

Abstract
Transforming growth factor (TGF)-β, type I receptor (TβRI) and c-Jun N-terminal kinases (JNK) phosphorylate Smad3 differentially to create 2 isoforms phosphorylated (p) at the COOH-terminus (C) or at the linker region (L) and regulate hepatocytic fibrocarcinogenesis. This study aimed to compare the differences between how hepatitis B virus (HBV) infection affected hepatocytic Smad3 phosphorylated isoforms before and after anti-viral therapy. To clarify the relationship between Smad3 phosphorylation and liver disease progression, we studied 10 random patients in each stage of HBV-related fibrotic liver disease (F1–4) and also 10 patients with HBV-associated HCC. To examine changes in phosphorylated Smad3 signalling before and after anti-HBV therapies, we chose 27 patients with chronic hepatitis B who underwent baseline and follow-up biopsies at 52 weeks from the start of nucleoside analogue treatments (Lamivudine 100 mg daily or Telbivudine 600 mg daily). Fibrosis stage, inflammatory activity and phosphorylated Smad3 positivity in the paired biopsy samples were compared. Hepatocytic pSmad3C signalling shifted to fibrocarcinogenic pSmad3L signalling as the livers progressed from chronic hepatitis B infection to HCC. After nucleoside analogue treatment, serum alanine aminotransferase (ALT) and HBV-DNA levels in 27 patients with HBV-related chronic liver diseases were decreased dramatically. Decrease in HBV-DNA restored pSmad3C signalling in hepatocytes, while eliminating prior fibrocarcinogenic pSmad3L signalling. Oral nucleoside analogue therapies can suppress fibrosis and reduce HCC incidence by successfully reversing phosphorylated Smad3 signalling; even liver disease progressed to cirrhosis in chronic hepatitis B patients.
Keywords: HBV, pSmadC, pSmadL, TGF-β
Introduction
Chronic inflammatory responses triggered by hepatitis virus infection subsequently develop liver fibrosis, characterized by extracellular matrix (ECM) accumulation that distorts the hepatic architecture by forming a fibrous scar [1]. Ultimately, nodules of regenerating hepatocytes become enclosed by scar tissue, an event defining cirrhosis. Once infected by hepatitis viruses B and C (HBV and HCV), the incidence from chronic hepatitis to cirrhosis, even hepatocellular carcinoma (HCC), increases [2]. Most HCC cases have a natural history of unresolved inflammation and severe fibrosis (or cirrhosis), irrespective of the underlying cause of liver disease [3]. Approaches to understanding the way in which human HCC develops from chronic liver diseases should therefore focus on molecular mechanisms shared in common between fibrosis and carcinogenesis/fibrocarcinogenesis caused by chronic inflammation.
HBV causes chronic infection in 350–400 million people worldwide [4]. Chronic HBV infection can lead to the development of HCC, cirrhosis and decompensated liver disease. HBV is known to integrate into the host genomes, which can then contribute to HCC development. In addition, cells with integrated HBV-DNA express viral gene products which ultimately cause carcinogenesis [5]. Previous research has identified that persistent high-level expression of hepatitis B virus X (HBx) protein in the transgenic mouse liver results in hyperplasia, which would lead to HCC, without preceding inflammation [6–8]. These observations suggest that HBV infection and chronic inflammation induce fibrocarcinogensis synergistically.
Hepatitis B is a treatable disease, and two types of therapy are available: interferon and nucleoside analogues. Lamivudine, a nucleoside analogue, suppresses HBV replication through inhibition of reverse-transcriptase and DNA polymerase activities [9]. Four other nucleoside and nucleoside analogues have been licensed: Adefovir (in 2002) [10], Entecavir (in 2005) [11], Telbivudine (in 2006) [12] and, most recently, Tenofovir disoproxil fumarate (in 2008) [13]. These anti-viral drugs act primarily by inhibiting reverse transcription of the pregenomic RNA to HBV DNA. When effective suppression of viral replication in chronic hepatitis B patients has been achieved, alanine aminotransferase (ALT) levels decrease markedly and liver histology improves significantly. Anti-viral therapies with interferon or a nucleoside analogue are useful in reduction of the incidence of HBV-related decompensated liver disease and the rate of HCC [14–16].
The canonical transforming growth factor (TGF)-β pathway involves receptor-activated Smads (R-Smads; Smad2 and Smad3) signalling through direct serine phosphorylation of COOH termini by TGF-β binding to its type 1 receptor (TβRI). The phosphorylation of R-Smads induces their association with the shared partner Smad4. This complex accumulates in the nucleus, where Smad3 and Smad4, but not Smad2, bind directly to DNA [17–21]. Smad3 contains two conserved polypeptide segments, the MH1 and MH2 domains, joined by a unconserved linker region [19]. The linker domain can undergo regulatory phosphorylation by other kinases, including mitogen-activated protein kinases and cyclin-dependent kinases. C-terminally phosphorylated Smad3C transmits a cytostatic TGF-β/activin signalling, whereas proinflammatory cytokines and HBx transmit mitogenic and fibrogenic signals by up-regulating c-Myc and plasminogen activator inhibitor (PAI)-1 transcription through the c-jun N-terminal kinsase (JNK)-dependent pSmad3L pathway [22–24]. JNK-mediated pSmad3L and TβRI-mediated pSmad3C signals oppose each other; most importantly, the balance can shift from cell growth to growth inhibition. Linker phosphorylation of Smad3 indirectly inhibits its COOH-terminal phosphorylation and subsequently suppresses pSmad3C signalling. TGF-β signalling confers a selective advantage on tumour cells by shifting from the cytostatic TβRI/pSmad3C pathway to the fibrocarcinogenic HBx/JNK/pSmad3L pathway during hepatic carcinogenesis [25–27]. Thus, multiple levels of cross-talk exist between Smad3 and JNK signalling pathways.
