

Abstract
Recent evidence suggests that type V collagen plays a role in organizing collagen fibrils, thus maintaining fibril size and spatial organization uniform. In this study we sought to characterize the importance of type V collagen morphological disorganization and to study the relationship between type V collagen, active remodeling of the pulmonary vascular/parenchyma (fibroblastic foci), and other collagen types in usual interstitial pneumonia (UIP). We examined type V collagen and several other collagens in 24 open lung biopsies with histological pattern of UIP from patients with idiopathic pulmonary fibrosis (IPF). We used immunofluorescence, morphometry, and three-dimensional reconstruction to evaluate the amount of collagen V and its interaction with the active remodeling progression in UIP, as well as types I and III collagen fibers. Active remodeling progression was significantly related to type V collagen density (p<0.05), showing a gradual and direct increase to minimal, moderate, and severe fibrosis degree in UIP and in the three different areas: normal, intervening, and mural-organizing fibrosis in UIP. Parenchymal changes were characterized by morphological disorganization of fibrillar collagen with diverse disarray and thickness when observed by three-dimensional reconstruction. We concluded that in the different temporal stages of UIP, vascular/parenchyma collagen type V is increased, in disarray, and is the most important predictor of survival.
Keywords: idiopathic pulmonary fibrosis, usual interstitial pneumonia, type V collagen, morphometry, three-dimensional reconstruction, survival
Usual interstitial pneumonia (UIP), the histological pattern of idiopathic pulmonary fibrosis (IPF), is characterized by an excessive and disordered deposition of matrix proteins caused by the proliferation and activation of fibroblasts. The resulting severe architectural changes of the alveolar walls cause the loss of functional alveolar capillary units, leading to respiratory insufficiency (Fukuda et al. 1995). To better understand the mechanisms of fibrosis formation and develop new therapeutic approaches, an exact understanding of the abnormalities of matrix remodeling in the lung is crucial. Structural and functional features of the fibrillar collagens I and III in the interstitium of the normal lung and their inappropriate accumulation in the fibrotic lung have been previously described (Raghu et al. 1985; Shahzeidi et al. 1994; Zhang et al. 1994; Mariani et al. 1996). In contrast, the physiologic functions and structural morphological changes of other fibrillar collagen types in lungs and their abnormal deposition patterns in fibrosis are less well understood.
In general, the fibrogenesis process secondary to tissue injury is characterized by a large amount of collagen deposition on the extracellular matrix (ECM) and morphologic disorganization of fibrillar collagen of different sizes and thickness (Hay 1991; Nishiyama et al. 1994; Walchii et al. 1994). The pathogenetic mechanism for this abnormal morphology is still not well delineated.
Type V collagen was first isolated from human placenta by Burgeson et al. (1976) and is found in lesser proportions in the pulmonary interstitium. It is an ECM protein with structural and biosynthetic properties strikingly different from those of type I and type III collagens (Mayne and Burgeson 1993). Type V collagen plays a basic function in the formation of fibrillar collagen mesh and has an important role in fibrinogenesis control or fiber size regulation (Hay 1991; Mayne and Burgeson 1993). Type V collagen is essential not only to regulate the diameters of other collagen fibers but also for maintaining the integrity of connective tissues, thus exhibiting different functions on a dependence of distribution and molecular isoforms (Mayne and Burgeson 1993). The molecules of type V collagen also contribute to the linking between stromal collagen and basement membrane, being important for cellular adhesion and matrix-repairing process (Mayne and Burgeson 1993; Adachi et al. 1997). The association among types I, III, and V collagens is important for the preservation and integrity of the connective tissues present in the vessels (Hay 1991). This increased interaction between different collagen types and consequent formation of the mixed heterotypical fibers can strongly influence the architecture and function of the ECM (Linsenmayer et al. 1990).
We have recently reported a severe vascular remodeling and interstitial fibrosis in rabbit lungs after immunization with human type V collagen, very similar to that observed in lung involvement found in diffuse connective tissue diseases (Teodoro et al. 2004). Curiously, the phenomenon was induced only by type V collagen immunization and did not occur after the administration of type I or type III collagen (Hay 1991), which suggests that these three proteins may play distinct roles in the fibrotic process, modulating the biomechanical properties of fibrils.
We postulate that if the collagen fibrils are in disarray and combine to make larger than normal lung tissues, then the expression and spatial organization of collagen V should be abnormal.
In order to characterize the importance of type V collagen in parenchyma and vessels and to explore the quantitative relationship between this factor and active remodeling (fibroblastic foci) degree as well as the relationship between three-dimensional (3D) reconstruction of type V collagen and other collagen types, we studied this collagen in UIP.
Materials and Methods
Patient Selection
Pulmonary specimens were obtained from 24 patients with IPF, according to the criteria outlined in the American Thoracic Society/European Respiratory Society (ATS/ERS) International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias (American Thoracic Society/European Respiratory Society 2002), by surgical lung biopsy. Only specimens from cases that fulfilled these consensus criteria were included.
We excluded specimens of any other possible etiology (e.g., pneumoconiosis) and/or with histological features suggestive of an alternative diagnosis (e.g., eosinophilic pneumonia). We also excluded biopsy specimens obtained from patients with a concomitant systemic disease (e.g., collagen vascular disease), extensive honeycomb changes (end-stage lung disease), a dual histological pattern (two different patterns at two different biopsy sites), and/or morphological features not consistent with a specific histological pattern (e.g., bronchocentric distribution in an otherwise classical case of UIP). After excluding specimens with histological and clinical evidence of diffuse alveolar damage, nonspecific interstitial pneumonia, desquamative interstitial pneumonia, lymphoid interstitial pneumonia, or respiratory bronchiolitis, all included patients exhibited clinical, radiological, and physiological alterations consistent with IPF and were given the definitive pathological diagnosis of UIP. In other words, the apparently clear-cut diagnosis of our patients with UIP was obtained by clinical, radiological, and histological consensus criteria.
