

Abstract
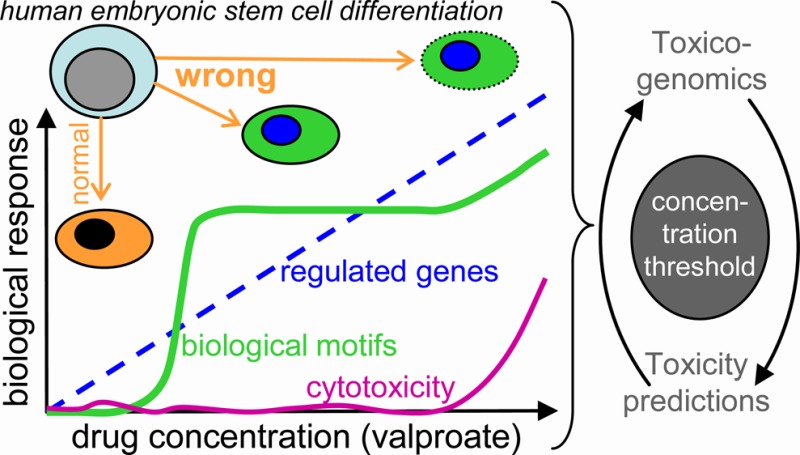
Information on design principles governing transcriptome changes upon transition from safe to hazardous drug concentrations or from tolerated to cytotoxic drug levels are important for the application of toxicogenomics data in developmental toxicology. Here, we tested the effect of eight concentrations of valproic acid (VPA; 25–1000 μM) in an assay that recapitulates the development of human embryonic stem cells to neuroectoderm. Cells were exposed to the drug during the entire differentiation process, and the number of differentially regulated genes increased continuously over the concentration range from zero to about 3000. We identified overrepresented transcription factor binding sites (TFBS) as well as superordinate cell biological processes, and we developed a gene ontology (GO) activation profiler, as well as a two-dimensional teratogenicity index. Analysis of the transcriptome data set by the above biostatistical and systems biology approaches yielded the following insights: (i) tolerated (≤25 μM), deregulated/teratogenic (150–550 μM), and cytotoxic (≥800 μM) concentrations could be differentiated. (ii) Biological signatures related to the mode of action of VPA, such as protein acetylation, developmental changes, and cell migration, emerged from the teratogenic concentrations range. (iii) Cytotoxicity was not accompanied by signatures of newly emerging canonical cell death/stress indicators, but by catabolism and decreased expression of cell cycle associated genes. (iv) Most, but not all of the GO groups and TFBS seen at the highest concentrations were already overrepresented at 350–450 μM. (v) The teratogenicity index reflected this behavior, and thus differed strongly from cytotoxicity. Our findings suggest the use of the highest noncytotoxic drug concentration for gene array toxicogenomics studies, as higher concentrations possibly yield wrong information on the mode of action, and lower drug levels result in decreased gene expression changes and thus a reduced power of the study.
Introduction
Many new test systems for neurodevelopmental disturbances are currently being developed.1−4 In addition to classical endpoints, toxicogenomics methods have been used to characterize the assays and to classify toxicants. For regulatory purposes, the descriptive data from, e.g., transcriptomics or metabolomics approaches need to be converted to quantifiable measures allowing one to compare and predict the hazard of chemicals and drugs.1,3,5 First attempts to determine benchmark concentrations on the basis of transcriptomics data have already been undertaken in vivo.(6,7) Their application to in vitro test systems is expected to yield important information about low-dose toxicant effects.
The developing central nervous system is one of the most frequent targets of systemic toxicity.8,9 Moreover, testing of nervous system development and possible long-term effects is particularly challenging. Animal testing for example according to OECD guidelines 414 (2-generation reproduction) or 426 (developmental neurotoxicity) is time-consuming and labor intensive. The testing capacities are by far not sufficient to test all compounds in vivo that should be studied for reproductive toxicity or developmental neurotoxicity (DNT) in the context of REACH. Moreover, toxicological risk assessment needs to be adapted to the modern needs of the pharmaceutical as well as the chemical industry.10 Between 1999 and 2011, 19% of the 279 new approved drugs in Europe were reported to have postapproval safety-issues, and 5 drugs were withdrawn from the market.11 The chemical industry, however, is confronted with the REACH initiative asking them to provide more detailed toxicological data regarding new compounds as well as on chemicals already on the market. Consequently the National Research Council of the USA has published their vision of Toxicology for the 21st century in 200712−14 in which they favor the development of mostly human based in vitro test systems, which can be used for high-throughput screening and for high content endpoints such as for transcriptomics approaches.
Cultures of differentiating pluripotent stem cells, such as human embryonic stem cells (hESC) or human induced pluripotent stem cells,15 offer unique possibilities of studying the very early steps of human development that lead to the formation of germ layers and primordial tissues. This opportunity was seized by the European Commission-funded research consortium for the use of embryonic stem cell-based novel alternative tests (ESNATS) for the prediction of toxicity of drug candidates (www.esnats.eu). Within this context, several tests have been established that recapitulate critical processes of early neuronal development in vitro.(4,16−23) Recently, two positive control compounds of developmental neurotoxicity, valproic acid (VPA) and methylmercury, have been tested in four test systems. Both test compounds induced characteristic gene expression alterations.24 The use of two compound concentrations indicated that the number of altered genes and the extent of deregulation strongly depend on the concentration of the test compound. The highest concentration used in this previous study was a benchmark concentration (BMC) that reduced overall cell viability by not more than 10%. This procedure was chosen to avoid testing of cytotoxic concentrations that might generate unspecific gene expression patterns due to cell death. However, our previous study also showed that exposure to a concentration that reduces viability by exactly 10% is difficult to perform. Reduction of viability slightly differs from experiment to experiment. In principle, this may cause unspecific cell death associated gene expression signatures. However, it has not yet been tested if cytotoxic concentrations really cause a death associated expression signature and how the transcriptome changes are affected qualitatively by concentrations deviating from the BMC.
Comparisons between test systems and test compounds in this earlier study24 suggested that different toxicants may regulate the same master transcription factors. As such factors define cell identity25 and they show coordinated regulation in different pathologies,26,27 it may be assumed that they also play a role in developmental toxicity, and the threshold of their activation may determine the threshold of teratogenicity.
To address the above issues, we used the UKN1 test system for more detailed experiments.24,28 In this assay, hESC differentiate to neuroepithelial precursor cells (NEP) within 6 days, and they were exposed to VPA during the entire period of differentiation. We used in this study eight concentrations of VPA covering a range from completely nontoxic to strongly cytotoxic and analyzed genome wide expression patterns. VPA was chosen as a well-studied positive control compound of developmental neurotoxicity.29,30 The data were analyzed by classical biostatistical approaches, but we also developed quantitative measures of developmental disturbance, based on systems biology considerations. For instance, we quantified not only alterations of individual gene ontologies (GO), as pioneered by the Piersma lab,29,31 but we also addressed superordinate cell biological processes to gain overall insight. We report that concentrations at the uppermost limit of the noncytotoxic range may be a reasonable compromise for gene array classification studies because with such concentrations, (i) the genes that indicate a teratogenic mode of action are sufficiently deregulated, and (ii) dilution by unspecific cytotoxicity associated genes only start to emerge.
Materials and Methods
Materials
Gelatin, putrescine, selenium, progesterone, apotransferin, glucose, insulin, and valproic acid were obtained from Sigma (Steinheim, Germany). Accutase was from PAA (Pasching, Austria). FGF-2 (basic fibroblast growth factor) and noggin were obtained from R&D Systems (Minneapolis, MN, USA). Y-27632, SB-43154, and dorsomorphine dihydrochloride were from Tocris Bioscience (Bristol, UK). MatrigelTM was from BD Biosciences (Massachusetts, USA). All cell culture reagents were from Gibco/Invitrogen (Darmstadt, Germany) unless otherwise specified.
