

Abstract
The discovery of secondary metabolites produced by microorganisms (e.g., penicillin in 1928) and the beginning of their industrial application (1940) opened new doors to what has been the main medication source for the treatment of infectious diseases and tumors. In fact, approximately 80 years after the discovery of the first antibiotic compound, and despite all of the warnings about the failure of the “goose that laid the golden egg,” the potential of this wealth is still inexorable: simply adjust the focus from “micro” to “nano”, that means changing the look from microorganisms to nanograms of DNA. Then, the search for new drugs, driven by genetic engineering combined with metagenomic strategies, shows us a way to bypass the barriers imposed by methodologies limited to isolation and culturing. However, we are far from solving the problem of supplying new molecules that are effective against the plasticity of multi- or pan-drug-resistant pathogens. Although the first advances in genetic engineering date back to 1990, there is still a lack of high-throughput methods to speed up the screening of new genes and design new molecules by recombination of pathways. In addition, it is necessary an increase in the variety of heterologous hosts and improvements throughout the full drug discovery pipeline. Among numerous studies focused on this subject, those on polyketide antibiotics stand out for the large technical-scientific efforts that established novel solutions for the transfer/engineering of major metabolic pathways using transposons and other episomes, overcoming one of the main methodological constraints for the heterologous expression of major pathways. In silico prediction analysis of three-dimensional enzymatic structures and advances in sequencing technologies have expanded access to the metabolic potential of microorganisms.
Keywords: environmental samples, pharmacology, PKSs, new drugs
Introduction
Despite efforts in the search for antibiotics in general, new discoveries do not translate into new classes of rapidly applicable pharmaceuticals. Over the past 30 years, only two novel classes of antibiotics have been launched on the market: the oxazolidinone linezolid and the cyclic lipopeptide daptomycin (Fischbach and Walsh, 2009). This small number of novel compounds demonstrates the inadequacy of traditional approaches, prompting the search for new strategies, among which metagenomics, bioinformatics and molecular engineering emerge as the most promising alternatives to balance this constant battle for human and environmental health.
Polyketides are natural metabolites that comprise the basic chemical structure of various anticancer, antifungal, and anticholesteremic agents; antibiotics; parasiticides and immunomodulators. As a testimony to its importance, the annual sales of pharmaceuticals derived from polyketides routinely reach 20 billion dollars (Weissman, 2009).
Since the discovery of the antibiotic properties of polyketides (i.e., streptomycin in 1950), the search for new compounds has led pharmaceutical companies to isolate tens of millions of antibiotic producing strains though cultivable soil microorganisms remain the main source of antibiotics and other active compounds. However, traditional methods for searching new drugs, which involve the cultivation of microorganisms isolated from soil, are not so promising currently. Some estimates of the high rate of rediscovery of known antibiotics produced by this technique are quite pessimistic: they were reaching 99.9% (Zaehner and Fieldler, 1995; Charusanti et al., 2012). On the other hand, some studies draw an interesting parallel between the discovery of certain new compounds not only in new species, but also in known species, reflecting both the under-exploitation of a minority of compounds produced by certain microorganisms and the constant acquisition of new polyketide synthase (PKS) pathways by unrelated species through horizontal gene transfer (Takagi and Shin-Ya, 2011).
As an alternative to these barriers, an emerging method for obtaining biomolecules with new or improved pharmacological properties is the direct study of genetic material from complex environmental samples called Metagenomics. By using this approach, it is possible to recover and express genes from uncultivable bacteria (which can be correspond to 99% of the total) because metagenomics is based on the genomic analysis of microorganism communities without the need to culture them in the laboratory. Therefore, metagenomics consists of extracting DNA directly from the environment and constructing a genomic library from this mixture (Knight et al., 2003; Committee On Metagenomics, 2007). Then, these materials can be mining for genes of biotechnological interest, both for genetic homology as functional screening. This technique has been the source of many new bioactive compounds; among them we can cite psymberin, norcardamine, violacein and turbomycin A (Banikand and Brady, 2010; Kim et al., 2010; Iqbal et al., 2012).
How an important tool for the discovery of new genetic pathways from metagenomics, bioinformatics analyses aid in the screening of promising sequences through the identification of possible genes, and functional inferences are based on homology and on simulations of the structural conformation of the expressed protein (Smith and Waterman, 1981; Fischer et al., 2009). Using metagenomics, it is possible to recover and study new PKS gene sequences, which can be used in cloning, combinatorial biosynthesis, and heterologous expression experiments to obtain new molecules.
This study presents an analysis of the current view in the search for new antibiotics with greater focus on the advances in the polyketides field and a discussion of the delicate dual role hero/villain of new bioactive compounds in the emergence of multidrug-resistant pathogens. For didactic purposes and considering that this fascinating group of biomolecules arouses the attention of researchers worldwide, some aspects of PKS biochemical pathway organization and a discussion of the main obstacles for developing this research field are presented. Lastly, we present recent results obtained in our laboratory related to this exploration.
Secondary Metabolite-Producing Microorganisms
Secondary metabolites are natural compounds produced by certain groups of bacteria, fungi and plants. These compounds are usually active against microorganisms at low concentrations due to their ability to inhibit essential primary metabolic processes. These metabolites are called “secondary” because they are not essential for cell survival, but their production often confers a competitive advantage to the microorganism (Martin and Demain, 1980).
Many secondary metabolites of biotechnological interest are produced by a group of filamentous gram-positive bacteria of the Actinomycetales order. The genus Streptomyces, which is in the Streptomycetaceae family, is the most well known in this order (Lechevalier and Lechevalier, 1981), and it mainly includes bacteria found in the soil and aquatic environments. These microorganisms are able to utilize different carbon sources and secrete a variety of hydrolytic enzymes that degrade insoluble organic materials such as cellulose and chitin (McCarthy and Williams, 1992; Hsiao and Kirby, 2008).
These microorganisms exhibit colony morphological differentiation that is coordinated with physiological differentiation and secondary metabolite synthesis. The life cycle of these microorganisms begins with spore fixation to their substrate and germination, resulting in filaments that are often “multinucleated” (i.e., a mixture of nucleic acids from more than one microorganism, and they lack a nuclear membrane because they occur in bacteria), which are called hyphae. In response to specific signals, which usually include nutritional deficiency, there is the formation of another type of mycelium called aerial mycelium, which emerges from the surface of the colony. Concomitantly, the presence of secondary metabolites is detected in the substrate. The aerial mycelium is formed of dividing septa with double cell walls, and each septum has a genome that will result in a spore for the dispersion and colonization of new habitats (Charter, 1993).
This genus has the ability to synthesize secondary metabolites with highly diverse chemical structures including a wide variety of antibiotics, and many of these compounds have important applications in medicine and agriculture (Vining, 1990; Piel, 2011). Among the filamentous actnomycetes, 75% of all metabolites are produced by the genus Streptomyces, the major source of the antibiotics sold worldwide (Padilla, 1998; Solecka et al., 2012). Streptomyces have a large linear chromosome of approximately 8 Mb, with high C+G content (i.e., 69 to 74%) (Gladek and Zabrzewsk, 1984; Gustafsson et al., 2004; Kinashi, 2011). Streptomyces have both linear and circular plasmids with sizes ranging from 2 kb to 1 Mb, and they may be involved in conjugation events (Bibb et al., 1978; Kinashi, 2011), biosynthesis (Hopwood, 1978), and antibiotic resistance (Kinashi et al., 1987; Jenke-Kodama and Dittmann, 2009).
Many factors can potentially determine the onset of antibiotic production in Streptomyces. The existence of a complex regulatory network enables the organisms to respond to a variety of physiological and environmental signals (Romero et al., 2011).
Morphological differentiation and the onset of antibiotic production are related to the type and availability of carbon and nitrogen sources (Martin and Demain, 1980), and to low molecular weight diffusible substances, which act as autoregulators and determine the initiation of antibiotic production and morphological differentiation. One example of an autoregulator is the A-factor, a γ-butyrolactone whose presence at nanomolar concentrations promotes the formation of aerial mycelium and the production of streptomycin in S. griseus (Horinouchi and Beppu, 1992). In S. coelicolor A3 (strain 2), a model-organism actinobacteria for molecular genetic studies whose complete genome has been sequenced, the genes that express γ-butyrolactones are located outside the gene cluster (Bentley et al., 2002) and are pleiotropic regulators of other specific biosynthetic pathway regulators (Hopwood et al., 1995).
Antibiotics Produced by Microorganisms: Polyketides
Polyketides are natural products containing multiple β-hydroxyketone or β-hydroxyaldehyde (-H2C(=O)CH2CH(OH)CH2C(=O)-) functional groups. The PKSs are diverse enzymatic complexes that catalyze polyketide carbon skeleton assembly from coenzyme-A fatty esters that are derived from short-chain fatty acids such as acetate, propionate and butyrate. The fatty acid synthases (FASs) are homologous to PKSs and synthesize lipids involved in a wide variety of essential cellular functions; however, PKSs have a much more complex biosynthetic routine and are only present in restricted taxonomic groups (Hopwood, 2004). Although both polyketides and fatty acids are derived from acetyl-CoA or other acyl-CoAs, in polyketides, more than one monomer type may be used to construct different sizes of aromatic or reduced chains (Figure 1). These chains can be as small as 6 to 8 carbons long (e.g., 6-methyl salicylic acid; 6-MSA), or as large as 164 carbon atoms (e.g., maitotoxin). In fatty acids, only groups with two carbons are assimilated from malonyl-CoA to form saturated molecules with chain sizes ranging from 16 to 20 carbons long (Hutchinson and Fujii, 1995; Weissman, 2009).
Figure 1.

