol. Immunoprecipitation was performed with protein G magnetic beads and 2.5 μg anti-p-C/EBP-β antibody or rabbit isotype control IgG in 1.7 ml siliconized microcentrifuge tubes. The immunoprecipitation reaction mixture was incubated overnight at 4°C on a rotating wheel. Immunoprecipitated material was washed 3 times with ChIP washing buffer, and the beads were resuspended in AM2 elution buffer. After reverse cross-linking buffer was added, the chromatin was collected with tubes placed in a magnetic stand. Cross-links were reversed by incubating samples for 15 min at 95°C in a thermocycler. Recovered materials were treated with proteinase K, and DNA was extracted with a DNA extraction kit (Omega). Then, a PCR assay was performed with the DNA templates immunoprecipitated DNA with isotype control rabbit IgG or the p-C/EBP-β antibody and the input DNA. Also, a water-only control was used to ensure that the PCR system was not contaminated. The PCR primers specific for fragments of the COX-1 promoter are listed in Table 2.
TABLE 2.
Primers for ChIP assay
| Primer namea (orientation) | Sequence |
|---|---|
| −404∼−384 (sense) | TTGGATCTGAGGGTAGTGGGT |
| −213∼−193 (antisense) | GTGCCCATTAGGAAGATAGGA |
| −194∼−173 (sense) | ACAATTCTGGTACTACTGGGTT |
| +20∼+37 (antisense) | GTTTCTAACTCTGCCCCG |
Nucleotide +1 represents the site of transcription initiation of the COX-1 promoter.
Flow cytometric analysis.
PAMs were infected with strain JXwn06 at an MOI of 2. At 24 hpi, cells were fixed with 4% paraformaldehyde for 10 min and permeabilized with 0.1% Triton X-100 for 3 min. Cells were then immunostained with COX-1 antibody for 3 h after being blocked with Fc-block (BD Pharmingen) in PBS for 30 min at 4°C. After being washed three times with PBS, cells were stained with goat anti-rabbit IgG-phycoerythrin (PE) for 30 min. Cells were then analyzed by flow cytometry using an Accuri C6 flow cytometer (BD Biosciences).
Confocal microscopy.
To determine the localization of p-C/EBP-β in PAMs, cells were seeded in a chamber slide (Lab-Tek) and infected with JXwn06 at an MOI of 2. At 4 h after infection, cells were fixed with 4% paraformaldehyde and permeabilized with 0.5% Triton X-100 for 3 min. Cells were then stained with primary rabbit polyclonal anti-p-C/EBP-β antibody (diluted 1:100) for 1 h at room temperature (RT) after being blocked with 10% horse serum in PBS for 1 h at RT. After being washed three times with PBS, cells were incubated with goat anti-rabbit IgG–R-phycoerythrin (Southern Biotech) for 30 min. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI) for 5 min. Immunofluorescence was observed using a Nikon A1 confocal microscope.
Statistical analysis.
Results are presented as means ± standard errors of the means (SEMs) for at least three independent experiments. Statistical analysis was performed using GraphPad Prism software, and a Student's t test P value of <0.05 was considered statistically significant. Pearson's analysis was applied to establish correlations.
RESULTS
HP-PRRSV significantly induces PGE2 production in vitro and in vivo.
Atypical PRRS caused by HP-PRRSV is characterized by high fever and high mortality. Therefore, we first investigated if there is a relationship between the high body temperature and survival time for HP-PRRSV-infected pigs. We statistically analyzed the relationship between the temperature at day 6 postinfection and the length of survival (in days) of 9 pigs inoculated with 1 ml HP-PRRSV JXwn06 (105.2 TCID50s virus/ml). As expected, there existed a significant inverse correlation (r = −0.67, P = 0.02) between the high body temperature and length of survival of the HP-PRRSV-infected pigs (Fig. 1A). Since PGE2 is critical for fever induction, we next evaluated the ability of HP-PRRSV to induce PGE2 production in vitro. PAMs were infected with HP-PRRSV JXwn06 at an MOI of 2, and then the supernatants of infected cells were collected and analyzed for PGE2 production. Our results showed that the PGE2 secretion induced by HP-PRRSV JXwn06 was gradually increased and remarkably higher than that of the no-infection controls (7-fold increase at 36 hpi) (Fig. 1B). Similar results were also obtained with strain CH-1a (the first isolate of the type 2 North American strain in China) (Fig. 1B), indicating that PRRSV-induced PGE2 production is not strain dependent. However, the amount of PGE2 induced by strain JXwn06 was significantly higher than that induced by CH-1a (a 1.4-fold increase at 36 hpi), suggesting that differences regarding the induction of PGE2 exist between the HP-PRRSV strain and the typical type 2 North American strain.
FIG 1.

PRRSV induces PGE2 production in vitro and in vivo. (A) Relationship of pig temperature at day 6 after HP-PRRSV strain JXnw06 infection and pig survival (in days). (B) PAMs were incubated with HP-PRRSV JXnw06 or type 2 PRRSV strain CH-1a at an MOI of 2, and then cell culture supernatants were collected at 0, 4, 8, 12, 24, and 36 h postincubation for PGE2 analysis using an ELISA kit. (C and D) Pigs were infected intranasally with 1 ml (105.2 TCID50s) HP-PRRSV strain JXnw06; plasma was collected at days 0, 3, and 7 postinfection and treated with indomethacin, and BALF was collected when the pigs were dying. PGE2 in plasma (C) and BALF (D) was quantified by ELISA. Data are means ± SEMs from three independent experiments. ctrl, control. Differences were evaluated by Student's t test (*, P < 0.05; **, P < 0.01).
To investigate whether HP-PRRSV has the ability to induce PEG2 induction in vivo, pigs were challenged with HP-PRRSV JXwn06, and plasma samples were collected at 0, 3, and 7 days postinfection and then evaluated for PGE2 production. As shown in Fig. 1C, pigs challenged with HP-PRRSV displayed significantly elevated levels of PGE2 in plasma at 3 and 7 days postinfection. Bronchoalveolar lavage fluid (BALF) was also collected from the necropsied dying pigs for analyzing PGE2 production. Our results showed that PGE2 production significantly increased compared to that in the control pigs, with a 2.8-fold increase (Fig. 1D). Collectively, these data indicate that HP-PRRSV infection results in remarkable increases of PGE2 production both in vivo and in vitro, and there was an association between high temperature and earlier death for pigs.
COX-1 is upregulated by HP-PRRSV in PAMs.