Neuroepithelial Differentiation
Human embryonic stem cells (hESC) (H9 from WiCells, Madison, Wi, USA) were differentiated according to the protocol published by Chambers and colleagues32 with the following modifications. Instead of using 500 μM noggin, we used the combination of 35 μM noggin and 600 nM dorsomorphine together with 10 μM SB-431642 for dual SMAD inhibition as described earlier.28,33 This was used to prevent BMP and TGF signaling, and thus to achieve a highly selective neuroectodermal lineage commitment. For handling details, see Supporting Information of.28 All differentiations were performed in 6-well plates containing 2 mL of medium each.
Experimental Exposure and Resazurin Viability Assay
Treatment with valproic acid (VPA) was done with the indicated concentrations from 25 μM to 1000 μM VPA dissolved in PBS. DoD0 medium was prepared as indicated in ref (28) and supplemented with the indicated concentrations of VPA. All concentrations were prepared from a 1 M stock solution (water). Medium supplemented with VPA was changed to DoD1, 2, and 4. In order to determine cytotoxicity, a resazurin assay was performed on DoD6 exactly as described previously.17,24
Affymetrix Gene Chip Analysis
After the resazurin assay, the medium was removed, and the cells were lysed in RNA protect solution (Quiagen). Affymetrix chip-based DNA microarray analysis (Human Genome U133 plus 2.0 arrays) was performed exactly as described earlier. (24)
Biostatistics
The following analyses were performed using the statistical programming language R-version 3.0.1. For the normalization of the entire set of 30 Affymetrix gene expression arrays, the extrapolation strategy (RMA+) algorithm34 was used that applies background correction, log2 transformation, quantile normalization, and a linear model fit to the normalized data in order to obtain a value for each probe set (PS) on each array. As reference, the normalization parameters obtained in earlier analyses24 were used. After normalization, at each concentration the difference between gene expression and corresponding controls was calculated (paired design). Replicates of controls were averaged before subtracting from corresponding exposed samples.
Differential expression was calculated using the R package limma.35 Here, the combined information of the complete set of genes is used by an empirical Bayes adjustment of the variance estimates of single genes. This form of a moderated t test is abbreviated here as the limma t test. The resulting p-values were multiplicity-adjusted to control the false discovery rate (FDR) by the Benjamini–Yekutieli procedure.36 As a result, for each concentration, a gene list was obtained, with corresponding estimates for log fold change and p-values of the limma t test (unadjusted and FDR-adjusted).
Data Display Algorithms
Heat maps were used to visualize matrices of gene expression values. Color encodes the magnitude of the values, ranging from blue (low) to yellow (high). Principal component analysis (PCA) plots were used to visualize expression data in two dimensions, representing the first two principal components, that is, the two orthogonal directions of the data with highest variance. The percentages of the variances covered are indicated in the figures. The software R, version 3.0.1, was used for all calculations and display of PCA and heat maps (R_Development_Core_Team 2012).37 The calculation and display of toxicity curves was done using GraphPad Prism 5.0 (Graphpad Software, La Jolla, USA). The Venn diagrams for the comparison of gene expression, gene ontology (GO) terms, and transcription factor binding sites (TFBS) between test systems were constructed according to Chow and Rodgers.38 The size of circles and areas was chosen proportional to the number of elements included if possible, i.e., if there is no empty section (zero in Venn diagram).
k-Means Clustering
Before clustering, we applied some adjustments to the data. First, we filtered out all of the PS with too small variation. For this, we used a standard deviation cutoff of 0.25. Second, we standardized the expression measurements for each PS, by subtracting its mean and dividing with its standard deviation. Standardization was required to capture better the temporal patterns of the genes. The clustering itself was performed using the k-means function from R.
Definition of Quantification Indices
For the index of overrepresented GOs, the significantly regulated genes were identified for each drug concentration. Then, the sum of oGO among up-regulated PS (at a given drug concentration), of oGO among down-regulated PS, and among all regulated PS was calculated. This sum was divided by 2 (to account for redundancies in this sum) to arrive at the used index number. For the gene regulation index, all genes were sorted according to their fold change relative to untreated controls. Then, for each condition, the top 100 genes were selected (highest fold changes, irrespective of the direction of regulation). For each of these genes, the negative log of the FDR corrected p-value was determined, and then, the sum of these 100 values was formed. The teratogenicity index is expressed by two coordinates in the plane formed by the index of oGOs and the gene regulation index.
Gene Set Enrichment Analysis
The gene ontology group enrichment was performed using R-version 3.0.1 with the topGO package,39 and only results from the biological process ontology were kept. The category was considered only if it was annotated with more than 100 genes, and the minimal enrichment p-value across all concentrations was below 0.001.
A new biostatistical method for measuring activation profiles of GO groups across all concentrations (up- or down-regulation) was applied. The method uses a segmentation test40 to identify significant enrichment with up- or down-regulated genes (restricted by p-value <0.05), comparing each concentration with the controls. The large number of enrichment tests is FDR-adjusted according to the two-step procedure by Benjamini et al.41 to limit the FDR to 1%. Each GO group obtains a corresponding activation profile. The profile 0++++++ means that the corresponding gene set is significantly enriched with up-regulated genes at the second lowest concentration and for all higher concentrations. Analogously, a gene set with profile 00000-- is enriched with down-regulated genes only at the two highest concentrations.
Transcription factor binding site enrichment (TFBS) was performed using the PRIMA algorithm (http://acgt.cs.tau.ac.il/prima/)42 provided in the Expander software suite (version 6.04;43http://acgt.cs.tau.ac.il/expander/) as described in ref (24). A transcription factor was included in heat maps or Venn diagrams only if the minimal enrichment p-value across all concentrations was below 0.001.
Results
Establishment of a Concentration Dependent Gene Expression Database for Valproic Acid
In order to investigate concentration dependent gene expression changes induced by VPA, a recently established hESC-based in vitro system, UKN1, was applied. This test system recapitulates the stage of human neuroectodermal induction leading to the formation of neural ectodermal progenitor cells (NEP).28,32 These developmental steps take place during a six days process, which is covered by exposure to the test compound (Figure 1A). In order to define a concentration range for gene array analysis, viability was tested in a concentration range from 25 to 1000 μM VPA. To this end, differentiating cells were treated with the indicated concentrations of VPA, and a resazurin viability assay was performed. Concentrations up to 550 μM VPA appeared to be noncytotoxic, whereas viability was decreased by 20–30% with 800 and 1000 μM (Figure 1B). Higher concentrations could not be used, as otherwise mRNA extractions were not feasible anymore. On the basis of the cytotoxicity tests, these eight concentrations (25, 150, 350, 450, 550, 650, 800, and 1000 μM) of VPA and untreated controls were analyzed by DNA microarray (DMA) analyses (Affymetrix). Exposure conditions exactly the same as those for the cytotoxicity experiments were used (Figure 1A).
Figure 1.

Transcriptome changes of neurally differentiating stem cells induced by increasing VPA concentrations. (A) Differentiation scheme of the cell model used in this study. Embryonic stem cells (hESC) were converted in six days of differentiation (DoD6) to a pure population of neuroepithelial precursors (NEP). Adapted from ref (24). (B) Viability assay using resazurin reduction. The cells were differentiated and treated as indicated in A. On DoD6, resazurin reduction was measured, and viability is given as a percentage of untreated controls. Data are the means ± SD of three experiments. (C) Cells as treated in B were used for whole transcriptome analysis. The result is displayed in the form of a two-dimensional principal component (PC) analysis diagram. Each point represents one experiment (= data from one microarray), and the color coding indicates the concentration of VPA (in μM) used in the experiment. (D) Heat map of differentially regulated genes from 3 independent experiments. All data sets (columns) were sorted by similarity clustering. The absolute gene expression levels (log2-scaling) of the 1000 transcripts with highest variance were color-coded for display.