Representative polyketide structures including prominent bioactives. Reproduced from Methods in Enzymology, 2009, Vol. 459, cap. 1, Pages 3–16 with permission of Elsevier Ltd. (Weissman, 2009).
Three classes of bacterial PKSs have been identified to date: type I, type II and type III (Shen et al., 1994). The type I PKSs (i.e., modular/iterative PKSs) are large enzymes, consisting of multi-domains that contain a series of active sites for the various stages of polyketide synthesis. Examples of natural products derived from these multicatalytic enzymes include the macrolides (erythromycin) and polyenes (nystatin). The type II PKSs (i.e., aromatic PKSs) are composed of separate mono and bi-functional enzymes that interact during synthesis to form a polyketide structure. This structure is then enzymatically converted to the cyclic form to produce polycyclic aromatic compounds such as tetracycline or doxorubicin (Hutchinson and Fujii, 1995).
The type III PKSs, which are also called chalcone synthases, are relatively small proteins that are mainly involved in the production of important plant compounds such as flavonoids and stilbenes (phenolic), and they are also found in bacteria and fungi. Unlike other PKSs, PKS III polyketide intermediates are not linked enzymatically but through free coenzyme-A thioesters (Brachmann et al., 2007).
Among the three PKSs, the most well-studied pathways for combinatorial biosynthesis are the type I PKSs, which allowed for the development of databases specific for this family and an efficient system for the comparison of enzymatic domains capable of predicting the majority of substrates through software based on homology (Yadav et al., 2003; Starcevic et al., 2008). This level of knowledge has not been achieved yet for type II PKSs, and due to their ecological and biotechnological importance, it is essential that similar prediction methods be developed to help guide proteomics and molecular engineering research.
One of the foremost scientists involved in studying PKSs was Professor Charles Richard Hutchinson who dedicated a part of his life to conducting detailed studies on these important classes of enzymes (The Journal of Antibiotics, 2011; Olano, 2011). The death of Dr. Hutchinson on January 5, 2010 was undoubtedly a great loss to academic research in this field. The legacy of Dr. Hutchinson (Olano, 2011) and the results of 40 years dedicated to PKS studies involving the search for new drugs, particularly against cancer (ironically, the disease that killed him at age 66) include the study of bacterial and fungal polyketides. Among the type I PKSs, Hutchinson and colleagues studied the erythromycin, geldanamycin, herbimycin, lasalocid, megalomycin, midecamycin, rapamycin and rifamycin synthesis pathways. In the type II PKS family, these researchers studied the elloramycin, doxorubicin, daunorubicin, fredericamycin, jadomycin and tetracenomycin synthesis pathways.
As shown in Figure 2, the modular PKS I (Figure 2A) has noniterative action. The reaction begins with an activation module (i.e., the loading module), that binds through a thioester bond of the initial precursor with the acyl carrier protein (ACP) by the action of the first acyltransferase (AT) site. Next, there is the transfer of the activated substrate to the ACP of the next module in a reaction known as the “ping-pong” mechanism. The enzymes of this module move the reaction process forward by adding a new monomer to the ketone chain and modify the group by oxidation-reduction reactions and transferase activity. This newly formed molecule will subsequently be the substrate for the next module. The final step of the reaction is the action of thioesterase (TE), which releases the ACP-bound polyketide molecules from the termination module. It is important to note that this is not the only PKS I group reaction type, there are cis AT, trans AT and iterative PKS I (one or two modules rotate the reaction steps between each other, and the same gene gives rise to multiple copies of the polypeptide, which interacts in the reactions) (Weissman, 2009).
Figure 2.

Scheme of PKS catalytic reactions and dynamics. Reproduced from Methods in Enzymology, 2009, Vol. 459, cap. 1, Pages 3–16 with permission of Elsevier Ltd. (Weissman, 2009).
Both PKS II (Figure 2B) and PKS III (Figure 2C) are iterative. However, PKS II action in the initial ketone chain synthesis involves the interaction of three enzymatic subunits: KSα, KSβ and ACP. First, the initial precursor (mostly acetyl-CoA) forms a thioester bond with the ACP. The reaction is catalyzed by KSα, but it can also spontaneously occur in vitro. Next, the first chain is constructed by a Claisen condensation reaction (i.e., a reaction between two esters or an ester and a ketone group in the presence of a strong base or a catalytic site, producing a β-keto ester or β-diketone, respectively) of a new substrate (i.e., malonyl-CoA and methyl-malonyl-CoA), which is catalyzed by KSα and KSβ subunits over the beginning of the chain still bound to the ACP (Weissman, 2009).
The Claisen condensation reaction is repeated numerous times with the elongating chain always anchored to the ACP and with the constant action of the same enzymes hence the name iterative. During ACP-anchored synthesis, various additional enzymatic units (e.g., cyclases and aromatases) act on this chain producing aromatic and cyclic groups, or they reduce the ketone groups. After the release of the polyketide from the ACP, numerous other enzymes, such as oxidases, transferases and hydrolases, act on the chain to make post-synthetic modifications (Weissman, 2009).
In turn, PKS III is surprisingly composed of only one KS domain, and the initial precursor is derived by the direct binding of free acetyl-CoA to the KS catalytic site, which performs the entire Claisen condensation reaction alone. The catalytic site redox potential and the instability of some chemical groups in the elongation chain will induce spontaneous chain cyclization (Brachmann, 2007; Weissman, 2009).
All biosynthetic genes necessary for the synthesis of a given antibiotic from primary metabolites occur in gene clusters occupying between 15 and 120 kb. These clusters contain genes that confer antibiotic auto-resistance and regulatory genes that coordinate the expression of other genes in the cluster in addition to structural genes encoding all enzymes of the secondary metabolic pathway (Fernandez-Moreno et al., 1991). This was first demonstrated through the cloning of the entire actinorhodin biosynthetic gene cluster, which was found on a 35 kb chromosomal DNA fragment of the producer Streptomyces coelicolor A3 (strain 2) and its expression in the non-producer and actinorhodin-sensitive strain S. parvulus, which produced actinorhodin and became resistant (Malpartida and Hopwood, 1984). Gene knockout experiments, complementation of blocked mutants and heterologous expression allowed for the biosynthetic gene clusters to be studied in detail, leading to the isolation and characterization of entire biosynthetic pathways (Hutchinson, 1997).
Multidrug- and Pandrug-Resistant Bacteria
It has been long known that microorganisms have several mechanisms of drug resistance, which arise from natural selection of the “gene pool” of constantly evolving microbial populations either by gene mutations (vertical evolution) or by the acquisition of genes from other non-related species through mobile genetic elements such as plasmids, phages and transposons (i.e., horizontal gene transfer). Figure 3A shows some of the major mechanisms of bacterial resistance to known antibiotics. Historically, the inappropriate use of pharmacological compounds in the clinical setting has placed artificial selection on the microbiota, accelerating the spread of resistance among pathogen and commensal microorganisms. As observed in Figure 3B, the emergence of resistant pathogens in a hospital environment typically occurs shortly after the introduction of a new compound (Schmieder and Edwards, 2012).
Figure 3.