COX-1 and COX-2 were demonstrated to play a key role in the PGE2 synthesis pathway. To explore if HP-PPRSV induces COX-1/2, real-time PCR was performed to determine the kinetics of COX-1/2 mRNA expression in PAMs infected with HP-PRRSV JXwn06. As shown in Fig. 2A, COX-1 expression was significantly induced in PAMs by HP-PRRSV JXwn06 infection, with 2.1-, 5.6-, and 5.7-fold increases at 16, 24, and 36 h postinfection, respectively, and the upregulation of COX-1 by HP-PRRSV JXwn06 occurred in a dose-dependent manner (Fig. 2B). The induction of COX-1 by HP-PRRSV was likely replication dependent because the UV-inactivated HP-PRRSV did not induce COX-1 expression (Fig. 2A). The kinetics of COX-1 mRNA expression induced by PRRSV strain CH-1a (the first type 2 PRRSV isolate in China) was similar to that induced by HP-PRRSV (data not shown). In contrast, COX-2 expression was not significantly induced by HP-PRRSV JXwn06 (Fig. 2C). Actually, COX-2 was slightly downregulated by HP-PRRSV to a level as low as 0.7-fold of the level of regulation for the control group (Fig. 2C). To further confirm the expression of COX-1/2 in PAMs, we next examined the expression of COX-1/2 at the protein level by Western blotting. As shown in Fig. 2D, COX-1 was gradually upregulated by HP-PRRSV JXwn06 infection (Fig. 2D). However, we did not observe any significant changes in COX-2 protein level before 24 h after HP-PRRSV infection but observed a slight downregulation at 24 hpi (Fig. 2D). The induction of COX-1 was also confirmed by flow cytometry (Fig. 2E). The mean fluorescence intensity of the stained COX-1 in HP-PRRSV-infected PAMs was significantly increased compared to that in noninfected PAMs, whereas there were no significant changes in the fluorescence intensity of the stained COX-2. Collectively, these results demonstrate that HP-PRRSV infection significantly upregulates COX-1 expression.
FIG 2.

COX-1 is significantly upregulated by HP-PRRSV infection in PAMs. (A) Cells were inoculated with medium alone, HP-PRRSV strain JXnw06 (MOI, 2), or UV-inactivated HP-PRRSV JXnw06 (UV-JXnw06). COX-1 expression was analyzed using real-time PCR at 0, 4, 8, 12, 24, and 36 h postincubation. Results were normalized to those for cyclophilin and are shown as the fold induction compared to that for medium alone. (B) PAMs were infected with HP-PRRSV at an MOI of 0.25, 0.5, 1, or 2, and the level of COX-1 mRNA was analyzed at 24 hpi using real-time PCR; results were normalized to those for cyclophilin and are shown as the fold induction compared to that for medium alone. (C) COX-2 expression was analyzed as described for panel A. (D) COX-1 and COX-2 protein levels in PAMs infected with HP-PRRSV were analyzed using Western blotting at 0, 4, 8, 12, 24, and 36 hpi. β-Actin was set up as a loading control. (E) The levels of COX-1 and COX-2 expression in PAMs infected with HP-PRRSV (MOI, 2) were assessed by flow cytometry at 24 hpi. Levels of COX-1 and COX-2 expression are presented as the mean fluorescence intensity (MFI). Data are means ± SEMs from at least three independent experiments. Differences were evaluated by Student's t test (*, P < 0.05; **, P < 0.01; ns, not significant).
HP-PRRSV induces PGE2 production through COX-1.
To investigate whether COX-1 plays a role in the induction of PGE2 by HP-PRRSV infection, PAMs were pretreated for 1 h with sc-560 (a specific inhibitor of COX-1) or celecoxib (a specific inhibitor of COX-2) before HP-PRRSV infection. Twenty-four hours later, the expression of PGE2 was analyzed by ELISA (Fig. 3A and B). As shown in Fig. 3A, sc-560 significantly inhibited the induction of PGE2 by HP-PRRSV infection, showing an ∼60% reduction at a concentration of 10 nM compared to that for the no-treatment control, while celecoxib induced an ∼30% reduction at a concentration of 40 nM. The inhibition of PGE2 by sc-560 was dose dependent, and an ∼75% reduction compared to that for the no-treatment control was observed at a concentration of 50 nM (Fig. 3B). To rule out the possibility that sc-560 affects HP-PRRSV replication, which might be responsible for the reduction of PGE2, PAMs were also collected for virus load analysis (Fig. 3C). As shown in Fig. 3C, sc-560 had no detectable effect on HP-PRRSV replication. These data suggest that induction of COX-1 by HP-PRRSV is required for the production of PGE2.
FIG 3.
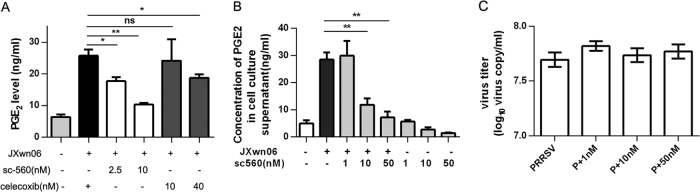
PRRSV induces PGE2 production through COX-1. (A) PAMs were pretreated with COX-1 inhibitor sc-560 (2.5 nM, 10 nM) or COX-2 inhibitor celecoxib (10 nM, 40 nM) for 1 h and then infected with HP-PRRSV strain JXnw06 (MOI, 2). Cell supernatants were harvested at 24 hpi and analyzed for PGE2 production using ELISA. (B) PAMs were pretreated with COX-1 inhibitor sc-560 at different dosages (1 nM, 10 nM, and 50 nM) for 1 h and then infected with HP-PRRSV strain JXnw06 (MOI, 2) in the absence or presence of sc-560. PGE2 production in cell supernatants was detected at 24 hpi by ELISA. (C) Intracellular PRRSV RNA was evaluated at 24 hpi using quantitative real-time PCR. “P” represents PRRSV infection. Data are means ± SEMs from at least three independent experiments. Differences were evaluated by Student's t test. (*, P < 0.05; **, P < 0.01; ns, not significant).
PRRSV induces COX-1 expression through the MEK-ERK1/2 pathway.
