

Abstract
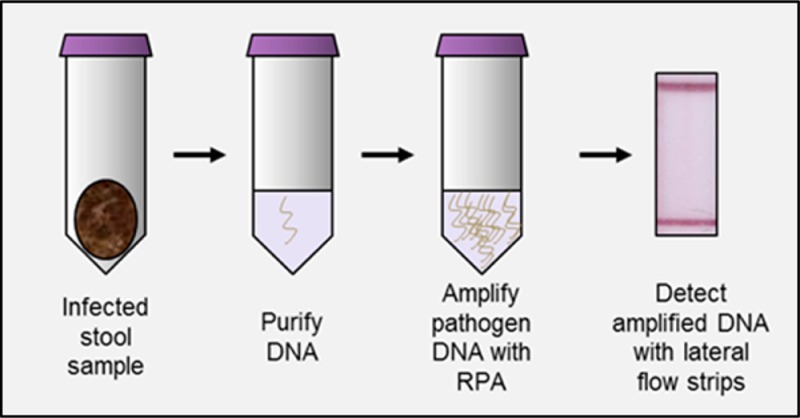
Diarrheal diseases cause more morbidity and mortality around the world than human immunodeficiency virus (HIV), malaria, or tuberculosis. Given that effective treatment of persistent diarrheal illness requires knowledge of the causative organism, diagnostic tests are of paramount importance. The protozoan parasites of the genus Cryptosporidium are increasingly recognized to be responsible for a significant portion of diarrhea morbidity. We present a novel nucleic acid test to detect the presence of Cryptosporidium species in DNA extracted from stool samples. The assay uses the isothermal amplification technique recombinase polymerase amplification (RPA) to amplify trace amounts of pathogen DNA extracted from stool to detectable levels in 30 min; products are then detected visually on simple lateral flow strips. The RPA-based Cryptosporidium assay (RPAC assay) was developed and optimized using DNA from human stool samples spiked with pathogen. It was then tested using DNA extracted from the stool of infected mice where it correctly identified the presence or absence of 27 out of 28 stool samples. It was finally tested using DNA extracted from the stool of infected patients where it correctly identified the presence or absence of 21 out of 21 stool samples. The assay was integrated into a foldable, paper and plastic device that enables DNA amplification with only the use of pipets, pipet tips, and a heater. The performance of the integrated assay is comparable to or better than polymerase chain reaction (PCR), without requiring the use of thermal cycling equipment. This platform can easily be adapted to detect DNA from multiple pathogens.
Despite advances in diagnosis and treatment, diarrheal illness remains one of the leading causes of morbidity and mortality in the developing world.1,2 Parasitic infections are typically responsible for episodes of persistent diarrhea, which in turn can lead to dehydration, wasting, and frequently death.3Cryptosporidium spp. are increasingly being found to be responsible for these persistent diarrheal episodes, accounting for 20% of diarrheal morbidity in children in both developed and developing countries.4Cryptosporidium is a particular threat for individuals with HIV, affecting them more than any other diarrheal parasite.2
Current diagnostic methods for cryptosporidiosis are suboptimal leading to misdiagnosis or inappropriate treatment. In many cases, because it requires specialized tests, clinicians do not even test for Cryptosporidium in high risk populations.5 The traditional approach to identify stool parasites relies heavily on microscopic analysis of stool smears. Even with a highly trained laboratory technician using appropriate methods, microscopic identification of stool parasites using acid fast staining has a high limit of detection (∼50 000–500 000 oocysts per gram of stool).6 The limit of detection of microscopy is higher than that associated with many clinically significant infections, where the number of organisms can range from as few as 103 oocysts per gram of stool to more than 107 oocysts per gram of stool.7 Fluorescent stains, such as Auramine O, are more sensitive than acid fast staining; however, the frequency of false positive tests led the CDC to recommend that diagnosis by fluorescence microscopy be confirmed with a secondary test such as an immunofluorescence antibody (IFA) test or an enzyme linked immunosorbent assay.8 Both IFA tests and fluorescence staining require the use of a fluorescence microscope, limiting their usefulness in low resource settings. Enzyme-linked-immunosorbent-assays (ELISA) and lateral flow tests that rely on antibodies have been developed to detect parasite antigens; however, their reported sensitivity in the field varies widely. In a multicenter, blinded study the four leading commercial assays demonstrated clinical sensitivities between 47.2% and 68.8%.9
The gold standard for Cryptosporidium detection is widely considered to be PCR, with a limit of detection (LOD) of ≤103 oocysts per gram of stool.10 Because of the increased sensitivity associated with polymerase chain reaction (PCR) as compared to microscopic methods, the rate of detection of Cryptosporidium and other intestinal parasites is nearly twice as high with nucleic acid-based tests.11 Despite these advantages, PCR still requires the use of thermal cycling equipment. Because of the high cost associated with thermal cyclers ($3 000–$10 000), there is a high investment burden on clinics or laboratories wishing to conduct PCR. For this reason PCR assays are typically only available in reference laboratories and are seldom used for initial diagnosis.