Mechanisms of bacterial resistance to antibiotics (A) and Antibiotics resistance evolution showing the rapid development of resistance to multiple classes of antibiotics (B). Reproduced from Future Microbiology, January 2012, Vol. 7, No. 1, Pages 73–89 with permission of Future Medicine Ltd. (Schmieder and Edwards, 2012).
Despite this situation, the emergence of genes related to antibiotic resistance preceded the clinical use of antibiotics by humans and should not be understood as the result of this selection but as its raw material. Microbial antibiotic resistance and pathogenicity are not modern phenomena. Studies using metagenomic DNA samples extracted from Beringian permafrost sediments that were protected from contamination by layers of volcanic tephra (C14 dated at 30,000 years old, belonging to the Pleistocene) revealed the presence of several gene clusters encoding resistance to β-lactam, tetracyclines, glycopeptide antibiotics and the presence of sequence variants similar to the modern vancomycin resistance element VanA (D’Costa et al., 2011). This demonstrates the natural course of the emergence of these genes through evolution, suggesting that their role in the antagonistic relations within the microbial community precedes the effects of human action.
If the constant adaptation of microorganisms to drugs in the circulation has required a constant search for new formulations, then the emergence of multidrug resistant bacteria classes has made the discovery of novel chemical molecules a priority. Some promising approaches have emerged, such as prospecting in unexplored bacterial niches, database searching for synthetic molecules (Fischbach and Walsh, 2009) and the use of metagenomics.
The first class corresponds to the methicillin-resistant Staphylococcus aureus (MRSA), and this group is identified as the cause of 19,000 annual deaths in the United States and an annual increase in health costs on the order of 3 to 4 billion dollars. As an aggravating factor, the prevalence of MRSA increases the likelihood of vancomycin resistant S. aureus (VRSA), further challenging disease treatment in hospitals (Fischbach and Walsh, 2009).
The second class includes gram-positive multidrug-resistant (MDR; with resistance to certain drugs) and pandrug-resistant (PDR; with resistance to all drugs) bacteria, which are less prevalent than MRSA, which are albeit related to incurable clinical pictures. The strains Acinetobacter baumannii, Escherichia coli, Klebsiella pneunominae, and Pseudomonas aeruginosa are MDR/PDR to penicillin, cephalosporin, carbapenem, monobactam, quinolone, aminoglycoside, tetracycline and polymyxin (Fischbach and Walsh, 2009).
The third class is comprised of Mycobacterium tuberculosis strains that are MDR and extensively drug resistant (XDR), and they are a growing problem in developing countries, requiring treatment for two years with antibiotics that cause severe side effects (Fischbach and Walsh, 2009).
This situation brings up the discussion of the misuse of drugs, which for years has accelerated this resistance process that is caused by generations of microbes that are resurgent from treatments that are insufficient in duration and dose. This concern has also led to the creation of stricter rules for the purchase of antibiotics such as the recent decision by the Brazilian National Health Surveillance Agency (Agência Nacional de Vigilância Sanitária - ANVISA) for the mandatory submission of a prescription to purchase antibiotics, thereby curbing self-medication.
Pharmaceutical Market and the Discovery Pipeline
Over the past 30 years, only two novel classes of antibiotics have been launched on the market: the oxazolidinone linezolid and the cyclic lipopeptide daptomycin. As an aggravating factor, telavancin (a glycopeptide), retapamulin (a pleuromutilin), and tigecycline (last generation tetracycline) were the only antibiotic active ingredients approved as entirely new molecules by the U.S. Food and Drug Administration (FDA), and they were launched on the market between 2005 and 2010. In addition, there are few antibiotics in circulation that are still considered to be safe in terms of retaining their active capacity against MDR micropathogens (Hamad, 2010).
This decline is also marked by an uneven distribution of molecules within the different classes of antibiotics with a predominance of polyketide compounds, which included one third of all drugs brought to market from 2005 to 2007 (Weissman, 2009). Figure 4A illustrates the revenues earned in billions of dollars for each of the main active ingredients of drugs marketed worldwide in 2009, particularly for the cephalosporin class of polyketides, which accounted for 11.9 billion dollars. However, the crisis scenario is marked by the slow process between prospection and the arrival of the molecule on the consumer market. Figure 4B illustrates the distribution profile of molecules in the pre-implementation phase in 2010, and it shows that beyond the actual discovery process of new bioactive compounds, the major bottleneck of the process is the pre-clinical period (Hamad, 2010).
Figure 4.

New drug distribution by chemical family and research stage. A. Revenue in billions of dollars for the most commonly used antibiotics. B. Production pipeline of new drugs. Reproduced from Nature Reviews Drug Discovery, September 2010, Vol. 9, No. 9, Pages 675–676 with permission of Nature Publishing Group Ltd. (Hamad, 2010).
Another factor that greatly contributes to the retention time of each drug at every stage is the nature of its application and urgency in the demand for certain clinical groups as shown in Figure 5 (Kaitin, 2010).
Figure 5.

Retention time averages of new molecules according to the pathological area, which were obtained from 2003 to 2007; * The anti-infective category excludes AIDS antiviral agents (which are in their category). Reproduced from Clinical Pharmacology & Therapeutics, February 2010, Vol. 87, Pages 356–361 with permission of Nature Publishing Group Ltd. (Kaitin, 2010).
As observed in Figure 5, HIV drugs have been the fastest implemented, and this is, in part, due to the speed at which current drugs become ineffective in the face of new prevalent serotypes. These classes of drugs and antineoplastic agents have the shortest regulation time (approval phase), which reflects a clear reduction in the bureaucratic process involving the release of these drugs, ensuring a more dynamic exchange between the academic/technical/scientific sector and the community.
Another aspect to be considered is the process of generating new drugs, which identifies the institutions that contribute the most to their generation. Figure 6, for example, illustrates the origin of the 252 new drugs approved by the FDA from 1998 to 2007.
Figure 6.

Public and private initiative contributions to the discovery of new drugs in different countries for the period between 1998 and 2007. Reproduced from Nature Reviews Drug Discovery, November 2010, Vol. 9, Pages 867–882 with permission of Nature Publishing Group Ltd. (Kneller, 2010).
As observed in the graph in Figure 6, the United States is responsible for most of the drugs produced, and universities represented an important focus for the generation of new products for the biotechnology private sector, surpassing the performance of large pharmaceutical companies (Kneller, 2010). These data reinforce the essential role of universities in creating new drugs, clearly demonstrating how the public and private sector could invest to boost new drugs production.
In Brazil, the regulation of new drug implementation priorities is the responsibility of the National Drug Policy (“Política Nacional de Medicamentos” - PNM) and the National Health Surveillance Agency (ANVISA). The PNM was established in 1998 and is responsible for regulating the list of drugs considered essential and the safety standards for medicine, human resources and technical/scientific development. Though the list of essential drugs already existed, it had not been updated for approximately 16 years. The PNM was responsible for reformulating the list in 1999, and it was subsequently updated in 2002 and 2006. The ANVISA, was created in 1999 with the priority of addressing issues of health surveillance (Vidotti et al., 2008).
An important health step in Brazil, where 70% of the population has access to public health, was the passing of the generic drug policy and the breaking of drug patents in 1999. Four years after the passing of this policy, generic drugs already represented 12% of the total drug sales in the country. Of the remaining drug sales, 68% corresponded to similar drugs (i.e., non-patented), and 20% corresponded to drugs protected by patents (Vidotti et al., 2008). This practice allowed a more democratic access to drugs considered essential to health.
The importance of generic and similar drugs is undeniable because they represent the most attractive options in an industry where it is common practice for drugs with the same formulation to differ only in brands and prices. Wadt (2003) analyzed new drugs brought to market in Brazil between 1998 and 2001 (Figure 7).
Figure 7.