To dissect the signaling pathways involved in HP-PRRSV-induced COX-1 production, PAMs were pretreated with the inhibitors of the key signaling molecules, including NF-κB, MEK, p38 MAPK, PI3K, and PKC, for 1 h before HP-PRRSV infection. Twenty-four hours later, the levels of COX-1 expression were analyzed by real-time PCR (Fig. 4A) and Western blot assay (Fig. 4B). As shown in Fig. 4A and B, GF-109203X (a PKC inhibitor) and PD98059 (an MEK inhibitor) significantly inhibited the induction of COX-1 production by HP-PRRSV infection. Consistent with the inhibition of COX-1 induction, PGE2 induction by HP-PRRSV was also significantly impaired by GF-109203X (a PKC inhibitor) and PD98059 (an MEK inhibitor) (Fig. 4C). An inhibitor of p38 MAPK (SB202190) had no effect on COX-1 mRNA expression (Fig. 4A). However, SB202190 remarkably downregulated COX-1 expression at the protein level (Fig. 4B) and subsequently impaired PEG2 secretion (Fig. 4C). These results suggest that the PKC, p38 MAPK, and MEK pathways may be involved in HP-PRRSV-induced COX-1 and PGE2 production and the inhibition of the MEK pathway has the most potent effect on PGE2 production (Fig. 4C).
FIG 4.

The MEK-ERK1/2 pathway is required for PRRSV-induced COX-1 production. PAMs were pretreated for 1 h with candidate inhibitors of signaling molecules, including GF-109203X (GF; a PCK inhibitor; 2 μM), LY294002 (LY; a PI3K inhibitor; 5 μM), PD98059 (PD; an MEK inhibitor; 10 μM), BAY11-7082 (BAY; an NF-κB inhibitor; 1 μM), or SB202190 (SB; a p38 MAPK inhibitor; 10 μM), and then infected with HP-PRRSV strain JXnw06 (MOI, 2) in the absence or presence of inhibitors. COX-1 expression was then examined by real-time PCR (A) and Western blotting (B) at 24 hpi, and the PGE2 level in the supernatant was detected using ELISA (C). (D) PAMs were inoculated with PRRSV (MOI, 2); cells were harvested at 0, 4, 8, 12, 24, and 36 h postincubation; and p-ERK1/2 and ERK1/2 were determined by immunoblotting with β-actin as a reference control. (E) PAMs were pretreated with PD98059 at a concentration of 0 μM, 5 μM, 10 μM, or 25 μM for 1 h and then infected with HP-PRRSV strain JXnw06 (MOI, 2) in the absence or presence of the inhibitor. At 24 h postinfection, COX-1, p-ERK1/2, and ERK1/2 were examined by Western blotting with α-tubulin as a reference control. Data are means ± SEMs from three independent experiments. Differences were evaluated by Student's t test. (*, P < 0.05; **, P < 0.01).
ERK1 and ERK2 (ERK1/2) have been suggested to play a key role in the RAF-MEK-ERK signaling cascade. We therefore examined whether ERK1 and ERK2 (ERK1/2) were phosphorylated during the course of HP-PRRSV infection. PAMs were collected at 0, 4, 8, 16, 24, and 36 hpi for Western blot analysis using an antibody specific for p-ERK1/2 and an antibody specific for ERK1/2 (Fig. 4D). As expected, ERK1/2 phosphorylation started to increase at 4 h postinfection and continued increasing toward 24 h postinfection. The total ERK1/2 level remained unchanged. To determine the role of ERK1/2 in PRRSV-induced COX-1 production, PAMs were pretreated with PD98059 (MEK inhibitor) before HP-PRRSV infection. Twenty-four hours later, COX-1 and p-ERK1/2 were analyzed using Western blotting. As shown in Fig. 4E, PD98059 significantly inhibited HP-PRRSV-induced ERK1/2 phosphorylation, which is in accordance with the decrease of COX-1 expression (Fig. 4E). Together, these data indicate that the MEK-ERK1/2 signaling pathway is required for HP-PRRSV-induced COX-1 and PGE2 production.
Cloning of the porcine COX-1 promoter and mapping an essential HP-PRRSV-responsive region.
To investigate the mechanism underlying the transcriptional regulation of COX-1 production, a 476-bp fragment of the 5′ flanking region of the porcine COX-1 gene was cloned (Fig. 5A). Using the bioinformatics approach (http://alggen.lsi.upc.es), several putative transcriptional regulatory motifs were found in the 5′ flanking region of the COX-1 gene, including C/EBP-β (bases −232 to −239), NF-κB (bases −388 to −378), and SP-1 (bases −66 to −55 and −6 to +3) (Fig. 5A). To evaluate the porcine COX-1 promoter activity and determine the region responsive to PRRSV infection, serial deletions starting from the 5′ end of the promoter were generated and are schematically shown in Fig. 5B. These constructs were transfected into Marc-145 cells, and the luciferase activities were assessed with or without PRRSV infection. Our results showed that the promoter construct pGL3−405/71-luc showed a 39% upregulation after PRRSV infection, while there were no significant changes for the luciferase activity of pGL3−215/+71, pGL3−111/+71, and pGL3−37/+71 (Fig. 5C). This observation suggests that regulatory elements might exist in the region from bases −405 to −215 of the COX-1 promoter and the responsive element could be the putative C/EBP-β (bases −232 to −239). Previous studies demonstrated that C/EBP-β is downstream of ERK1/2 (26, 27). Mutation of the C/EBP-β site (pGL3−405/71-C/EBP-β MUT-luc) significantly impaired PRRSV-induced COX-1 promoter activation (Fig. 5D), suggesting that the putative C/EBP-β motif in the porcine COX-1 promoter (Fig. 5A) could be the possible responsive element. Furthermore, we performed a chromatin immunoprecipitation assay to analyze the DNA-protein interaction between the COX-1 promoter and p-C/EBP-β. As shown in Fig. 5E, a p-C/EBP-β regulatory binding element at the site from bases −404 to −193 was found in the porcine COX-1 promoter. These findings were in agreement with those of the bioinformatics analysis and the results of the luciferase assay presented in Fig. 5A and C, confirming that the p-C/EBP-β regulatory motif exists in the porcine COX-1 promoter. Therefore, the transcriptional regulatory motif C/EBP-β (bases −232 to −239) (Fig. 5A) might be the element in the COX-1 promoter responsive to PRRSV infection.
FIG 5.