Baseline Characterization
Median age of patients with IPF (15 males and 9 females) was 63.5 ± 7.6 (mean ± SEM) years. A baseline assessment of severity of dyspnea was made using the level of dyspnea (LOD) scale (Watters et al. 1986) (Table 1).
Table 1.
Baseline characteristics of idiopathic pulmonary fibrosis
| Variables | Values |
| Age at biopsy (years) | 63.5 ± 7.6 |
| Sex (F/M) | 15/9 |
| Dyspnea | |
| LOD | 10 ± 4 |
| Spirometry | |
| FEV1 (%pr) | 81 ± 5 |
| FVC (%pr) | 71 ± 4 |
| FEV1/FVC | 80 ± 6 |
| TLC (%pr) | 79 ± 4 |
| RV (%pr) | 93 ± 11 |
| RV/TLC (%pr) | 40 ± 3 |
| DLCO (%pr) | 60 ± 7 |
| DLCO/VA(%pr) | 63 ± 7 |
| PaO2 (mmHg) | 59 ± 2 |
| PaCO2 (mmHg) | 33 ± 08 |
| HRCT score | |
| Alveolar | 1.87 ± 0.90 |
| Interstitial | 1.90 ± 0.56 |
| Total | 3.77 ± 0.73 |
| Clinical score (CS) | |
| Total CS | 53 ± 16 |
Data are represented as mean ± SEM.
LOD, level of dyspnea; pr, predicted; TLC, total lung capacity; RV, residual volume; HRCT, high-resolution computerized tomography.
Physiological Testing
Pulmonary function tests included FEV1, FVC, FEV1/FVC ratio × 100, total lung capacity (TLC), residual volume (RV), and carbon monoxide transfer factor (DLCO). TLC, RV, and RV/TLC percentages were measured by the helium dilution method with a Master Screen apparatus (Erich Jaeger GmbH; Wuerzburg, Germany), DLCO and DLCO/VA by the single breath-holding helium dilution method (Quanjer et al. 1993). Lung function measurements (Table 1) were expressed as percentages of predicted values. In all patients, arterial PaO2 and PaCO2 were also measured at rest.
High-resolution Computerized Tomography (HRCT)
HRCT examinations were performed using 1.0- or 1.5-mm-thick sections taken at 1-cm intervals throughout the entire lungs during inspirations in the supine position and through the caudal 10 cm of the lungs at 2- to 3-cm increments in the prone position. Two thoracic radiologists prospectively and independently scored all lobes or HRCT for ground-glass opacity (CT alveolar) and interstitial opacity (CT interstitial) on a scale of 0-5; the mean score for each lobe and for the entire lung was calculated (Kazerooni et al. 1997) (Table 1).
Clinical Scoring
Overall clinical severity was assessed using a previously developed clinical and HRCT examination composite score (Watters et al. 1986). Total clinical and HRCT score ranged from 0 to 100 points (100 being the most severe disease) based on variables including the LOD scale, HRCT, and pulmonary function test results (Table 1).
Pathological Review of the Specimens
Pulmonary specimens were reviewed by three pathologists blinded to their clinical features. Each specimen was assigned a histological diagnosis according to the criteria outlined in the ATS/ERS multidisciplinary consensus classification of the idiopathic interstitial pneumonias (American Thoracic Society/European Respiratory Society 2002). In open lung biopsy specimens, UIP lungs were defined by alternating areas of normal parenchyma, alveolar collapse, honeycombing, and severe mural-organizing fibrosis defined as sites of active remodeling overlying fibrous airspace walls, thus showing temporal heterogeneity or overlying normal rigid pulmonary structures (e.g., interlobular septa) in a form of fibroblast foci and granulation tissue (Katzenstein and Myers 1998). Further demographic data and radiological characteristics are shown in Table 1.
Non-UIP lungs were used as controls and were obtained from five individuals who had died due to violent causes (three men and two women; mean age 60 ± 3.6 years). Tissue specimens were obtained from areas of non-pneumonia and non-emphysematous lung.
Parenchymal Remodeling
Semiquantitative analysis of the following alternating areas of active parenchymal remodeling (histological patterns) associated with UIP was performed in (1) alveolar collapse with relatively unaffected lung tissue or with mild interstitial thickening by fibrosis (minimal activity fibrosis) (Figure 1A), (2) moderate mural-organizing fibrosis with fibroblast foci (moderate activity fibrosis) (Figure 1B), and (3) severe mural-organizing fibrosis with honeycombing and foci of actively proliferating fibroblasts and myofibroblasts (severe activity fibrosis) (Figure 1C) (Parra et al. 2005).
Figure 1.

(A) Usual interstitial pneumonia (UIP) minimal fibrosis characterized by alveolar collapse with relatively unaffected lung tissue or with mild interstitial thickening by fibrosis (hematoxylin and eosin [H&E]). (B) UIP moderate fibrosis: pulmonary parenchyma showed high degree of inflammatory activity and moderate mural-organizing fibrosis with fibroblast foci (H&E). (C) UIP severe fibrosis: severe mural-organizing fibrosis with honeycombing and foci of actively proliferating fibroblasts and myofibroblasts (H&E).