Evaluation of new drugs launched in Brazil from 1998 to 2001. Source: Wadt (2003). Reproduced with permission of author.
From the 285 “new” drugs approved by the ANVISA, 84 were considered non-innovative (red) because they had the same formulations as drugs previously released. Of the remaining drugs, 47 were not targeted in the author’s study (yellow), and 154 drugs (in blue) contained a new chemical molecule in their formulation (Wadt, 2003).
Historically, pharmaceutical corporations in Brazil, both national and transnational, have focused on the modification of pharmaceuticals, relegating the discovery of new formulations to the background with scarce resources. Fortunately, this situation is beginning to change with the increased investment and involvement of companies in all of the developmental and production stages of new drugs, which seems to have a close relationship with changes in the regulatory policies for this sector, a production capacity expansion and a favorable economic environment coupled with the consolidation of university research. In fact, an increase in the creation of study groups and a greater interaction between these and the pharmaceutical companies have expanded the research capacity for new products, culminating in the first drug developed entirely in Brazil in 2004: the anti-inflammatory alpha-humulene (Vidotti et al., 2008) that is derived from the plant species Cordia verbenacea, which is known by its popular name of “erva-baleeira.” Thus, it is clear that the worldwide market for drugs is still dominated by large pharmaceutical and biotechnology companies with active academic participation in the development of new formulations, and that the United States is the largest producer of pharmaceuticals.
Other developed countries share the responsibility for the remainder of the new formulations. Although still in its early stages, Brazil has made progress in the technical/scientific development of new drugs; however, there is still much to be done to ensure democratic access to medications, and investment in academic research seems to be a promising path that has already been well established in developed countries, showing the successful implementation of partnership strategies between universities and public/private initiatives.
Metagenomics as a Tool for Discovery of Novel Polyketides
The recent widespread dissemination of pathogenic bacteria resistant to antibiotics and the increasing need for bioactive molecules with new or improved pharmacological properties promoted an increased interest in the discovery of new antibiotics (Demain and Sanchez, 2009).
Traditionally, the discovery of novel bioactive compounds with biotechnological importance is performed by analyzing natural sources, such as soil microorganisms, for the presence of desired activities. Since the discovery of streptomycin in the 50s, which is produced by a soil-isolated actinobacteria, microbiologists and pharmacists around the world have invested great efforts in the culture of various microorganisms in the search for new strains able to synthesize antibiotics. The search for new antibiotics has led pharmaceutical companies to isolate tens of millions of producing strains. To date, cultivable soil microorganisms still represent the main source of antibiotics and other active compounds.
Currently, traditional methods for the searching for new drugs involving the cultivation of soil microorganisms are not as promising, and this is mainly due to the high rediscovery rate of known antibiotics, which reaches 99.9% (Zaehner and Fieldler, 1995; Charusanti et al., 2012). However, progress in molecular microbial ecology has demonstrated that the genetic diversity of microorganisms in nature is much greater than that reflected in the lineage collections and databases. It is estimated that only a small portion of these, approximately 1%, is cultivable by standard culture media techniques (Amann et al., 1995; Committee On Metagenomics, 2007).
The vast majority of microorganisms have needs that are too complex to be reproduced in artificial environments including microorganisms that have synergisms/syntrophisms with other organisms, and those requiring amino acid supplementation, unusual carbohydrates, growth factors and a whole set of physical-chemical factors produced by the metabolism of other microorganisms such as optimal pH and moisture, harmful metabolic waste reduction, and the creation of anaerobic or aerobic environment among many other interactions.
Metagenomics is a method based on the genetic and functional analysis of the microbiota without culturing, and it directly acts on the nucleic acid pool in the environment. In one of its applications, the methodology makes use of the PCR amplification of conserved gene regions followed by amplicon cloning and sequencing, creating so-called gene diversity libraries (Committee On Metagenomics, 2007; Gomes et al., 2010).
Some regions that are highly conserved across all genomes, such as genes corresponding to ribosomal RNA synthesis (i.e., rDNA and rRNA), particularly the 16S rDNA (prokaryotes) and 18S rDNA (eukaryotes), allow the classification of organisms thanks to the intercalation of variable and non-variable sequences.
Studies employing the amplification of 16S and 18S rDNA from DNA samples extracted directly from the environment are dramatically changing the traditionally accepted phylogenetic definitions of the main microbial groups (Bull et al., 2000).
Other gene regions also enable tracking this diversity and are often used to determine the abundance of a specific group of enzymes or regulators among the other proteins present in a given community. These regions are important to infer the ecological role played by proteins in maintaining the ecosystem studied and identifying environments rich in biotechnologically interesting enzymes (Metsä-Ketelä et al., 1999).
Another metagenomics methodological approach is the extraction of high-molecular-weight and pure DNA and the cloning of this material in vectors that can carry large inserts such as BACs and YACs (i.e., bacterial artificial chromosomes and yeast artificial chromosomes, respectively) and cosmids and fosmids, thus creating metagenomic libraries that are searchable for genes of ecological and/or biotechnological interest, enabling the discovery of previously unknown biosynthetic pathways. One major barrier to the implementation of this technique is obtaining high quality DNA due to contamination with co-extracted environmental materials such as fulvic and humic acids (Rajendhran and Gunasekaran, 2008). This need became significant because of the fact by which pathways antibiotics are produced, once they can be extremely complex, reaching up to 100 kb of genomic extension and with more than 40 coordinated genes (Fischbach et al., 2008), which turns out to create difficulty to obtaining the complete pathways by cloning of small inserts.
Currently, there are some kits (e.g., kit Ultra Clean Mega Soil DNA from Mo Bio) and even diversified non-commercial extraction methods (Liles et al., 2008) that, in part, allow for the circumvention of these difficulties; however, the physical-chemical nature of the environment directly influences their performance, requiring adjustments for each condition studied (Massini, 2009; Schuch, 2007).
The use of metagenomics as a search strategy for the discovery of genes of interest significantly increases the potential success of these approaches because it covers a diversity that is not constrained by laboratory culturing. Because the wide diversity of the soil microbiota consists of a large genetic and biological pool that can be mined for the discovery of novel genes, entire metabolic pathways and their products (Cowan et al., 2005; Baltz, 2006), metagenomics offers a way to access an environment’s microbial diversity in its entirety (Figure 8).
Figure 8.

Some metagenomic contributions to the discovery of new drugs.
Use of Bioinformatics to Predict Protein Function
How an important partner of metagenomic, the Bioinformatic analyses give support to identification of new genes for several medical and industrial important pathways. Since the second half of the 90s and the advent of automated DNA sequencers, there has been an explosion in the number of nucleotide sequences that are stored and analyzed, requiring a constant improvement in the computational resources. This demand has created an interface between computer science, molecular biology and the areas of statistics, mathematics and software engineering, giving rise to a new scientific field: Bioinformatics (Prosdocimi et al., 2001).
Currently, bioinformatics is an indispensable tool for processing the large volume of data generated by increasingly sophisticated and elegant methodological approaches for assessing diversity, transforming them into data that are comprehensible to the scientific community.
Blast (Basic Local Alignment Search Tool), one of the most widespread bioinformatic tools for sequence analysis is a search engine based on the biological homology of DNA and amino acid sequences. Blast is a variation of the Smith and Waterman Algorithm (Smith and Waterman, 1981). The Blast algorithm allows for sequence comparison in five different ways: Blastp, Blastn, Blastx, tBlastn, and tBlastx according to the user’s needs and nature of the database to be queried (Embrapa, 2007).
The Blastp software compares amino acid sequences (query) against a protein sequence database (subject). The Blastn software compares nucleotide sequence input against a database of nucleotide sequences. Blastx compares nucleotide sequences, which are translated to all possible reading frames, against a database of protein sequences. The tBlastn software compares an amino acid sequence input against a database of nucleotide sequences translated into all possible reading frames. The tBlastx software compares the six possible reading frames of a sequence against the six possible nucleotide sequence translations found in the National Center for Biotechnology Information (NCBI) database (Embrapa, 2007).
A powerful ally in the characterization of enzymatic groups has been the use of phylogenomics, which represents a set of methodologies that explores the relationships among biological sequences (i.e., DNA and proteins), thus allowing protein function prediction based on known orthologs/paralogs genes (Sjölander, 2004).
The use of global and local alignment, the construction of phylogenetic trees, and different database searches for homologous genes, motifs and conserved enzymatic domains using bioinformatic tools are part of the phylogenomic approach. The data obtained are then cross-referenced with biological information in the literature, and this is aided by the use of biostatistics indexes.
Use of Molecular Biology for the Production of New Polyketides
One of the most promising approaches for the discovery of novel bioactive compounds is related to the use of metagenomics to search for new biosynthetic genes for cloning and expression in a heterologous or homologous host that does not contain the specific pathway (Iqbal et al., 2012).
Consequently, for the expression of the genetic material of a new enzyme or enzymatic group recovered by metagenomics, it is necessary to introduce this material into a living organism capable of executing the new genetic information, resulting in the translation of the enzyme (or group of enzymes) into its active conformation. This means that the success of research aimed at producing new molecules by this strategy is restricted by the availability of a heterologous/homologous host compatible with the correct expression of the exogenous gene or even a native host with modified pathways to not interfere with detection of the new product. In the case of PKS II-related genes from Streptomyces, there are available native heterologous hosts (i.e., Streptomyces lividans, S. albus, and S. coelicolor) and non-native heterologous hosts developed from mutant strains of Escherichia coli that have been specifically created to expression genes from this group (Martin and Demain, 1980; WANG et al., 2000; Baltz, 2006).
The availability of these strains is a key point in the search for new drugs because the particularities of Streptomyces gene sequences significantly reduce compatibility with other possible hosts that are commonly employed for the heterologous expression of other groups. One of the most limiting factors for the expression of exogenous proteins in the group in question is related to their own set of preferred codons (codon usage), which is present in most of the Streptomyces genome. Codon usage can act as a limiting factor for the exchange between the native host’s gene of interest and the heterologous host that will be used for expression because if a codon set predominates in a genome, it is natural that the majority of tRNAs (RNA transporters) available in the organism will correspond to this preferred codon. Thus, the translation of an exogenous protein with low frequency preferred codons in a candidate host will not occur correctly, or the protein will be expressed at low levels due to the lack of cistrons (tRNAs) corresponding to the rare triplets.
Using a two-dimensional distribution map of preferred codon usage in the most studied living organisms, Gustafsson and colleagues (2004) established considerations on groups of organisms that share preferred codons because these groups can be deduced based on their spatial proximity in a multivariate statistical chart (Figure 9).
Figure 9.