Cloning and sequencing of the porcine COX-1 promoter and mapping of an essential PRRSV-responsive region. (A) Cloning and sequence analysis of the 476-bp porcine COX-1 promoter. The putative transcription factor binding sites are shown in boxes; the positions of the putative regulatory motifs are relative to the transcription initiation site. (B) Schematic representation of the truncated COX-1 promoter in a basic luciferase (LUC) expression vector (pGL3) (numbers indicate base positions). Black rectangles, translation initiation. (C) Marc-145 cells were transfected with 2 μg of each constructed plasmid for 24 h, and then cells were infected with or without the CH-1a strain of virus (MOI, 2). At 24 h postinfection, cells were harvested for luciferase activity analysis. (D) Effect of mutation in the putative C/EBP-β region on the COX-1 promoter activity induced by PRRSV. Marc-145 cells were transfected with 2 μg of the constructed mutant promoter for 24 h, and then cells were infected with or without CH-1a virus (MOI of 2). At 24 h postinfection, cells were harvested to determine luciferase activity. (E) p-C/EBP-β and COX-1 promoter interaction. PAMs were incubated with HP-PRRSV strain JXnw06 (MOI, 2) for 6 h, and a ChIP assay was then performed as described in Materials and Methods. Data are means ± SEMs from three independent experiments. Differences were evaluated by Student's t test. (*, P < 0.05).
Phosphorylation of C/EBP-β is induced by HP-PRRSV infection.
Previous studies showed that C/EBP-β was involved in COX-2 regulation (28–30). However, little is known about the role of C/EBP-β in COX-1 expression. To further investigate whether C/EBP-β is engaged in the HP-PRRSV-induced COX-1 response, C/EBP-β and p-C/EBP-β levels in PAMs infected with HP-PRRSV were analyzed. As shown in Fig. 6A, C/EBP-β phosphorylation in the whole-cell lysates started to increase at 2 h postinfection and was maximized at 6 h postinfection, while the level of C/EBP-β remained unchanged. To further verify that PRRSV can activate C/EBP-β, we analyzed the p-C/EBP-β level in subcellular fractions after PRRSV infection. Our results showed that p-C/EBP-β was gradually increased in nuclei starting at 2 hpi, while cytoplasmic p-C/EBP-β was increased at 2 to 3 hpi and then decreased (Fig. 6A), suggesting that phosphorylated C/EBP-β is translocated into the nucleus. The translocation of p-C/EBP from the cytoplasm to the nucleus was further confirmed by confocal microscopy analysis. Our results displayed that p-C/EBP-β (red in Fig. 6B) mainly existed in the nucleus at 4 h after PRRSV infection. To investigate whether MEK/ERK1/2 signaling pathway activation is essential for C/EBP-β phosphorylation during PRRSV infection, PAMs were pretreated with different doses of PD98059 before PRRSV infection. At 24 h after PRRSV infection, C/EBP-β phosphorylation was analyzed by Western blotting. As shown in Fig. 6C, p-C/EBP-β expression induced by PRRSV infection was significantly impaired by the MEK/ERK1/2 inhibitor PD98059 at a concentration of 25 μM, suggesting that the activation of the MEK/ERK1/2 signaling pathway is required for the phosphorylation of C/EBP-β. To further evaluate the role of C/EBP-β in COX-1 induction by PRRSV infection, PAMs were transfected with specific siRNA targeting C/EBP-β at 12 h before PRRSV infection. COX-1 expression at 24 hpi was then analyzed by Western blotting. As shown in Fig. 6D, knockdown of C/EBP-β remarkably decreased the level of COX-1 expression induced by PRRSV, which correlated well with the impairment of C/EBP-β expression. Taken together, these results suggest that activation of C/EBP-β is involved in COX-1 induction by HP-PRRSV infection.
FIG 6.

C/EBP-β is activated by HP-PRRSV infection. (A) Western blotting was performed to detect C/EBP-β and p-C/EBP-β expression. PAMs were inoculated with HP-PRRSV strain JXnw06 (MOI, 2), and cells were harvested for C/EBP-β and p-C/EBP-β analysis at 0, 2, 3, 4, 6, and 8 hpi. Total cell lysates (top) and nuclear and cytoplasmic protein fractions (middle and bottom) were separated and detected. TE, NE, and CE, total, nuclear, and cytoplasmic extracts, respectively. (B) p-C/EBP-β localization was observed using confocal microscopy. PAMs were infected with HP-PRRSV strain JXnw06 (MOI, 2) for 4 h. p-C/EBP-β was detected by indirect immunofluorescence staining using antibody against p-C/EBP-β protein (red). The nucleus (blue) was stained with DAPI. (C) Effect of an MEK inhibitor (PD98059) on p-C/EBP-β expression in PAMs infected with HP-PRRSV. PAMs were pretreated with the MEK inhibitor (PD98059) at a dosage of 0, 5, 10, or 25 μM for 1 h and then infected with HP-PRRSV strain JXnw06 (MOI, 2). At 24 h postinfection, cells were harvested for p-C/EBP-β analysis by Western blotting using a specific antibody against p-C/EBP-β. (D) Effect of C/EBP-β knockdown on COX-1 protein expression. PAMs were transfected with siRNA targeting C/EBP-β for 12 h and then infected with HP-PRRSV strain JXnw06 (MOI, 2). At 24 h postinfection, C/EBP-β knockdown efficiency and COX-1 protein expression were determined by Western blotting.
Collectively, these data indicate that HP-PRRSV induces C/EBP-β activation and then the phosphorylated C/EBP-β migrates to nuclei and binds the COX-1 promoter region to initiate COX-1 transcription.
DISCUSSION
In the present study, we investigated the ability of HP-PRRSV to induce PGE2 production. We demonstrated that HP-PRRSV infection could induce PGE2 secretion, which correlated well with the COX-1 production induced by HP-PRRSV infection, suggesting that COX-1 could be the critical regulatory factor in the production of PGE2 in response to HP-PRRSV. The addition of an MEK/ERK1/2 inhibitor impaired COX-1 expression and subsequently reduced the level of PGE2 secretion in PAMs infected with HP-PRRSV, indicating that the induction of COX-1 and PGE2 is dependent on MEK/ERK1/2 activation. Furthermore, HP-PRRSV infection could activate C/EBP-β. Pretreatment with a specific inhibitor of ERK1/2 (PD98059) inhibited PRRSV-induced C/EBP-β phosphorylation and p-C/EBP-β translocation into the nucleus. Silencing of C/EBP-β by specific siRNA decreased PRRSV-induced COX-1 expression and subsequently suppressed PGE2 synthesis. Using a ChIP assay, we further demonstrated that p-C/EBP-β bound to the region from bases −404 to −193 in the porcine COX-1 promoter.