A number of isothermal nucleic acid amplification techniques have been developed to enable performance of nucleic acid testing outside of reference laboratories. Nucleic acid sequence based amplification, loop-mediated amplification, rolling circle amplification, strand displacement amplification, and recombinase polymerase amplification among others have been explored.12,13 Recombinase polymerase amplification (RPA) offers significant advantages over other isothermal amplification techniques because of its speed and low temperature requirements. RPA is an isothermal process that functions efficiently between 25 and 42 °C.13 Because the RPA enzymes function well between room temperature and body temperature, it is theoretically possible to completely alleviate the need for heating equipment. The reaction enzymes are stable in dried formulation and can be safely stored without refrigeration for point-of-care use for up to a year.14 Perhaps most significantly, with simple primer modifications, it is possible to detect RPA amplicons using commercially available lateral flow strips. Using this technology, we developed an RPA-based Cryptosporidium assay (RPAC) to detect DNA from Cryptosporidium spp. using nucleic acid isolated from stool samples.
Materials and Methods
Ethics Statement
All human stool samples were collected from normal healthy volunteers according to Rice University IRB approved protocol 11-101E. Informed, written consent was given by all human volunteers. Animal testing was completed in Galveston, TX at the University of Texas Medical Branch in compliance with the Animal Welfare Act (Public Law 89-544) and university protocols (IACUC approval no. 1005021A).
Study Design
The objective of this study was to develop, optimize, and evaluate the performance of a new RPAC assay to detect Cryptosporidium in stool samples. Controlled laboratory experiments were first performed to optimize assay parameters and evaluate assay performance using samples spiked with oocysts spanning a clinically relevant range of concentrations. Assays were performed in triplicate or quadruplicate, as described below. In all cases, positive and negative test results were objectively determined using a predetermined signal-to-background ratio (SBR) threshold; no data were excluded from analysis.
Once optimized, experiments were performed to assess the performance of the RPAC assay using stool samples from 18 animals infected with Cryptosporidium and 10 healthy controls. The sample size of the pilot animal study was based on the availability of banked samples. Finally, the RPAC assay was performed using banked stool samples from 10 patients with cryptosporidiosis and 10 healthy volunteers. The sample size of the pilot preclinical study was based on the number of banked stool samples available from state health authorities. Positive or negative RPAC assay results were determined using the same predetermined threshold; results of the RPAC assay were compared to the gold standard of PCR which was performed in duplicate. The RPAC assay and PCR testing of animal and human samples were performed at separate institutions by different operators, with the RPAC assay operator blinded to whether specimens were from healthy or infected subjects. No data were excluded from analysis.
RPAC Assay Development and Optimization
The RPAC assay was initially developed using the TwistAmp Basic kit (TwistDX, U.K.). TwistAmp Basic reactions were performed according to the manufacturer’s recommended protocols with the amplified products detected via gel electrophoresis.
A 208 base pair DNA target sequence specific to Cryptosporidium spp. was selected on the 18S RNA gene as an RPA target. A number of forward and reverse primers were screened for their ability to efficiently amplify the 18S gene target (data not shown). The forward primer (RPAF6, 5′-GTGGCAATGACGGGTAACGGGGAATTAGGG-3′) and reverse primer (RPAR7, 5′-AATTGATACTTGTAAAGGGGTTTATACTTAACTC-3′) were ultimately selected based on their ability to consistently amplify the targeted sequence. All primers and probes were purchased from Integrated DNA Technologies (Coralville, IA).
RPA reactions were mixed in sterile 1.5 mL screw-top microcentrifuge tubes according to the manufacturer’s instructions, then incubated at 37 °C for 30 min (optimal time identified from experiments with times ranging from 10 to 40 min; data not shown). The reaction was stopped and products were purified using the Qiagen MiniElute PCR Purification Kit according to the manufacturer’s recommended protocol (Germantown, MD). Amplified products were electrophoresed on a 3% agarose gel and read with a Bio Rad Gel Doc XR+ gel reader.
As a proof of concept, we went on to develop a lateral flow RPAC assay to detect Cryptosporidium spp. using commercially available lateral flow strips (HybriDetect MGHD1, Milenia Biotec, Germany). The lateral flow strips contained a sample pad with dried gold nanoparticles that were conjugated to rabbit anti-FITC antibodies; streptavidin was immobilized at the detection line on the lateral flow strip. Dual-labeled RPA products with a 5′FITC label on one strand and a 5′biotin label on the complementary strand attached to the anti-FITC gold. The DNA-gold conjugates were then captured at the streptavidin detection line. The strips also contained a control line functionalized with antirabbit antibody that captured any anti-FITC gold nanoparticles not captured at the detection line. This is shown schematically in Figure 2A.
Figure 2.
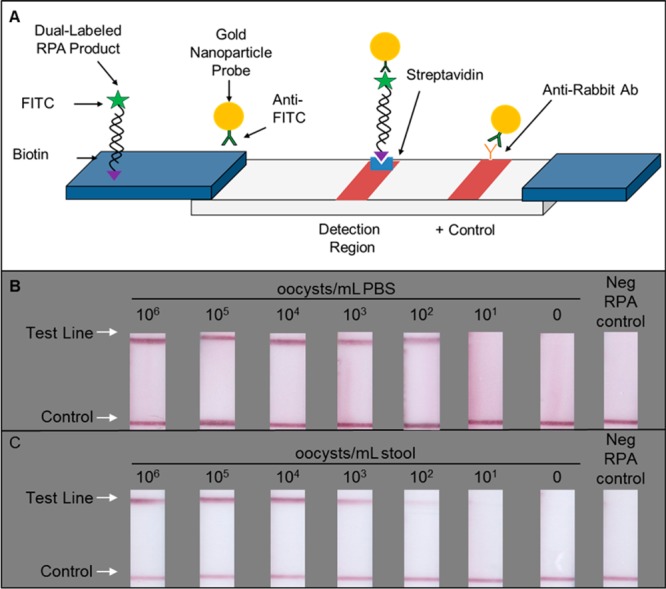
Lateral flow detection of RPA product. Dual labeled amplicons can be detected visually using lateral flow strips (A). Anti-FITC conjugated gold nanoparticles dried in the sample pad bind to the FITC label on RPA amplicons. Gold nanoparticles wick down the strip where amplicon bound nanoparticles are captured at the streptavidin detection line and those nanoparticles not bound to amplicons are captured at the positive control line. RPA products from DNA extracted from as few as 102 oocysts/mL PBS (B) can be detected visually. RPA products from DNA extracted from as few as 102 oocysts/mL stool (C) can be detected visually.
