

Abstract
Chronic hepatitis B virus (HBV) infection is a major risk factor for liver cirrhosis and hepatocellular carcinoma. Nevertheless, the molecular mechanism of HBV replication remains elusive. SIRT1 is a class III histone deacetylase that is a structure component of the HBV cccDNA minichromosome. In this study, we found by using microarray-based gene expression profiling analysis that SIRT1 was upregulated in HBV-expressing cells. Gene silencing of SIRT1 significantly inhibited HBV DNA replicative intermediates, 3.5-kb mRNA, and core protein levels. In contrast, the overexpression of SIRT1 augmented HBV replication. Furthermore, SIRT1 enhanced the activity of HBV core promoter by targeting transcription factor AP-1. The c-Jun subunit of AP-1 was bound to the HBV core promoter region, as demonstrated by using a chromatin immunoprecipitation assay. Mutation of AP-1 binding site or knockdown of AP-1 abolished the effect of SIRT1 on HBV replication. Finally, SIRT1 inhibitor sirtinol also suppressed the HBV DNA replicative intermediate, as well as 3.5-kb mRNA. Our study identified a novel host factor, SIRT1, which may facilitate HBV replication in hepatocytes. These data suggest a rationale for the use of SIRT1 inhibitor in the treatment of HBV infection.
INTRODUCTION
Hepatitis B virus (HBV) infection remains a major health problem worldwide despite the availability of an effective prevention vaccine. HBV is a hepatotropic, noncytopathic, 3.2-kb partially double-stranded DNA virus (1). Upon viral entry into a hepatocyte, the capsid dissociates, and the genomic relaxed circular DNA (rcDNA) is converted into a covalently closed circular DNA (cccDNA) molecule. The cccDNA is found as a viral minichromosome and acts as the major transcriptional template for the virus. Four major RNA species (3.5-, 2.4-, 2.1-, and 0.7-kb viral RNA transcripts) produced from this viral minichromosome are then transported to and translated in the cytoplasm to produce the viral proteins, namely, viral surface proteins, core protein, viral polymerase, and X protein (2).
The control of HBV replication is one of the major strategies to reduce the morbidity and mortality associated with chronic HBV infection. Thus far, no treatment can completely eliminate the infection in all patients with chronic hepatitis B. The chemotherapeutic treatments (i.e., lamivudine, adefovir, entecavir, and telbivudine) inhibit the HBV replication by targeting the viral DNA polymerase. However, long-term treatment leads to the development of problematic drug-resistant virus. Alpha interferon is also useful for treating HBV infection but has substantial side effects. Therefore, identification and characterization of the viral and host factors responsible for HBV infection will provide valuable information for the development of anti-HBV therapeutics.
The sirtuin family (SIRT1 to SIRT7) are mammalian homologues of the yeast silent information regulator 2 (Sir2), which are class III histone deacetylases (HDACs) that utilize NAD+ as a cofactor for their functions (3). SIRT1, the most widely studied sirtuin, plays an important role in the regulation of many cellular functions, including stress responses, cell proliferation, DNA repair and apoptosis (4), and carcinogenesis (5). In addition to its enzymatic activity on histone substrates, SIRT1 deacetylates various nonhistone proteins, such as p53 (6), Ku70 (7), nuclear factor κB (NF-κB) (8), and forkhead transcription factor (FOXO) (9). Emerging evidence suggests that SIRT1 is also involved in virus infection. The HIV-1 Tat protein is also a substrate for the deacetylase activity of SIRT1 (10). SIRT1 has been found in regulating Tat-induced HIV-1 transactivation (11). During HBV infection, SIRT1 was found to recruit to HBV cccDNA minichromosome (12).
In this study, we investigated the role of SIRT1 in HBV replication. Our data showed that SIRT1 was significantly upregulated in HBV-expressing cell lines and that the silencing or overexpression of SIRT1 regulated HBV replication. Further, SIRT1 enhanced HBV core promoter activity through transcription factor AP-1. Finally, SIRT1 inhibitor sirtinol also markedly suppressed HBV DNA replication. Our data suggested a role for SIRT1 in HBV replication and identified it as a novel host factor in the HBV replication process.
MATERIALS AND METHODS
Materials.
Lentivirus plasmids vector pLKO.1-puro containing MISSION short-hairpin RNA (shRNA) targeting SIRT1 (SHC001) or nontargeting shRNA (SH2421) were obtained from Sigma-Aldrich. AP-1 shRNA (targeting c-Jun subunit) or nontargeting shRNA (shCont) was cloned into a modified pLentilox-3.7 lentivirus plasmid vector containing a blasticidin-resistant gene. The sequence of c-Jun targeting shRNA is 5′-CGGACCTTATGGCTACAGTAAC-3′, and the sequence of shCont is 5′-GCAACAAGATGAAGAGCACCAA-3′. SIRT1 was constructed by in-frame insertion of full-length SIRT1 into pcDNA3.1 that contains a Flag tag at the C terminus. pCH9/3091 was obtained from Lin Lan (The Third Military Medical University, China). pGEM-HBV1.3 was a gift from U. Protzer (University of Heidelberg, Heidelberg, Germany). Rabbit anti-SIRT1 monoclonal antibody (ab32441) was obtained from Abcam. Rabbit c-Jun polyclonal antibody (catalog no. 9165) was obtained from Cell Signaling Technology. Rabbit anti-hepatitis B virus core antigen (HBcAg) polyclonal antibody (B0586) was obtained from Dako (Denmark). Rabbit anti-β-actin monoclonal antibody (sc-1616-R) was obtained from Santa Cruz Biotechnology (USA), and sirtinol (CAS 410536-97-9) was obtained from Calbiochem (USA).
Cell culture and transfection.