It is extremely important to understand the molecular mechanisms involved in the process of reversibility of human fibrocarcinogenesis, and to discover the way forward to new therapeutic strategies. To clarify the relationship between Smad3 phosphorylation and liver disease progression, we studied 10 random patients at each stage of HBV-related fibrotic liver disease (F1–4) and 10 patients with HBV-associated HCC. Hepatocytic cytostatic pSmad3C signalling shifted to fibrocarcinogenic pSmad3L signalling as the patients progressed from chronic hepatitis B infection to HCC. Conversely, sustained suppression of HBV DNA replication during 52 weeks of an oral nucleoside analogue therapy could reduce fibrocarcinogenic potential by switching phospho-R-Smad signalling from the pSmad3L to the pSmad3C pathway.
Patients and methods
Patients and follow-up
To clarify the relationship between Smad3 phosphorylation and liver disease progression, we studied 10 random patients at each stage of HBV-related liver disease (F1–4) and 10 patients with HBV-associated HCC in the department of gastroenterology and hepatology at Kansai Medical University Hospital, Osaka, Japan. All patients were seropositive for hepatitis B surface antigen (Abbott Laboratories, Chicago, IL, USA) and seronegative for anti-HCV antibody (Ortho Diagnostics, Tokyo, Japan).
To examine the changes in Smad3 phosphorylation after anti-HBV-treatment, we chose 27 patients with chronic HBV infection who underwent baseline and follow-up biopsies after 52 weeks' administration of a nucleoside analogue between 2007 and 2009, in the department of hepatology at the First Hospital of Jilin University, Changchun, China. Sixteen patients received Lamivudine (100 mg daily) and 11 patients received Telbivudine (600 mg daily). These patients underwent annual laboratory evaluation, including HBV-DNA and ALT tests.
Informed consent was obtained from all subjects. The study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki. We also obtained approval for this study from our institutional ethics committees.
Domain-specific antibodies against the phosphorylated Smad3
Two polyclonal anti-phospho-Smad3 sera, pSmad3L (Ser208/213) and pSmad3C (Ser423/425), were raised against the phosphorylated linker and C-terminal regions of Smad3 by immunization of rabbits with synthetic peptides. Relevant anti-sera were affinity-purified using phosphorylated peptides as described previously [22].
Histology and immunohistochemical analysis
Formalin-fixed, paraffin-embedded liver tissues were processed, and 4-μm-thick serial sections were cut and stained with haematoxylin and eosin (H&E) or Azan staining to detect fibrosis. Inflammatory activity and fibrotic stage were graded histologically according to Desmet et al.'s classification by a blinded pathologist, using the following scales: (i) a 0–4 scale for fibrosis – 0, no fibrosis; 1 – minimal portal fibrosis; 2 – portal fibrosis with septa formation; 3 – localized bridging fibrosis; and 4 – extensive bridging fibrosis; and (ii) a 0–3 scale for inflammation: 0 – no inflammation; 1 – mild inflammation; 2 – moderate inflammation; and 3 – severe inflammation.
Immunohistochemical analysis was performed as described previously [28]. Primary antibodies used in this study included the affinity-purified rabbit polyclonal anti-pSmad3L (2 μg/ml) and anti-pSmad3C (0·5 μg/ml) antibodies. Anti-pSmad3C antibody cross-reacted weakly with C-terminally phosphorylated Smad2: to block binding of anti-pSmad3C antibody to phosphorylated domains in Smad2, anti-pSmad3C antibody was adsorbed with 1 μg/ml C-terminally phosphorylated Smad2 peptide.
For immunohistochemical analyses, sections exposed to primary antibodies were then incubated with peroxidase-labelled polymer conjugated with goat anti-rabbit immunoglobulin (Ig)G (Dako, Glostrup, Denmark). Finally, sections were developed with 3, 3′-diaminobenzidine tetrahydrochloride (DAB; Vector Laboratories, Burlingame, CA, USA), counterstained with Mayer's haematoxylin (Merck, Darmstadt, Germany), and mounted under coverslips. We counted and scored pSmad3 positivity in hepatocytes as follows: 0, no positivity; 1, <25%; 2, 25–50%; 3, 51–75%; 4, > 75%. Scoring of pathological changes in the liver was performed by a blinded pathologist.
Statistical analyses
The two-tailed paired Mann–Whitney U-test and mean ± standard error of the mean (s.e.m.) were used to identify significant differences in hepatocytic Smad3 phosphorylation and the grades of inflammatory activity and fibrotic stages. P-values < 0·05 were considered statistically significant.
Results
A representative chronic hepatitis B patient medicated with an oral nucleoside analogue therapy
Histological evaluation, which is useful in assessing the stage of chronic hepatitis B infection, includes separate considerations of fibrosis and inflammatory activity [29]. To investigate domain-specific phosphorylation-mediated Smad3 signalling in vivo, we generated antibodies specific to each phosphorylation site, determined the distribution of pSmad3L and pSmad3C in liver specimens, and semi-quantitatively scored hepatocytic Smad3 phosphorylation from 0 to 4 [22,25,26,30]. First, we show one case at Kansai Medical University hospital.
A 32-year-old man with chronic hepatitis B infection received Entecavir therapy (0·5 mg daily), and the serum HBV-DNA level decreased to below detection level after 24 weeks' administration. The sequential laboratory data showed that the serum ALT level decreased and remained at the normal level. Fifty-two weeks later, we performed a follow-up biopsy to evaluate whether his disease had regressed or progressed (Fig. 1a). Fibrotic stage decreased from F3 to F1 (Fig. 1b), as inflammatory activity reduced from A3 to A1 (Fig. 1c). To support this, pSmad3L decreased in hepatocytic nuclei after anti-HBV therapy, while phosphorylation at Smad3C recovered in the hepatocytic nuclei throughout liver lobules (Fig. 1d).
Figure 1.