Dual labeled RPA products are generated with the TwistAmp nfo kit (TwistDx, U.K.) using an unlabeled forward primer, a biotin labeled reverse primer, and a TwistAmp LF probe. The TwistAmp LF probe has a 3′ blocker and an internal abasic site that replaces a nucleotide. The LF probe binds to the single-stranded, antisense DNA generated by the biotin labeled reverse primer. In turn, an endonuclease cuts the probe’s internal abasic site, unblocking the end of the probe and allowing it to act as a primer. A polymerase then extends the probe and generates a dual labeled RPA product that can be detected using a lateral flow strip. Generation of these dual-labeled products required a biotin labeled reverse primer (5′-Biotin-AATTGATACTTGTAAAGGGGTTTATACTTAACTC-3′) as well as the addition of a FITC labeled probe (5′-FITC-ACAGGGAGGTAGTGACAAGAAATAACAATA-idSp-AGGACTTTTTGGTTTTGTA-3SpC-3′).
As with the basic reactions, the reactions using lateral flow detection were incubated for 30 min at 37 °C. After incubation, two microliters of amplified products were added directly to 98 μL of running buffer (supplied with the HybridDetect lateral flow strips), briefly vortexed, and 10 μL of the diluted products were added to a Milenia HybridDetect lateral flow strip. The sample-end of each strip was then placed into a well of a 96-well plate containing 100 μL of running buffer. After 3 min, the strips were removed and scanned using a flatbed scanner.
Image Analysis to Assess Test Results
Positive test results contain two visible lines: a control line next to the absorbent pad indicating the test ran successfully and a second line next to the sample pad indicating the presence of Cryptosporidium. Generally the distinction between a positive and negative test result was visually apparent – a negative test result had only a single control band visible. Occasionally it was difficult to determine whether a faint test band qualified as a positive or negative test result. To resolve this issue, we determined a signal-to-background ratio (SBR) threshold to differentiate a positive test result from a negative test result. This was done by scanning images of 10 negative control strips. Using a custom-built Matlab script (Mathwork, Natick, MA), the signal-to-background ratio (SBR) of each test line was calculated by dividing the average signal intensity of the test line by the average signal intensity of the area surrounding the test line. For the 10 negative controls, we calculated the average SBR and standard deviation. A positive SBR threshold was set at the average of the 10 negative controls plus 3 times their standard deviation.
DNA Extraction from PBS Spiked with Oocysts
The RPAC assay was evaluated using DNA extracted from PBS solutions containing Cryptosporidium parvum oocysts in PBS purchased from Waterborne, Inc. (New Orleans, LA). Oocysts was serially diluted to create 450 μL solutions with 106, 105, 104, 103, 102, 101 oocysts/mL PBS. Total nucleic acids were extracted from each solution using a modified protocol for Qiagen’s QIAamp DNA mini kit (Qiagen, Germantown, MD). After total nucleic acids were extracted, they were used as a template for the RPAC assay described above. Negative controls containing PBS without oocysts also underwent the modified extraction protocol as well as the RPAC assay.
Briefly, the modified nucleic acid extraction protocol consists of the following steps. Each tube containing sample was centrifuged for 5 min at 4000g. The supernatant was removed, and 180 μL of buffer ATL and 20 μL of proteinase K was added to each tube (all buffers supplied with the QIAamp kits). The tubes were vortexed continuously for 1 min and then set to incubate in a heat block at 55 °C for 2.5 h. During incubation, the samples were briefly vortexed every 30–45 min. After incubation, 200 μL of buffer AL was added to each tube. Samples were vortexed 15 s then set to incubate in a heat block at 70 °C for 10 min. After incubation, 200 μL of ethanol was added to each tube, and the total nucleic acids were purified using a QIAamp DNA Mini Spin Column (Qiagen, Germantown, MD) according to the manufacturer’s instructions.
DNA Extraction from Stool Spiked with Oocysts
Uninfected stool samples were collected from healthy volunteers in Houston, TX according to Rice University IRB approved protocol 11-101E. Volunteer stool samples were used fresh within 1 day. DNA from oocysts spiked into fresh stool samples was extracted using the Autogen Quickgene DNA tissue kits (Holliston,MA) and the Autogen QuickGene-Mini80 DNA extraction device. A total of 250 μL of stool diluted with PBS (50% stool, 50% PBS) was incubated with the supplied tissue lysis buffer and proteinase K for 1 h at 80 °C. The stool samples were then centrifuged at 8 000g for 5 min. The supernatant was removed, added to a tube containing 180 μL of the second supplied lysis buffer, vortexed for 15 s, and incubated for 10 min at 80 °C. Lastly, 240 μL of ethanol was added to the lysate and vortexed for 15 s. The lysate was added to a DNA binding column and washed three times using the supplied washing buffer. Nucleic acids were eluted in 200 μL of the supplied elution buffer or water.