HepG2 and Huh-7 were maintained in modified Eagle medium (MEM) containing 10% fetal calf serum. HepG2.2.15 and HepG2-HBV1.1 cells (stably HBV-transfected HepG2 cells) were maintained in MEM containing 10% fetal calf serum and 500 μg of G418/ml. Primary human hepatocytes (PHH) were maintained in hepatocyte medium (catalog no. 5210; Sciencell). All cells were maintained in a humidified incubator at 37°C with 5% CO2. Transfection was carried out using Lipofectamine 2000 (Invitrogen).
Closed circular HBV DNA preparation.
The HBV sequence was amplified from pGEM-HBV1.3 by PCR using the sense primer P1 (5′-CCGGAATGCTCTTCTTTTTCACCTCTGCCTARTCATC-3′) and the antisense primer P2 (5′-CCGGAATGCTCTTCAAAAAGTTGCATGGTGCTGGTG-3′). The PCR product was purified and digested with BspQI. To circularize the HBV genome, the digested or gel-purified HBV DNA was ligated with T4 DNA ligase. Afterward, the ligation product was extracted and resuspended in Opti-MEM.
Southern blot analysis.
HBV replicative intermediates were obtained as follows. Cells from one 35-mm-diameter dish were lysed with 0.5 ml of lysis buffer (10 mM Tris-HCl [pH 8.0], 1 mM EDTA, 1% NP-40, 2% sucrose) at 37°C for 15 min. Cell debris and nuclei were removed by centrifugation; the supernatant was treated with 40 U of DNase I/ml and 10 mM MgCl2 for 4 h and then mixed with 200 μl of 35% polyethylene glycol 8000 containing 1.5 M NaCl. After incubation on ice for 1 h, viral nucleocapsids were pelleted by centrifugation at 11,000 × g for 5 min at 4°C, followed by overnight digestion at 45°C in 500 μl of digestion buffer containing 0.5 mg of proteinase K (TaKaRa)/ml, 0.5% sodium dodecyl sulfate (SDS), 150 mM NaCl, 25 mM Tris-HCl (pH 8.0), and 10 mM EDTA. The digestion mixture was extracted twice with phenol, and DNA was precipitated with ethanol and dissolved in TE (10 mM Tris-HCl [pH 8.0], 1 mM EDTA) buffer and separated on 0.9% agarose gels. DNA samples were transferred onto nylon membranes (Roche, Germany). After UV cross-linking and prehybridization, the membrane was hybridized with digoxigenin-labeled HBV-specific probe generated by using a random-primed labeling kit (Roche, Germany). The signal was detected by exposing on an X-ray film.
Quantitative real-time PCR (qPCR).
The 3.5-kb mRNA was detected according to the method of Levrero and coworkers (13). Absolute quantification of HBV replicative intermediates were carried out using SYBR green (Roche, Germany). Relative quantification of AP1 was carried out by using EvaGreen Supermix (Bio-Rad) with β-actin mRNA as an endogenous control. Values represent the means ± the standard deviations (SD) of three independent experiments. The expression values of target genes were calculated by using the 2−ΔΔCT method.
Western blotting.
The protein concentration was determined by using a BCA protein assay (Bio-Rad). Total proteins (30 μg) were separated by SDS-PAGE and transferred onto a nitrocellulose membrane. The membrane was then blocked with 5% nonfat milk for 1 h and incubated with the relevant primary antibodies (anti-SIRT1, 1:5,000; anti-c-Jun, 1:1,000; anti-core protein, 1:1,000; anti-β-actin, 1:2,000) overnight at 4°C. Blots were developed by using ECL Western blot reagents.
ChIP.
Chromatin immunoprecipitation (ChIP) assays were performed on genomic DNA samples from HepG2.2.15 cells and HepG2 cells using antibody specific to c-Jun according to the manufacturer's protocol (Millipore, Darmstadt, Germany). The region of the core promoter of HBV, including the c-Jun sites, was amplified from the immunoprecipitated DNA samples by PCR using the sense primer P1 (5′-CACCAGGTCTTGCCCAAGGTCTTAC-3′) and the antisense primer P2 (5′-ACAAACAGTCTTTGAAGTATGCCTC-3′).
Detection of HBsAg and HBeAg.
The levels of HBV surface antigen (HBsAg) and HBeAg in culture medium were assessed by using an enzyme-linked immunosorbent assay (ELISA) according to the manufacturer's protocol (KHB, Shanghai, China). Each experiment was performed in triplicate and independently repeated three times.
Luciferase assay.
The luciferase report vectors (pGL3-Cp, pGL3-Xp, pGL3-Sp1, and pGL3-Sp2) were cotransfected with pcDNA3.1-SIRT1 or pcDNA3.1 into HepG2 cells. pRL-TK was cotransfected with reporter plasmid to normalize the transfection efficiency. After transfection for 36 h, the cells were lysed for luciferase activity measurement by using a dual-luciferase reporter assay system (Promega, USA) according to the manufacturer's instructions. The luciferase activity was determined by GloMax microplate luminometer (Promega).
Cell proliferation assay.
The effect of sirtinol treatment on cell proliferation was measured by using a (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt) (MTS) assay according to the manufacturer's instructions (Promega).
Statistical analysis.
Results are expressed as means ± the SD. Differences between selected groups were evaluated using the Student t test and the Fisher exact test. A difference was considered significant when P < 0.05. All statistical analyses were performed using SPSS for Windows 10.0.
RESULTS
Expression of SIRT1 in HBV-expressing cells.