Graphical spatial representation of codon usage of organisms often studied in the academic sector, correlating the degree of subject compatibility according to codon preference. Reproduced from Trends in Biotechnology, July 2004, Vol. 22, No.7, Pages 346–353 with permission of Elsevier Ltd. (Gustafsson et al., 2004).
Thus, based on the graph, it can be inferred that Saccharomyces cerevisiae would be the most suitable candidate for the expression of genes from the Arabidopsis plant and genes from the Caenorhabditis elegans nematode worm. At the same time, there is a closer relationship between the bacterial generas Escherichia coli and Bacillus. However, all seven organisms cited share more codon usage with each other than with Streptomyces (they are distant from all others in the map’s spatial conformation).
The codon usage pattern of Streptomyces has been defined by the authors as the most extreme studied because it is known that for the Streptomyces coelicolor species, the majority of codons have a C or G as a terminal nucleotide, reflecting the high G/C nitrogenous bases content (approximately 71%) in the species analyzed. The codon usage profile for this genus explains the difficulty in expressing genes derived from Streptomyces using organisms from another genus as hosts (Gustafsson et al., 2004).
However, if other organisms are used for expression, the efficiency conditions for translation of the protein of interest can be improved by molecular engineering. In the specific case of the expression of enzymes involved in type II polyketide biosynthesis, the literature reports the use of Escherichia coli by two different approaches.
One approach consists of the modification of the codons present in the gene sequence for the set of triplets optimized as the most abundant in E. coli and thus for which the heterologous host machinery is fully functional (Baltz, 2006). Another approach consists of changes throughout the heterologous host genome (Escherichia coli) that increase the occurrence of rare codons, which limit the expression of genes derived from Streptomyces. The advantage of such mutant E. coli strains is the versatile use of the same organism for the expression of different polyketide genes coupled with the rapid growth of E. coli compared to Streptomyces. However, even if these mutants prove suitable for the enzymatic expression and production of simple polyketide structures, the expression level of these metabolites is greater in the heterologous hosts of the Streptomyces group (Baltz, 2006; Peirú et al., 2008).
Methods of Molecular Engineering for Expression of Large Gene Clusters
A promising method for the generation of novel bioactive compounds explores the use of the combinatorial biosynthesis of enzymes from different PKS pathways even from pigments and searching for the rational design of new drugs by homologous/heterologous biosynthesis (Shen et al., 1999; Rix et al., 2002). For homologous expression involving the substitution of native pathways for others engineered with the addition or silencing (knockout) of a given gene or the exchange of well conserved regions between close organisms, an interesting alternative is the use of cross recombination (i.e., crossing-over). This recombination occurs due to common regions between the host chromosome and the recombinant element, which allows for the recognition and exchange of fragments by crossing-over events such that the insertion of exogenous material takes place at specific sites and is not limited by the DNA fragment size. However, there is not always a similar region in the host organism to guide gene insertion. In many situations, a desirable feature of a good host is to lack (naturally or artificially) certain pathways, facilitating the detection of successful mutants through phenotypic change. In such cases, even when dealing with the exchange between homologues, it is often necessary to add a region of homology so that the fragment may be integrated into the genome at the desired position (Fu et al., 2008; Smolke, 2010).
For a long time, one of the biggest challenges for the heterologous expression of PKS gene clusters was the manipulation and efficient cloning of large DNA fragments. While using the classical methods of engineering (i.e., PCR amplification, restriction digestion, and ligation experiments), it is possible to manipulate sequences measuring 15–20 Kb in length. This practice is inefficient and tediously time-consuming when cloning larger fragments. Therefore, the method is no longer recommended because more effective approaches for large inserts have been reported (Rivero-Muller et al., 2007; Fu et al., 2008; Stevens et al., 2010).
In this context, systems based on mechanisms such as RED/ET λ phage recombination joined with large molecular engineering constructs have been elegantly designed to facilitate the transposition of large inserts. The RED/ET regions and transposons are integrative DNA elements called episomes, which are, by definition, autonomous replication elements that, unlike common vectors such as most plasmids and cosmids, have the ability to integrate into the host genome. These systems can be adapted using commercially available kits (e.g., Red/ET Recombination System - Genebridges, Germany) that include vectors containing genes for special enzyme expression (or the purified enzyme) and specific inverted sequences (ITs). Construct adaptation involves the addition of homology arms (HAs) measuring approximately 40–50 bp for each recombination step between the large fragments and often involves the use of selective markers that are not inherited in the final inserted fragment (Sambrook and Russell, 2001; Rivero-Muller et al., 2007).
Another promising possibility is the use of transposons (e.g., the MycoMar transposon from Drosophila mauritiana), which may be combined with RED/ET recombination sites and have as an advantage the transposition of entire pathways in homologous and heterologous hosts, with 60 kb inserts already reported as generating stable mutants with high recombination rates (Fu et al., 2008). An advantage of this method compared to that previously mentioned is the possibility of inserting a fragment without the need to include a homologous sequence.
Metagenomic Studies Conducted at the LBMP
In one of the first metagenomic studies performed at the Microorganisms and Plants Biochemistry Laboratory (“Laboratório de Bioquímica de Micro-organismos e Plantas; LBMP”) at the Technology Department of São Paulo State University (“Universidade Estadual Paulista - UNESP”) Jaboticabal Campus, Silveira and colleagues (2006) compared environmental DNA samples obtained from two areas (i.e., under an eucalyptus arboretum and under native forest) to verify the biodiversity of microbial communities by 16S rDNA sequencing. The results showed that 72% of the analyzed sequences were novel and came from bacteria that had not been sequenced yet, suggesting an important source of genetic sequences potentially useful as biotechnological tools. The microbiota composition differed between the two environments. Under the native forest, microorganisms belonging to the phylum Bacteroidetes and Planctomycetes appeared in greater quantity, while under the eucalyptus arboretum, the predominant sequences were classified as Actinobacteria, Firmicutes, and Verrucomicrobia. An interesting observation was that the population of actinobacteria was greater in the area under the eucalyptus arboretum. These microorganisms are of great importance in biotechnology as producers of bioactive molecules, and this finding indicates that this soil can be a promising source for the mining of new genes related to antibiotic production (Silveira et al., 2006).
That study led to the construction of a metagenomic library from the soil studied with high molecular weight inserts to search for genes involved in polyketide biosynthesis. The library contains 9,320 clones and was constructed in cosmid vectors with cloned inserts measuring approximately 20 to 45 kb. From this library, assessments were made to detect the presence of genes involved in the antibiotic biosynthesis from PKS I (Schuch, 2007) and PKS II (Gomes, 2008).
Besides the metagenomic library constructed from soil under eucalyptus litter, the LBMP team also constructed libraries from mangrove soil, sugarcane straw soil, and from a petroleum-degrading consortium, while others are still under construction. These collections have been mined to search for several genes of industrial interest including catalases, amylases, lipases, peptidases, cellulases, antibiotics pathway genes, xylose-isomerases, and phosphatases among others using different mining approaches, such as the use of hybridization probes, degenerate primers, and biochemical assays.
Likewise, other diversity libraries were constructed (i.e., 16S rDNA and ITS) including those obtained from activated sludge (Val-Moraes et al., 2011), native forest soil and commercial monocultures (Pereira et al., 2006) and other sources. The accessed diversity will be employed for the construction of an EcoChip, which may eventually be used to rapidly identify organisms present in different habitats, thus enabling comparative studies between communities under the variables of time and space. As previously mentioned, metagenomic DNA is a promising source of new genes that encode molecules with potential biotechnology application, and the search for new antibiotic pathways is an important line of research developed in our laboratory. By way of illustration, the following items describe the methodological strategies and the main results obtained by our team in the search for new polyketide pathways.
Metagenomic Library Construction and the Search for Genes Involved in Type I PKS Polyketide Biosynthesis