Testing the Specificity of the RPAC Assay
The RPAC assay was tested for specificity against a number of other organisms that present similarly in the clinic. Purified nucleic acids prepared from cultures of Clostridium difficile, Salmonella enterica, Giardia intestinalis, and Blastocystis hominis were purchased from ATCC (Manassas, VA). DNA was also extracted from a stool sample that tested positive for Fasciola by PCR and microscopy. A volume of 10 μL of DNA (100 ng DNA per μL) from each extraction was used as a template in an RPA reaction with the lateral flow RPAC assay. A positive control containing DNA extracted from 106Cryptosporidium oocysts/mL PBS (8.4 ng DNA per μL) and a negative control containing no template DNA were also tested. DNA extractions from non-Cryptosporidium organisms contained an excess concentration of DNA to ensure that RPAC assay negativity was not due to lack of DNA.
Testing the RPAC Assay with Stool Samples from Infected Animals
To test the performance of the RPAC assay using stool from infected animals, SCID-beige mice were infected orally with 1 × 106Cryptosporidium parvum oocysts (Iowa strain) by gavage. After 4 days of infection the mice were treated daily for 10 days with 1000 mg/kg of Paromomycin, 100 mg/kg of pyrazolopyrimidine, or placebo. Negative controls included uninfected animals. Stool pellets were collected at various time points and stored at −20 °C until DNA was extracted and tested for Cryptosporidium infection by real time qPCR. For DNA extractions 25 mg of stool from each mouse was used. DNA was extracted and purified using the QIAamp DNA Stool Mini Kit (Qiagen, Valencia, CA). The purity and concentration of DNA was determined by spectrophotometry using a Nanodrop (Thermo Scientific, Wilmington, DE).
The parasite burden was determined by real time qPCR using the Applied Biosystems 7500 Real-Time PCR Systems (Life Technologies, Grand Island, NY), the Platinum SYBR Green qPCR SuperMix-UDG Kit (Life Technologies, Grand Island, NY), and primers for the C. parvum CP23 gene (CP-F, CAATCAGCAACCAAGCTCAA and CP-R, TTGTTGAGCAGCAGGTTCAG). The conditions for PCR were 5 min 95 °C × 1 cycle, 15 s at 95 °C, 1 min at 66 °C × 60 cycles. To test the specificity of the primers, an additional dissociation stage was added at the end of the reaction for dissociation curve analysis. A standard curve was generated from serial dilutions of DNA from a known number of parasites and was included in each reaction plate. Extracted DNA samples from 18 infected and 10 uninfected mice were blinded for RPA testing, and results were compared to that of PCR.
Testing the RPAC Assay with Stool Samples from Infected Patients and Healthy Controls
Human stool samples from 10 infected patients were generously provided by the Texas Department of State Health Services Lab under a material transfer agreement. All stool samples were stored in liquid zinc-polyvinyl alcohol (Zn-PVA) and were previously confirmed to be infected with Cryptosporidium by a regional reference lab using the acid-fast staining method. DNA was extracted from each sample. The extracted DNA was tested for the presence of the Cryptosporidium CP23 gene using PCR with amplified products detected via gel electrophoresis and quantified via qPCR. Extracted DNA from these 10 samples along with 11 samples from healthy volunteers was tested using the RPAC assay; samples were coded so that the individual performing RPA did not know whether the specimen came from a patient or a healthy volunteer.
Evaluating the RPAC Assay in a Paper and Plastic Device
We previously described a paper and plastic foldable device designed to facilitate RPA use in low-resource settings.15 This RPA device was used to perform the RPAC assay using DNA from the 10 infected human samples and the 11 uninfected control samples.
Briefly, the devices were constructed of five components cut with a laser cutter. Components consisted of an acetate base layer, a double-sided adhesive layer for alignment, a cellulose wicking strip patterned with melted wax, a cellulose master mix pad, and a glass fiber pad for holding magnesium acetate. All components were purchased from Grafix (Maple Heights, OH), GE Healthcare (Waukesha, WI), or Millipore (Billerica, MA). Devices were assembled by stacking components.
Once assembled, the devices were used as a platform for the lateral flow RPAC assay. First, the reagents were added to their respective pads: the master mix pad received 37.5 μL of master mix containing rehydration buffer, water, primers, and probes while the magnesium acetate pad received 2.5 μL of magnesium acetate. The wicking strip was placed into a tube containing extracted DNA to capture 10 μL of solution containing template DNA. The wick was then folded down to bring the template DNA into contact with the master mix pad. Lastly, the reaction was initiated by folding the device in half to bring the magnesium acetate pad into contact with the sample wick and master mix pads. The sealed device was then incubated on a heat block at 37 °C for 30 min.
After incubation, the devices were removed from the heat block and peeled open. A volume of 2 μL were taken from the master mix pad with a pipet and diluted with 98 μL of running buffer. A volume of 10 μL of each dilution were added to the sample-end of a Milenia lateral flow detection strip which was then placed into an individual well of a 96 well plate containing 100 μL of running buffer. After 3 min, the strips were removed and scanned using a flatbed scanner.