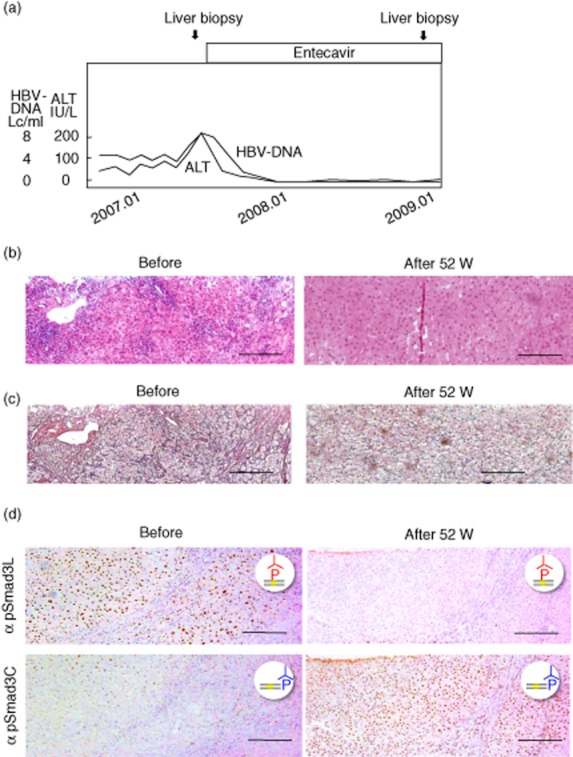
Representative data of an anti-hepatitis B virus (HBV)-treated patient. (a) A 32-year-old man with chronic hepatitis B infection received Entecavir therapy (0·5 mg daily), and the serum HBV-DNA and alanine aminotransferase (ALT) were detected after 24 weeks' treatment. (b,c) Fifty-two weeks later, liver biopsies were used to evaluate the fibrotic stage (c) and inflammatory activity (b). (d) Immunochemistry of pSmad3L and pSmad3C before and after anti-HBV therapy.
Liver histology improved significantly after 52 weeks of Lamibudine or Telbibudine therapy in chronic hepatitis B patients
China has one of the world's highest rates of hepatitis B infection, and a nationwide HBV survey showed that 7·2% of the Chinese population aged 1–59 years were hepatitis B surface antigen (HBsAg) carriers [31]. We further analysed statistically the changes of fibrotic stage in liver specimens obtained by a paired liver biopsy during nucleoside analogue therapy at the First Hospital, Jilin University. Table 1 shows the clinical background and positivity for pSmad3C and pSmad3L before and after 52 weeks' anti-HBV therapy.
Table 1.
Clinicopathological features and Smad3L/C phosphorylation in liver specimens from patients with hepatitis B virus (HBV)-related chronic liver disease.
| No. | Gender | Age | Therapy | ALT (IU/l) |
HBV-DNA (LC/ml) |
Fibrosis staging |
Inflammatory activity grade |
pSmad3L staining |
pSmad3C staining |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Before | After 52 W | Before | After 52 W | Before | After 52 W | Before | After 52 W | Before | After 52 W | Before | After 52 W | ||||
| 1 | F | 32 | T | 42 | 32 | 10·0 | 0 | 3 | 1 | 2 | 1 | 4 | 1 | 0 | 1 |
| 2 | F | 37 | L | 34 | 24 | 6·2 | 5·8 | 1 | 0 | 1 | 1 | 4 | 1 | 2 | 2 |
| 3 | M | 20 | L | 71 | 23 | 10·1 | 3·8 | 1 | 0 | 1 | 1 | 4 | 3 | 1 | 3 |
| 4 | M | 42 | T | 66 | 21 | 10·1 | 6·6 | 1 | 0 | 2 | 0 | 4 | 3 | 4 | 4 |
| 5 | F | 42 | T | 63 | 19 | 8·9 | 0 | 1 | 0 | 1 | 1 | 4 | 3 | 3 | 3 |
| 6 | M | 44 | L | 133 | 12 | 8·9 | 0 | 3 | 1 | 3 | 1 | 4 | 3 | 3 | 3 |
| 7 | M | 19 | L | 67 | 24 | 6·4 | 4·9 | 3 | 1 | 2 | 1 | 4 | 4 | 1 | 1 |
| 8 | M | 26 | L | 218 | 24 | 9·6 | 0 | 4 | 2 | 3 | 1 | 4 | 4 | 0 | 1 |
| 9 | M | 46 | T | 239 | 23 | 10·0 | 0 | 2 | 1 | 3 | 1 | 3 | 0 | 4 | 4 |
| 10 | M | 26 | L | 36 | 26 | 9·3 | 4·2 | 0 | 0 | 1 | 0 | 3 | 0 | 1 | 2 |
| 11 | M | 38 | T | 62 | 25 | 8·5 | 0 | 3 | 2 | 3 | 1 | 3 | 1 | 3 | 3 |
| 12 | M | 19 | T | 21 | 25 | 10·0 | 4·7 | 0 | 0 | 1 | 0 | 3 | 3 | 3 | 2 |
| 13 | F | 20 | T | 51 | 21 | 10·1 | 4·3 | 2 | 1 | 2 | 1 | 3 | 3 | 3 | 4 |
| 14 | M | 38 | L | 92 | 32 | 10·3 | 4·1 | 2 | 1 | 2 | 1 | 3 | 3 | 2 | 3 |
| 15 | F | 42 | L | 43 | 20 | 6·4 | 0 | 3 | 2 | 2 | 1 | 3 | 3 | 1 | 2 |
| 16 | M | 30 | T | 60 | 24 | 7·0 | 0 | 2 | 1 | 2 | 0 | 2 | 1 | 3 | 4 |
| 17 | F | 30 | T | 32 | 16 | 12·5 | 5·6 | 1 | 1 | 2 | 0 | 2 | 1 | 3 | 4 |
| 18 | M | 29 | L | 66 | 21 | 10·1 | 0 | 1 | 0 | 1 | 0 | 2 | 2 | 4 | 4 |
| 19 | M | 38 | L | 169 | 21 | 10·0 | 0 | 1 | 1 | 2 | 1 | 2 | 2 | 2 | 2 |
| 20 | M | 45 | L | 98 | 21 | 10·2 | 0 | 3 | 3 | 2 | 1 | 2 | 2 | 2 | 2 |
| 21 | M | 48 | L | 64 | 65 | 7·2 | 7·0 | 2 | 2 | 2 | 1 | 2 | 2 | 3 | 3 |
| 22 | M | 36 | T | 32 | 16 | 12·5 | 5·6 | 1 | 1 | 2 | 1 | 2 | 3 | 3 | 3 |
| 23 | M | 27 | T | 116 | 24 | 9·2 | 0 | 1 | 0 | 2 | 0 | 1 | 0 | 0 | 3 |
| 24 | F | 44 | L | 39 | 23 | 5·7 | 0 | 3 | 1 | 2 | 1 | 1 | 0 | 0 | 3 |
| 25 | M | 42 | T | 23 | 20 | 10·0 | 12·5 | 1 | 0 | 1 | 1 | 1 | 1 | 3 | 3 |
| 26 | M | 44 | L | 121 | 21 | 9·9 | 3·5 | 4 | 3 | 3 | 2 | 0 | 0 | 1 | 4 |
| 27 | M | 45 | T | 112 | 26 | 8·9 | 0 | 3 | 2 | 3 | 1 | 0 | 0 | 4 | 4 |
T = Telbivudine; L = Lamivudine; ALT: alanine aminotransferase; M = male; F = female; W = weeks. Fibrosis stages include F0 (no fibrosis), F1 (mild fibrosis), F2 (moderate fibrosis) and F3 (severe fibrosis) and F4 (cirrhosis). Inflammatory activity grades include A0 (no activity), A1 (mild activity), A2 (moderate activity) and A3 (severe activity). Hepatocytic Smad3 phosphorylation is scored as follows: 0 = no positivity; 1 = under 25% of nuclei showing positivity; 2 = 25–50%; 3 = 50–75%; 4 = more than 75%.