The type I PKS, which is also known as modular, constitutes one of the main pathways studied in the search for new bioactive compounds. In a study involving the search for new type I polyketide antibiotics (Schuch, 2007), the goal was to recover and study PKS gene sequences from metagenomic DNA. For this purpose, a cosmid metagenomic library was built to search for type I PKS genes. A cosmid was subcloned and fully sequenced yielding a 29.6 kb contig. Analysis of the sequence contained in the insert revealed the presence of 21 open reading frames (ORFs), and one of which corresponded to a typical PKS module measuring 6,498 bp. Computational and phylogenetic analyzes were conducted to better understand the PKS gene found and the other ORFs present in the metagenomic DNA insert.
For metagenomic library construction, soil samples were collected under an eucalyptus arboretum that is planted on the UNESP campus in the city of Jaboticabal, State of Sao Paulo, Brazil in February 1969. This soil was characterized by molecular phylogeny in a previous study (Silveira et al., 2006). Twenty single samples were collect at random in a zigzag pattern at a depth of 0–20 cm, covering the entire area. The samples were pooled and homogenized, resulting in a composite sample. The samples were immediately transported to the laboratory for DNA extraction. Metagenomic DNA was extracted from 1 g of soil using the FastDNA® Spin kit for soil (MP Biomedicals, Irvine, CA, USA). A cosmid library was constructed using the pWEB-TNC™ Cosmid Cloning Kit (Epicentre Biotechnologies Madison, WI, USA). Metagenomic DNA analysis showed that the sample had sufficient quality and integrity for cloning. A library of 9,320 clones was constructed in cosmid vectors from 10 μg of metagenomic DNA.
The library was mined for the presence of biosynthesis genes for modular polyketides biosyntheses (PKSI). The mining for PKS genes was performed by PCR using cosmid DNA obtained from pools of 96 clones using the alkaline lysis method as the template (Sambrook and Russell, 2001). As previously described (Courtois et al., 2003), two sets of degenerate primers that were complementary to highly conserved regions in type I PKS genes were used. The PCR-positive pools were subjected to individual cosmid DNA extraction and PCR amplification to identify positive single clones. In total, 15 PCR-positive pools were amplified, and from these, three individual clones were identified. The three amplicons were sequenced, and the sequences were compared to available database sequences using the Blastn tool. One of the amplicons showed homology with PKS sequences and was selected for subcloning and sequencing. The mining of the metagenomic library by PCR has proven to be an efficient method, allowing for the identification of a clone whose insert contains genes possibly involved in polyketide biosynthesis.
Clone B7P37, whose amplicon showed homology with PKS sequences, had its cosmid subcloned and fully sequenced. Sequencing of the selected cosmid was performed using the shotgun sequencing technique as previously described (Silakowski et al., 1999). Briefly, fragments measuring approximately 1.5 to 2 kb in length were separately cloned into pUC19DNA/SmaI (Fermentas) vectors. The plasmid DNA was obtained by the alkaline lysis method (Sambrook and Russell, 2001), and the clones of the sub-library were subjected to automated DNA sequencing. The raw electropherogram data were analyzed by PhredPhrap (Ewing and Green, 1998; Ewing et al., 1998) and Consed (Gordon et al., 1998) to generate sequences in FASTA format to evaluate quality and alignment. Phred values above 40 were considered high quality. The ORFs were determined using the ORF Finder software (NCBI). Gene annotation was performed from the ORFs amino acid sequences by searching for similarities to proteins present in different databases using the Blastp local alignment tool. A search for functional domains was also performed using the protein signatures database InterPro (Quevillon et al., 2005). For PKS catalytic domain annotation, the PKS/NRPS annotation tool was used (Bachmann and Ravel, 2009). Global nucleotide and amino acid sequence alignments were performed using the software Clustal X (Larkin et al., 2007).
The metagenomic DNA insert obtained was 29,697 bp long. The mean GC content was 64%. Twenty-one ORFs were identified (Figure 10). The potential roles of the genes contained in the ORFs were determined by comparison with similar sequences present in the databases and functional domain analysis (Tables 1 and 2). All possible genes found in the sequenced region displayed typical characteristics of bacteria, and when subjected to database homology searches, each of the translated ORFs showed greater similarity to bacterial proteins.
Figure 10.
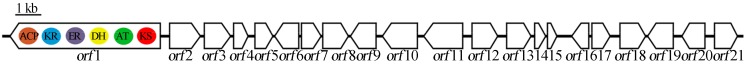
ORFs map of the sequenced metagenomic DNA insert for the clone B7B37. ORF1: PKSI biosynthesis gene cluster.
Table 1.
ORF1 PKS biosynthesis gene cluster.
| Protein (gene) | length (pb) | Proposed function (protein domains with their positions in the sequence of aa) |
|---|---|---|
| PKS (pks) | 6.498 | KS (32-458), AT (558-852), DH (912-1069), ER (1399-1706), KR (1729-1908), ACP (2004-2070) |
Table 2.
Probable role of the ORFs shown in Figure 10.
| Protein | length (aa) | Similar protein in the database | Bacteria of similar protein | Sim/Id(%) |
|---|---|---|---|---|
| ORF1 | 2148 | polyketide synthase | Chondromyces crocatus | 50/36 |
| ORF2 | 400 | efflux RND transporter protein | Xanthobacter autotrophicus | 66/48 |
| ORF3 | 357 | efflux ABC transporter permease | Brevundimonas sp. | 68/50 |
| ORF4 | 177 | ABC transporter related | Xanthobacter autotrophicus | 76/58 |
| ORF5 | 231 | oxidoreductase | Gemmata obscuriglobus | 51/35 |
| ORF6 | 330 | NAD-dependent epimerase/dehydratase | Reinekea sp. | 61/44 |
| ORF7 | 233 | peptide aspartate b-dioxygenase | Legionella pneumophila | 59/40 |
| ORF8 | 369 | alkane 1-monooxygenase | Acidiphilium cryptum | 53/37 |
| ORF9 | 336 | delta 9 acyl-lipid fatty acid desaturase | Rhodopirellula baltica | 65/49 |
| ORF10 | 494 | glycerol kinase | Mesorhizobium loti | 79/68 |
| ORF11 | 507 | FAD dependent oxidoreductase | Methylobacterium sp. | 68/58 |
| ORF12 | 354 | hypothetical protein | Marinobacter algicola | 58/37 |
| ORF13 | 327 | hypothetical protein | Ruegeria pomeroyi | 62/43 |
| ORF14 | 135 | hypothetical protein | Symbiobacterium thermophilum | 42/28 |
| ORF15 | 147 | hypothetical protein | Chromohalobacter salexigens | 50/39 |
| ORF16 | 200 | hypothetical protein | Sulfitobacter sp. | 64/44 |
| ORF17 | 242 | ABC-type transporter | Polaromonas sp. | 52/33 |
| ORF18 | 326 | ABC-type transporter | Methylobacterium nodulans | 57/42 |
| ORF19 | 378 | monooxygenase | Agrobacterium radiobacter | 56/41 |
| ORF20 | 306 | aldo/keto reductase | Thiomonas intermedia | 71/56 |
| ORF21 | 314 | hypothetical protein | Nitrobacter hamburgensis | 69/49 |
(aa: amino acid, sim: similarity, id: Identities).
ORF 1 measured 6,948 bp, corresponded to a PKS module, and presented homology with myxobacteria type I PKS (36% identity and a 50% Blastp similarity with Chondromyces crocatus CndA protein, its closest neighbor). The determination of catalytic domains present in the module was performed using the PKS/NRPS annotation tool (Bachmann and Ravel, 2009), and the enzymatic domains module organization was determined as follows: KS-AT-DH-ER-KR-ACP (Table 1).
The sequenced gene cluster contains other genes that may be involved in the biosynthesis of a possible polyketide. ORFs 2, 3, 4, 17 and 18 correspond to membrane transporters associated with the ABC transporter superfamily. These transporters perform active transport through the membranes of different types of molecules such as sugars, amino acids, oligopeptides, ions, and drugs. Numerous polyketide-producing microorganisms have at least one ABC transporter gene within the antibiotic biosynthesis gene cluster, which often confers drug resistance (Méndez and Salas, 1998).
The ORF 5 sequence showed homology to members of the reductases/short-chain dehydrogenase family that use NAD or NAD(P) as a cofactor. The ORF 6 sequence showed homology to members of the NAD-dependent epimerases/dehydratases family. ORF 7 appeared to be a member of the aspartyl/asparaginyl beta-hydroxylase family of proteins that catalyze oxidative reactions in a variety of metabolic processes. ORFs 8 and 9 showed homology with the fatty acid desaturase family that catalyzes the insertion of a double bond in the delta position of fatty acids. ORF 10 belongs to the carbohydrate kinase family. ORF 11 belongs to a family that includes several FAD-dependent oxidoreductases. ORF 12 belongs to the periplasmic binding protein-like II family. ORF 13 showed homology with the Tat (twin-arginine translocation) pathway signal sequence protein family. ORFs 14, 15 and 16 were classified as putative proteins with unknown functions. ORF 19 belongs to the monooxygenases family, which incorporates a hydroxyl group on substrates is found in several metabolic pathways. ORF 20 was classified as belonging to the aldo-keto reductase family, and ORF 21 was classified as belonging to the quinoprotein amine dehydrogenase family.
ORF1 Domain Analysis
Phylogenetic analyzes were conducted to locate the KS and AT domains of the metagenomic clone within a phylogenetic tree constructed with domains from 19 PKS gene clusters that are already characterized and available in the PKSDB database (Yadav et al., 2003). These 19 PKS gene clusters contained 182 KS domains and 188 AT domains. Through phylogenetic analysis of the KS domain, it was observed that the metagenomic domain sequence was grouped outside the large monophyletic group that includes the actinomycetales, suggesting that the clone does not belong to this order (Figure 11). However, this hypothesis cannot be completely supported considering that functional limitations may have led some genes to evolve differently from other genes that encode typical KS domains (Ginolhac et al., 2004). For example, KS domains that catalyze the entry of an amino acid chain (hybrid group) and extender modules (KSQ) are grouped separately. In the obtained phylogenetic tree, the metagenomic KS domain appears as a sibling group of domains that correspond to extender modules. The SEARCHPKS tool (Yadav et al., 2003) also classified the metagenomic KS as an extender module.
Figure 11.