Principal component analysis (PCA) illustrates a convincing concentration progression model (Figure 1C). The data of three independent experiments per concentration clustered closely together and moved into the direction of the first principal component with increasing concentrations. An exception was the concentration of 650 μM VPA, which showed a high degree of variability (Figure 1C). This concentration was in the range of beginning cytotoxicity. Accordingly, we observed high variability in the resazurin assay between the different experiments (Figure 1B). Therefore, this concentration was excluded from the further analysis shown in the main figures, but evaluations with 650 μM are available in the Supporting Information (Table S1). Further, we generated heat maps based on z-scores and corresponding dendrograms (Figure S1, Supporting Information) calculated by hierarchical clustering (Figure 1D). This analysis confirmed the results from the PCA (Figure 1C) that the different experimental replicates cluster closely together, apart from 650 μM that was already identified as an outlier.
For the following analysis of differential gene expression, the Benjamini-Yekutieli false discovery rate adjustment (BY-FDR) of the p-values was performed independently for each data set of a given drug concentration. This method has the advantage that the results do not vary if one concentration is added to or left out from the analysis. However, this approach can introduce a bias since BY-FDR is more liberal in the case of large numbers of differentially expressed genes. To get an overview over the extent of the bias, the number of differentially up-regulated and down-regulated genes was calculated for three types of adjustment (no adjustment, per-concentration FDR adjustment, and overall FDR adjustment) (Figure S2, Supporting Information). As the differences between per-concentration and overall adjustment were not substantial, the analysis throughout the manuscript is based on per concentration adjustment. One reason for the small differences is that genes were selected not only based on p-value cut-offs but also required a >2-fold change.
After FDR correction, no significantly altered PS were found for the two lowest tested VPA concentrations of 25 and 150 μM (Figure 2A). Between 350 and 1000 μM VPA, the numbers of up- or down-regulated probe sets (PS) gradually increased (Figure 2A and Table S1, Supporting Information). From these data, it is not clear, whether genes regulated, e.g., at 450 μM, were not at all regulated at lower concentrations or whether they were regulated but did not pass our stringent selection threshold (p < 0.05 and fold change > 2). Therefore, we selected the PS that were regulated by 450 μM VPA and determined their average fold change (FC). Therefore, we determined the FC of the same set of PS at lower and higher concentrations (Figure 2B). As a complementary approach, we selected all PS regulated significantly (p < 0.05) at 450 μM, with or without a 2-fold cutoff of the FC. Then, we examined the distribution of the p-values of all these PS at lower drug concentrations (Figure S3, Supporting Information). From these additional statistical analyses, we conclude that a subgroup of PS is only regulated at higher drug concentrations and not at all at low exposure; another subgroup is regulated concentration dependently, i.e., to a lower extent at low drug concentrations and to a higher extent at high concentrations of VPA.
Figure 2.

Concentration-dependence of expression changes and biological grouping of probe sets after exposure to VPA. (A) Number of differentially expressed probe sets (PS) compared to the untreated control (p < 0.05, BY-corrected; fold change (FC) >2). Left panel (red) represents the amount of up-regulated PS and the right panel (green) the amount of down-regulated PS. (B) The PS identified in A to be regulated by 450 μM VPA (up, n = 554; down, n = 110) were examined for their regulation at all drug concentrations. The average FC of this set of up-regulated PS is shown in the left panel for different VPA concentrations; the data for the down-regulated PS are in the right panel. (C) Significantly up- and down-regulated PS and their potential upstream regulators were identified by bioinformatic approaches. The numbers of oGOs (left panel) and TFBS (right panel) are displayed for 350, 550, and 1000 μM VPA. Red represents the GO (left panel) and TFBS (right panel) overrepresented among up-regulated PS and green from down-regulated PS. (D) The oGOs determined in C were classified in superordinate cell biological processes by expert knowledge. The identified processes are given in the middle; the left panel refers to up- and the right panel down-regulated PS.
Next, we analyzed the significantly (p-value <0.05 after FDR correction) regulated PS for overrepresented gene ontologies terms (oGO). For this, we used the differentially regulated PS of 350 (low), 550 (medium), and 1000 (high) μM VPA. This GO enrichment analysis and comparison across drug concentrations revealed that also on the level of oGOs the number increased with increasing VPA concentrations (Figure 2C and Table S2, Supporting Information). Next, the identified oGOs were further assigned to superordinate cell biological processes as detailed in Supporting Information (Table S2). For the up-regulated PS, it became clear that all identified processes were already changed with low VPA concentration. In addition, only a few more oGOs were identified additionally with higher concentrations (Figure 2C), and these additional oGOs all fell into the same superordinate cell biological processes (Figure 2D). Surprisingly, this finding applies also to stress pathways and the process apoptosis/death. For example, cell death processes were overrepresented already at low concentrations, and there was no new oGO emerging in this category at very high concentrations. For the down-regulated PS, few superordinate cell biological processes were identified, and these processes only start to be down-regulated with medium VPA concentration. They remained changed up to the highest concentrations (Figure 2D). Especially, neuronal pathways were down-regulated from 550 μM on. In summary, these data suggest that key biological processes start to be altered already at low and medium drug concentrations, and hardly any further processes are changed at high (cytotoxic) concentrations.
To investigate this finding by a different approach, we generated Venn diagrams from the data on concentration progression. This data display illustrated that most genes altered at lower concentrations are also altered at higher concentrations (Figure 3A and Table S3, Supporting Information). More details on the concentration progression are shown in dendrograms of up- and down-regulated genes (Figure S1, Supporting Information): at higher concentrations, additional genes were deregulated, and many of the genes deregulated at low concentrations to a small extent show lower p-values (stronger deregulation) at higher concentrations. Over 50% of the differentially expressed PS were induced with high drug concentrations only (Figure 3B and Table S4, Supporting Information). This situation was different for overrepresented transcription factor binding sites (TFBS) (Figure 3B and Table S4, Supporting Information). A relatively large fraction of all TFBS found in this study at any concentration was already overrepresented at low drug levels of 350 μM. This was particularly obvious for the overrepresented TFBS of down-regulated genes, illustrated in Figure 3B. In summary, the regulation of PS and TFBS showed a characteristically different behavior: while many PS were regulated at high VPA concentrations only, the corresponding TFBS were already changed at low concentrations.
Figure 3.

Overlap of differentially expressed PS and TFBS at increasing VPA concentrations. (A) The number of significantly up (upper panel)- and down-regulated (lower panel) PS (cut off: p-value <0.05, BY corrected and FC > 2) for each concentration were compared to the PS of the next higher concentration. The overlap between the concentrations is displayed as Venn diagrams. (B) Comparison of up- and down-regulated PS (and of TFBS overrepresented among these PS) between the indicated concentrations. Overlaps are displayed as Venn diagrams with absolute numbers of altered elements in the circles, and the number of nonaltered elements in the lower right corner. The percentage number refers to elements affected by 1000 μM only, relative to all affected elements.
Also, oGO between different VPA concentrations were compared in a Venn diagram to visualize the degree of overlap. This analysis showed that oGO groups behave more similarly to TFBS than to PS (Figure 4 and Table S5, Supporting Information): For up- and down-regulated genes, most oGO groups started to be overrepresented at 350 μM, and relatively few oGO groups are added at higher concentrations. These findings are illustrated at higher detail in dendrograms (Figure S1, Supporting Information). A relatively large fraction of oGO groups overrepresented at 350 μM overlaps with oGO groups overrepresented at 550 and 1000 μM (n = 55). For down-regulated genes, oGO groups began to emerge at slightly higher concentrations, with only 6 oGO groups being overrepresented at 350 μM. The overall features remained the same: 22 of the 25 GO overrepresented at 1000 μM were already overrepresented at 550 μM (Figure 4B). Again, we also classified the oGOs in superordinate cell biological processes. Also, on these higher levels, the results were confirmed. Taken together, these analyses illustrate different progression models for genes on the one side and oGO groups and TFBS on the other. Increasing VPA concentrations lead to substantial numbers of additionally up- or down-regulated genes. However, most oGO groups and TFBS were already overrepresented at 350 or 450 μM with relatively little added at higher concentrations.