Results
Performance of the RPAC Assay in Solution with Spiked Stool Samples
Using total nucleic acids extracted from PBS containing oocysts, amplified products from as few as 103 oocysts/mL PBS were detectable via gel electrophoresis (Figure 1A). When using nucleic acids extracted from stool, amplified products from as few as 104 oocysts/mL stool are detectable via gel electrophoresis (Figure 1B).
Figure 1.

Gel detection of RPA products. Amplified products were detected using gel electrophoresis stained with ethidium bromide. Using DNA extracted from oocysts spiked into PBS, RPA products from as few as 103 oocysts/mL PBS (A) are visible on the gel. Using DNA extracted from oocysts spiked into uninfected stool samples from healthy volunteers, RPA products from as few as 104 oocysts/mL stool (B) are visible on the gel.
As seen in Figure 2B, RPA products amplified from as few as 102 oocysts/mL PBS could be detected visually using lateral flow strips (objective determination of a positive versus a negative test result outlined previously in the Materials and Methods section). It should be noted that the performance using lateral flow strips is 1–2 orders of magnitude more sensitive than detection of products by gel electrophoresis. Similarly, RPA lateral flow reactions were performed using template DNA extracted from stool spiked with oocysts. As shown in Figure 2C, products amplified from as little as 102–103 oocysts/mL stool could be detected visually.
RPAC Assay Consistency
The assay was repeated multiple times using aliquots from the same sample in order to assess the intrasample variability of the RPAC assay. Nucleic acids were extracted from stool samples spiked with varying concentrations of oocysts (102–106 oocysts/mL stool). Three separate RPAC assays were performed on aliquots from each of the DNA extractions in order to assess the variability in assay results between samples. For every concentration tested, all three RPAC assay repeats yielded the same result, consistently detecting as few as 102 oocysts/mL stool (Supporting Information).
Sample-to-sample consistency was also determined by repeatedly (n = 4) creating serial dilutions of stool with varying concentrations of spiked oocysts. Nucleic acids from each spiked stool sample were extracted and tested using the RPAC assay and PCR. The RPAC assay performed well when benchmarked against PCR, consistently demonstrating equal or better performance (Table 1). Calculated oocysts per reactions were based on starting concentration of oocysts per mL of stool, amount of stool used in each extraction, elution volume, and volume of DNA elution used per reaction according to details described in the Materials and Methods sections.
Table 1. Testing the RPAC Assay on Different Dilution Seriesa.
| dilution
series 1 |
dilution
series 2 |
dilution
series 3 |
dilution
series 4 |
||||||
|---|---|---|---|---|---|---|---|---|---|
| concentration (oocysts/mL) | calculated oocysts per reaction | RPAC | PCR | RPAC | PCR | RPAC | PCR | RPAC | PCR |
| 105 | 625 | + | + | + | + | + | + | + | + |
| 104 | 63 | + | + | + | + | + | + | + | + |
| 103 | 6 | + | – | + | + | + | + | + | + |
| 102 | ≤1 | + | – | – | – | + | – | – | – |
| 0 | 0 | – | – | – | – | – | – | – | – |
To assess sample-to sample reliability, PCR and the RPAC assay were performed using DNA extracted from four separate dilution series of stool samples each containing the various concentrations of parasites typically found in stool. The RPAC assay demonstrated comparable or better performance compared with PCR.
Specificity of the RPAC Assay
The RPAC assay was tested for specificity using DNA extracted from a number of other intestinal pathogens that cause illness with similar clinical presentation to Cryptosporidium. The RPAC assay yielded a positive result on the lateral flow strip only for the sample containing Cryptosporidium; the test line was visually negative for all other organisms tested (Figure 3).
Figure 3.
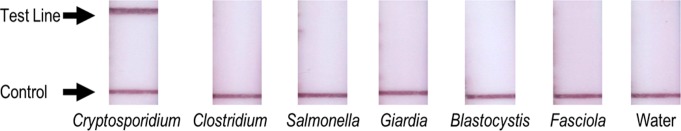
Testing the RPAC assay for specificity. RPA products detected using lateral flow RPAC assay yield visually positive results only when tested using DNA extracted from PBS spiked with Cryptosporidium; results are visually negative for all other organisms tested.
Performance of the RPAC Assay with Stool Samples from Infected Animals and Healthy Controls
Once the RPAC assay was optimized using spiked samples, it was tested using DNA extracted from fecal pellets of 18 Cryptosporidium-infected and 10 uninfected mice; quantitative PCR to detect Cryptosporidium DNA was used as a reference standard. Visual readout of the lateral flow strips correctly identified the presence of Cryptosporidium DNA in each of the infected samples. The RPAC assay correctly identified the absence of Cryptosporidium in 9 out of the 10 uninfected samples (Table 2). The RPAC assay was falsely positive for one uninfected mouse sample. This sample was retested using the RPAC assay; results of the second test were negative.