Serum HBV-DNA decreased in most patients and was undetectable in 52% (14 of 27) patients after anti-HBV treatment (Fig. 2a). The HBV-DNA copies decreased below 4 log copies/ml (LC/ml) in 59% (16 of 27) patients. Only one patient exhibited deterioration in liver function after the development of Lamibudine resistance. The fibrotic stage for liver biopsies also regressed significantly in the livers of post-treatment biopsies. At least −1 point improvement in the fibrotic score was observed in 74% patients (20 of 27) and −2 point improvement was observed in 19% patients (five of 27) after anti-HBV treatment (Fig. 2c). The mean change in fibrotic score regressed by −0·96 point in the livers of chronic hepatitis B patients after anti-HBV therapy. At baseline, 82% (22 of 27) of patients in this study had elevated ALT levels (>34 IU/l). After 52 weeks' treatment, 96% (26 of 27) of patients had normal serum ALT (Fig. 2b). As the serum ALT decreased, the inflammatory activity on follow-up biopsy decreased in 89% (24 of 27) patients (Fig. 2d). These data indicated that regression of liver fibrosis was associated with cessation of chronic inflammation after anti-HBV viral therapy.
Figure 2.
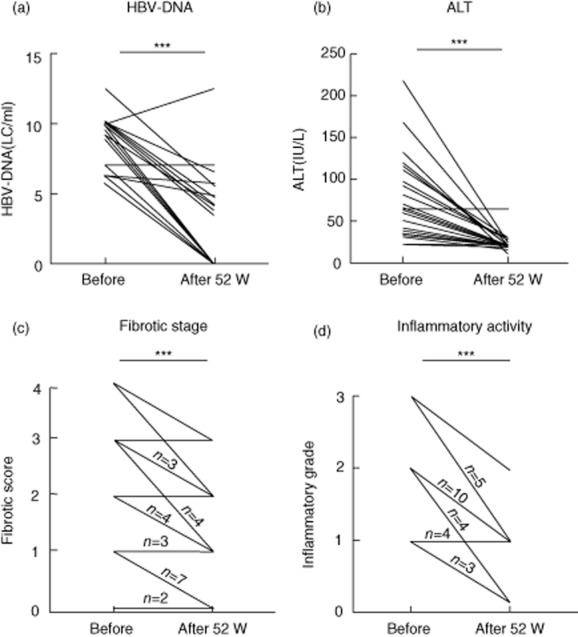
Serum hepatitis B virus (HBV) clearance and liver function improvement were observed in HBV-infected patients with nucleoside analogue treatment. Serum HBV-DNA (a) and alanine aminotransferase (ALT) (b) was measured before and after 52 weeks' treatment of Lamibudine or Telmibudine. Fibrotic score (c) and inflammatory grade (d) and are determined histologically according to Desmet's classification. ***P < 0·001 using a two-tailed paired Mann–Whitney U-test, one-way analysis of variance (anova). The lines with numbers represent the number of patients, and the lines with no numbers represent only one patient.
An oral DNA analogue treatment restored the pSmad3L to the basal pSmad3C signalling in the hepatocytes of chronic hepatitis B patients
To investigate domain-specific phosphorylation-mediated Smad3 signalling in vivo, we generated two types of antibodies against the C-terminal and linker regions of phosphorylated Smad3. Using these antibodies we have reported that Smad3, phosphorylated at the linker region, and Smad3, phosphorylated at the C-terminal region, exist as separate molecules and transmit distinct signals. To clarify the relationship between Smad3 phosphorylation and liver disease progression, we studied 10 random patients at each stage of HBV-related fibrotic liver disease (F1–4) and 10 patients with HBV-associated HCC at Kansai Medical University. As HBV-infected livers progressed from chronic hepatitis through cirrhosis to HCC, pSmad3L gradually increased (Fig. 3a). Conversely, in severely fibrotic liver (F3 and F4) and in HCC, positivity for pSmad3C was lower than that in livers with mild fibrosis (F1 and F2) (Fig. 3b).
Figure 3.
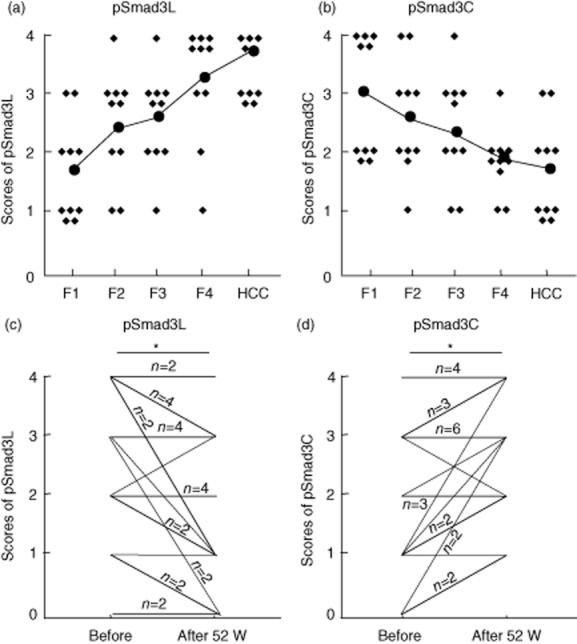
Increased pSmad3L and impaired pSmad3C signalling as chronic hepatitis B infection progresses. (a,b) The phosphorylation of Smad3L and Smad3C in liver biopsies obtained from 10 random patients in each stage of hepatitis B virus (HBV)-related liver disease (F1–4) and also 10 patients with HBV-associated HCC. (c,d) Positivity of pSmad3L and pSmad3C were compared before and after treatment with nucleoside analogue. *P < 0.05 using a two-tailed paired Mann-Whitney test, one-way analysis of variance (ANOVA). The lines with numbers represent the number of patients; the lines with no numbers represent only one patient.