PRRSV, in particular, HP-PRRSV, can cause high fever. However, the mechanism by which HP-PRRSV leads to high fever is unknown. Here, we found that HP-PRRSV could significantly induce PGE2 production in infected pigs (Fig. 1B to D). PGE2 plays an important role in pulmonary physiology, pathological symptoms, and fever induction (18, 31–33). Previous work demonstrated that PGE2 released from local tissue, such as lung, enters the brain's hypothalamus through blood vessels and activates EP3 in neurons in POA, leading to thermogenesis to cause fever (19). Accordingly, an increased body temperature was shown to be temporally associated with the increase of PGE2 in plasma and was correlated with pig death (Fig. 1A and C). This correlation suggested that PGE2 might contribute to the pathological symptoms in the lung and fever. A previous study showed that fever is a hallmark among the evoked defensive mechanisms and an initial barrier to infecting pathogens (11). High fever could also lead to potential damage to the body (34). Interestingly, the typical type 2 PRRSV isolate CH-1a induced less PGE2 in PAMs than the HP-PRRSV isolate (Fig. 1B), and this might partially explain the mild fever induced by infection with the CH-1a strain.
Here, we found that COX-1 was upregulated in PAMs after HP-PRRSV infection (Fig. 2 and 3). Preconditioning with a COX-1-specific inhibitor reduced the production of PGE2 in HP-PRRSV-infected PAMs (Fig. 3), confirming that COX-1 contributes to PGE2 induction. COX is the rate-limiting enzyme in the PGE2 synthesis cascade. COX-2 is normally absent from cells, and when induced, the COX-2 protein level increases on a scale of hours after a single stimulus (35). COX-1, on the other hand, is often referred to as a housekeeping enzyme because it is constitutively expressed in almost all tissues and performs an important function in mediating various normal physiological processes (29, 36). However, in some cases, COX-1 can be upregulated and play a role in PGE2 production (37). For example, COX-1 was found to promote PGE2 production and control the thermoregulatory response to influenza A virus infection at 4 dpi in mice (38). Mice deficient in gelatinase B (matrix metallopeptidase 9 [MMP-9]) develop a compensatory mechanism involving the COX-1–PGE2 pathway in macrophages, resulting in enhanced early vascular permeability (39). Mounting evidence has shown that the pattern of expression of the two COX isoforms is far more complex than the general concept of constitutive (COX-1) and inducible (COX-2) expression (37, 40). Influenza virus stimulates the production of PGE2 via various signaling molecules, including calcium-dependent phospholipase A2 (cPLA2), COX-1, and COX-2 (41–43). In our work, COX-1 inhibition by sc-560 at 50 nM did not completely block PGE2 expression (Fig. 3), suggesting that other PGE2 synthesis mechanisms may also be involved in PRRSV-induced PGE2 production. Whether cPLA2 plays a role in PRRSV-induced PGE2 production needs to be investigated in the future.
We also observed that PAMs exhibited high baseline levels of COX-2 expression and PGE2 production in BALF. The level of PGE2 in BALF was much higher than that in plasma. PGE2 is normally present at a higher level in lung than in other tissue, suggesting that PGE2 might play a beneficial role in lung under normal conditions (44). COX-2 could be strongly upregulated following respiratory syncytial virus and parainfluenza virus type 3 (PI3) infections (45). In addition to the induction of COX-2 in inflammatory lesions, COX-2 is present constitutively in the brain, spinal cord, kidney, and lung (46–48). However, we showed here that the level of COX-2 expression was decreased in PRRSV-infected PAMs. Interestingly, early studies reported that the induction of COX-2 and downregulation of COX-1 expression by lipopolysaccharide (LPS) induced prostaglandin E2 production in astrocytes, but the enhancement of COX-1 inhibited COX-2 production in HCT-116 carcinoma cells (49), suggesting that there might exist an inverse relationship between COX-1 and COX-2 production. Indeed, inhibition of COX-1 could upregulate COX-2 (50). Actually, in our study, COX-1 upregulation occurred before the decrease in COX-2. Whether the reduction of COX-2 during PRRSV infection is involved in PGE2 production remains unknown and should be further investigated.
Subsequently, using several inhibitors of signal transduction pathways, we demonstrated that the augmentation of COX-1 and PGE2 by HP-PRRSV in PAMs was dependent on the MEK/ERK1/2 pathway. The MEK/ERK1/2 pathway seems to be required for induction of PGE2 production by some stimuli. For example, activation of the p44/p42 ERK signal but not the p38 signal plays a pivotal role in phosphatidylserine (PS) liposome-induced PGE2 in microglia (51), and proteinase-activated receptor 2 (PAR2) stimulation can trigger PGE2 formation via the MEK/ERK/cPLA-2/COX-1 pathway in human lung epithelial cells (52, 53). In addition to the MEK/ERK1/2 pathway, PKC and p38 pathway inhibitors also significantly depressed PGE2 production (about 50%), suggesting that both the PKC and p38 signaling pathways may also play a role in PGE2 production induced by PRRSV. ERK is downstream of PKC, and inhibition of PKC signal transduction with GF-109203X downregulated ERK activation (data not shown), leading to the suppression of the COX-1 and PGE2 production induced by PRRSV. Interestingly, the p38 inhibitor only slightly decreased HP-PRRSV-induced COX-1 mRNA expression but significantly suppressed its secretion, suggesting that the p38 pathway might be involved in the COX-1 posttranscription modulation induced by HP-PRRSV. Further studies are needed to determine the underlying mechanism.
In the present study, we cloned and characterized the COX-1 promoter (bases −405 to +71) and found that there was a putative C/EBP-β-bound element located at positions −232 to −239. A ChIP assay further demonstrated that p-C/EBP-β bound the region from bases −404 to −193 in the COX-1 promoter. Studies showed that COX-1 could be regulated at the transcriptional level with lipopolysaccharide infection (54, 55). The fine regulation of p-C/EBP-β on the COX-1 promoter in HP-PRRSV-infected PAMs needs to be investigated in the future.
In conclusion, our results demonstrated that HP-PRRSV infection could induce PGE2 production in vivo and in vitro. Our data further showed that upregulation of PGE2 in PAMs was via the MEK/ERK1/2-p-C/EBP-β-COX-1 pathway (Fig. 7). Further research on this aspect will provide new insights into the molecular mechanism controlling the PGE2 production induced by PRRSV infection, which, in turn, should contribute substantially to a better understanding of HP-PRRSV pathogenesis.
FIG 7.