To investigate how HBV alters gene expression in liver cells, the gene expression profile was compared in HepG2 and HepG2.2.15 cell lines. HepG2.2.15 is an HBV stably transfected cell line constitutively producing HBV (14). Array-based transcriptional mapping revealed that HBV replication significantly upregulated the expression of SIRT1 in HepG2.2.15 cells. To verify whether SIRT1 was specifically upregulated by HBV replication, we first examined the mRNA and protein levels of SIRT1 in a panel of cell lines, including primary human hepatocytes (PHH), HepG2, HepG2.2.15, and HepG2-pHBV1.1, which is another HepG2 cell line stably expressing HBV established in our lab (14), and human hepatoma HepG2 cells transiently transfected with HBV expressing plasmid pCH9/3091 (containing a 1.1-unit length HBV genome driven by a cytomegalovirus promoter) or pGEM-HBV1.3 (containing a 1.3-unit length HBV genome driven by an endogenous viral promoter). Both mRNA and protein levels of SIRT1 were upregulated in HBV-expressing cells relative to control cells, whereas SIRT1 was expressed at a very low level in PHH cells (Fig. 1A and B). To confirm the induction of SIRT1 by HBV, PHH cells were infected with recombinant adenoviruses expressing HBV (Ad-HBV) and control vector (Ad-Vector). Consistently, HBV replication in PHH cells enhanced the expression of SIRT1 (Fig. 1B). In addition, the expression of SIRT1 was also examined in the HepAD38 cell line, in which HBV replication can be regulated by tetracycline. The core protein was detected to confirm that HBV gene expression was indeed regulated by tetracycline (Fig. 1C). The mRNA and protein levels of SIRT1 in HepAD38 cells without tetracycline were much higher than in HepAD38 cells with tetracycline (Fig. 1C and D). Taken together, these data revealed that HBV replication upregulated SIRT1 expression, suggesting that SIRT1 might play a role in the HBV replication process.
FIG 1.
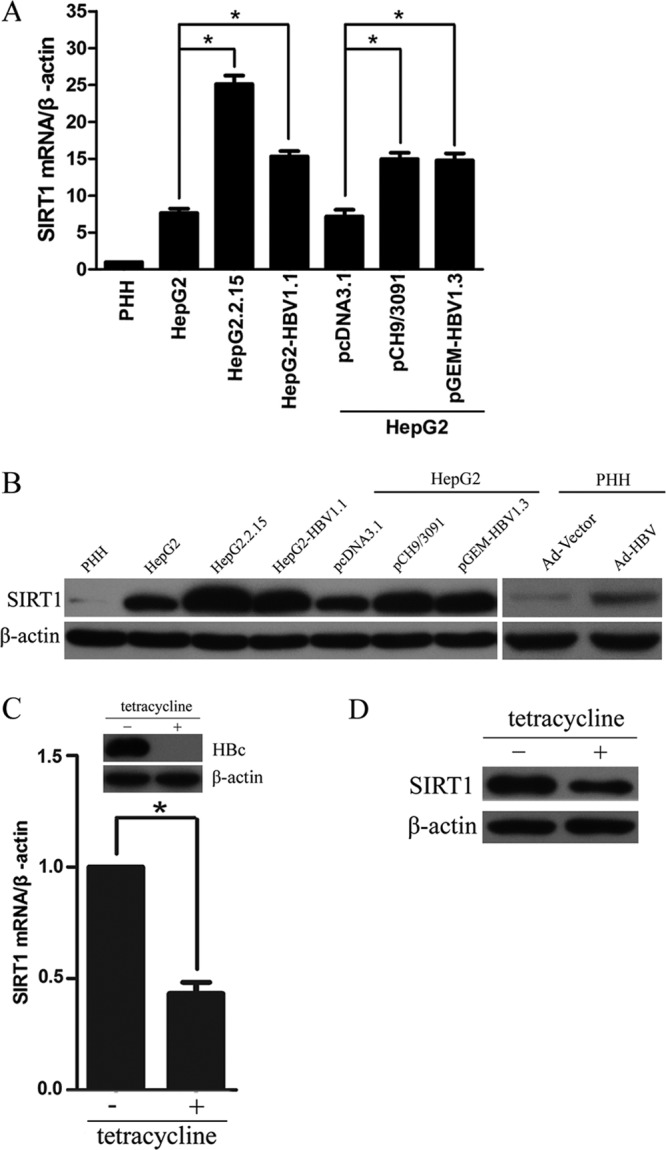
Expression of SIRT1 in HBV-expressing cells. (A) The SIRT1 mRNA levels in PHH, HepG2, HepG2.2.15, HepG2-HBV1.1, and HepG2 cells transiently transfected with plasmid pCH9/3091 or pGEM-HBV1.3 were analyzed by real-time PCR. *, P < 0.01. (B) The SIRT1 protein levels in HepG2, HepG2.2.15, HepG2-HBV1.1, and HepG2 cells transiently transfected with plasmid pCH9/3091 or pGEM-HBV1.3 and in PHHs infected with adenoviruses expressing HBV (Ad-HBV) and control vector (Ad-Vector) were analyzed by Western blotting. β-Actin was used as a loading control. (C and D) Expression of SIRT1 in HepAD38 cells. Cells were cultured in medium with tetracycline (no HBV replication) or without tetracycline (HBV replication) and subjected to real-time PCR analysis (C) and Western blotting (D).
Silencing of SIRT1 suppressed the replication of HBV.