Therefore, we compared the phosphorylation of Smad3 in patients of HBV before and after nucleoside analogue treatment. The phosphorylation level of Smad3L in the heptocytic nuclei decreased, whereas pSmad3C positivity in hepatocytic nuclei recovered after anti-HBV treatment (Fig. 3c,d). These data indicate successful recovery from the pSmad3L to the pSmad3C signalling after oral nucleoside analogue therapy.
Furthermore, we analysed the relationship between pSmad3L and pSmad3C with HBV-DNA, ALT, fibrotic score and inflammatory activity. The results showed that the phosphorylation level of Smad3L decreased after nucleoside analogue treatment, which was in keeping with the levels of HBV-DNA, ALT, fibrosis stage and inflammatory activity. More importantly, phosphorylation of Smad3C increased with the improvement of liver pathology after treatment with the nucleoside analogue (Fig. 4).
Figure 4.
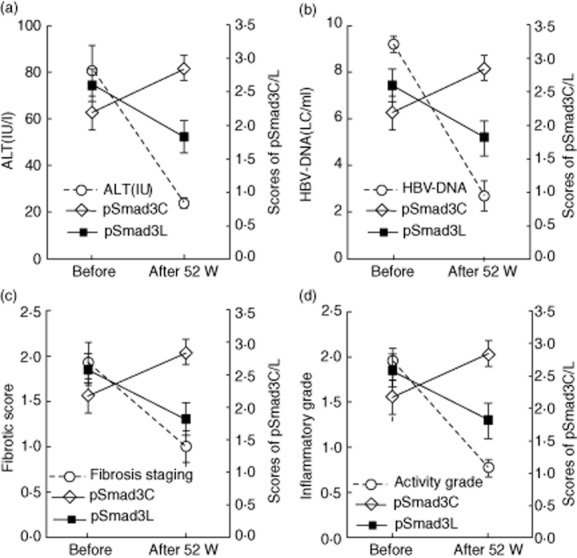
The relationship between pSmad3L and pSmad3C with ALT, hepatitis B virus (HBV)-DNA, fibrotic score and inflammatory grade. The relativity between pSmad3L/pSmad3C and ALT (a), hepatitis B virus (HBV)-DNA (b), fibrosis score (c) and inflammatory grade (d).
Discussion
Most studies report that, if patients can achieve sustained suppression of HBV replication, anti-viral therapies towards HBV hepatitis are beneficial in preventing liver cirrhosis and HCC [16]. In this study, we performed a detailed histological analysis of 27 liver biopsies from patients with chronic hepatitis B infection who had received 52 weeks of Lamivudine or Telbivudine treatment. The results demonstrate that hepatocytic proper pSmad3C signalling shifted to fibrocarcinogenic pSmad3L signalling as the livers progressed from chronic hepatitis B infection to HCC, and suppression of liver inflammation and regression of fibrosis by a nucleoside analogue resulted in the reduction of pSmad3L and an increase of pSmad3C signalling (Fig. 5). This study offers a molecular mechanism for the therapeutic efficacy of Lamivudine and Telbivudine for the prevention of liver disease in chronic hepatitis B patients.
Figure 5.
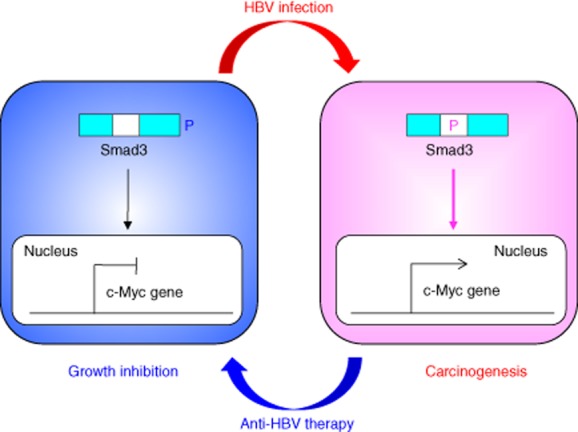
Reversibility of phosphor-Smad3 signalling between growth inhibition and carcinogenesis in chronic hepatitis B infection.During hepatitis B virus (HBV)-related chronic liver diseases, chronic inflammation together with HBV infection, shifts hepatocytic phospho-Smad signalling from growth inhibition pSmad3C to carcinogenic pSmad3L, increasing the risk of hepatocellular carcinoma (HCC). Chronic inflammation caused by HBV infection responds effectively to anti-viral therapy by successfully switching phospho-Smad signalling from carcinogenic pSmad3L to growth inhibition pSmad3C.