Figure 4.

Overlap of GOs affected by low, medium and high VPA concentrations. (A) Up-regulated and (B) down-regulated PS were analyzed for oGO, and then the identified oGO were compared between drug concentrations. The overlaps of oGO at VPA concentrations of 350 μM (low), 550 μM (medium), and 1000 μM are displayed as Venn diagrams. Right: The oGOs from specified areas of the diagrams (see legend; 550 ∧ 1000 means overlap area of 550 μM and 1000 μM circles) were grouped according to superordinate cell biological processes, and the absolute numbers within these groups are displayed.
Unbiased k-Means Clustering of PS Concentration–Response Profiles
The previous analysis focused on the differentially expressed PS for each concentration compared to the control. This informs only to a limited extent on the concentration–response behavior of the individual PS. Therefore, we performed an unbiased k-means clustering of the concentration–response data sets for each gene. Using this method, we grouped the responses into clusters of 5 different up-regulation responses and 5 types of down-regulations (Figure 5A and Table S6, Supporting Information). The number of PS falling into each cluster and the number of oGOs among these PS are given in Figure 5B. Most concentration–response courses showed a monotonic pattern, but clusters 1, 5, and 6 contained PS with nonmonotonic regulation patterns. Together, the latter clusters comprised 6.8% of all PS (n = 9624) considered in the cluster analysis.
Figure 5.

Unbiased k-means clustering of concentration–response curves. (A) Display of the 10 clusters generated from different VPA concentration–response curves of the gene expression data sets. (B) The number of PS in each cluster (solid bars) and oGOs within each cluster (hatched bars) are displayed. Red, up-regulated; green, down-regulated; gray, nonmonotonic. (C, D) All oGOs containing less than 1000 genes are displayed for clusters 5 and 10.
Clusters 5 and 10 were chosen for further investigation, as they contained the genes that were regulated at high drug concentrations (800 and 1000 μM) only. The two clusters contained a low number of differentially regulated PS. Nevertheless, a relatively high number of GO terms was overrepresented among these genes (Figure 5B and Table S7, Supporting Information). The oGO identified were relatively heterogeneous, and no dominant superordinate cell biological process was identified. In particular, there was no overrepresentation of genes relating to cell death, cell stress, or degeneration (Figure 5C,D). This result was well in line with our earlier data. To further follow up on this, and to investigate which types of responses were triggered at which drug concentration, we chose in the following analyses a more targeted approach to specifically investigate the concentration dependency of the regulation of biological processes.
Gene Ontology Activation Profiles to Identify Biological Processes Related to Cytotoxic Drug Concentrations
All analyses in the previous paragraphs (Figure 2 to Figure 5) are driven by single gene results. First, genes were combined to clusters with respect to similar expression trajectories across the whole concentration range. Then functional gene groups (e.g., GO groups) were analyzed for overrepresentation with genes from these identified clusters. A meaningful alternative is to use a GO group (or any other functional gene group representing, e.g., a biological process) as the starting point for the analysis. Therefore, we applied a new biostatistical method that analyzes the activity (up- or down-regulation) of genes annotated to a GO group across all concentrations. The method uses a segmentation test40 to identify significant enrichment with up- or down-regulated genes (restricted by p-value <0.05), comparing each concentration with the controls. Each GO group obtains its own single characteristic activation profile. For example, the profile 0++++++ means that the corresponding gene set is significantly enriched with up-regulated genes at the second lowest concentration and for all higher concentrations but not at the lowest concentration. Analogously, a gene set with profile 00000-- is enriched with down-regulated genes only at the two highest concentrations but not at the five lower concentrations. The result of all GO activation profiles is given in Supporting Information (Table S8). For visualization of the GO activation profiles, we calculated a quantitative GO activation score and plotted the concentration–response profiles for some exemplary GOs (Figure 6A). Next, we classified the oGOs identified with this method in superordinate biological processes as described before.
Figure 6.

Concentration-dependent regulation of GOs and of superordinate cell biological processes. The test system was exposed to various concentrations of VPA for 6 days. Then, transcriptome analysis was performed, and differentially expressed genes were identified as in Figure 1. The data sets were examined for overrepresentation of any GO, and GO-based concentration–response profiles were generated as detailed in Table S8 (Supporting Information). Examples for transcriptome activation responses on the level of GO are shown. (A) Quantitative GO activation score was calculated by multiplying the percentage of genes within the GO that was found to be significantly regulated with the average fold change of these regulations. Positive values reflect oGO among up-regulated genes, and negative values indicate oGO among down-regulated genes. The names of the example GOs are indicated in the figures. Note the different concentrations at which a GO is first turned on and the different types of concentration–response behavior at high concentrations. (B). All GO profiles among up- and down-regulated genes were sorted, and the following three groups were selected for further analysis: low (GO found to be up-regulated at all VPA concentrations starting from 150 μM, clusters 0++++++ and 00+++++); intermediate (GO found to be up-regulated at all VPA concentrations higher than 350 μM, clusters 000++++ and 0000+++); and high (GO found to be up-regulated only at 800 and 1000 μM, clusters 00000++ and 000000+). Then, the GO were assigned to superordinate cell biological processes (e.g., migration and adhesion), and we calculated how many of all oGO in the low–medium–high groups belonged to the biological processes. For easier overview, a color code was applied with no color for values <10%, yellow for 10–20%, orange for 20–30%, and red for >30%.
The concentration dependence of the regulation of superordinate biological processes indicated three major groups: those mainly affected at high concentrations (00000++ and 000000+), those activated already at medium concentrations and then being maintained (000++++ and 0000+++), and finally those represented already among low concentrations and remaining up-regulated at higher concentrations (0++++++ and 00+++++) (Figure 6B). These findings agreed well with those obtained by other analytical approaches, such as unbiased k-means clustering (Figure 5) or comparison of the concentration dependence of oGO and PS (Figures 2 and 4). Application of the GO profiler analysis to the concentration data illustrates that only two key biological processes (catabolism and cell division) were overrepresented exclusively at cytotoxic concentrations (800 and 1000 μM) without being enriched in the noncytotoxic range (Figure 6B and Table S8, Supporting Information). GO groups involved in catabolism of DNA and proteins were overrepresented for the up- and down-regulated genes at cytotoxic concentrations. Moreover, cell division associated genes were overrepresented for the down-regulated genes only at 800 and 1000 μm VPA. The superordinate biological processes representing best the known mechanism of action of VPA, protein acetylation and epigenetic processes, were found (as expected) to start at noncytotoxic concentrations (Figure 6B and Table S8, Supporting Information). Another process clearly activated at noncytotoxic concentrations is migration and adhesion (Figure 6B and Table S8, Supporting Information). Other biological processes that are of interest for the mechanism of action of VPA are development and differentiation, metabolism, signal transduction, and response to external compound. GO groups belonging to these processes were overrepresented in the noncytotoxic but also the cytotoxic concentration range.