To elucidate the functional role of SIRT1 in HBV replication, we transduced HepG2.2.15 and HepG2-HBV1.1 with lentiviruses containing shRNAs targeting SIRT1 or scramble control shRNA. Two independent shRNAs (shSIRT1-1 and shSIRT1-2) showed efficient knockdown of SIRT1 (>90%) in both cell lines compared to scramble shRNA (shCont)-transduced cells (Fig. 2A). The effect of SIRT1 knockdown on HBV replication was first examined in HepG2.2.15 and HepG2-HBV1.1 cells. Downregulation of SIRT1 resulted in decreased levels of HBV DNA replicative intermediates, as evidenced by both real-time PCR (Fig. 2B) and Southern blotting (Fig. 2C). The 3.5-kb mRNA is composed of two precore (pcRNA) and pregenomic (pgRNA) mRNAs. pgRNA serves as the template for reverse transcription and genomic DNA formation, as well as for the translation of the two proteins: HBc and Pol. pcRNA serves as a template to translate the precore, which is further processed into secreted HBeAg. We therefore investigated whether SIRT1 affected this important 3.5-kb mRNA expression. Real-time PCR result revealed that 3.5-kb mRNA level was significantly reduced in SIRT1-silencing cells. Quantification of 3.5-kb mRNA levels showed a 50 to 70% decrease after SIRT1 knockdown (Fig. 2D). In addition, HBV core protein was further determined by Western blotting, and HBsAg and HBeAg expression levels in cell culture supernatant were analyzed by ELISA. Similar to its effect on HBV DNA replicative intermediates and 3.5-kb mRNA, downregulation of SIRT1 also inhibited core protein expression, as well as HBsAg and HBeAg secretions (Fig. 2E and F). On the other hand, the effect of SIRT1 silencing on HBV replication was further analyzed in Huh-7 cells transiently transfected with plasmid pGEM-HBV1.3 or closed circular HBV genome. Concordantly, gene silencing of SIRT1 significantly inhibited HBV DNA level (Fig. 2G and H). These results suggested that SIRT1 knockdown could repress HBV replication.
FIG 2.

Gene silencing of SIRT1 inhibited HBV replication. (A) Western blotting of SIRT1 protein expression in cells transduced with lentiviruses expressing scramble control (shCont) or SIRT1-targeting shRNAs (shSIRT1-1 and shSIRT1-2). Cells were harvested 5 days after viral transduction. β-Actin was used as a loading control. (B to F) Effects of SIRT1 knockdown on HBV replication in HepG2.2.15 and HepG2-HBV1.1 cells. HBV replicative intermediates were extracted from nucleocapsid 5 days after viral transduction. (B) Real-time PCR of HBV DNA. The results are expressed as the number of HBV DNA copies per cells. *, P < 0.001. (C) Southern blot analysis. M, ladder; rcDNA, intracellular HBV relax circle; dsDNA, double-stranded DNA; ssDNA, single-stranded DNA. (D) The 3.5-kb mRNA levels were analyzed by qPCR using selective primers and β-actin primers (equal loading of each RNA samples) at 4 days after lentiviral transduction. *, P < 0.001. (E) Core protein was determined by using Western blotting. β-Actin was used as a loading control. (F) Expression of HBsAg and HBeAg antigen detected using ELISA 5 days after lentiviral transduction. *, P < 0.05. (G) Huh-7 cells were transduced with lentiviruses expressing indicated shRNAs 2 days after transfection with pGEM-HBV1.3. The HBV replicative intermediates were then analyzed by real-time PCR. (H) HBV replicative intermediates extracted from Huh-7 cells transfected with the closed circular HBV genome were analyzed by real-time PCR (left panel) and Southern blotting (right panel). The PCR product amplified from pGEM-HBV1.3 was purified and digested with BspQI. The digested HBV DNA was then ligated to form closed circular HBV DNA.
Overexpression of SIRT1 promoted HBV replication.
The results described above suggested endogenous SIRT1 may facilitate the replication of HBV. To further test this hypothesis, HepG2.2.15 and HepG2-pHBV1.1 cells were transfected with vector expressing SIRT1. SIRT1 was overexpressed markedly in both cell lines (Fig. 3A). Real-time PCR (Fig. 3B) and Southern blot (Fig. 3C) analyses showed that overexpression of SIRT1 increased the level of HBV DNA replicative intermediates significantly. Compared to cells transfected with control vector pcDNA3.1, HepG2.2.15 cells ectopically expressing SIRT1 increased the level of 3.5-kb mRNA nearly 2-fold (Fig. 3D). Furthermore, the overexpression of SIRT1 also resulted in the upregulation of HBsAg and HBeAg expression (Fig. 3E and F).
FIG 3.

SIRT1 overexpression promoted HBV replication. (A) Western blotting of SIRT1 proteins in cells transfected with pcDNA3.1 vector expressing Flag-SIRT1. β-Actin was used as a loading control. (B and C) SIRT1 overexpression enhanced HBV DNA level. HBV intermediates extracted from nucleocapsid 5 days after transfection were subjected to real-time PCR (B) and Southern blotting (C). (D) Ectopic expression of SIRT1 increased the 3.5-kb mRNA level. Total RNA was extracted from HepG2.2.15 and HepG2-HBV1.1 cells after transfection for 4 days. *, P < 0.001. (E and F) HBsAg and HBeAg ELISA were used to screen culture supernatants 5 days after transfection. *, P < 0.05.
Effect of SIRT1 on the promoter activity of HBV genes.