Without treatment, the 5-year survival rates of patients with compensated and decompensated chronic hepatitis B infection is 55–84% [32,33] and 14% [34], respectively. Although liver transplantation is the accepted therapeutic option for decompensated cirrhosis, this treatment is restricted by the lack of sufficient donor organs and prohibitive cost. Oral nucleoside therapy treatment can achieve recovery of liver fibrosis and disappearance of ascites or jaundice even in decompensated HBV-infected liver cirrhosis patients [35–37]. Suzuki et al. reported that a −0·6 improvement was observed in hepatitis B e antigen-negative chronic hepatitis B patients after 1 year of Lamivudine treatment [38]. In this study, we found that fibrosis regressed by −1 point after 52 weeks' treatment of anti-HBV treatment in the livers of chronic HBV patients. Conversely, the fibrosis regression rate was −0·28 points/year in HCV-infected patients after sustained virological response (SVR) [37]. These results indicate that treatment with nucleoside analogues resulted in a three-to-four times faster fibrosis regression rate in HBV-infected patients compared with that of interferon-treated HCV-infected patients. One reason why the fibrosis regression rate was different between HBV and HCV is that during HCV-related hepatocarcinogenesis, JNK-activated chronic inflammation confers a selective advantage on preneoplastic hepatocytes by shifting Smad3 signalling from pSmad3C to the oncogenic pSmad3L pathway. Conversely, HBx oncoprotein participates directly in hepatocarcinogenesis by shifting hepatocytic Smad3-mediated signalling from tumour suppression to oncogenesis in patients with chronic hepatitis B infection. Both HBV and HCV trigger changes in gene expression, which is mediated by genetic or epigenetic alternations. The contribution of HBV to HCC involves the expression of HBx; for HCV, the core protein, and non-structural (NS) proteins NS3 and NS5A contribute to oncogenic transformation [39]. Such differences may result in faster progression or regression rates of fibrocarcinogenesis in HBV patients compared with those in HCV patients before and after anti-viral therapy.
In this study, Lamibudine or Telbibudine was used as the anti-HBV therapy. We could not find any difference in anti-HBV effects or occurrence of side effects between these two drugs (data not shown). At present, Entecavir and Tenofovir are recommended as the primary oral agents for hepatitis B infection, because of their strong anti-viral effects and low resistance rate [40]. Further study is necessary for Entecavir, because the toxic effects of an oral nucleoside therapy are much milder than those of interferon therapy. It is strongly recommended that both chronic hepatitis B patients and liver cirrhosis patients use this oral nucleoside therapy even if their disease progresses to decompensated liver cirrhosis. Moreover, anti-viral therapy may result in a possible improvement in survival even in HBV-related HCC patients, because remaining liver function is an important factor in selecting further treatment for HCC.
Although both HBV and HCV are known to cause HCC via promoting inflammatory reactions, patients with HBV tend to develop HCC at a younger age than HCV patients, sometimes up to a decade earlier [41]. In the study by Rabe et al., the median age of HCC presentation in HBV-infected patients was 55 (range 31–76) versus 66 (range 42–82) in HCV patients [42]. HCV patients show significantly more evidence of liver dysfunction in contrast to their HBV counterparts. Wong et al. found that on presentation HCV patients, compared to HBV patients, have significantly greater rates of cirrhosis and worse liver function [43]. HCV patients may reflect the fact that oncogenesis in HCV patients almost always occurs after the development of fibrosis, followed by cirrhosis, and finally HCC, illustrating the greater amount of liver damage that has to occur prior to hepatocarcinogenesis in HCV patients. Both HBV and HCV cause HCC via promoting inflammatory reactions and genetic mutations in the liver, although HCV is thought to contribute to greater oxidase DNA damage than HBV [44]. During HCV-related chronic liver disease progression, proinflammatory cytokines together with somatic mutations convert hepatocytic proper TGF-β/pSmad3C signalling into carcinogenic pSmad3L signalling [25]. HBV, a DNA virus, can cause HCC in non-cirrhotic livers and is known to integrate into the genomes of liver cells, which can then contribute to HCC development in several different ways [45]. The integration leads to rearrangements of the host DNA and possibly mutations in key regulatory cellular genes that possibly provide a selective growth advantage in the targeted cells [44]. We have shown Smad3 phosphorylation profiles in HBV-infected human liver, concluding that HBx oncoprotein contributes directly to hepatocarcinogenesis by shifting hepatocytic Smad3-mediated signalling from tumour suppression to oncogenesis [26].
Monitoring the phosphorylation status of signalling molecules is a key step in dissecting their pathways. Clinical analyses of pSmad3L and pSmad3C in liver disease progression have provided substantial mechanistic insight. After achievement of SVR, interferon therapy could restore Smad signalling from pSmad3L to the pSmad3C pathway shown by normal hepatocytes. In contrast, patients with advanced liver fibrosis who developed HCC progressed to fibrosis despite improved inflammatory activity, because hepatocytes maintained high pSmad3L and low pSmad3C signalling. In this study we have demonstrated that most patients recovered pSmad3 signalling, but some patients still showed high levels of pSmad3L in their livers, and serum HBV-DNA even decreased below detection level. One reason why pSmad3L levels remained high may be that the duration of this follow-up study was too short. Chronic HBV infection and liver inflammation no longer play critical roles in HCC development in later cirrhotic liver after preneoplastic hepatocytes have acquired oncogenic signalling caused by genetic alteration and epigenetic changes. These clinical observations support roles for pSmad3C as a tumour suppressor and pSmad3L as a promoter during carcinogenesis.
Liver fibrosis is a dynamic process in fibrogenesis and fibrolysis. Persistence of inflammation leads to ECM accumulation and cirrhosis. Conversely, suppression of inflammation leads to regression of fibrosis and reduces the risk of HCC. Although the importance of Smad expression and activity has been shown in several types of cancer, few studies have investigated the relationship between phosphorylated Smad3 isoforms and cancer progression. We have shown that patients with chronic hepatitis B or chronic hepatitis C infection and high levels of pSmad3L develop HCC more easily. In addition, this study, together with our previous research, has identified that HBV-DNA reduction by an oral nucleoside analogue suppresses fibrosis and reduces HCC incidence by successfully switching Smad3 signalling from fibrocarcinogenic pSmad3L to pSmad3C in chronic hepatitis B patients, and the fibrocarcinogenic regression rate was much faster than that observed in chronic hepatitis C patients after SVR. Thus, the assessment of Smad3 phosphorylation in hepatocytes is a predictive biomarker for liver fibrosis progression and HCC incidence after anti-viral therapy.
Acknowledgments
Financial support for this study was provided by the National Basic Research Program of China (973 Program-2010CB945300, 2013CB944900), the National Natural Science Foundation of China (30972738, 31021061, 81130058, 81270484) and the Hundred Talents Program of the Chinese Academy of Sciences.