Collagen Characterization
Immunofluorescence was used for the characterization of types I, III, and V collagens in 3-μm paraffin-embedded sections mounted on a methacryloxypropyltrimethoxysilane (Sigma; St Louis, MO) slide. Sections were dewaxed with xylene and dehydrated in graded ethanol. Antigen retrieval was performed through the enzymatic treatment of lungs with bovine pepsin (10,000 units of solid tissue; Sigma) in 4 mg/ml acetic acid buffer, pH 2.2, for 30 min at 37C, with a subsequent incubation with 5% milk in phosphate buffer, pH 7.0. Next, slides were incubated overnight with mouse polyclonal anti-human type I (1:100) and type V (1:2000) antibodies and monoclonal anti-human type III collagen antibody (1:100) (Miller and Rhodes 1982; Harlow and Lane 1988).
Formalin-fixed paraffin sections of normal human lung were used as positive controls and were stained simultaneously. For negative and autofluorescence control, sections were incubated with fetal bovine serum instead of the primary antibody (Schroeder et al. 1953). The same tissue treatment was carried out for immunofluorescence detection. Sections were incubated with the same primary antibody diluted with PBS plus BSA 1% overnight at 4C in a humid atmosphere. Sections were then incubated with a FITC-conjugated goat anti-mouse and anti-rabbit immunoglobulin (dilution 1:50; Sigma) as a secondary antibody and mounted with an aqueous mounting medium.
For the 3D reconstruction, double staining with fluorescein and rhodamine (rhodamine-conjugated goat anti-mouse IgG-R) (dilution 1:40; Santa Cruz Biotechnology, Santa Cruz, CA) was used for the characterization of the interaction of type I and type III collagens and type V and type I collagens in 20-μm paraffin sections. This was done according to a previously described method (Blumer et al. 2003). Sections not treated with protease were stained and examined to distinguish collagen fibers with normal structures from fibers displaying disorganized structures.
Type V Collagen Quantitation
We evaluated interstitial and vascular type V collagen using a CCD Sony DXC-101 camera system (Sony; Tokyo, Japan) applied to a Zeiss Axioplan microscope (Carl Zeiss; Jena, Germany) equipped for fluorescence, from which the images are shown on a monitor (Trinitron; Sony). By means of a digitizing system (Oculus TCX; Coreco Inc., St Laurent, Quebec, Canada) inserted in a computer (133 MHz Pentium), the images are software processed (Image ProPlus; Media Cybernetics, Silver Spring, MD). For the interstitial type V collagen at a magnification of X400, a total of 30 fields per case were analyzed in normal, intervening, and severe fibrosis areas. Thresholds for type V collagen fibers were established for each slide after enhancing the contrast to a point at which the fibers were easily identified as green bands. The area occupied by the fibers was determined by digital densitometric recognition by adjusting the threshold level of measurement up to the gray density of the collagen fibers. Bronchovascular bundles were carefully avoided during the measurements. Because inflation of the specimens was not controlled, the area occupied by the fibers was divided by the total area occupied by the alveolar septa. The results express the septal area fraction occupied by the collagen fibers. Vascular type V collagen was evaluated in pre- and intra-acinar arteries, which ranged from four to eight per biopsy specimen. Quantification of collagen fibers in artery walls was also performed by an image analysis system. A mean of six medium- to large-sized arteries in each biopsy specimen were analyzed at a magnification of × 200. Collagen V content was measured in each vascular wall and expressed as a relation between the quantity of collagen V divided by the total vascular area studied. Vascular area of each artery analyzed was carefully measured in the image analysis system using a cursor that allows the free determination of the area from the internal elastic membrane to the adventitial layer. Results express the amount of collagen V fibers (in area) per total area of vascular wall expressed as a fraction.
Collagen 3D Reconstruction
Laser-scanning confocal microscopy (LSCM) for the 3D image reconstruction was used to evaluate the interaction of types I and III and types I and V collagens in sections of mural-organizing fibrosis and pre-acinar artery in all cases. These sections were examined with a LSCM (LSM 410; Carl Zeiss) using the × 40 objective. Images were stored in the laser-scanning microscope, formatted, and stored in a computer (LSM Image Browser software, ver. 2,50.0929; Carl Zeiss) as tagged information file format files. A series of 45 or 50 optical sections were produced with a 0.5-μm distance. Images for 3D reconstruction of fibroblastic foci and pre-acinar artery in UIP tissue were obtained by this method.
Statistical Analysis
Differences among groups were assessed by Shapiro-Wilk's tests for normality, Levene's one-way analysis for homogeneity of variance, and t-test and ANOVA with Bonferroni post-hoc test for multiple comparisons were performed when appropriate. Kaplan-Meier analysis was used to examine the survival in the different histological patterns. A p value <0.05 was considered significant. Values were expressed as mean ± SD. All statistical procedures were carried out using SPSS software (Norusis 2002).
Results
Collagen Characterization in Non-UIP and UIP Lungs
Under hematoxylin-eosin (H&E) stain, non-UIP lungs showed preserved architecture and thin alveolar septa (Figure 2A). In contrast, UIP lung histological pattern was characterized by normal areas (Figure 3A), intervening areas (alveolar collapse) (Figure 3B), and dense fibrosis areas with “honeycomb” and mural-organizing fibrosis with fibroblastic foci (Figure 3C).
Figure 2.