Table 2. Performance of RPAC Assay Relative to qPCR for DNA Extracted from Stools of Infected and Uninfected Mice.
| RPAC assay positive | RPAC assay negative | |
|---|---|---|
| qPCR negative | 1 | 9 |
| qPCR positive | 18 | 0 |
Performance of the RPAC Assay with Stool Samples from Infected Patients and Healthy Controls
A total of 10 human stool samples clinically verified to contain Cryptosporidium by a reference laboratory and 11 stool samples from healthy volunteers presumed to be uninfected were deidentified and tested using the RPAC assay. The samples were also tested by real time quantitative PCR and by PCR with gel electrophoresis detection (Table 3). All stool samples from infected patients that were verified by a reference laboratory to contain Cryptosporidium using acid fast staining also tested positive by RPA with lateral flow detection. PCR with gel electrophoresis was positive for 6 of 10 samples, while qPCR was positive for 5 of 9 samples for which DNA was available. All stool samples from healthy volunteers tested negative by the RPAC assay and PCR (data not shown). While PCR is generally reported to be more sensitive than acid fast staining, we hypothesize that PCR may have given negative results in some acid-fast positive cases due to degradation of DNA during the 8 month interval between when acid fast staining was performed and when DNA was extracted.
Table 3. Performance of the RPAC Assay Compared to Various Gold Standards.
| sample number | acid fast staining | RPAC assay | RPAC device | PCR/gel | real-time qPCR (units = parasites/g stool) |
|---|---|---|---|---|---|
| 1 | + | + | – | – | 0 |
| 2 | + | + | – | – | 0 |
| 3 | + | + | + | – | 0 |
| 4 | + | + | + | + | 0 |
| 5 | + | + | + | – | 5.4 × 103 |
| 6 | + | + | + | + | 1.2 × 107 |
| 7 | + | + | + | + | 1.5 × 107 |
| 8 | + | + | + | + | 1.6 × 107 |
| 9 | + | + | + | + | 3.9 × 108 |
| 10 | + | + | + | + | DNA not available |
Performance of the RPAC Assay in a Paper and Plastic Device
We tested all 21 of the human stool samples with the RPAC assay using a previously described paper and plastic platform (Figure 4). Results were positive for 8 of 10 infected samples and negative for all of healthy volunteers (Table 3).
Figure 4.
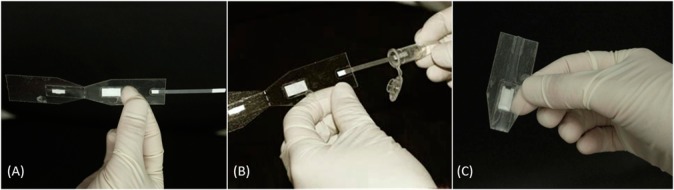
RPA testing using a paper and plastic foldable device. (A) The reagents are first added to their respective pads, (B) the wick is then dipped into the tube containing DNA extracted from the stool specimen, (C) and the device is folded to initiate and perform the RPA reaction.
Discussion
This paper describes a novel RPA-based assay for Cryptosporidium (RPAC assay); in laboratory evaluation, the RPAC assay was positive when tested with DNA extracted from stool samples spiked with as few as 100–1,000 oocysts per mL of stool (1–10 oocysts/reaction). The RPAC assay further demonstrated specificity when tested using a nucleic acid panel of five organisms that cause diarrheal illness with clinical signs and symptoms similar to cryptosporidiosis.
In preclinical testing, the RPAC assay properly detected Cryptosporidium in DNA extracted from 18/18 infected mouse stool samples and 10/10 infected human stool samples; all but one of the 21 negative controls tested negative. When the single false positive specimen was retested the RPAC assay accurately identified it as negative, indicating that the initial positive reading was likely due to amplicon carryover contamination during the RPAC assay setup. The likelihood of carryover contamination can be reduced in the future by implementing the RPAC assay on a compact, fully enclosed device that completely contains the reaction.
As a step toward demonstrating that the RPAC assay could be performed in a low-resource setting, we implemented the assay in a paper and plastic foldable device requiring only a micropipet, pipet tips, and a single temperature heater (Figure 4). When implemented in this device, the RPAC assay properly identified Cryptosporidium DNA in 8/10 infected human stool samples. The two samples which were falsely negative by RPAC assay in the paper and plastic device were also negative by PCR, suggesting a low concentration of target DNA available to amplify. It is important to note that this paper and plastic device only represents a step toward a fully integrated device. Future iterations of the device should provide sample to answer results without requiring that the device be unsealed to introduce amplified products onto lateral flow strips. While such products currently exist on the commercial market (i.e., Biohelix’s BESt Cassette), implementation on a paper and plastic substrate would significantly reduce their price and broaden their widespread usage.
A complicating factor in assessing the performance of the RPAC assay with clinical samples is the choice of an appropriate gold standard. While PCR is generally considered to be the most sensitive test for Cryptosporidium, it was negative in several of the clinical samples identified as positive by acid-fast staining. RPA is widely considered to be more robust than PCR and we attribute the lower apparent sensitivity of PCR to inhibitors found in fixatives compared to that of the RPAC assay. The difference could also be due to the different genetic sequences targeted by RPA and PCR.
As described in the Materials and Methods section, positive and negative results were objectively determined by scanning strips and using image analysis software to determine whether the signal at the test line exceeded a threshold. We recognize that a smaller signal-to-background ratio tended to correspond to a lower level of infection and that in settings without access to a scanner or image processing software, a faint test line might have been considered an indeterminate test result. That being said, of the 70 human and mouse results presented in this paper, only a single visual determination differed from the objective electronic determination.