Disclosure
The authors declare no conflicts of interest.
References
- 1.Friedman SL. Evolving challenges in hepatic fibrosis. Nat Rev Gastroenterol Hepatol. 2010;7:425–436. doi: 10.1038/nrgastro.2010.97. [DOI] [PubMed] [Google Scholar]
- 2.Tsukuma H, Hiyama T, Tanaka S, et al. Risk factors for hepatocellular carcinoma among patients with chronic liver disease. N Engl J Med. 1993;328:1797–1801. doi: 10.1056/NEJM199306243282501. [DOI] [PubMed] [Google Scholar]
- 3.Omata M, Yoshida H. Prevention and treatment of hepatocellular carcinoma. Liver Transpl. 2004;10:S111–114. doi: 10.1002/lt.20046. [DOI] [PubMed] [Google Scholar]
- 4.Lee WM. Hepatitis B virus infection. N Engl J Med. 1997;337:1733–1745. doi: 10.1056/NEJM199712113372406. [DOI] [PubMed] [Google Scholar]
- 5.Jiang ZS, Jhunjhunwala S, Liu JF, et al. The effects of hepatitis B virus integration into the genomes of hepatocellular carcinoma patients. Genome Res. 2012;22:593–601. doi: 10.1101/gr.133926.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim CM, Koike K, Saito I, Miyamura T, Jay G. HBx gene of hepatitis B virus induces liver cancer in transgenic mice. Nature. 1991;351:317–320. doi: 10.1038/351317a0. [DOI] [PubMed] [Google Scholar]
- 7.Yu DY, Moon HB, Son JK, et al. Incidence of hepatocellular carcinoma in transgenic mice expressing the hepatitis B virus X-protein. J Hepatol. 1999;31:123–132. doi: 10.1016/s0168-8278(99)80172-x. [DOI] [PubMed] [Google Scholar]
- 8.Koike K, Moriya K, Iino S, et al. High-level expression of hepatitis B virus HBx gene and hepatocarcinogenesis in transgenic mice. Hepatology. 1994;19:810–819. [PubMed] [Google Scholar]
- 9.Dienstag JL, Schiff ER, Wright TL, et al. Lamivudine as initial treatment for chronic hepatitis B in the United States. N Engl J Med. 1999;341:1256–1263. doi: 10.1056/NEJM199910213411702. [DOI] [PubMed] [Google Scholar]
- 10.Marcellin P, Chang TT, Lim SG, et al. Adefovir dipivoxil for the treatment of hepatitis B e antigen-positive chronic hepatitis B. N Engl J Med. 2003;348:808–816. doi: 10.1056/NEJMoa020681. [DOI] [PubMed] [Google Scholar]
- 11.Chang TT, Gish RG, de Man R, et al. A comparison of entecavir and lamivudine for HBeAg-positive chronic hepatitis B. N Engl J Med. 2006;354:1001–1010. doi: 10.1056/NEJMoa051285. [DOI] [PubMed] [Google Scholar]
- 12.Lai CL, Gane E, Liaw YF, et al. Telbivudine versus lamivudine in patients with chronic hepatitis B. N Engl J Med. 2007;357:2576–2588. doi: 10.1056/NEJMoa066422. [DOI] [PubMed] [Google Scholar]
- 13.Heijtink RA, Kruining J, de Wilde GA, Balzarini J, de Clercq E, Schalm SW. Inhibitory effects of acyclic nucleoside phosphonates on human hepatitis B virus and duck hepatitis B virus infections in tissue culture. Antimicrob Agents Chemother. 1994;38:2180–2182. doi: 10.1128/aac.38.9.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dienstag JL, Goldin RD, Heathcote EJ, et al. Histological outcome during long-term lamivudine therapy. Gastroenterology. 2003;124:105–117. doi: 10.1053/gast.2003.50013. [DOI] [PubMed] [Google Scholar]
- 15.Hadziyannis SJ, Tassopoulos NC, Heathcote EJ, et al. Long-term therapy with adefovir dipivoxil for HBeAg-negative chronic hepatitis B. N Engl J Med. 2005;352:2673–2681. doi: 10.1056/NEJMoa042957. [DOI] [PubMed] [Google Scholar]
- 16.Liaw YF, Sung JJ, Chow WC, et al. Lamivudine for patients with chronic hepatitis B and advanced liver disease. N Engl J Med. 2004;351:1521–1531. doi: 10.1056/NEJMoa033364. [DOI] [PubMed] [Google Scholar]
- 17.Heldin CH, Miyazono K, ten Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–471. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]
- 18.Wrana JL. Regulation of Smad activity. Cell. 2000;100:189–192. doi: 10.1016/s0092-8674(00)81556-1. [DOI] [PubMed] [Google Scholar]
- 19.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 20.Feng XH, Derynck R. Specificity and versatility in TGF-beta signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–693. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- 21.Guo X, Wang XF. Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Res. 2009;19:71–88. doi: 10.1038/cr.2008.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Furukawa F, Matsuzaki K, Mori S, et al. p38 MAPK mediates fibrogenic signal through Smad3 phosphorylation in rat myofibroblasts. Hepatology. 2003;38:879–889. doi: 10.1053/jhep.2003.50384. [DOI] [PubMed] [Google Scholar]
- 23.Mori S, Matsuzaki K, Yoshida K, et al. TGF-beta and HGF transmit the signals through JNK-dependent Smad2/3 phosphorylation at the linker regions. Oncogene. 2004;23:7416–7429. doi: 10.1038/sj.onc.1207981. [DOI] [PubMed] [Google Scholar]
- 24.Matsuura I, Denissova NG, Wang G, He D, Long J, Liu F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature. 2004;430:226–231. doi: 10.1038/nature02650. [DOI] [PubMed] [Google Scholar]
- 25.Matsuzaki K, Murata M, Yoshida K, et al. Chronic inflammation associated with hepatitis C virus infection perturbs hepatic transforming growth factor beta signaling, promoting cirrhosis and hepatocellular carcinoma. Hepatology. 2007;46:48–57. doi: 10.1002/hep.21672. [DOI] [PubMed] [Google Scholar]