To investigate the mechanism by which SIRT1 acts on HBV, we examined the effect of SIRT1 on the activity of HBV SpI, SpII, core, and X promoters using luciferase reporter assay. Overexpression of SIRT1 significantly enhanced core promoter activity but had no effect on SpI, SpII, and X promoter activities (Fig. 4A). In contrast, downregulation of SIRT1 suppressed nearly 50% of the core promoter activity (Fig. 4B). Transcription factors, such as nuclear factor κB (NF-κB), the activator protein-1 (AP-1) family, and PPARα, have been shown to play important roles on HBV transcription (15). To further examine the mechanism of transcriptional inhibition of SIRT1 on the HBV core promoter, a panel of transcription factors, including AP-1, COUP-TF1, CREB1, CREB2, HNF4, NF-κB, p53, p56, and TR2, was screened by real-time PCR (Fig. 4C). AP-1 can be formed through either homodimerization of Jun proteins (c-Jun, JunB, and JunD) or heterodimerization of Jun and Fos proteins (c-Fos, FosB, Fra-1, and Fra-2). In the present study, we found that the mRNA and protein levels of c-Jun subunit of AP-1 were decreased in SIRT1-silencing HepG2.2.15 cells (Fig. 4C and D). On the other hand, the overexpression of SIRT1 significantly increased the c-Jun protein level (Fig. 4E). We further checked the sequence of the HBV core promoter and found a binding site for AP-1 analyzed by software predicting the transcription binding site (data not shown). To determine the interaction between AP-1 and HBV core promoter, we performed a chromatin immunoprecipitation (ChIP) assay. The data indicated HBV core promoter fragment containing AP-1 binding site could be detected in anti-c-Jun antibody-immunoprecipitated candidates in HepG2.2.15 cells, whereas it was undetectable in HepG2 cells (Fig. 5A). Importantly, the enhancement of HBV replication induced by SIRT1 was abolished when the binding site of AP-1 in HBV core promoter was mutated (Fig. 5B and C). To confirm that AP-1 was involved in the SIRT1-mediated regulation of HBV replication, we cotransfected HepG2.2.15 cells with vector expressing SIRT1 and plasmid expressing shRNA targeting c-Jun. The knockdown efficiency of c-Jun was confirmed by Western blot analysis (Fig. 5D). A dual-luciferase assay showed that augmenting the HBV core promoter activity mediated by SIRT1 was abolished when c-Jun was suppressed (Fig. 5E). Importantly, silencing of c-Jun subunit of AP-1 also abolished enhancement of HBV replication induced by SIRT1, as evidenced by real-time PCR and Southern blot analysis (Fig. 5F and G). Taken together, these results suggested that SIRT1 is a positive regulator of AP-1 and that SIRT1 may upregulate the activity of HBV core promoter via AP-1.
FIG 4.

SIRT1 regulates HBV core promoter activity. (A) Effect of SIRT1 overexpression on four HBV promoters. Different luciferase reporter vectors were cotransfected with pcDNA3.1-Flag-SIRT1 into HepG2 cells. The plasmid RL-TK was cotransfected to normalize the transfection efficiency. The luciferase activity was measured at 48 h posttransfection. *, P < 0.001. (B) The activity of core promoter was suppressed in SIRT1-silencing cells. The HepG2 cells were transduced with lentivirus expressing the indicated shRNAs 10 h after the transfection of plasmids (pGL3-Cp and RL-TK). The luciferase activity was measured at 72 h posttransduction. *, P < 0.001. (C) Real-time PCR analysis of gene expression of various transcription factors related to HBV replication. *, P < 0.01. (D) The expression of c-Jun subunit of AP-1 in SIRT1-silencing cells was analyzed by using Western blotting. β-Actin was used as a loading control. (E) SIRT1 overexpression induced c-Jun expression. HepG2.2.15 cells were transfected with vector pcDNA3.1-Flag-SIRT1 before analysis. β-Actin was used as a loading control.
FIG 5.

Transcription factor AP-1 was involved in SIRT1-mediated regulation of HBV replication. (A) ChIP assays were performed to confirm the interaction between AP-1 and HBV core promoter region. (B and C) Mutated AP-1 binding site abolished SIRT1-induced promotion of HBV replication. The HepG2 cells were cotransfected with plasmids pGEM-HBV1.3 or pGEM-HBV1.3 MUT (containing mutated AP-1 binding site) and pcDNA3.1-Flag-SIRT1. The effect of SIRT1 on HBV replication was then analyzed by real-time PCR (B) and Southern blot analysis (C). *, P < 0.01. (D) Western blotting of SIRT1, c-Jun, and β-actin in HepG2.2.15 cells. The plasmid pcDNA3.1-Flag-SIRT1 was cotransfected with shRNA targeting c-Jun subunit of AP-1 (shc-Jun) or scramble control shRNA (shCont), respectively, for 72 h before analysis. (E) c-Jun knockdown abolished SIRT1-induced upregulation of core promoter. The HepG2 cells were harvested for dual-luciferase assay after cotransfection with plasmids as indicated. *, P < 0.01. (F and G) Effect of c-Jun silencing on HBV replication in cells overexpressing SIRT1. HepG2.2.15 or HepG2-HBV1.1 cells were cotransfected with indicated plasmids, respectively. HBV replicative intermediates were extracted and then subjected to real-time PCR (F) and Southern blot analysis (G). *, P < 0.01.
Effect of a SIRT1 inhibitor, sirtinol, on HBV replication.
In order to ascertain the biological significance of SIRT1 in HBV replication process, we determined the consequence of SIRT1 inhibition by an inhibitor (sirtinol) on HBV replicative cells. Sirtinol was found to inhibit SIRT1 transcriptional activity directly without affecting the other classes of histone deacetylases. The HepG2.2.15 cells and HepG2-HBV1.1 cells were treated with sirtinol, and the cytotoxicity of sirtinol was first studied using an MTS assay. The concentrations causing 50% cytotoxicity were 100 μM in HepG2.2.15 cells and 150 μM in HepG2-HBV1.1 cells (Fig. 6A). Sirtinol treatment (25 or 50 μM in HepG2.2.15 cells; 12.5 or 25 μM in HepG2-HBV1.1 cells) for 48 h resulted in a significantly decreased level of HBV DNA replicative intermediates, as shown by real-time PCR (Fig. 6B) and Southern blot (Fig. 6C) analysis in a dose-dependent manner. Concordantly, 3.5-kb mRNA levels in both two cell lines were inhibited under sirtinol treatment (Fig. 6D). In contrast, treatment with the class I/class II HDAC inhibitor trichostatin A (TSA) enhanced HBV DNA replicative intermediates (data not shown). These data suggested that class I/class II histone deacetylases upregulate HBV replication, possibly through the epigenetic regulation of cccDNA minichromosome, whereas the SIRT1 inhibitor may have anti-HBV potential.