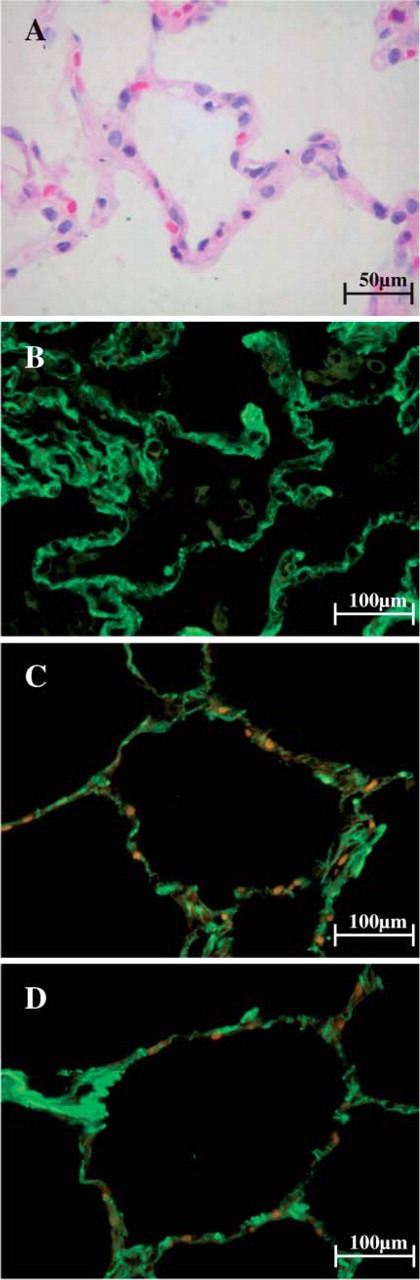
Morphological features of non-UIP lungs when stained with H&E and observed at optical microscope are characterized by preserved architecture and thin alveolar septa (A). After staining with fluorescein for types I, III, and V collagen fibers and observed at fluorescent microscope, alveolar walls of non-UIP lungs show fine green birefringence for type I collagen (B) and fine and minimal green birefringence for type III and type V collagen (C,D). Orange and red areas indicate the presence of elastic fibers and red cells, respectively.
Figure 3.
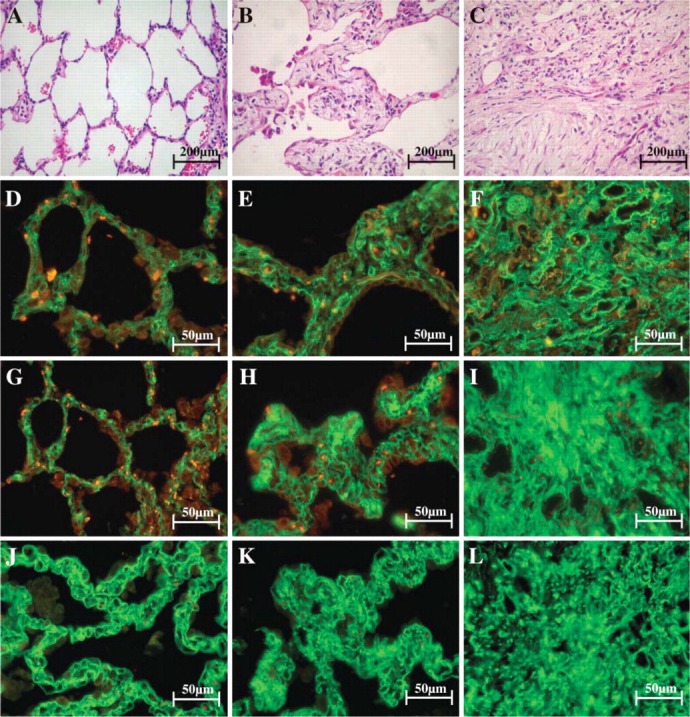
Morphological features of non-UIP lungs when stained with H&E and observed at optical microscope are characterized by normal areas (A), intervening areas (B), and dense fibrosis areas with mural-organizing fibrosis (C). After staining with fluorescein for types I (D-F), III (G-I), and V (J-L) collagen fibers and observed at fluorescent microscope, strong green birefringence of types I and III collagen in alveolar wall and type V collagen in basement membrane of alveolar capillaries in tissue sections are shown (D,G,J), coincident with the maintenance of the architecture of both (A). In contrast, the intervening (E,H,K) and several fibrosis areas (F,I,L) show distortion of architecture and a diffuse increase of types I, III, and V collagen birefringence. Orange and red areas indicate the presence of elastic fibers and red cells, respectively.