Phylogenetic analysis of the KS domain. The reconstruction was computed for 182 protein sequences using the distance method (i.e., NJ and JTT matrix) with 500 resamplings. The group named hybrid PKS/NRPS includes the KS domains that are preceded by NRPS (non-ribosomal protein synthesis). KSQ and KS extender module domains with an ACP-KS-AT-AT-ACP organization were grouped and named the KSQ/2AT group. The KS domains that were tightly clustered were named after the produced polyketide. The ORF 1 sequence obtained from B7P37 clone from the metagenomic library is shown in bold.
The sequence of the metagenomic KS active site was analyzed in relation to the functionality of the domain, and the presence of cysteine in the active site and the conserved sequence DTACSSSLVA, which is present in the KS functional domain, was observed (Figure 12). In KS extender modules, there is a substitution of a cysteine in the active site for a glutamine (KSQ) in Streptomyces systems and a serine (KSS) or tyrosine (KSY) in myxobacteria systems (Moffitt and Neilan, 2003; Katz, 2009). Therefore, it can be stated that the metagenomic KS is functional; however, it is not possible to predict whether it corresponds to an extender module.
Figure 12.

Alignment of the KS and KR domains of ORF 1 with different homologous KS and KR domains. Protein sequences were aligned using the ClustalX software. Only the region containing the active site of the enzyme is shown. The number corresponds software the amino acid position of the first residue listed in the first motif. The amino acids in bold are conserved residues or important motifs for the function of each domain. The active-site residues in the KS sequence are marked with an asterisk. The important motifs for the functioning of the KR residue are underlined.
Substrate recognition, which is defined by the AT domain of the PKS module, is the most important factor that determines the structure and diversity of a polyketide (Ginolhac, 2004). In the AT domains phylogenetic tree, specific domains for malonyl-CoA and methylmalonyl-CoA were grouped into two distinct clusters (Figure 13). The metagenomic AT domain was grouped outside of any malonyl-CoA or methylmalonyl-CoA clusters. For this reason, it was not possible to make any prediction of substrate incorporation based on the phylogenetic analysis.
Figure 13.

Phylogenetic analysis of the AT domain. The AT domains located outside the two major groups for malonyl-CoA and methylmalonyl-CoA remain unresolved by phylogenetic analysis. The sequence obtained from the metagenomic library is shown in bold.
Among the first 25 results obtained for substrate prediction using the SEARCHPKS tool (Yadav et al., 2003), 19 coincided with methylmalonyl-CoA, which was ranked in the first position, 2 coincided with ethylmalonyl-CoA, 2 coincided with malonyl-CoA, 2 coincided with glyceryl 2-CoA and 1 coincided with methylmalonyl-CoA/propionyl-CoA. Moreover, the YADAV prediction for the metagenomic AT domain provided similar values for the different substrates (between 6×10−54 and 3×10−46).
PKS modules may contain one or more inactive domains. These inactive domains can be generally identified by analysis of conserved regions corresponding to the active site. The consensus region of the active site of a KR is GxGxxGxxxA (Bachmann and Ravel, 2009). The corresponding region of the metagenomic KR domain is GxSxxGxxxA, indicating that this KR is most likely non-functional (Figure 12). An inactive KR domain leads to superfluous DH and KR domains.
In many cases, the colinearity between the genetic organization and sequence of enzymatic transformations involving type I PKSs is so strong that various structural features of a product can be predicted with confidence from gene sequence analysis. In the case of metagenomic PKS, prediction was not possible, and this can be explained by the fact that the metagenomic approach allows for the study of gene sequences with completely unknown functions. To definitively prove the involvement of the discovered genes in polyketide biosynthesis, studies on the expression and inactivation of these genes are needed. This work demonstrates that metagenomic DNA represents a promising source of new genes that can be used in combinatorial biosynthesis experiments aimed at the identification of new molecules.
Mining for PKS II using metagenomic libraries
Another study conducted by the LBMP for the mining and characterization of new PKS pathways involved the search for conserved KS sequences of aromatic polyketides by PCR amplification as a detection strategy for metagenomic clone candidates of host polyketides pathways; this was followed by sequencing and bioinformatic analysis of the inserts found in PCR-positive clones (Gomes, 2008, 2011).
The search for new PKS pathways exploits the highly diffusible characteristic of these metabolites among the most diversified groups of microorganisms, persisting (thanks to molecular engineering intervention) after more than half a century of “harassment” from intense scientific research as the most promising source of new pharmacological chemical structures. A decisive factor for the spread of polyketide pathways is the fact that these small molecules will often confer adaptive advantages to their host and can be propagated by plasmids. The mechanism that maintains polyketide evolution is based on the high maintenance cost of these pathways in the organisms that possess them. The moment that these molecules do not confer adaptive advantage, their evolutionary lifetime will be shortened (Fischbach et al., 2008). This negative selection (i.e., increased metabolic expenditure) is coupled with the positive selection conferred by successful pathways, and creates an effective screening process for innovative bioactive molecules, greatly expanding the range of possibilities for the PKS research field.
In addition to this strong natural selection, the use of combinatorial biosynthesis can increase the ability to produce new polyketides by many orders of magnitude. This approach takes advantage of the fact that although each biochemical pathway typically results in a specific type of molecules, there will be enzymes common to all pathways within the same PKS family that are derived from homologous genes and conserved regions, even though they not are identical to each other.
This conservation of properties among homologous enzymes allows for a certain compatibility among enzymatic groups from different clusters. As with aromatic polyketide antibiotics, in which production of the final molecule is the result of the interaction of various enzymes belonging to a cluster, combinatorial biosynthesis allows for the assembly of molecules through enzymatic recombination between known producing organisms and novel genes obtained through metagenomic mining, which may be understood as a strategy of accelerating the evolutionary processes, resulting in new bioactive molecules.
To understand how promising this approach is, one must consider the immense number of complex peptides, polyketides, and other known chemical scaffolds that are widely diversified by the presence and arrangement of structures by processes including glycosylation, methylation, halogenation, acyl group attachment, and hydroxylation, which have evolved over billions of years in actinobacteria and fungi (Baltz, 2006).
The study of the most PKS II conserved unit, the “minimal pks”, has been seen as a promising for the production of new molecules by enzymatic combination (Shen et al., 1999). The peculiarities of the enzymes from different clusters from the specific selection of precursor units to compatibility with other enzymes involved in processes, including cyclases, ketoreductases, and aromatases, is decisive for the rational development of new molecules.
Shen and colleagues (Shen et al., 1999) described an interesting study in which recombinant organisms were produced from the introduction of a minimal pks derived from spore pigment PKS II. This resulted in a variety of new carbon skeletons that were superior to those found in experiments that used a minimal pks from a natural antibiotics producer. With regard to the production of antibiotics by molecular engineering and the discovery of new PKS II pathways through metagenomics, the challenge is to identify the most promising pathway to obtain an interesting compound and making the right choice of which enzymes will be recombined, predicting their capacity for joint activity.
In this context, the methodology used in this study explored the homology concept to infer the catalytic functions of the putative enzymes through in silico analysis. Together, the strategies used allowed the recovery of an unknown pathway, which is described in detail through the identification of conserved enzymatic domains and the cross-referencing of information with previously published data. Figure 14 is a comparison between the biosynthetic grouping obtained by metagenomics and some PKS IIs involved in pigments or antibiotics synthesis. There is a greater similarity within each compared cluster in the composition and arrangement of these genes and, on a smaller scale, between the genes of the two groups.
Figure 14.

Comparative analysis of the B5-37 metagenomic contig and several PKS II clusters. A: PKSs pigments. 1: act spore pigment cluster (actinorhodin pigment) from Streptomyces coelicolor; 2: whiE spore pigment cluster from S. coelicolor; 3: sch spore pigment cluster from S. halstedii; 4: cur spore pigment cluster from S. curacoi; 5: tcm tetracenomycin pigment cluster from S. glaucescens (Yu et al., 1998); 6: melanin cluster from S. avermitilis (Omura et al., 2001) B: Gene grouping obtained for the metagenomic contig. 7: B5-37 metagenomic cluster (this work). C: Polyketides antibiotics gene grouping. 8: antibiotic cluster 1 from S. avermitilis; 9. antibiotic cluster 2 from S. avermitilis (Omura et al., 2001); 10: actinorhodin antibiotic cluster (Baerson and Rimando, 2007); 11: antrachinona antibiotic cluster from Photorhabdus luminescens (Brachmann et al., 2007); 12: antibiotic A-74528 cluster (Zaleta-Rivera et al., 2010). Note: Images are not scaled to a size that is relative to the original clusters.
This characteristic is quite common for PKSs with a certain degree of compatibility between enzymes from different pathways, making them excellent raw material for molecular engineering and combinatorial biosynthesis (Hopwood, 1997).
As observed, the grouping has an enzymatic arrangement that is similar to the physical arrangement of clusters related to spore pigments. However, there is a greater complexity of this gene cluster, particularly for ORFs that are related to modification enzymes that act after synthesis and modeling; this seems to be more representative of enzymes involved in antibiotic synthesis. However, similar to the data obtained for the KSα phylogeny, the comparative results do not allow for the discrimination of the cluster as antibiotic or pigment. Moreover, the presence of homology to carbohydrate degradation genes may indicate a cluster involved in lignin, cellulose, and chitin degradation. There have been reports of PKS II-derived clusters that have undergone differentiation and became active in this process (Hsiao and Kirby, 2008).
Figure 15 shows a map of the putative genes found (i.e., ORFs 1–22/Blastp homology) that were confirmed by the presence of related enzymatic domains (domains A-U/PRODOM database).
Figure 15.

Distribution of ORFs (1–22) and Domains (A–U) along the studied contig.
Phenetic Analysis of Alpha-Ketoacyl Synthases (KSAs)
The KSA enzymatic subunit is related to the choice of elongation precursor for the biosynthesis of PKS II antibiotics such that differences in its sequence may reflect subclasses of different molecules (Metsä-Ketelä et al., 1999). Therefore, a phenetic analysis was performed between the likely metagenomic KSA (ORF 15) and other sequences available in the database; among these, the KSAs of PKS II pigments and antibiotics were selected. The use of phenetics allows for the detection of similarities among the metagenomic KSAs and other diversified pathways by comparing conserved KSA gene regions (Figure 16).
Figure 16.