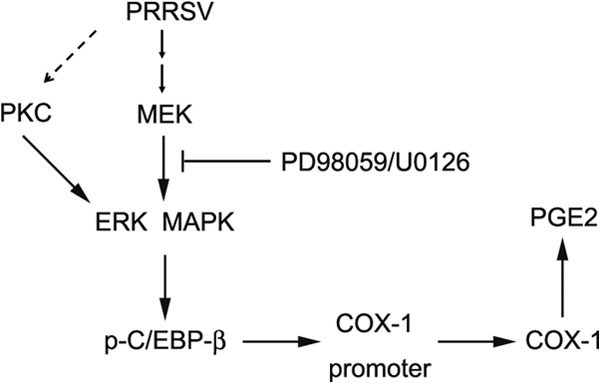
Model of COX-1-mediated PGE2 production in response to HP-PRRSV infection. HP-PRRSV engages an ERK1/2-dependent pathway, which leads to the activation of ERK1/2 and, subsequently, the phosphorylation and activation of C/EBP-β. The other signals resulting from PKC in combination with ERK-MAPK might also be involved in the induction of COX-1 by HP-PRRSV. p-C/EBP-β is critical for the transactivation of COX-1, a key enzyme for the synthesis of PGE2. The disruption of ERK1/2 diminished the enhanced COX-1 induced by HP-PRRSV.
ACKNOWLEDGMENTS
This work was supported by a Faculty Starting Grant and the State Key Laboratory of Agrobiotechnology (grant 2013SKLAB1-5 and grant 2013SKLAB06-2), China Agricultural University, Beijing, China.
Footnotes
Published ahead of print 18 December 2013
REFERENCES
- 1.Fu Y, Quan R, Zhang H, Hou J, Tang J, Feng WH. 2012. Porcine reproductive and respiratory syndrome virus induces interleukin-15 through the NF-kappaB signaling pathway. J. Virol. 86:7625–7636. 10.1128/JVI.00177-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balasuriya UB, MacLachlan NJ. 2004. The immune response to equine arteritis virus: potential lessons for other arteriviruses. Vet. Immunol. Immunopathol. 102:107–129. 10.1016/j.vetimm.2004.09.003 [DOI] [PubMed] [Google Scholar]
- 3.Zhou Y, Yang X, Wang HN, Zhang A, Zhang Z, Kang R, Zeng F, Li H. 2012. Molecular characterization of a complete genome and 12 Nsp2 genes of PRRSV of southwestern China. Food Environ. Virol. 4:102–114. 10.1007/s12560-012-9083-z [DOI] [PubMed] [Google Scholar]
- 4.Kim DY, Calvert JG, Chang KO, Horlen K, Kerrigan M, Rowland RR. 2007. Expression and stability of foreign tags inserted into nsp2 of porcine reproductive and respiratory syndrome virus (PRRSV). Virus Res. 128:106–114. 10.1016/j.virusres.2007.04.019 [DOI] [PubMed] [Google Scholar]
- 5.Liu XD, Chen HB, Tong Q, Li XY, Zhu MJ, Wu ZF, Zhou R, Zhao SH. 2011. Molecular characterization of caveolin-1 in pigs infected with Haemophilus parasuis. J. Immunol. 186:3031–3046. 10.4049/jimmunol.0902687 [DOI] [PubMed] [Google Scholar]
- 6.Guo XK, Zhang Q, Gao L, Li N, Chen XX, Feng WH. 2013. Increasing expression of microRNA 181 inhibits porcine reproductive and respiratory syndrome virus replication and has implications for controlling virus infection. J. Virol. 87:1159–1171. 10.1128/JVI.02386-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wernike K, Hoffmann B, Dauber M, Lange E, Schirrmeier H, Beer M. 2012. Detection and typing of highly pathogenic porcine reproductive and respiratory syndrome virus by multiplex real-time RT-PCR. PLoS One 7:e38251. 10.1371/journal.pone.0038251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sivier V, Odelin MF, Gonthier R, Pozzetto B. 2001. Viral respiratory infections as cause of fever in hospitalized aged patients during a winter season. Rev. Med. Interne 22:1180–1187. (In French.) 10.1016/S0248-8663(01)00489-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rasoul-Rockenschaub S, Zielinski CC, Muller C, Tichatschek E, Popow-Kraupp T, Kunz C. 1990. Viral reactivation as a cause of unexplained fever in patients with progressive metastatic breast cancer. Cancer Immunol. Immunother. 31:191–195. 10.1007/BF01744736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guilherme L, Kalil J. 2007. Rheumatic fever: from innate to acquired immune response. Ann. N. Y. Acad. Sci. 1107:426–433. 10.1196/annals.1381.045 [DOI] [PubMed] [Google Scholar]
- 11.Blatteis CM, Li S, Li Z, Feleder C, Perlik V. 2005. Cytokines, PGE2 and endotoxic fever: a re-assessment. Prostaglandins Other Lipid Mediat. 76:1–18. 10.1016/j.prostaglandins.2005.01.001 [DOI] [PubMed] [Google Scholar]
- 12.Dinarello CA, Wolff SM. 1982. Molecular basis of fever in humans. Am. J. Med. 72:799–819. 10.1016/0002-9343(82)90548-4 [DOI] [PubMed] [Google Scholar]
- 13.Li S, Ma X, Ma L, Wang C, He Y, Yu Z. 2013. Effects of ectopic HER-2/neu gene expression on the COX-2/PGE2/P450arom signaling pathway in endometrial carcinoma cells: HER-2/neu gene expression in endometrial carcinoma cells. J. Exp. Clin. Cancer Res. 32:11. 10.1186/1756-9966-32-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shimamoto C, Fujiwara S, Kato M, Ito S, Katsu K, Mori H, Nakahari T. 2005. Inhibition of ACh-stimulated exocytosis by NSAIDs in guinea pig antral mucous cells: autocrine regulation of mucin secretion by PGE2. Am. J. Physiol. Gastrointest. Liver Physiol. 288:G39–G47. 10.1152/ajpgi.00060.2004 [DOI] [PubMed] [Google Scholar]
- 15.Lemieux LI, Rahal SS, Kennedy CR. 2003. PGE2 reduces arachidonic acid release in murine podocytes: evidence for an autocrine feedback loop. Am. J. Physiol. Cell Physiol. 284:C302–C309. 10.1152/ajpcell.00024.2002 [DOI] [PubMed] [Google Scholar]
- 16.Oka K, Oka T, Hori T. 1998. PGE2 receptor subtype EP1 antagonist may inhibit central interleukin-1beta-induced fever in rats. Am. J. Physiol. 275:R1762–R1765 [DOI] [PubMed] [Google Scholar]
- 17.Chen X, Landgraf R, Pittman QJ. 1997. Differential ventral septal vasopressin release is associated with sexual dimorphism in PGE2 fever. Am. J. Physiol. 272:R1664–R1669 [DOI] [PubMed] [Google Scholar]