Development of a Measure of Teratogenicity to Quantitatively Compare Drug Concentrations
The distinct concentration–response behavior of the regulation of genes vs GO and superordinate cell biological processes suggested that these features may be used to develop a quantitative measure of teratogenic activity. For this purpose, the effect of each drug concentration was defined by two measures, the gene regulation index and the GO overrepresentation index. When these values were plotted onto a coordinate system formed by the two indices, it became evident that the distance from the origin was a useful measure of teratogenicity (Figure 7A). This way of data presentation also clearly revealed that VPA concentrations between 125 and 450 μM resulted in a progressive deviation of normal differentiation (increasing numbers of oGO among deregulated PS). At higher concentrations, the extent of gene deregulation increased, but oGOs remained fairly constant. The concentrations marking the distinct increase of teratogenicity according to this presentation correlate well with the VPA plasma concentrations associated with human or animal birth defects. The curve formed by the teratogenicity index data (Figure 7A) differed fundamentally from the cytotoxicity curve (Figure 1B). The latter showed strong and progressive changes at high drug concentrations, while the former did not change at high VPA levels but rather at medium levels. Thus, it seems feasible to define a specific teratogenicity measure that yields clearly different information from plain cytotoxicity in the same assay.
Figure 7.
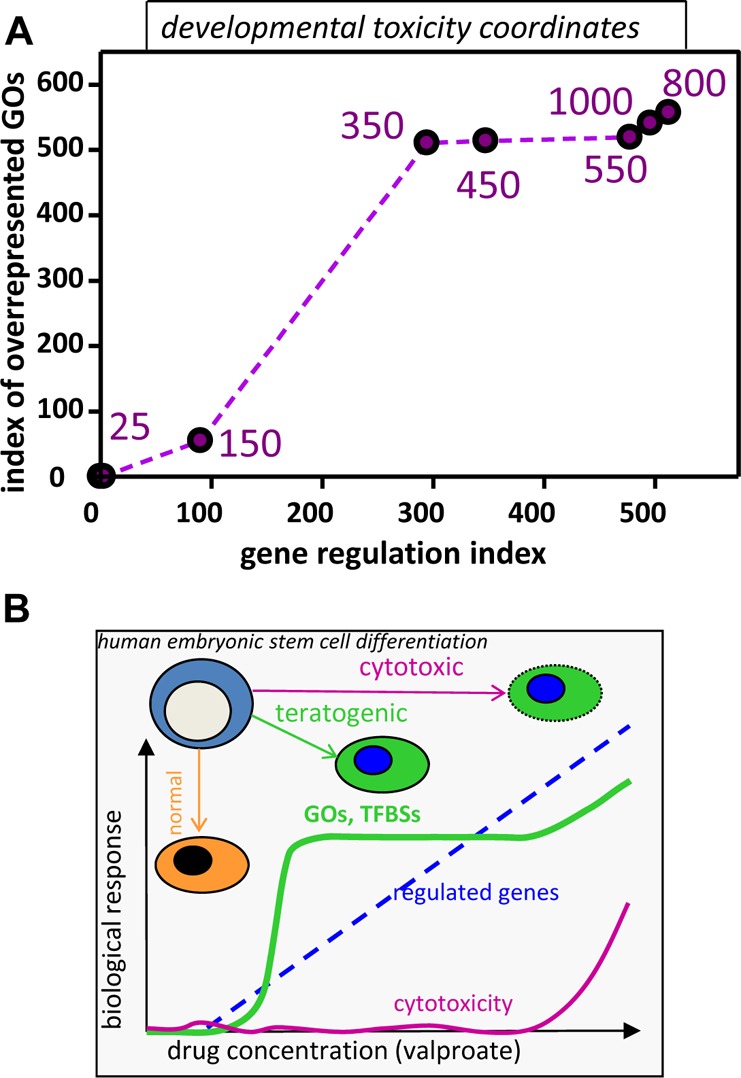
Toxicological implications of transcriptomics responses at different drug concentrations. (A) Transcriptome data were obtained for multiple drug (VPA) concentrations as in Figure 1. To obtain a measure of the deviation of the transcriptome from normal (teratogenicity measure), two indices were calculated and used to define the two dimensions of a developmental toxicity plane. The first dimension reflects the extent of gene regulation. The index is the sum of the negative logarithms of the p-values of the 100 most regulated genes. The second dimension reflects the extent of coordinated changes in biological processes reflected by GOs. The index is proportional to the number of overrepresented GOs in the gene sets. The purple numbers indicate the concentrations of VPA associated with the data points. (B) Summary of overall findings on concentration-dependent transcriptome deviations: drug concentrations were chosen in a way to allow either normal neuroectodermal differentiation of human embryonic stem cells (hESC) or disturbed differentiation (teratogenic concentration range) or cytotoxicity. Over this large concentration range (25–1000 μM), the number of deregulated genes increased continuously, once a certain threshold concentration was reached (125 μM). In contrast to this, superordinate biological regulations, as indicated by enriched GOs (gene ontologies) or TFBS (transcription factor binding sites), increased steeply in the teratogenic range and then more or less reached a plateau. At cytotoxic concentrations, only few additional GOs and TFBS were overrepresented. Thus, cytotoxicity, overall gene regulation, and coordinate regulation of biological processes showed largely different concentration dependencies. There were clear lower thresholds, and the extent of transcriptome regulation as such, without further bioinformatic analysis, did not appear to be a good measure for teratogenicity.
Discussion
In this study, we used a stem cell based test system that recapitulates neural differentiation of hESC. In order to elucidate the optimal concentration for toxicogenomics, we challenged this test system with increasing concentration of the well-known neurodevelopmental toxicants VPA. We found that the number of differentially regulated PS continuously increased but that up-targets like TFBS and functional gene ontologies reach saturation already at the lower border of intermediate concentrations. In addition, we show that with clearly cytotoxic concentrations only few more superordinate biological processes occur. Therefore, we conclude that the most sensitive concentration for DNT toxicogenomic testing would be the highest nontoxic concentration of a compound. However, small deviations from this concentration will not disrupt the results on the basis of oGO and TFBS. We present here the development of the transcriptomics-based teratogenicity index that exactly reflects this behavior for the comparison of deregulated PS and oGOs (Figure 7A). Although transcriptomics is frequently applied in toxicology, it has remained unclear how it is best used for toxicological statements and predictions. Basic issues, such as the question of appropriate concentrations, timing of exposure, and timing of sampling, have not been addressed in a systematic and quantitative manner. Only very few studies, mainly in the field of carcinogenesis, have attempted to correlate the extent or type of overall transcriptome changes with toxicological predictions in a quantitative manner. For instance, Thomas and colleagues6,7 have correlated benchmark doses based on GO terms and classical pathology with cancer development. We are not aware of any such study in developmental toxicology, and our study is a first attempt in this direction, even though only a single model compound, VPA,18,24,28 has been studied. Given the astonishing dearth of data in the field, we feel that our study is an important beginning but should not be overinterpreted. Further data are required to see whether the response features observed here are representative of a larger group of developmental toxicants.
Not every cell system is equally suited for studies on design principles of gene regulation.44 Our test system provided the required homogeneity and synchronicity to perform such analysis. Previously others and we have shown that the differentiation protocol delivers a >90% cell population of the same cell type.28,32 Moreover, genome wide mRNA analysis demonstrated a monotonic progression model of deregulated genes. A very high fraction of genes deregulated at a lower concentration were also deregulated at the next higher concentration. Moreover, the next higher concentration usually yielded additional genes that have not yet been deregulated at the lower concentration. These are good conditions to study the control mechanisms and principles of the cellular response to increasing concentrations. To examine whether all gene regulation was monotonic, we took several approaches. Unsupervised clustering indicated that there is a smaller subfraction of PS that behaved differently. To follow up on this, we selected the PS that were regulated by an intermediate drug concentration (450 μM) and determined their average fold change (FC). Then, we determined the FC of the same set of PS at lower and higher concentrations. At the lowest concentration (25 μM), there was no FC at all. This could indicate that the genes were not regulated in any consistent way. The FC-curves showed overall a sigmoid shape. The linear middle part supports the suggestion that the selected PS were regulated increasingly more with increasing concentration, and this may have affected the number that was detected after filtering for significance and FC. The flattening of the curve on the lower and upper ends suggests that most likely additional mechanisms are involved, i.e., regulation of different genes at different concentrations, as observed here by other methods. As a complementary approach, we selected the PS that were significantly regulated at 450 μM (p < 0.05) and calculated the p-values for the same PS at low concentrations. Most of the PS lost significance at 150 μM and nearly all at 25 μM. The situation was somewhat different, when the initial selection of PS included a FC cutoff of 2. Under this condition, all PS selected at 450 μM still reached a p < 0.05 significance level at 350 μM, and most of them still did so at 150 μM. At 25 μM, still about 10% of the PS were significant. This suggests, that some PS that we considered here as regulated only at high drug concentrations but not at low drug exposure were indeed already affected at low concentrations but that the change was too small to be picked up as significant. This feature may have to be considered for future systems biology based quantitative models of gene regulation.