FIG 6.

Effect of SIRT1 inhibitor sirtinol on HBV replication. (A) Cell viability in HepG2.2.15 and HepG2-HBV1.1 cells after sirtinol treatment 48 h was examined by using MTS assay. (B and C) Sirtinol treatment inhibited HBV DNA level. HBV DNA in HepG2.2.15 and HepG2-HBV1.1 cells treated with different concentration of sirtinol for 48 h were extracted and subjected to real-time PCR (B) and Southern blot analysis (C). (D) The HBV 3.5-kb mRNA levels in HepG2.2.15 and HepG2-HBV1.1 cells treated with sirtinol were analyzed by using real-time PCR. *, P < 0.001.
DISCUSSION
SIRT1 displays a wide array of cellular functions, ranging from stress response (16) to DNA repair (17), metabolism in multiple tissues (18), and tumor development (19). SIRT1 is upregulated in HBV-related HCC tissues, where it is essential for tumor cell growth, suggesting a potential role of SIRT1 in HBV-related HCC carcinogenesis (5). In the present study, we examined the effect of SIRT1 on HBV replication and found that the suppression of SIRT1 possessed antiviral property.
HBV cccDNA, the template for transcription of all viral mRNAs, accumulates as a stable episome organized into minichromosome by histone and nonhistone proteins. A recent study by Belloni et al. (12) found that transcription of the HBV minichromosome is regulated by epigenetic changes of cccDNA-bound histones. Cellular histone acetyltransferases (CBP, p300, and PCAF/GCN5) and the histone deacetylases (HDAC1 and SIRT1) are all recruited in vivo onto the cccDNA. These researchers further demonstrated that high recruitment of HDAC1 and SIRT1 onto cccDNA correlated with a decline in HBV replication. In contrast to those findings, our results indicated that SIRT1 was upregulated in HBV-infected cells. The overexpression of SIRT1 augmented HBV replication and suppression of SIRT1 by RNA interference, and inhibitor suppressed HBV replication significantly in cells stably or transiently expressing HBV. Unfortunately, the Belloni study did not determine the expression of SIRT1 in HBV-infected cells and the direct effect of SIRT1 on HBV replication, which precluded a direct comparison between the two studies. In view of the discrepancy observed, we also examined the effect of class I/class II histone deacetylase inhibitor TSA on HBV replication. Consistent with the findings of Belloni et al., both real-time PCR and Southern blotting results showed that TSA treatment enhanced HBV replicative intermediates. This suggested that class I/class II histone deacetylase, not SIRT1, play a major role in the epigenetic regulation of cccDNA minichromosome. However, direct evidence support for this hypothesis requires further investigation. Based on our findings, SIRT1 functions as a positive regulator of HBV replication through the regulation of HBV core promoter activity. Nevertheless, together, these data suggest that SIRT1 plays a functional role in the HBV replication process.
We identified a new molecular mechanism by which SIRT1 regulated HBV replication in addition to the epigenetic regulation of the histone cccDNA minichromosome. We found that SIRT1 regulates HBV core promoter activity when knocked down or overexpressed. This finding is also supported by the observation that SIRT1 regulated 3.5-kb mRNA, core protein, and e antigen levels, which are all transcribed from the core promoter. Further analysis of the transcription factors showed that the activator protein (AP-1) was possibly involved in the regulation of the HBV core promoter by SIRT1. AP-1 is a heterodimeric transcription factor composed of proteins belonging to the c-Fos, c-Jun, ATF, and JDP families (20). It regulates the expression of multiple genes essential for cell proliferation, differentiation, and apoptosis (21). AP-1 has been also implicated in virus infection. An AP-1 binding site was found in the enhancer/core element of the HIV-1 promoter, which controls the ability of HIV-1 to establish latent infection (22). Human serum protein enhances HIV-1 replication and upregulates the transcription factor AP-1 (23). In terms of HBV, AP-1 was shown to be involved in the upregulation of the HBx protein expression by insulin (24) and by E6 protein of human papillomavirus type 16 (HPV-16 E6) (25). In contrast, HBx protein could also enhance AP-1 activation through interaction with Jab1 (26). In the present study, an AP-1 binding site was found in the core promoter region, and AP-1 was also proved to bind to HBV core promoter. Activation of AP-1 by SIRT1 may responsible, at least in part, for the facilitation of HBV replication mediated by SIRT1. In contrast to our finding, Zhang et al. found SIRT1 deacetylated and suppressed AP-1 transcriptional activity by using HEK293 cell model (27). The discrepancy between these two studies may partially depend on different cell models and experimental conditions. We used HepG2.2.15 cells that originated from human hepatocarcinoma cell with HBV replication, which were totally different from HEK293 cells. The HBV replication in cells may cause certain stress or stimulation and finally lead to substrate of SIRT1 or regulation of certain target of SIRT1 differed from cells under normal conditions. Although the mechanism by which SIRT1 promoted HBV replication remains to be further investigated, our findings suggest that the activation of AP-1 is probably involved.