- 18.Oka T. 2004. Prostaglandin E2 as a mediator of fever: the role of prostaglandin E (EP) receptors. Front. Biosci. 9:3046–3057. 10.2741/1458 [DOI] [PubMed] [Google Scholar]
- 19.Steiner AA, Ivanov AI, Serrats J, Hosokawa H, Phayre AN, Robbins JR, Roberts JL, Kobayashi S, Matsumura K, Sawchenko PE, Romanovsky AA. 2006. Cellular and molecular bases of the initiation of fever. PLoS Biol. 4:e284. 10.1371/journal.pbio.0040284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang YH, Lu J, Elmquist JK, Saper CB. 2003. Specific roles of cyclooxygenase-1 and cyclooxygenase-2 in lipopolysaccharide-induced fever and Fos expression in rat brain. J. Comp. Neurol. 463:3–12. 10.1002/cne.10743 [DOI] [PubMed] [Google Scholar]
- 21.Vigano T, Habib A, Hernandez A, Bonazzi A, Boraschi D, Lebret M, Cassina E, Maclouf J, Sala A, Folco G. 1997. Cyclooxygenase-2 and synthesis of PGE2 in human bronchial smooth-muscle cells. Am. J. Respir. Crit. Care Med. 155:864–868. 10.1164/ajrccm.155.3.9117018 [DOI] [PubMed] [Google Scholar]
- 22.Font-Nieves M, Sans-Fons MG, Gorina R, Bonfill-Teixidor E, Salas-Perdomo A, Marquez-Kisinousky L, Santalucia T, Planas AM. 2012. Induction of COX-2 enzyme and down-regulation of COX-1 expression by lipopolysaccharide (LPS) control prostaglandin E2 production in astrocytes. J. Biol. Chem. 287:6454–6468. 10.1074/jbc.M111.327874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen JX, Berry LC, Christman BW, Meyrick B. 2003. Glutathione mediates LPS-stimulated COX-2 expression via early transient p42/44 MAPK activation. J. Cell. Physiol. 197:86–93. 10.1002/jcp.10353 [DOI] [PubMed] [Google Scholar]
- 24.Zauli G, Pandolfi A, Gonelli A, Di Pietro R, Guarnieri S, Ciabattoni G, Rana R, Vitale M, Secchiero P. 2003. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) sequentially upregulates nitric oxide and prostanoid production in primary human endothelial cells. Circ. Res. 92:732–740. 10.1161/01.RES.0000067928.83455.9C [DOI] [PubMed] [Google Scholar]
- 25.Jana B, Kozlowska A, Koszykowska M, Majewski M. 2009. Expression of cyclooxygenase-2 in the inflammatory changed porcine uterus. Polish J. Vet. Sci. 12:1–8 [PubMed] [Google Scholar]
- 26.Studzinski GP, Wang X, Ji Y, Wang Q, Zhang Y, Kutner A, Harrison JS. 2005. The rationale for deltanoids in therapy for myeloid leukemia: role of KSR-MAPK-C/EBP pathway. J. Steroid Biochem. Mol. Biol. 97:47–55. 10.1016/j.jsbmb.2005.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Belmonte N, Phillips BW, Massiera F, Villageois P, Wdziekonski B, Saint-Marc P, Nichols J, Aubert J, Saeki K, Yuo A, Narumiya S, Ailhaud G, Dani C. 2001. Activation of extracellular signal-regulated kinases and CREB/ATF-1 mediate the expression of CCAAT/enhancer binding proteins beta and -delta in preadipocytes. Mol. Endocrinol. 15:2037–2049. 10.1210/me.15.11.2037 [DOI] [PubMed] [Google Scholar]
- 28.Liu YW, Wang SA, Hsu TY, Chen TA, Chang WC, Hung JJ. 2010. Inhibition of LPS-induced C/EBP delta by trichostatin A has a positive effect on LPS-induced cyclooxygenase 2 expression in RAW264.7 cells. J. Cell. Biochem. 110:1430–1438. 10.1002/jcb.22682 [DOI] [PubMed] [Google Scholar]
- 29.Lai CS, Lee JH, Ho CT, Liu CB, Wang JM, Wang YJ, Pan MH. 2009. Rosmanol potently inhibits lipopolysaccharide-induced iNOS and COX-2 expression through downregulating MAPK, NF-kappaB, STAT3 and C/EBP signaling pathways. J. Agric. Food Chem. 57:10990–10998. 10.1021/jf9025713 [DOI] [PubMed] [Google Scholar]
- 30.Caivano M, Gorgoni B, Cohen P, Poli V. 2001. The induction of cyclooxygenase-2 mRNA in macrophages is biphasic and requires both CCAAT enhancer-binding protein beta (C/EBP beta) and C/EBP delta transcription factors. J. Biol. Chem. 276:48693–48701. 10.1074/jbc.M108282200 [DOI] [PubMed] [Google Scholar]
- 31.Zhang G, Lee LY. 2007. Prostaglandin E2 enhances the sensitizing effect of hyperthermia on pulmonary C-fibers in rats. Respir. Physiol. Neurobiol. 156:241–249. 10.1016/j.resp.2006.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brigham KL, Serafin W, Zadoff A, Blair I, Meyrick B, Oates JA. 1988. Prostaglandin E2 attenuation of sheep lung responses to endotoxin. J. Appl. Physiol. 64:2568–2574 [DOI] [PubMed] [Google Scholar]
- 33.Clark JG, Kostal KM, Marino BA. 1982. Modulation of collagen production following bleomycin-induced pulmonary fibrosis in hamsters. Presence of a factor in lung that increases fibroblast prostaglandin E2 and cAMP and suppresses fibroblast proliferation and collagen production. J. Biol. Chem. 257:8098–8105 [PubMed] [Google Scholar]
- 34.Hasday JD, Shah N, Mackowiak PA, Tulapurkar M, Nagarsekar A, Singh I. 2011. Fever, hyperthermia, and the lung: it's all about context and timing. Trans. Am. Clin. Climatol. Assoc. 122:34–47 [PMC free article] [PubMed] [Google Scholar]
- 35.Brant KA, Fabisiak JP. 2009. Nickel and the microbial toxin, MALP-2, stimulate proangiogenic mediators from human lung fibroblasts via a HIF-1alpha and COX-2-mediated pathway. Toxicol. Sci. 107:227–237. 10.1093/toxsci/kfn208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miyauchi M, Hiraoka M, Oka H, Sato S, Kudo Y, Ogawa I, Noguchi K, Ishikawa I, Takata T. 2004. Immuno-localization of COX-1 and COX-2 in the rat molar periodontal tissue after topical application of lipopolysaccharide. Arch. Oral Biol. 49:739–746. 10.1016/j.archoralbio.2004.04.004 [DOI] [PubMed] [Google Scholar]