One limitation of the RPAC assay, like all DNA-based assays, is their inability to distinguish between viable oocysts and nonviable oocysts. While the ability to discern viable, infectious oocysts from nonviable oocysts would be useful to assess the efficacy of water treatment methodologies, the RPAC assay was developed with clinical use in mind where the presence of any oocysts is of concern. The mRNA-based platform NASBA identifies only viable oocysts and has demonstrated comparable sensitivity as the RPAC assay;16,17 however, RPA offers several advantages over NASBA including lower temperature requirements, speed of reaction, ease of detection of products, ease of transport, etc. Using industry standard water preconcentration techniques, the RPAC assay could still prove useful for water quality assessment approaches that seek to determine whether oocysts are present within a sample.
The primary limitations of our study involve the pilot sample size and complexity of sample preparation. Future field studies with a larger number of samples will be necessary to fully characterize the assay performance. Given the robustness of oocysts and the inhomogeneity of stool samples, DNA extraction from stool samples at the point-of-care is a difficult challenge. Govindarajan et al. have described a field-deployable DNA extraction device that does not require electricity and can be used at the point-of-care for viscous samples.18 This device or a similar type of device could be designed to process fresh stool samples at the point-of-care.
Conclusion
The RPAC assay provides a sensitive nucleic acid test to diagnose one of the most common causes of persistent diarrhea. Using RPA reagents that are stable enough for use in field studies, Cryptosporidium testing could finally be accomplished without the need for expert microscopy or costly thermal cyclers. This device could greatly impact the approach to studies of the epidemiology of cryptosporidiosis and greatly advance clinical care.
Acknowledgments
We thank Cathy Snider and Charles Stager, Ph.D. for providing clinically infected stool samples. Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number U54AI057156. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Supporting Information Available
Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
This paper was published ASAP on February 12, 2014, with missing funding information in the Acknowledgements section. The corrected version was reposted February 14, 2014.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Lozano R.; Naghavi M.; Foreman K.; Lim S.; Shibuya K.; Aboyans V.; Abraham J.; Adair T.; Aggarwal R.; Ahn S. Y.; Alvarado M.; Anderson H. R.; Anderson L. M.; Andrews K. G.; Atkinson C.; Baddour L. M.; Barker-Collo S.; Bartels D. H.; Bell M. L.; Benjamin E. J.; Bennett D.; Bhalla K.; Bikbov B.; Bin Abdulhak A.; Birbeck G.; Blyth F.; Bolliger I.; Boufous S.; Bucello C.; Burch M.; Burney P.; Carapetis J.; Chen H.; Chou D.; Chugh S. S.; Coffeng L. E.; Colan S. D.; Colquhoun S.; Colson K. E.; Condon J.; Connor M. D.; Cooper L. T.; Corriere M.; Cortinovis M.; de Vaccaro K. C.; Couser W.; Cowie B. C.; Criqui M. H.; Cross M.; Dabhadkar K. C.; Dahodwala N.; De Leo D.; Degenhardt L.; Delossantos A.; Denenberg J.; Des Jarlais D. C.; Dharmaratne S. D.; Dorsey E. R.; Driscoll T.; Duber H.; Ebel B.; Erwin P. J.; Espindola P.; Ezzati M.; Feigin V.; Flaxman A. D.; Forouzanfar M. H.; Fowkes F. G. R.; Franklin R.; Fransen M.; Freeman M. K.; Gabriel S. E.; Gakidou E.; Gaspari F.; Gillum R. F.; Gonzalez-Medina D.; Halasa Y. A.; Haring D.; Harrison J. E.; Havmoeller R.; Hay R. J.; Hoen B.; Hotez P. J.; Hoy D.; Jacobsen K. H.; James S. L.; Jasrasaria R.; Jayaraman S.; Johns N.; Karthikeyan G.; Kassebaum N.; Keren A.; Khoo J.-P.; Knowlton L. M.; Kobusingye O.; Koranteng A.; Krishnamurthi R.; Lipnick M.; Lipshultz S. E.; Ohno S. L.; Mabweijano J.; MacIntyre M. F.; Mallinger L.; March L.; Marks G. B.; Marks R.; Matsumori A.; Matzopoulos R.; Mayosi B. M.; McAnulty J. H.; McDermott M. M.; McGrath J.; Mensah G. A.; Merriman T. R.; Michaud C.; Miller M.; Miller T. R.; Mock C.; Mocumbi A. O.; Mokdad A. A.; Moran A.; Mulholland K.; Nair M. N.; Naldi L.; Narayan K. M. V.; Nasseri K.; Norman P.; O’Donnell M.; Omer S. B.; Ortblad K.; Osborne R.; Ozgediz D.; Pahari B.; Pandian J. D.; Rivero A. P.; Padilla R. P.; Perez-Ruiz F.; Perico N.; Phillips D.; Pierce K.; Pope C. A. 3rd; Porrini E.; Pourmalek F.; Raju M.; Ranganathan D.; Rehm J. T.; Rein D. B.; Remuzzi G.; Rivara F. P.; Roberts T.; De Leon F. R.; Rosenfeld L. C.; Rushton L.; Sacco R. L.; Salomon J. A.; Sampson U.; Sanman E.; Schwebel D. C.; Segui-Gomez M.; Shepard D. S.; Singh D.; Singleton J.; Sliwa K.; Smith E.; Steer A.; Taylor J. A.; Thomas B.; Tleyjeh I. M.; Towbin J. A.; Truelsen T.; Undurraga E. A.; Venketasubramanian N.; Vijayakumar L.; Vos T.; Wagner G. R.; Wang M.; Wang W.; Watt K.; Weinstock M. A.; Weintraub R.; Wilkinson J. D.; Woolf A. D.; Wulf S.; Yeh P.