Figure 2 and Figure 3 show the collagenous fibers in alveolar walls of the non-UIP and UIP lungs stained with fluorescein and observed under fluorescent microscope (Figures 2B-2D and Figures 3D-3L). In tissue sections, non-UIP lungs (Figures 2B-2D) and normal areas in UIP (Figures 3B, 3G, and 3J) showed strong green birefringence of type I and type III collagens in alveolar wall and type V collagen in the basement membrane of alveolar capillaries, coincident with the maintenance of the architecture in both. In contrast, the intervening areas (Figures 3E, 3H, 3K) showed architectural distortion and a diffuse increase of types I, III, and V collagen birefringence. In the severe fibrosis areas (Figures 3F, 3I, and 3L) an interaction among types I, III, and V collagens coincided with increased birefringence expression, especially in active fibrogenesis areas (fibroblastic foci). Fibroblastic foci (Figure 4A) showed the thick reticular green birefringence of type I (Figure 4D) contrasting with the thin reticular of type III (Figure 4G) and punctuate pattern of type V collagen (Figure 4J). Equally important alterations of these collagen system fibers were observed in the terminal bronchiole (Figures 4E, 4H, and 4K) and pre-acinar artery (Figures 4F, 4I, and 4L) in severe fibrosis areas. Both types I and III collagens were located in large amounts in the adventitia of terminal bronchioles (Figures 4E and 4H) and to a lesser degree in the medial and adventitial layers of pre-acinar arteries (Figures 4F and 4I). Increased type I and type III collagen expression also coincided with increased expression of type V collagen in the basement membrane of the terminal bronchiole (Figure 4K) and pre-acinar arteries (Figure 4L). This correlated with the alveolar thickening associated with fibrocellular proliferation and the consequent distortion of the bronchioles and their pre-acinar arteries by the remodeling seen with H&E preparations (Figures 4B and 4C).
Figure 5.

Graphic representation of the confidence interval for interstitial and vessel area fractions of collagen type V as a function of the UIP histological patterns (A), for septal area fraction of type V collagen as a function of the control (non-UIP lung) and three different areas of UIP (B), and for interstitial and vessel area fractions of collagen type V as a function of patients' status (C). Kaplan-Meier survival curve for patients with UIP grouped by activity histological patterns. Significant difference is shown (log rank 437.45, p=0.005) between these groups (D). Note: (A) UIP histological patterns (activity). The three grades of fibrosis observed in UIP: 1 = minimal fibrosis, 2 = moderate fibrosis, and 3 = severe fibrosis presented a significant association with quantitation of type V collagen. (B) UIP histological areas. The three areas were observed in UIP: normal areas, alveolar collapse areas, and dense fibrosis areas with fibroblastic foci areas.
Figure 4.

Morphological features of UIP lungs when stained with H&E and observed at optical microscope are characterized by fibroblastic foci (A), terminal bronchiole (B), and pre-acinar artery (C). After staining with fluorescein for types I (D-F), III (G-I), and V (J-L) collagen fibers and observed at fluorescent microscope, fibroblastic foci show the thick reticular green birefringence of type I (D) contrasting with the thin reticular green birefringence of collagen III (G) and coincident with increase of punctuate green birefringence of collagen V (J). Types I and III collagen were localized in great amounts in the adventitia of terminal bronchioles (E,H) and to a lesser degree in medial and adventitial layer of pre-acinar arteries (F,I). Increased types I and III collagen expression also coincide with increased expression of collagen V localized in basement membrane of terminal bronchiole (K) and pre-acinar artery (L). Orange and red areas indicate the presence of elastic fibers and red cells, respectively.
Type V Collagen Density in Parenchyma Remodeling
Table 2 and Figure 5A show the progressive, gradual, and significant (p<0.05) increase of type V collagen density, directly related to minimal, moderate, and severe activity fibrosis degrees present in UIP. In this regard, high values of type V collagen coincided with severe activity fibrosis histological pattern (0.22 ± 0.02), moderate activity fibrosis with an intermediate value of type V collagen (0.16 ± 0.01), and minimal activity fibrosis with a minimal value of type V collagen (0.14 ± 0.05).
Table 2.
Descriptive and formal analysis of type V collagen fibers in interstitium and vessels stratified by usual interstitial pneumonia (UIP) histological patterns in cases of open lung biopsies of patients with UIP
| Controls and histological patterns of UIP (activity) | ||||
| Type V collagen | Controls | Minimal fibrosis | Moderate fibrosis | Severe fibrosis |
| Interstitiuma | 0.10 ± 0.01 | 0.14 ± 0.05 | 0.16 ± 0.01 | 0.22 ± 0.02 |
| Vesselsb | 0.04 ± 0.02 | 0.09 ± 0.02 | 0.16 ± 0.04 | 0.33 ± 0.07 |
UIP minimal fibrosis X UIP moderate fibrosis (p=0.002), UIP moderate fibrosis X UIP severe fibrosis (p=0.005).
UIP minimal fibrosis X UIP moderate fibrosis (p=0.06); UIP moderate fibrosis X UIP severe fibrosis (p<0.001).
t-Test, one-way ANOVA, and Dunnett 3 multiple comparison post hoc tests were used for analysis of variance of type V collagen fibers in interstitium, vessels, and their distribution in UIP histological patterns, histological areas, and controls (non-UIP lungs). Level of significance 0.05.
Measurements of type V collagen in the three different areas in UIP are shown in Table 3 and Figure 5B. Morphological changes of the type V collagen fibers in UIP showed significant differences in normal, intervening, and dense fibrosis areas when compared to non-UIP lungs (p<0.05). In decreasing order, mural-organizing fibroses (0.20 ± 0.03) were those with greater amounts of type V collagen fibers, followed by those with intervening (0.17 ± 0.03) and normal (0.13 ± 0.04) areas and non-UIP lungs (0.10 ± 0.01).
Type V Collagen Density in Vascular Remodeling
Measurements of type V collagen in vessels of UIP are shown in Table 2 and Figure 5C. Morphological changes of type V collagen fibers in vessels showed significant differences in minimal, moderate, and severe activity fibrosis degrees present in UIP when compared to non-UIP lungs (p<0.05). In decreasing order, those showing severe activity fibrosis (0.33 ± 0.07) were those with greater amounts of type V collagen fibers, followed by those with moderate activity fibrosis (0.16 ± 0.04), minimal activity fibrosis (0.09 ± 0.02), and non-UIP lungs (0.04 ± 0.02).