Comprehensive biostatistical analyses including the establishment of a novel gene ontology activation profiler (GOAP) suggest the following concept of three concentration ranges. A range of tolerance is observed up to 25 μm VPA. At 25 μM, not a single up- or down-regulated gene with p < 0.05 and a fold change higher than two was obtained. This observation is in agreement with the general concept that toxicity based on, e.g., enzyme inhibition or induction, or receptor activation, acts by threshold mechanisms.45,46 In the cytotoxic concentration range (800 μM and 1000 μM), we expected that an apoptosis signature may occur, but we observed the induction of apoptosis, death, and stress associated oGO groups already at the intermediate concentration range. Also, GO analysis of the genes that are exclusively up- or down-regulated with the cytotoxic concentrations revealed no additional death related oGOs. In addition, GOAP analysis revealed that GO that are related to catabolism and metabolism are induced with cytotoxic concentrations. Among the down-regulated PS, cell division related GOs were induced. A range of deregulations occurred between 150 and 550 μM VPA. Analysis of GOs overrepresented among PS in this range gave evidence for developmental disturbances, cell migration, and the down-regulation of neuronal pathways. This confirmed massively disturbed neurodevelopment with these concentrations.28,47 Therefore, we defined the range from 150 μM to 550 μM VPA as the deregulated/teratogenic concentration range (Figure 7B). These findings are in good agreement with human data that indicate that VPA plasma concentrations above 500 μM are associated with strongly increasing risk for malformations.48,49 The newly established GOAP method revealed additional and more sensitive answers for this range of teratogenic concentrations. Between 150 and 550 μM, a number of GO groups became overrepresented that reflect the specific mode of action of VPA, namely, protein acetylation and epigenetic processes. Down-regulated PS comprises genes involved in all kind of epigenetic processes16 like several histone acetyl transferases (e.g., KAT2B, KAT6A, KAT5), polycomb proteins (e.g., SUZ12), lysine methyl transferases (e.g., EHMT2), and HDACs themselves (e.g., HDAC 2, 5, and 9).
Regarding GO regulation patterns over the concentration range, we also identified GO groups that showed, e.g., a 00+++00 nonmonotonic pattern. These GO groups contained some PS with nonmonotonic behavior. Altogether, they were below 10%. We could verify the main monotonic regulation pattern by another analysis targeting upstream regulators of transcriptional changes. Overrepresented TFBS were searched in the deregulated PS. We found that most TFBS are already induced with teratogenic concentrations, and only few additional TFBS were induced with cytotoxic concentrations (Figure 7B). These comprised AP-2 and Pax5 for up-regulated and IPF1 and PBX for down-regulated PS.
On the basis of differentially expressed PS and altered functional processes, we developed a teratogenicity index that could distinguish between DNT-relevant (the teratogenic range) and cytotoxic concentrations. For this purpose, the extent of transcriptome deregulation was captured by measures for deregulation of individual genes and a measure for biological processes (oGO). This information was displayed in a two-dimension plot. This graphical display exactly represented the situation described in greater detail before. Although cytotoxicity only starts at 800 μM VPA, the functional processes were significantly changed already with 350 μM and the teratogenicity index only slightly changed with 800 μM and 1000 μM. Therefore, we present here an index that distinguishes between noncytotoxic, teratogenic, and cytotoxic concentrations and could help in the future to improve the experimental design of toxicogenomic studies. In particular, our data support the use of the highest noncytotoxic concentration of toxicogenomics studies, if no other information on the test compounds is available. At this concentration, a maximum power is reached (number of deregulated PS) without compromising the biological information obtained (e.g., altered oGO or TFBS). Accidental use of slightly too high concentrations would be expected to have only minor effects on the overall outcome.
In conclusion, the data from this concentration progression study suggest a steady increase of deregulated genes. However, they mostly belong to the same biological processes and seem to be controlled by the same TF that are already deregulated at the intermediate concentrations of 350 and 450 μM VPA (Figure 7B). This is consistent with the recently identified superenhancers.26,27 Superenhancers are unusual enhancers, which are found at genes responsible for cell type identity and occupied by key transcription factors for this cell type. In ESC, e.g., these DNA sequences are mainly occupied by the key stemness factors Oct4, Nanog, and Sox2.27 It was also shown that these superenhancers play crucial roles in the development of many diseases.26 Therefore, it is very likely that they are also disturbed or differentially occupied by a toxic compound and play a crucial role in the interpretation of our upstream target analysis. In the future, it will be interesting to investigate whether deviations from the normal development on the level of TFBS and superordinate cell biological processes will be shared by diverse toxicants and whether the time of toxicant exposure has an effect on such findings.
Acknowledgments
We thank M. Kapitza and Tamara Rotshteyn for excellent technical support. For critical reading of the manuscript, we thank Stefanie Klima and Simon Gutbier.
Glossary
Abbreviations
- PS
probe sets
- DMA
DNA microarray
- BMC
benchmark concentration
- TFBS
transcription factor binding site
- GO
gene ontology
- oGO
overrepresented gene ontology term
- HDAC
histone acetyl transferase
- hESC
human embryonic stem cells
- NEP
neuroectoderm progenitor
Supporting Information Available
Names and regulations of the probe sets, gene ontologies, and TFBS. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by the European Commission’s FP7, ESNATS project and the Ph.D. schools RTG1331 and KoRS-CB at the university of Konstanz, Germany.
The authors declare no competing financial interest.
Author Status
# T.W. and E.R. share first authorship of this work.
Author Status
○ J.G.H and M.L. share senior authorship of this work.