At present, a number of SIRT1 inhibitors have been discovered. These include the 2-hydorxynaphthaldehyde derivative of sirtinol (28), the physiological sirtuin inhibitor nicotinamide, the coumarin derivative of splitomicin (29), cambinol (30), and tenovin and its derivatives (31). Thus far, two SIRT1 inhibitors, cambinol (30) and tenovin (31), have been tested in animal models of cancer. Sirtinol is a specific inhibitor for SIRT1 and does not inhibit class I and class II HDACs (32). In the present study, sirtinol exhibited robust anti-HBV activity by decreasing the expression of various viral macromolecules, i.e., HBV DNA and 3.5-kb mRNA. The antiviral mechanism of sirtinol is completely different from that of typical HBV reverse transcriptase/polymerase inhibitors. Therefore, this class of drug might represent a novel approach to selectively treat HBV infection and its associated diseases. However, the results are still preliminary, and the toxicity of SIRT1 inhibitor needs to be determined in animal models.
Briefly, we identified critical host genes that facilitate efficiently HBV replication using high-throughput gene profiling strategy. We show here that SIRT1 functions as a promoter gene during the HBV replication process, partially through the activation of transcription factor AP-1. Further study is needed to determine whether SIRT1 could be an anti-HBV target and to assess the possible therapeutic application of sirtinol in anti-HBV therapy.
ACKNOWLEDGMENTS
This study was supported by the National Natural Science Foundation of China (grant 81201282), the National Science and Technology Major Project (2013ZX10002002), the Chongqing Science and Technology Commission (cstc2013jcyjC10002), the Chongqing Natural Science Foundation (cstc2012jjA10047), and a Ph.D. program through the Ministry Education of China (program 20125503120004).
Footnotes
Published ahead of print 11 December 2013
REFERENCES
- 1.Summers J, Mason WS. 1982. Replication of the genome of a hepatitis B–like virus by reverse transcription of an RNA intermediate. Cell 29:403–415. 10.1016/0092-8674(82)90157-X [DOI] [PubMed] [Google Scholar]
- 2.Locarnini S. 2004. Molecular virology of hepatitis B virus. Semin. Liver Dis. 24(Suppl 1):3–10. 10.1055/s-2004-832922 [DOI] [PubMed] [Google Scholar]
- 3.Imai S, Armstrong CM, Kaeberlein M, Guarente L. 2000. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403:795–800. 10.1038/35001622 [DOI] [PubMed] [Google Scholar]
- 4.Michan S, Sinclair D. 2007. Sirtuins in mammals: insights into their biological function. Biochem. J. 404:1–13. 10.1042/BJ20070140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen J, Zhang B, Wong N, Lo AW, To KF, Chan AW, Ng MH, Ho CY, Cheng SH, Lai PB, Yu J, Ng HK, Ling MT, Huang AL, Cai XF, Ko BC. 2011. Sirtuin 1 is upregulated in a subset of hepatocellular carcinomas where it is essential for telomere maintenance and tumor cell growth. Cancer Res. 71:4138–4149. 10.1158/0008-5472.CAN-10-4274 [DOI] [PubMed] [Google Scholar]
- 6.Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. 2001. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 107:149–159. 10.1016/S0092-8674(01)00527-X [DOI] [PubMed] [Google Scholar]
- 7.Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, Gorospe M, de Cabo R, Sinclair DA. 2004. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science 305:390–392. 10.1126/science.1099196 [DOI] [PubMed] [Google Scholar]
- 8.Bouras T, Fu M, Sauve AA, Wang F, Quong AA, Perkins ND, Hay RT, Gu W, Pestell RG. 2005. SIRT1 deacetylation and repression of p300 involves lysine residues 1020/1024 within the cell cycle regulatory domain 1. J. Biol. Chem. 280:10264–10276. 10.1074/jbc.M408748200 [DOI] [PubMed] [Google Scholar]
- 9.Haigis MC, Sinclair DA. 2010. Mammalian sirtuins: biological insights and disease relevance. Annu. Rev. Pathol. 5:253–295. 10.1146/annurev.pathol.4.110807.092250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pagans S, Pedal A, North BJ, Kaehlcke K, Marshall BL, Dorr A, Hetzer-Egger C, Henklein P, Frye R, McBurney MW, Hruby H, Jung M, Verdin E, Ott M. 2005. SIRT1 regulates HIV transcription via Tat deacetylation. PLoS Biol. 3:e41. 10.1371/journal.pbio.0030041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Niu D, Zhang J, Ren Y, Feng H, Chen WN. 2009. HBx genotype D represses GSTP1 expression and increases the oxidative level and apoptosis in HepG2 cells. Mol. Oncol. 3:67–76. 10.1016/j.molonc.2008.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belloni L, Pollicino T, De Nicola F, Guerrieri F, Raffa G, Fanciulli M, Raimondo G, Levrero M. 2009. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc. Natl. Acad. Sci. U. S. A. 106:19975–19979. 10.1073/pnas.0908365106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Belloni L, Allweiss L, Guerrieri F, Pediconi N, Volz T, Pollicino T, Petersen J, Raimondo G, Dandri M, Levrero M. 2012. IFN-α inhibits HBV transcription and replication in cell culture and in humanized mice by targeting the epigenetic regulation of the nuclear cccDNA minichromosome. J. Clin. Invest. 122:529–537. 10.1172/JCI58847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Z, Liu X, Chen J, Su H, Luo Q, Ye J, Tang N, Zhang W, Chen W, Ko BC, Huang A. 2010. Heparin sulfate d-glucosaminyl 3-O-sulfotransferase 3B1 plays a role in HBV replication. Virology 406:280–285. 10.1016/j.virol.2010.07.030 [DOI] [PubMed] [Google Scholar]