During follow-up of the 24 patients, 15 died and the clinical diagnoses were UIP. Survival curves comparing clinical, functional, and morphological parameters in UIP were analyzed. The most important predictor of survival in UIP was type V collagen density grade in the different histological patterns (log rank 437.45, p=0.005, Figure 5D).
Table 3.
Descriptive and formal analysis of type V collagen fibers stratified by UIP histological areas in cases of open lung biopsies of controls (non-UIP lungs)
| Controls (non-UIP lungs) and histological areas of UIP | ||||
| Control | Normal areas | Collapse areas | Fibroblastic foci areas | |
| Type V collagena | 0.10 ± 0.01 | 0.13 ± 0.04 | 0.17 ± 0.03 | 0.20 ± 0.03 |
Control lungs X UIP normal areas (p=0.05); UIP normal areas X UIP collapse areas (p=0.009), UIP collapse areas X UIP fibroblastic foci areas (p=0.002).
Type V Collagen 3D Reconstruction
UIP parenchymal changes were characterized by the morphological disorganization of fibrillar collagen with diverse disarray and thickness. The resulting pattern combining greenish type I and reddish type III birefringence represents the interaction of both fibrillar collagens in major fibrogenesis areas, especially in fibro-blastic foci and pre-acinar arteries of UIP. In these different areas and structures, type I and type III collagens were characterized by increase in fiber thickness. In fibroblastic foci, abundant type I and type III collagen were expressed and disarrayed. A similar characterization was observed in the basement membrane of pre-acinar arteries (Figures 6A and 6B). In addition, greenish type V collagen showed major thickness and dramatic morphological disorganization when compared with reddish type I collagen. Both collagen types were substantially present in fibroblastic foci and pre-acinar arteries walls (Figures 6C and 6D), thus representing the interaction between fibrillar collagen types. These features were similar and independent from the fibrosis degree present in UIP.
Figure 6.
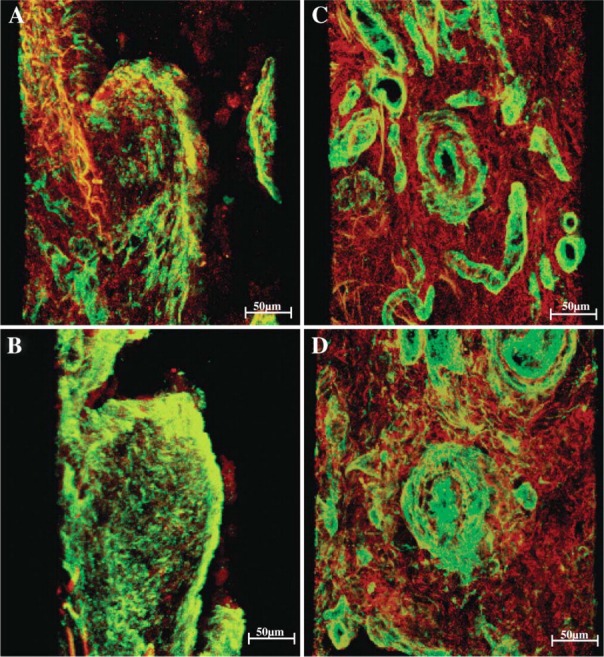
Three-dimensional image reconstruction shows double staining, greenish type I and reddish type III birefringence represent the interaction of both fibrillar collagens in major fibrogenesis areas of UIP. In the fibroblastic foci (A), abundant collagen types I and III were expressed and disarrayed. Similar characterization was observed in basement membrane of pre-acinar arteries (B). In addition, greenish type V collagen showed major thickness and dramatic morphological disorganization when compared with reddish type I collagen. Both collagen types were present substantially in fibroblastic foci (C) and pre-acinar arteries walls (D).
Discussion
In this study we evaluated the importance of interstitial and vascular type V collagen and explored the quantitative relationship between this factor, active remodeling (fibroblastic foci) degree, and survival as well as the relationship between spatial organization of type V and other collagen types and survival in patients with IPF. We demonstrated that, at the different temporal stages of UIP, collagen fibrils are increased, in disarray, and combined to make larger than non-UIP lungs, creating an abnormal spatial organization and expression of collagen V. Parenchymal overexpression of type V collagen density was the most important predictor of survival in UIP.
Collagen is a major constituent of ECM and perhaps the most important molecule in the stabilization of tissue structure, growth, and repair (Birk and Linsenmayer 1994). Type V collagen is present in the walls of bronchi and bronchioles, as well as in the walls of pulmonary vessels; these findings are consistent with reports of type V collagen production by smooth muscle and endothelial cells and of its presence in the basement membrane of capillary and arterial walls (Konomi et al. 1984).
Previous immunofluorescent studies of collagen polymorphism in fibrotic lung have suggested that the deposition of collagen III is a phenomenon observed in the early stages of fibrosis, whereas type I collagen is the predominant collagen deposited at later stages (Shahzeidi 1994; Kenyon et al. 2003).
Active pulmonary remodeling in UIP is mainly dependent on mural-organizing fibrosis (fibroblastic foci), histologically defined as the fibroblastic proliferation immersed in an edematous ECM with deposit of collagen fibers (Fulmer et al. 1980; Kirk et al. 1984), thus representing tissue cicatrization. Although the role of type I and type III fibrillar collagen as support proteins in the collagen-remodeling events is well known, questions arise concerning type V collagen expression in fibrogenesis. Would type V collagen increase and the morphological disorganization of its fibers with different sizes and thickness indicate a different remodeling event with a difficult resolution and a different type of fibrosis?