Supplementary Material
References
- Pallocca G.; Fabbri M.; Sacco M. G.; Gribaldo L.; Pamies D.; Laurenza I.; Bal-Price A. (2013) miRNA expression profiling in a human stem cell-based model as a tool for developmental neurotoxicity testing. Cell. Biol. Toxicol. 29, 239–257. [DOI] [PubMed] [Google Scholar]
- Bal-Price A. K.; Coecke S.; Costa L.; Crofton K. M.; Fritsche E.; Goldberg A.; Grandjean P.; Lein P. J.; Li A.; Lucchini R.; Mundy W. R.; Padilla S.; Persico A. M.; Seiler A. E.; Kreysa J. (2012) Advancing the science of developmental neurotoxicity (DNT): testing for better safety evaluation. Altex 29, 202–215. [DOI] [PubMed] [Google Scholar]
- Vojnits K.; Ensenat-Waser R.; Gaspar J. A.; Meganathan K.; Jagtap S.; Hescheler J.; Sachinidis A.; Bremer-Hoffmann S. (2012) A tanscriptomics study to elucidate the toxicological mechanism of methylmercury chloride in a human stem cell based in vitro test. Curr. Med. Chem. 19, 6224–6232. [PubMed] [Google Scholar]
- Klaric M.; Winkler J.; Vojnits K.; Meganathan K.; Jagtap S.; Ensenat-Waser R.; Hescheler J.; Sachinidis A.; Bremer-Hoffmann S. (2013) Current status of human pluripotent stem cell based in vitro toxicity tests. Front. Biosci. 5, 118–133. [DOI] [PubMed] [Google Scholar]
- Ramirez T.; Daneshian M.; Kamp H.; Bois F. Y.; Clench M. R.; Coen M.; Donley B.; Fischer S. M.; Ekman D. R.; Fabian E.; Guillou C.; Heuer J.; Hogberg H. T.; Jungnickel H.; Keun H. C.; Krennrich G.; Krupp E.; Luch A.; Noor F.; Peter E.; Riefke B.; Seymour M.; Skinner N.; Smirnova L.; Verheij E.; Wagner S.; Hartung T.; van Ravenzwaay B.; Leist M. (2013) Metabolomics in toxicology and preclinical research. Altex 30, 209–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas R. S.; Clewell H. J. III; Allen B. C.; Wesselkamper S. C.; Wang N. C.; Lambert J. C.; Hess-Wilson J. K.; Zhao Q. J.; Andersen M. E. (2011) Application of transcriptional benchmark dose values in quantitative cancer and noncancer risk assessment. Toxicol. Sci. 120, 194–205. [DOI] [PubMed] [Google Scholar]
- Thomas R. S.; Wesselkamper S. C.; Wang N. C.; Zhao Q. J.; Petersen D. D.; Lambert J. C.; Cote I.; Yang L.; Healy E.; Black M. B.; Clewell H. J. III; Allen B. C.; Andersen M. E. (2013) Temporal concordance between apical and transcriptional points of departure for chemical risk assessment. Toxicol. Sci. 134, 180–194. [DOI] [PubMed] [Google Scholar]
- van Thriel C.; Westerink R. H.; Beste C.; Bale A. S.; Lein P. J.; Leist M. (2012) Translating neurobehavioural endpoints of developmental neurotoxicity tests into in vitro assays and readouts. Neurotoxicology 33, 911–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler S.; Basketter D.; Creton S.; Pelkonen O.; van Benthem J.; Zuang V.; Andersen K. E.; Angers-Loustau A.; Aptula A.; Bal-Price A.; Benfenati E.; Bernauer U.; Bessems J.; Bois F. Y.; Boobis A.; Brandon E.; Bremer S.; Broschard T.; Casati S.; Coecke S.; Corvi R.; Cronin M.; Daston G.; Dekant W.; Felter S.; Grignard E.; Gundert-Remy U.; Heinonen T.; Kimber I.; Kleinjans J.; Komulainen H.; Kreiling R.; Kreysa J.; Leite S. B.; Loizou G.; Maxwell G.; Mazzatorta P.; Munn S.; Pfuhler S.; Phrakonkham P.; Piersma A.; Poth A.; Prieto P.; Repetto G.; Rogiers V.; Schoeters G.; Schwarz M.; Serafimova R.; Tahti H.; Testai E.; van Delft J.; van Loveren H.; Vinken M.; Worth A.; Zaldivar J. M. (2011) Alternative (non-animal) methods for cosmetics testing: current status and future prospects-2010. Arch. Toxicol. 85, 367–485. [DOI] [PubMed] [Google Scholar]
- Basketter D. A.; Clewell H.; Kimber I.; Rossi A.; Blaauboer B.; Burrier R.; Daneshian M.; Eskes C.; Goldberg A.; Hasiwa N.; Hoffmann S.; Jaworska J.; Knudsen T. B.; Landsiedel R.; Leist M.; Locke P.; Maxwell G.; McKim J.; McVey E. A.; Ouedraogo G.; Patlewicz G.; Pelkonen O.; Roggen E.; Rovida C.; Ruhdel I.; Schwarz M.; Schepky A.; Schoeters G.; Skinner N.; Trentz K.; Turner M.; Vanparys P.; Yager J.; Zurlo J.; Hartung T. (2012) A roadmap for the development of alternative (non-animal) methods for systemic toxicity testing - t4 report*. Altex 29, 3–91. [DOI] [PubMed] [Google Scholar]
- Mol P. G.; Arnardottir A. H.; Motola D.; Vrijlandt P. J.; Duijnhoven R. G.; Haaijer-Ruskamp F. M.; de Graeff P. A.; Denig P.; Straus S. M. (2013) Post-approval safety issues with innovative drugs: a European cohort study. Drug Saf. 36, 1105–1115. [DOI] [PubMed] [Google Scholar]
- Andersen M. E.; Clewell H. J.; Carmichael P. L.; Boekelheide K. (2011) Can case study approaches speed implementation of the NRC report: “toxicity testing in the 21st century: a vision and a strategy?”. Altex 28, 175–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krewski D.; Westphal M.; Al-Zoughool M.; Croteau M. C.; Andersen M. E. (2011) New directions in toxicity testing. Annu. Rev. Public Health 32, 161–178. [DOI] [PubMed] [Google Scholar]
- Leist M.; Hartung T.; Nicotera P. (2008) The dawning of a new age of toxicology. Altex 25, 103–114. [PubMed] [Google Scholar]
- Leist M.; Bremer S.; Brundin P.; Hescheler J.; Kirkeby A.; Krause K. H.; Poerzgen P.; Puceat M.; Schmidt M.; Schrattenholz A.; Zak N. B.; Hentze H. (2008) The biological and ethical basis of the use of human embryonic stem cells for in vitro test systems or cell therapy. Altex 25, 163–190. [PubMed] [Google Scholar]
- Weng M. K.; Zimmer B.; Poltl D.; Broeg M. P.; Ivanova V.; Gaspar J. A.; Sachinidis A.; Wullner U.; Waldmann T.; Leist M. (2012) Extensive transcriptional regulation of chromatin modifiers during human neurodevelopment. PLoS One 7, e36708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiegler N. V.; Krug A. K.; Matt F.; Leist M. (2011) Assessment of chemical-induced impairment of human neurite outgrowth by multiparametric live cell imaging in high-density cultures. Toxicol. Sci. 121, 73–87. [DOI] [PubMed] [Google Scholar]
- Zimmer B.; Lee G.; Balmer N. V.; Meganathan K.; Sachinidis A.; Studer L.; Leist M. (2012) Evaluation of developmental toxicants and signaling pathways in a functional test based on the migration of human neural crest cells. Environ. Health Perspect. 120, 1116–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuegler P. B.; Baumann B. A.; Zimmer B.; Keller S.; Marx A.; Kadereit S.; Leist M. (2012) GFAP-independent inflammatory competence and trophic functions of astrocytes generated from murine embryonic stem cells. Glia 60, 218–228. [DOI] [PubMed] [Google Scholar]
- Zimmer B.; Schildknecht S.; Kuegler P. B.; Tanavde V.; Kadereit S.; Leist M. (2011) Sensitivity of dopaminergic neuron differentiation from stem cells to chronic low-dose methylmercury exposure. Toxicol. Sci. 121, 357–367. [DOI] [PubMed] [Google Scholar]