- 15.Quasdorff M, Protzer U. 2010. Control of hepatitis B virus at the level of transcription. J. Viral Hepat. 17:527–536. 10.1111/j.1365-2893.2010.01315.x [DOI] [PubMed] [Google Scholar]
- 16.Wang C, Chen L, Hou X, Li Z, Kabra N, Ma Y, Nemoto S, Finkel T, Gu W, Cress WD, Chen J. 2006. Interactions between E2F1 and SirT1 regulate apoptotic response to DNA damage. Nat. Cell Biol. 8:1025–1031. 10.1038/ncb1468 [DOI] [PubMed] [Google Scholar]
- 17.Wang RH, Sengupta K, Li C, Kim HS, Cao L, Xiao C, Kim S, Xu X, Zheng Y, Chilton B, Jia R, Zheng ZM, Appella E, Wang XW, Ried T, Deng CX. 2008. Impaired DNA damage response, genome instability, and tumorigenesis in SIRT1 mutant mice. Cancer Cell 14:312–323. 10.1016/j.ccr.2008.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brooks CL, Gu W. 2009. How does SIRT1 affect metabolism, senescence and cancer? Nat. Rev. Cancer 9:123–128. 10.1038/nrc2562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lim CS. 2007. Human SIRT1: a potential biomarker for tumorigenesis? Cell Biol. Int. 31:636–637. 10.1016/j.cellbi.2006.11.003 [DOI] [PubMed] [Google Scholar]
- 20.Forner A, Hessheimer AJ, Isabel Real M, Bruix J. 2006. Treatment of hepatocellular carcinoma. Crit. Rev. Oncol. Hematol. 60:89–98. 10.1016/j.critrevonc.2006.06.001 [DOI] [PubMed] [Google Scholar]
- 21.Shaulian E, Karin M. 2002. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 4:E131–E136. 10.1038/ncb0502-e131 [DOI] [PubMed] [Google Scholar]
- 22.Duverger A, Wolschendorf F, Zhang M, Wagner F, Hatcher B, Jones J, Cron RQ, van der Sluis RM, Jeeninga RE, Berkhout B, Kutsch O. 2012. An AP-1 binding site in the enhancer/core element of the HIV-1 promoter controls the ability of HIV-1 to establish latent infection. J. Virol. 87:2264–2277. 10.1128/JVI.01594-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perdomo MF, Hosia W, Jejcic A, Corthals GL, Vahlne A. 2012. Human serum protein enhances HIV-1 replication and upregulates the transcription factor AP-1. Proc. Natl. Acad. Sci. U. S. A. 109:17639–17644. 10.1073/pnas.1206893109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi BH, Park CJ, Rho HM. 1998. Insulin activates the hepatitis B virus X gene through the activating protein-1 binding site in HepG2 cells. DNA Cell Biol. 17:951–956. 10.1089/dna.1998.17.951 [DOI] [PubMed] [Google Scholar]
- 25.Liew CT, Li HM, Lo KW, Leow CK, Chan JY, Hin LY, Lau WY, Lai PB, Lim BK, Huang J, Leung WT, Wu S, Lee JC. 1999. High frequency of p16INK4A gene alterations in hepatocellular carcinoma. Oncogene 18:789–795. 10.1038/sj.onc.1202359 [DOI] [PubMed] [Google Scholar]
- 26.Tanaka Y, Kanai F, Ichimura T, Tateishi K, Asaoka Y, Guleng B, Jazag A, Ohta M, Imamura J, Ikenoue T, Ijichi H, Kawabe T, Isobe T, Omata M. 2006. The hepatitis B virus X protein enhances AP-1 activation through interaction with Jab1. Oncogene 25:633–642. 10.1038/sj.onc.1209093 [DOI] [PubMed] [Google Scholar]
- 27.Zhang R, Chen HZ, Liu JJ, Jia YY, Zhang ZQ, Yang RF, Zhang Y, Xu J, Wei YS, Liu DP, Liang CC. 2010. SIRT1 suppresses activator protein-1 transcriptional activity and cyclooxygenase-2 expression in macrophages. J. Biol. Chem. 285:7097–7110. 10.1074/jbc.M109.038604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grozinger CM, Chao ED, Blackwell HE, Moazed D, Schreiber SL. 2001. Identification of a class of small molecule inhibitors of the sirtuin family of NAD-dependent deacetylases by phenotypic screening. J. Biol. Chem. 276:38837–38843. 10.1074/jbc.M106779200 [DOI] [PubMed] [Google Scholar]
- 29.Freitag M, Schemies J, Larsen T, El Gaghlab K, Schulz F, Rumpf T, Jung M, Link A. 2011. Synthesis and biological activity of splitomicin analogs targeted at human NAD+-dependent histone deacetylases (sirtuins). Bioorg. Med. Chem. 19:3669–3677. 10.1016/j.bmc.2011.01.026 [DOI] [PubMed] [Google Scholar]
- 30.Heltweg B, Gatbonton T, Schuler AD, Posakony J, Li H, Goehle S, Kollipara R, Depinho RA, Gu Y, Simon JA, Bedalov A. 2006. Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res. 66:4368–4377. 10.1158/0008-5472.CAN-05-3617 [DOI] [PubMed] [Google Scholar]
- 31.Lain S, Hollick JJ, Campbell J, Staples OD, Higgins M, Aoubala M, McCarthy A, Appleyard V, Murray KE, Baker L, Thompson A, Mathers J, Holland SJ, Stark MJ, Pass G, Woods J, Lane DP, Westwood NJ. 2008. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell 13:454–463. 10.1016/j.ccr.2008.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ota H, Tokunaga E, Chang K, Hikasa M, Iijima K, Eto M, Kozaki K, Akishita M, Ouchi Y, Kaneki M. 2006. Sirt1 inhibitor, Sirtinol, induces senescence-like growth arrest with attenuated Ras-MAPK signaling in human cancer cells. Oncogene 25:176–185 [DOI] [PubMed] [Google Scholar]