We have also found an association between collagen type I and type V that led to heterotypic fibrils with controlled diameters, which are important in influencing the functional characteristic of a given tissue (Van der Rest et al. 1990; Birk 2001). Genetic alterations of collagen V molecules impair the control of matrix assembly and are involved in the remodeling process, which culminates in lung fibrosis in human connective tissue diseases (Teodoro et al. 2004; Malfait et al. 2005). Recent evidence suggests that mutations in both the α 1(V) and α2(V) chains of type V collagen in Ehlers-Danlos syndrome lead to morphological changes in tissues (Burrows et al. 1996; Nicholls et al. 1996; Fichard et al. 1997) through changes in both fibril and tissue structures.
These results suggest that a failure in the regulation of collagen V and morphological disorganization of the fibrils with different sizes and thickness could potentially play a significant contributory role in the complex mechanisms of disordered matrix protein deposition leading to lung fibrosis. To confirm this possibility derived from the observations described here, animal studies to quantitatively assess the chronological sequence and regulation of collagen V expression in lung fibrosis under controlled conditions will be needed.
Acknowledgments
This study was supported by the following Brazilian agencies: the National Council for Scientific and Technological Development (CNPq); the Foundation for the Support of Research of the State of São Paulo (FAPESP 2001/14566-9); and the Laboratories for Medical Research (LIM 17), Clinicas Hospital, School of Medicine, University of São Paulo.
Literature Cited
- Adachi E, Hopkinson I, Hayashi T. (1997) Basement-membrane stromal relationships: interactions between collagen fibrils and the lamina densa. Int Rev Cytol 173:73–156 [DOI] [PubMed] [Google Scholar]
- American Thoracic Society/European Respiratory Society (2002) International Multidisciplinary Consensus Classification of the Interstitial Pneumonia. Am J Respir Crit Care Med 65:277–304 [DOI] [PubMed] [Google Scholar]
- Birk DE. (2001) Type V collagen: heterotypic type I/V collagen interactions in the regulation of fibril assembly. Micron 32:223–237 [DOI] [PubMed] [Google Scholar]
- Birk DE, Linsenmayer TF. (1994) Collagen fibril assembly, deposition, and organization into tissue-specific matrices. In Extracellular Matrix Assembly and Structure, Yurchenco PD, Birk DE, Mecham RP, eds. San Diego, Academic Press, 91–128 [Google Scholar]
- Blumer R, Konakci KZ, Brugger PC, Blumer MJ, Moser D, Schoefer C, Lukas JR, et al. (2003) Muscle spindles and Golgi tendon organs in bovine calf extraocular muscle studied by means of double-fluorescent labeling, electron microscopy, and three-dimensional reconstruction. Exp Eye Res 77:447–462 [DOI] [PubMed] [Google Scholar]
- Burgeson RE, El Adli FA, Kaitila II, Hollister DW. (1976) Fetal membrane collagens: identification of two new collagen alpha chains. Proc Natl Acad Sci USA 73:2579–2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrows NP, Nicholls AC, Yates JR, Gatward G, Sarathachandra P, Richards A, Pope FM. (1996) The gene encoding collagen 1(V) (COL5A1) is linked to mixed Ehlers-Danlos syndrome type I/II. J Invest Dermatol 106:1273–1276 [DOI] [PubMed] [Google Scholar]
- Fichard A, Tillet E, Delacoux F, Garrone R, Ruggiero F. (1997) Human recombinant ά1(V) collagen chain. Homotrimeric assembly and subsequent processing. J Biol Chem 272:30083–30087 [DOI] [PubMed] [Google Scholar]
- Fukuda Y, Basset F, Ferrans VJ, Yamanaka N. (1995) Significance of early intra-alveolar fibrotic lesions and integrin expression in lung biopsy specimens from patients with idiopathic pulmonary fibrosis. Hum Pathol 26:53–61 [DOI] [PubMed] [Google Scholar]
- Fulmer JD, Bienkowski RS, Cowan MJ, Breul SD, Bradley KM, Ferrans VJ, Roberts WC, et al. (1980) Collagen concentration and rates of synthesis in idiopathic pulmonary fibrosis. Am Rev Respir Dis 122:289–301 [DOI] [PubMed] [Google Scholar]
- Harlow E, Lane D. (1988) Antibodies: A Laboratory Manual. Cold Spring Harbor, NY, Cold Spring Harbor Laboratory, 56–81 [Google Scholar]
- Hay ED. (1991) Matrix assembly. In Hay ED, ed. Cell Biology of Extracellular Matrix, 20th ed New York, Plenum, 221–249 [Google Scholar]
- Katzenstein AL, Myers JL. (1998) Idiopathic pulmonary fibrosis: clinical relevance of pathologic classification. Am J Respir Crit Care Med 157:1301–1315 [DOI] [PubMed] [Google Scholar]
- Kazerooni EA, Martinez FJ, Flint A, Jamadar DA, Gross BH, Spizarny DL, Cascade PN, et al. (1997) Thin-section CT obtained at 10-mm increments versus three-level thin-section CT for idiopathic pulmonary fibrosis: correlation with pathologic scoring. AJR Am J Roentgenol 169:977–983 [DOI] [PubMed] [Google Scholar]