- Zimmer B.; Kuegler P. B.; Baudis B.; Genewsky A.; Tanavde V.; Koh W.; Tan B.; Waldmann T.; Kadereit S.; Leist M. (2011) Coordinated waves of gene expression during neuronal differentiation of embryonic stem cells as basis for novel approaches to developmental neurotoxicity testing. Cell Death Differ. 18, 383–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuegler P. B.; Zimmer B.; Waldmann T.; Baudis B.; Ilmjarv S.; Hescheler J.; Gaughwin P.; Brundin P.; Mundy W.; Bal-Price A. K.; Schrattenholz A.; Krause K. H.; van Thriel C.; Rao M. S.; Kadereit S.; Leist M. (2010) Markers of murine embryonic and neural stem cells, neurons and astrocytes: reference points for developmental neurotoxicity testing. Altex 27, 17–42. [DOI] [PubMed] [Google Scholar]
- Meganathan K.; Jagtap S.; Wagh V.; Winkler J.; Gaspar J. A.; Hildebrand D.; Trusch M.; Lehmann K.; Hescheler J.; Schluter H.; Sachinidis A. (2012) Identification of thalidomide-specific transcriptomics and proteomics signatures during differentiation of human embryonic stem cells. PLoS One 7, e44228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug A. K.; Kolde R.; Gaspar J. A.; Rempel E.; Balmer N. V.; Meganathan K.; Vojnits K.; Baquie M.; Waldmann T.; Ensenat-Waser R.; Jagtap S.; Evans R. M.; Julien S.; Peterson H.; Zagoura D.; Kadereit S.; Gerhard D.; Sotiriadou I.; Heke M.; Natarajan K.; Henry M.; Winkler J.; Marchan R.; Stoppini L.; Bosgra S.; Westerhout J.; Verwei M.; Vilo J.; Kortenkamp A.; Hescheler J.; Hothorn L.; Bremer S.; van Thriel C.; Krause K. H.; Hengstler J. G.; Rahnenfuhrer J.; Leist M.; Sachinidis A. (2013) Human embryonic stem cell-derived test systems for developmental neurotoxicity: a transcriptomics approach. Arch. Toxicol. 87, 123–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loven J.; Hoke H. A.; Lin C. Y.; Lau A.; Orlando D. A.; Vakoc C. R.; Bradner J. E.; Lee T. I.; Young R. A. (2013) Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D.; Abraham B. J.; Lee T. I.; Lau A.; Saint-Andre V.; Sigova A. A.; Hoke H. A.; Young R. A. (2013) Super-enhancers in the control of cell identity and disease. Cell 155, 934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte W. A.; Orlando D. A.; Hnisz D.; Abraham B. J.; Lin C. Y.; Kagey M. H.; Rahl P. B.; Lee T. I.; Young R. A. (2013) Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmer N. V.; Weng M. K.; Zimmer B.; Ivanova V. N.; Chambers S. M.; Nikolaeva E.; Jagtap S.; Sachinidis A.; Hescheler J.; Waldmann T.; Leist M. (2012) Epigenetic changes and disturbed neural development in a human embryonic stem cell-based model relating to the fetal valproate syndrome. Hum. Mol. Genet. 21, 4104–4114. [DOI] [PubMed] [Google Scholar]
- Theunissen P. T.; Robinson J. F.; Pennings J. L.; de Jong E.; Claessen S. M.; Kleinjans J. C.; Piersma A. H. (2012) Transcriptomic concentration-response evaluation of valproic acid, cyproconazole, and hexaconazole in the neural embryonic stem cell test (ESTn). Toxicol. Sci. 125, 430–438. [DOI] [PubMed] [Google Scholar]
- Jergil M.; Kultima K.; Gustafson A. L.; Dencker L.; Stigson M. (2009) Valproic acid-induced deregulation in vitro of genes associated in vivo with neural tube defects. Toxicol. Sci. 108, 132–148. [DOI] [PubMed] [Google Scholar]
- Theunissen P. T.; Pennings J. L.; Robinson J. F.; Claessen S. M.; Kleinjans J. C.; Piersma A. H. (2011) Time-response evaluation by transcriptomics of methylmercury effects on neural differentiation of murine embryonic stem cells. Toxicol. Sci. 122, 437–447. [DOI] [PubMed] [Google Scholar]
- Chambers S. M.; Fasano C. A.; Papapetrou E. P.; Tomishima M.; Sadelain M.; Studer L. (2009) Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 27, 275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers S. M.; Mica Y.; Studer L.; Tomishima M. J. (2011) Converting human pluripotent stem cells to neural tissue and neurons to model neurodegeneration. Methods Mol. Biol. 793, 87–97. [DOI] [PubMed] [Google Scholar]
- Chang K., Harbron C., and South M. C.. RefPlus: A Function Set for the Extrapolation Strategy (RMA+) and Extrapolation Averaging (RMA++) Methods, R package 1.26.20. http://bioconductor.wustl.edu/bioc/html/RefPlus.html.
- Smyth G. K.; Michaud J.; Scott H. S. (2005) Use of within-array replicate spots for assessing differential expression in microarray experiments. Bioinformatics 21, 2067–2075. [DOI] [PubMed] [Google Scholar]
- Benjamini Y.; Yekutieli D. (2001) The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 29, 1165–1188. [Google Scholar]
- (2012) R: A Language and Environment for Statistical Computing, Institute for Statistics and Mathematics, Vienna, Austria.
- Chow S., and Rodgers P. (2005) Construction Area-Proportional Venn and Euler Diagrams with Three Circles, in Euler Diagrams Workshop, Paris. http://kar.kent.ac.uk/id/eprint/14285.
- Alexa A., and Rahnenführer J. (2010) topGO: Enrichment Analysis for Gene Ontology, R package 2.14.10. http://bioconductor.wustl.edu/bioc/html/topGO.html.
- Minguez P.; Al-Shahrour F.; Dopazo J. (2006) A function-centric approach to the biological interpretation of microarray time-series. Genome Inf. 17, 57–66. [PubMed] [Google Scholar]
- Benjamini Y.; Krieger A. M.; Yekutieli D. (2006) Adaptive linear step-up procedures that control the false discovery rate. Biometrika 93, 491–507. [Google Scholar]
- Elkon R.; Linhart C.; Sharan R.; Shamir R.; Shiloh Y. (2003) Genome-wide in silico identification of transcriptional regulators controlling the cell cycle in human cells. Genome Res. 13, 773–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulitsky I.; Maron-Katz A.; Shavit S.; Sagir D.; Linhart C.; Elkon R.; Tanay A.; Sharan R.; Shiloh Y.; Shamir R. (2010) Expander: from expression microarrays to networks and functions. Nat. Protoc. 5, 303–322. [DOI] [PubMed] [Google Scholar]
- Leist M.; Efremova L.; Karreman C. (2010) Food for thought ... considerations and guidelines for basic test method descriptions in toxicology. Altex 27, 309–317. [DOI] [PubMed] [Google Scholar]
- Dietrich D. R.; von Aulock S.; Marquardt H.; Blaauboer B.; Dekant W.; Kehrer J.; Hengstler J.; Collier A.; Batta Gori G.; Pelkonen O.; Lang F.; Nijkamp F. P.; Stemmer K.; Li A.; Savolainen K.; Wallace Hayes A.; Gooderham N.; Harvey A. (2013) Scientifically unfounded precaution drives European Commission’s recommendations on EDC regulation, while defying common sense, well-established science and risk assessment principles. Altex 30, 381–385. [DOI] [PubMed] [Google Scholar]
- Hengstler J. G.; Bogdanffy M. S.; Bolt H. M.; Oesch F. (2003) Challenging dogma: thresholds for genotoxic carcinogens? The case of vinyl acetate. Annu. Rev. Pharmacol. Toxicol. 43, 485–520. [DOI] [PubMed] [Google Scholar]
- Krug A. K.; Balmer N. V.; Matt F.; Schonenberger F.; Merhof D.; Leist M. (2013) Evaluation of a human neurite growth assay as specific screen for developmental neurotoxicants. Arch. Toxicol. 87, 2215–2231. [DOI] [PubMed] [Google Scholar]
- Wiltse J. (2005) Mode of action: inhibition of histone deacetylase, altering WNT-dependent gene expression, and regulation of beta-catenin--developmental effects of valproic acid. Crit. Rev. Toxicol. 35, 727–738. [DOI] [PubMed] [Google Scholar]
- Sztajnkrycer M. D. (2002) Valproic acid toxicity: overview and management. J. Toxicol. Clin. Toxicol. 40, 789–801. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.