- 37.Zidar N, Odar K, Glavac D, Jerse M, Zupanc T, Stajer D. 2009. Cyclooxygenase in normal human tissues—is COX-1 really a constitutive isoform, and COX-2 an inducible isoform? J. Cell. Mol. Med. 13:3753–3763. 10.1111/j.1582-4934.2008.00430.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carey MA, Bradbury JA, Rebolloso YD, Graves JP, Zeldin DC, Germolec DR. 2010. Pharmacologic inhibition of COX-1 and COX-2 in influenza A viral infection in mice. PLoS One 5:e11610. 10.1371/journal.pone.0011610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kolaczkowska E, Scislowska-Czarnecka A, Chadzinska M, Plytycz B, van Rooijen N, Opdenakker G, Arnold B. 2006. Enhanced early vascular permeability in gelatinase B (MMP-9)-deficient mice: putative contribution of COX-1-derived PGE2 of macrophage origin. J. Leukoc. Biol. 80:125–132. 10.1189/jlb.0106013 [DOI] [PubMed] [Google Scholar]
- 40.Giovanni G, Giovanni P. 2002. Do non-steroidal anti-inflammatory drugs and COX-2 selective inhibitors have different renal effects? J. Nephrol. 15:480–488 [PubMed] [Google Scholar]
- 41.Dieter P, Scheibe R, Kamionka S, Kolada A. 2002. LPS-induced synthesis and release of PGE2 in liver macrophages: regulation by CPLA2, COX-1, COX-2, and PGE2 synthase. Adv. Exp. Med. Biol. 507:457–462. 10.1007/978-1-4615-0193-0_71 [DOI] [PubMed] [Google Scholar]
- 42.Gresham A, Masferrer J, Chen X, Leal-Khouri S, Pentland AP. 1996. Increased synthesis of high-molecular-weight cPLA2 mediates early UV-induced PGE2 in human skin. Am. J. Physiol. 270:C1037–C1050 [DOI] [PubMed] [Google Scholar]
- 43.Mizumura K, Hashimoto S, Maruoka S, Gon Y, Kitamura N, Matsumoto K, Hayashi S, Shimizu K, Horie T. 2003. Role of mitogen-activated protein kinases in influenza virus induction of prostaglandin E2 from arachidonic acid in bronchial epithelial cells. Clin. Exp. Allergy 33:1244–1251. 10.1046/j.1365-2222.2003.01750.x [DOI] [PubMed] [Google Scholar]
- 44.Vancheri C, Mastruzzo C, Sortino MA, Crimi N. 2004. The lung as a privileged site for the beneficial actions of PGE2. Trends Immunol. 25:40–46. 10.1016/j.it.2003.11.001 [DOI] [PubMed] [Google Scholar]
- 45.Radi ZA, Meyerholz DK, Ackermann MR. 2010. Pulmonary cyclooxygenase-1 (COX-1) and COX-2 cellular expression and distribution after respiratory syncytial virus and parainfluenza virus infection. Viral Immunol. 23:43–48. 10.1089/vim.2009.0042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gilroy DW, Tomlinson A, Willoughby DA. 1998. Differential effects of inhibition of isoforms of cyclooxygenase (COX-1, COX-2) in chronic inflammation. Inflamm. Res. 47:79–85. 10.1007/s000110050285 [DOI] [PubMed] [Google Scholar]
- 47.Lipsky PE. 1999. Specific COX-2 inhibitors in arthritis, oncology, and beyond: where is the science headed? J. Rheumatol. Suppl. 56:25–30 [PubMed] [Google Scholar]
- 48.Soslow RA, Dannenberg AJ, Rush D, Woerner BM, Khan KN, Masferrer J, Koki AT. 2000. COX-2 is expressed in human pulmonary, colonic, and mammary tumors. Cancer 89:2637–2645. [DOI] [PubMed] [Google Scholar]
- 49.Bottone FG, Jr, Martinez JM, Alston-Mills B, Eling TE. 2004. Gene modulation by Cox-1 and Cox-2 specific inhibitors in human colorectal carcinoma cancer cells. Carcinogenesis 25:349–357. 10.1093/carcin/bgh016 [DOI] [PubMed] [Google Scholar]
- 50.Tanaka A, Araki H, Hase S, Komoike Y, Takeuchi K. 2002. Up-regulation of COX-2 by inhibition of COX-1 in the rat: a key to NSAID-induced gastric injury. Aliment. Pharmacol. Ther. 16(Suppl. 2):90–101. 10.1046/j.1365-2036.16.s2.22.x [DOI] [PubMed] [Google Scholar]
- 51.Zhang J, Fujii S, Wu Z, Hashioka S, Tanaka Y, Shiratsuchi A, Nakanishi Y, Nakanishi H. 2006. Involvement of COX-1 and up-regulated prostaglandin E synthases in phosphatidylserine liposome-induced prostaglandin E2 production by microglia. J. Neuroimmunol. 172:112–120. 10.1016/j.jneuroim.2005.11.008 [DOI] [PubMed] [Google Scholar]
- 52.Nagataki M, Moriyuki K, Sekiguchi F, Kawabata A. 2008. Evidence that PAR2-triggered prostaglandin E2 (PGE2) formation involves the ERK-cytosolic phospholipase A2-COX-1-microsomal PGE synthase-1 cascade in human lung epithelial cells. Cell Biochem. Funct. 26:279–282. 10.1002/cbf.1434 [DOI] [PubMed] [Google Scholar]
- 53.Sandee D, Sivanuntakorn S, Vichai V, Kramyu J, Kirtikara K. 2009. Up-regulation of microsomal prostaglandin E synthase-1 in COX-1 and COX-2 knock-out mouse fibroblast cell lines. Prostaglandins Other Lipid Mediat. 88:111–116. 10.1016/j.prostaglandins.2008.12.001 [DOI] [PubMed] [Google Scholar]
- 54.Son DS, Wilson AJ, Parl AK, Khabele D. 2010. The effects of the histone deacetylase inhibitor romidepsin (FK228) are enhanced by aspirin (ASA) in COX-1 positive ovarian cancer cells through augmentation of p21. Cancer Biol. Ther. 9:928–935. 10.4161/cbt.9.11.11873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lacroix S, Rivest S. 1998. Effect of acute systemic inflammatory response and cytokines on the transcription of the genes encoding cyclooxygenase enzymes (COX-1 and COX-2) in the rat brain. J. Neurochem. 70:452–466 [DOI] [PubMed] [Google Scholar]