- Kenyon NJ, Ward RW, McGrew G, Last JA. (2003) TGF-β1 causes airway fibrosis and increased collagen I and III mRNA in mice. Thorax 58:772–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk JM, Heard BE, Kerr I, Turner-Warwick M, Laurent GJ. (1984) Quantitation of types I and III collagen in biopsy lung samples from patients with cryptogenic fibrosing alveolitis. Coll Relat Res 4:169–182 [DOI] [PubMed] [Google Scholar]
- Konomi H, Hayashi T, Nakayasu K, Arima M. (1984) Localization of type V collagen and type IV collagen in human cornea, lung, and skin. Immunohistochemical evidence by anti-collagen antibodies characterized by immunoelectroblotting. Am J Pathol 116:417–426 [PMC free article] [PubMed] [Google Scholar]
- Linsenmayer TF, Fitch JM, Birk DE. (1990) Heterotypic collagen fibrils and stabilizing collagens: controlling elements in corneal morphogenesis?. Ann NY Acad Sci 580:143–160 [DOI] [PubMed] [Google Scholar]
- Malfait F, Coucke P, Symoens S, Loeys B, Nuytinck L, De Paepe A. (2005) The molecular basis of classic Ehlers-Danlos syndrome: a comprehensive study of biochemical and molecular findings in 48 unrelated patients. Hum Mutat 25:28–37 [DOI] [PubMed] [Google Scholar]
- Mariani TJ, Roby JD, Mecham RP, Parks WC, Crouch E, Pierce RA. (1996) Localization of type I procollagen gene expression in silica-induced granulomatous lung disease and implication of transforning growth factor-beta as a mediator of fibrosis. Am J Pathol. 148:151–164 [PMC free article] [PubMed] [Google Scholar]
- Mayne R, Burgeson R. (1993) Structure and function of collagen types. In Ulecham R, ed. Biology of Extracellular Matrix, 20th ed London, Academic Press, 1–37 [Google Scholar]
- Miller EJ, Rhodes RK. (1982) Preparation and characterization of the different types of collagen. Methods Enzymol 82:33–64 [DOI] [PubMed] [Google Scholar]
- Nicholls AC, Oliver JE, McCarron S, Harrison JB, Greenspan DS, Pope FM. (1996) An exon skipping mutation of a type V collagen gene (COL5A1) in Ehlers-Danlos syndrome. J Med Genet 33:940–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama T, McDonough AM, Bruns RR, Burgeson RE. (1994) Type XII and XIV collagens mediate interactions between banded collagen fibers in vitro and may modulate extracellular matrix deformability. J Biol Chem. 269:28193–28199 [PubMed] [Google Scholar]
- Norusis MJ. (2002) SPSS for Windows [10.0]. Chicago, SPSS Inc. [Google Scholar]
- Parra ER, David YR, da Costa LRS, Ab'Saber AM, Sousa R, Kairalla RA, de Carvalho CRR, et al. (2005) Heterogeneous remodeling of lung vessels in idiopathic pulmonary fibrosis. Lung 183:291–300 [DOI] [PubMed] [Google Scholar]
- Quanjer PH, Tammeling GJ, Cotes JE, Pedersen OF, Peslin R, Yernault J-C. (1993) Lung volumes and forced ventilatory flows. Report Working Party Standardization of Lung Function Tests, European Community for Steel and Coal. Official Statement of the European Respiratory Society. Eur Respir J Suppl 16:5–40 [PubMed] [Google Scholar]
- Raghu G, Striker LJ, Hudson LD, Striker GE. (1985) Extracellular matrix in normal and fibrotic human lungs. Am Rev Respir Dis 131:281–289 [DOI] [PubMed] [Google Scholar]
- Schroeder WA, Honnen L, Green FC. (1953) Chromatographic separation and identification of some peptides in partial hydrolysates of gelatin. Proc Natl Acad Sci USA 39:23–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahzeidi S, Jeffery PK, Laurent GJ, McAnulty RJ. (1994) Increased type I procollagen mRNA transcripts in the lung of mice during the development of bleomycin-induced fibrosis. Eur Respir J 7:1938–1943 [PubMed] [Google Scholar]
- Teodoro WR, Velosa AP, Witzel SS, Garippo AL, Farhat C, Parra ER, Sonohara S, et al. (2004) Architectural remodelling in lungs of rabbits induced by type V collagen immunization: a preliminary morphologic model to study diffuse connective tissue diseases. Pathol Res Pract 200:681–691 [DOI] [PubMed] [Google Scholar]
- Van der Rest M, Dublet B, Champliaud MF. (1990) Fibril-associated collagens. Biomaterials 11:28–31 [PubMed] [Google Scholar]
- Walchii C, Koch M, Chiquet M, Odermatt BF, Trueb B. (1994) Tissue-specific expression of the fibril-associated collagen XII and XIV. J Cell Sci 107:669–681 [DOI] [PubMed] [Google Scholar]
- Watters LC, King TE, Schwarz MI, Waldron JA, Stanford RE. (1986) A clinical, radiographic, and physiologic scoring system for the longitudinal assessment of patients with idiopathic pulmonary fibrosis. Am Rev Respir Dis 133:97–103 [DOI] [PubMed] [Google Scholar]
- Zhang K, Gharaee-Kermani M, McGarry B, Phan SH. (1994) In situ hybridization analysis of rat lung alpha 1(I) and alpha 2(I) collagen gene expression in pulmonary fibrosis induced by endotracheal bleomycin injection. Lab Invest 70:192–202 [PubMed] [Google Scholar]