-H.; Yip P.; Zabetian A.; Zheng Z.-J.; Lopez A. D.; Murray C. J. L. Lancet 2013, 380(9859), 2095–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White A. C. Cryptosporidium Species. In Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases, 7th ed.; Mandell G. L., Bennett J. E., Eds.; Elsevier: Philadelphia, PA, 2009; pp 3547–3560. [Google Scholar]
- Guerrant D. I.; Moore S. R.; Lima A. A. M.; Patrick P. D.; Schorling J. B.; Guerrant R. L. Am. J. Trop. Med. Hyg. 1999, 61(5), 707–713. [DOI] [PubMed] [Google Scholar]; Putignani L.; Menichella D. Interdiscip. Perspect. Infect. Dis. 2010, 2010, 753512. [DOI] [PMC free article] [PubMed] [Google Scholar]; Mondal D.; Haque R.; Sack R. B.; Kirkpatrick B. D.; Petri W. A. Jr. Am. J. Trop. Med. Hyg. 2009, 80(5), 824–826. [PMC free article] [PubMed] [Google Scholar]
- Fletcher S. M.; Stark D.; Harkness J.; Ellis J. Clin. Microbiol. Rev. 2012, 25(3), 420–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalmers R. M.; Campbell B. M.; Crouch N.; Charlett A.; Davies A. P. J. Med. Microbiol. 2011, 60(11), 1598–1604. [DOI] [PubMed] [Google Scholar]
- Weber R.; Bryan R. T.; Bishop H. S.; Wahlquist S. P.; Sullivan J. J.; Juranek D. D. J. Clin. Microbiol. 1991, 29(7), 1323–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushen O. Y.; Kohli A.; Pinkerton R. C.; Dupnik K.; Newman R. D.; Sears C. L.; Fayer R.; Lima A. A. M.; Guerrant R. L. Trans. R. Soc. Trop. Med. Hyg. 2007, 101(4), 378–384. [DOI] [PubMed] [Google Scholar]
- Center for Disease Control and Prevention. Laboratory Identification of Parasites of Public Health Concern. http://www.cdc.gov/mmwr/pdf/ss/ss6105.pdf (accessed August 8, 2012).
- Agnamey P.; Sarfati C.; Pinel C.; Rabodoniriina M.; Kapel N.; Dutoit E.; Garnaud C.; Diouf M.; Garin J. F.; Totet A.; Derouin F.; Natl A. C. J. Clin. Microbiol. 2011, 49(4), 1605–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Hove R.-J.; van Lieshout L.; Brienen E. A. T.; Perez M. A.; Verweij J. J. Diagn. Microbiol. Infect. Dis. 2008, 61(3), 280–283. [DOI] [PubMed] [Google Scholar]; Haque R.; Roy S.; Siddique A.; Mondal U.; Rahman S. M. M.; Mondal D.; Houpt E.; Petri W. A. Jr. Am. J. Trop. Med. Hyg. 2007, 76(4), 713–717. [PubMed] [Google Scholar]
- Amar C. F. L.; East C. L.; Gray J.; Iturriza-Gomara M.; Maclure E. A.; McLauchlin J. Eur. J. Clin. Microbiol. Infect. Dis. 2007, 26(5), 311–323. [DOI] [PubMed] [Google Scholar]
- Compton J. Nature 1991, 350(6313), 91–92. [DOI] [PubMed] [Google Scholar]; Notomi T.; Okayama H.; Masubuchi H.; Yonekawa T.; Watanabe K.; Amino N.; Hase T. Nucleic Acids Res. 2000, 28(12), e63. [DOI] [PMC free article] [PubMed] [Google Scholar]; Demidov V. V. Expert Rev. Mol. Diagn. 2002, 2(6), 542–548. [DOI] [PubMed] [Google Scholar]; Craw P.; Balachandran W. Lab Chip 2012, 12(14), 2469–2486. [DOI] [PubMed] [Google Scholar]; Asiello P. J.; Baeumner A. J. Lab Chip 2011, 11(8), 1420–1430. [DOI] [PubMed] [Google Scholar]; Walker G. T.; Fraiser M. S.; Schram J. L.; Little M. C.; Nadeau J. G.; Malinowski D. P. Nucleic Acids Res. 1992, 20(7), 1691–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piepenburg O.; Williams C. H.; Stemple D. L.; Armes N. A. PLoS Biol. 2006, 4(7), e204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forest M.RPA Storage Question; Crannell Z., Ed.; TwistDx: Cambridge, U.K., 2013. [Google Scholar]
- Rohrman B. A.; Richards-Kortum R. R. Lab Chip 2012, 12(17), 3082–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar Z. P.; Fritsch I. Anal. Chem. 2003, 75(15), 3890–3897. [DOI] [PubMed] [Google Scholar]
- Connelly J. T.; Nugen S. R.; Borejsza-Wysocki W.; Durst R. A.; Montagna R. A.; Baeumner A. J. Anal. Bioanal. Chem. 2008, 391(2), 487–495. [DOI] [PubMed] [Google Scholar]
- Govindarajan A. V.; Ramachandran S.; Vigil G. D.; Yager P.; Boehringer K. F. Lab Chip 2012, 12(1), 174–181. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.