- 26.Murata M, Matsuzaki K, Yoshida K, et al. Hepatitis B virus X protein shifts human hepatic transforming growth factor (TGF)-beta signaling from tumor suppression to oncogenesis in early chronic hepatitis B. Hepatology. 2009;49:1203–1217. doi: 10.1002/hep.22765. [DOI] [PubMed] [Google Scholar]
- 27.Nagata H, Hatano E, Tada M, et al. Inhibition of c-Jun NH2-terminal kinase switches Smad3 signaling from oncogenesis to tumor-suppression in rat hepatocellular carcinoma. Hepatology. 2009;49:1944–1953. doi: 10.1002/hep.22860. [DOI] [PubMed] [Google Scholar]
- 28.Yoshida K, Matsuzaki K, Mori S, et al. Transforming growth factor-beta and platelet-derived growth factor signal via c-Jun N-terminal kinase-dependent Smad2/3 phosphorylation in rat hepatic stellate cells after acute liver injury. Am J Pathol. 2005;166:1029–1039. doi: 10.1016/s0002-9440(10)62324-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Desmet VJ, Gerber M, Hoofnagle JH, Manns M, Scheuer PJ. Classification of chronic hepatitis: diagnosis, grading and staging. Hepatology. 1994;19:1513–1520. [PubMed] [Google Scholar]
- 30.Sekimoto G, Matsuzaki K, Yoshida K, et al. Reversible Smad-dependent signaling between tumor suppression and oncogenesis. Cancer Res. 2007;67:5090–5096. doi: 10.1158/0008-5472.CAN-06-4629. [DOI] [PubMed] [Google Scholar]
- 31.Liang X, Bi S, Yang W, et al. Epidemiological serosurvey of hepatitis B in China – declining HBV prevalence due to hepatitis B vaccination. Vaccine. 2009;27:6550–6557. doi: 10.1016/j.vaccine.2009.08.048. [DOI] [PubMed] [Google Scholar]
- 32.Weissberg JI, Andres LL, Smith CI, et al. Survival in chronic hepatitis B. An analysis of 379 patients. Ann Intern Med. 1984;101:613–616. doi: 10.7326/0003-4819-101-5-613. [DOI] [PubMed] [Google Scholar]
- 33.Realdi G, Fattovich G, Hadziyannis S, et al. Survival and prognostic factors in 366 patients with compensated cirrhosis type B: a multicenter study. The Investigators of the European Concerted Action on Viral Hepatitis (EUROHEP) J Hepatol. 1994;21:656–666. doi: 10.1016/s0168-8278(94)80115-0. [DOI] [PubMed] [Google Scholar]
- 34.de Jongh FE, Janssen HL, de Man RA, Hop WC, Schalm SW, van Blankenstein M. Survival and prognostic indicators in hepatitis B surface antigen-positive cirrhosis of the liver. Gastroenterology. 1992;103:1630–1635. doi: 10.1016/0016-5085(92)91188-a. [DOI] [PubMed] [Google Scholar]
- 35.Lai CL, Chien RN, Leung NW, et al. A one-year trial of lamivudine for chronic hepatitis B. Asia Hepatitis Lamivudine Study Group. N Engl J Med. 1998;339:61–68. doi: 10.1056/NEJM199807093390201. [DOI] [PubMed] [Google Scholar]
- 36.Honkoop P, de Man RA, Zondervan PE, Schalm SW. Histological improvement in patients with chronic hepatitis B virus infection treated with lamivudine. Liver. 1997;17:103–106. doi: 10.1111/j.1600-0676.1997.tb00789.x. [DOI] [PubMed] [Google Scholar]
- 37.Yamaguchi T, Matsuzaki K, Inokuchi R, et al. Phosphorylated Smad2 and Smad3 signaling: shifting between tumor suppression and fibro-carcinogenesis in chronic hepatitis C. Hepatol Res. 2013;43:1327–1342. doi: 10.1111/hepr.12082. [DOI] [PubMed] [Google Scholar]
- 38.Suzuki Y, Kumada H, Ikeda K, et al. Histological changes in liver biopsies after one year of lamivudine treatment in patients with chronic hepatitis B infection. J Hepatol. 1999;30:743–748. doi: 10.1016/s0168-8278(99)80123-8. [DOI] [PubMed] [Google Scholar]
- 39.Arzumanyan A, Reis HM, Feitelson MA. Pathogenic mechanisms in HBV-and HCV-associated hepatocellular carcinoma. Nat Rev Cancer. 2013;13:123–135. doi: 10.1038/nrc3449. [DOI] [PubMed] [Google Scholar]
- 40.Lok AS, McMahon BJ. Chronic hepatitis B: update 2009. Hepatology. 2009;50:661–662. doi: 10.1002/hep.23190. [DOI] [PubMed] [Google Scholar]
- 41.Barazani Y, Hiatt JR, Tong MJ, Busuttil RW. Chronic viral hepatitis and hepatocellular carcinoma. World J Surg. 2007;31:1243–1248. doi: 10.1007/s00268-007-9041-3. [DOI] [PubMed] [Google Scholar]
- 42.Rabe C, Pilz T, Klostermann C, et al. Clinical characteristics and outcome of a cohort of 101 patients with hepatocellular carcinoma. World J Gastroenterol. 2001;7:208–215. doi: 10.3748/wjg.v7.i2.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wong PY, Xia V, Imagawa DK, Hoefs J, Hu KQ. Clinical presentation of hepatocellular carcinoma (HCC) in Asian-Americans versus non-Asian-Americans. J Immigr Minor Health. 2011;13:842–848. doi: 10.1007/s10903-010-9395-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zemel R, Issachar A, Tur-Kaspa R. The role of oncogenic viruses in the pathogenesis of hepatocellular carcinoma. Clin Liver Dis. 2011;15:261–279. doi: 10.1016/j.cld.2011.03.001. vii–x. [DOI] [PubMed] [Google Scholar]
- 45.Yun C, Cho H, Kim SJ, et al. Mitotic aberration coupled with centrosome amplification is induced by hepatitis B virus X oncoprotein via the Ras-mitogen-activated protein/extracellular signal-regulated kinase-mitogen-activated protein pathway. Mol Cancer Res. 2004;2:159–169. [PubMed] [Google Scholar]