Phenetic comparison of KSA genes for various PKS clusters available in the literature and the metagenomic ORF. A: PKS II antibiotics; B: PKS II undefined; C: PKS II spore pigments. The tree was generated using the Phylip bioinformatic tool package (Neighbor Joining, JTT matrix, Bootstrapping 1000) and was generated for a nucleotide sequence alignment translated into amino acids.
It may be noted that there was a split of the sequences into two main groups except for that of the KSA from the fatty acid synthase (FAS) used in the outgroup and the KSA from Streptomyces resistomycificus. The first group (Figure 16A) contained KSAs from known aromatic polyketide antibiotic producers. The second group was divided into two branches: one containing KSAs related to spore pigment production (Figure 16C), and the other comprised three sequences (Figure 16B): the metagenomic KSA (metagenomic putative KS-alpha) and KSAs belonging to two strains of Streptomyces including S. hygroscopicus ATCC 53653 (ZP_05513869.1) and S. bingchenggensis BCW-1 (ADI05885.1), whose action forms were still not described. These analyzes suggest the possibility that the metagenomic cluster is involved in the synthesis of a new metabolite that is different from the known classes with a pathway involved in the synthesis of a new pigment. This result shows how metagenomics provides access to unknown gene diversity, providing a wide range of possible sources of enzymes for combinatorial biosynthesis of new pharmacological molecules.
Prediction of Protein Structure
There are few data concerning the three-dimensional structure and organization of PKS II enzymatic complexes, which undoubtedly limits aromatic polyketide rational design strategies. However, 3-D structural data of some enzymes involved in the pathway provide the use of strategies that aim to clarify how interprotein interactions occur (Castaldo et al., 2008). An important milestone in the early studies of pathway protein interactions was reached by the X-ray crystallization of KS protein and its heterodimeric form (KSA/CLF) from Streptomyces coelicolor (Keatinge-Clay et al., 2003), allowing spatial knowledge of the cavity formed by the subunits responsible for the catalysis of the decarboxylative condensation reactions that characterize the polyketides bonds. Together, KSA, CLF, and ACP form an enzymatic complex called minimal pks, which dictate the nature and chain length of the substrate. The noncovalent attraction between these proteins is responsible for the acquisition of its functional form and the architecture of the active site. Besides these proteins, the enzymatic complex often depends on interaction with other enzymes to become active. The cyclases, for example, seem to decisively act on the stability of the enzymatic complex, preventing the elimination of short polyketide chains in the active site to ensure a better performance of the pathway (Castaldo et al., 2008).
The use of structural modeling by homology allows for functional inferences that simulate the folding of proteins related to the genes found in addition to the visual comparison of metagenomic 3-D models obtained with 3-D structures that were previously determined by X-ray crystallography or nuclear magnetic resonance (NMR). For this reason, this strategy was used to characterize the ORFs studied.
For structural prediction, 3-D models were generated using the Swiss-model software (http://swissmodel.expasy.org, accessed in December 12 of 2010), and they were applied as model structures that were resolved by classical methods using model identity values higher than 30% (recommended for obtaining accurate models; Da Silveira et al., 2005). The models were checked for their predictive ability and correspondence with functional biological structures by evaluating spatial distribution parameters using the Prosa-web (https://prosa.services.came.sbg.ac.at/prosa.php, accessed in December 20 of 2010) Qmean, Verify3D and Procheck (http://swissmodel.expasy.org/workspace/index.php?func=tools_structureassessment1, accessed in December 20 of 2010) software packages.
A high-accuracy structural model (both in experimental and in silico modeling) should have approximately 90% of its amino acids located in the most favorable region of the Ramachandran plot (PROCHECK). However, it is assumed that at 85% and above, it is possible to obtain models with a good confidence index (Da Silveira et al., 2005). Table 3 lists the results obtained for the ORF model and the corresponding molds used by PHYRE. Only ORF 17 and its model did not achieve a value greater than 85%, and it showed the least identity in relation to the molds (30%).
Table 3.
Mold/model stereochemical evaluation
| 3D/L | Id.a | Percent of residues in Ramachandran Plot (Procheck)b | Score 3D Profile* | |||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||
| Most favorable region | Allowed regions | Generously allowed regions | Disallowed regions | Total | Ideal | Sideal | ||
| 1TQYA (394) | 57 | 87.5 | 11.3 | 0.6 | 0.6 | 155.98 | 180.22 | 0.86 |
| ORF15 (416) | 57 | 85.6 | 13.6 | 0.3 | 0.6 | 183.07 | 190.36 | 0.96 |
| 1TQYH (402) | 47 | 87.2 | 11.5 | 0.9 | 0.3 | 192.39 | 183.90 | 1.04 |
| ORF16 (402) | 47 | 86.2 | 12.2 | 0.6 | 0.9 | 163.75 | 183.90 | 0.89 |
| 1NQ4A (95) | 33 | 74.1 | 21.0 | 2.5 | 2.5 | 34.32 | 42.96 | 0.79 |
| ORF17 (85) | 33 | 73.6 | 18.1 | 2.8 | 5.6 | 25.24 | 38.40 | 0.65 |
result in percentage for the most similar model (multiple alignments were used), Id.: Identity.
Ramachandran plot (PROCHECK) based on the torsion angles Φ and Ψ (C-alpha and N and C-alpha and C-peptide).
, Data calculated from the analysis using the software Verify3 (http://nihserver.mbi.ucla.edu/Verify_3D/). Total score: provided. Ideal score: Exp−0.83 + 1,008 × ln(L), and Sideal, which is equivalent to sequence compatibility with their 3-D structure and is obtained by dividing the total score and ideal score with ideal values above 0.45 Sideal. L.: number of amino acid residues (Da Silveira et al., 2005).
ORF 15 showed the highest identity (57%), allowing for a high resolution structure as also observed by the Z-Score (Figure 17), which is a parameter obtained from the Prosa Web software that depicts how much a predictive structure has quality parameters comparable to those obtained for structures resolved by classical methods (i.e., X-Ray Crystallography and Nuclear Magnetic Resonance), which returned an expected value for X-Ray Crystallography (Figure 17B, graph on the right: region in light blue). Although its Ramachandran index (PROCHECK) returned 0.6% amino acids in the non-allowed region, it can be observed in the updated Ramachandran plot (RAMPAGE; Lovell et al., 2003) that the two residues are glycine (Figure 17B, center); in a way, this region is not prohibitive because of its short side chain (only one H). This result also reflects the high conservation of these proteins.
Figure 17.

Functional prediction for the model and ORF 15. Center: Ramachandran (Rampage) based on the torsion angles C-alpha and C-beta. Right: Z-score (Prosa Web).
The predicted model could also be used in enzyme-substrate docking studies that are suitable for substrate prediction, and this is highly desirable because KSAs are involved in the choice of the elongation substrate (i.e., malonate, methyl-, and ethyl- malonate among other derived substrates) and this specificity sometimes allows for differentiating pathways of distinct antibiotic classes (Metsä-Ketelä et al., 1999). Figure 18 shows the results of the Qmean scoring for ORF 15 and the model.
Figure 18.

3-D structure quality evaluation through Qmean. A: Mold 1TQYA; B: ORF 15.
Analysis of the scores given by the Qmean software allows for the evaluation of how well a particular 3-D structural model assumes the configuration expected for a given combination of specific amino acid residues, taking into account the interactions between them and the different chemical environments. This analysis is employed to evaluate both 3-D models obtained by classical methods and in silico-predicted models. The Qmean color scale indicates how close a model is to the ideal with the regions shown in blue indicating high-quality prediction (deviations are lower than 1 Å), and the red regions indicate lower predictive power (deviations above 3.5 Å). ORF 15 exhibited a structural pattern with a quality that resembles that obtained for the model structure, with most of the residues occupying an ideal conformation. As observed in Figure 18, the graphs to the right of each model, the overall pattern of scores evaluated by Qmean, were within normal range for both structures.
Figure 19 shows a comparative analysis obtained through the Procheck software for the secondary structure of ORF 15 and the mold 1TQYA.
Figure 19.

2-D structure comparison between ORF 15 and the mold 1TQYA.
As observed in Figure 19, ORF 15 differs from the model structure. ORF 15 is larger than the model and has two more beta sheet structures than the model (peptide region: 397–418). Moreover, ORF 15 differs in the length of the beta sheet structures in the peptide regions 260–270 and 304–307, where it is possible to observe larger beta sheets in the ORF 15 structure. These differences in the secondary structure will affect the overall protein structure, which can directly affect its functional characteristics by interfering with the redox potential and substrate affinity.
The KSA and CLF structures are subunits of the same enzyme. KS, which forms a KSA/CLF heterodimer in its quaternary structure, can appear as a single copy or, as found in the 1TQY model (Streptomyces coelicolor), as an octahedral structure that is formed by four identical monomers with each one constituted by a KSA/CLF dimer.
All of the structural prediction results obtained indicated good quality models that can be used in enzymatic contact analysis to predict the KS quaternary structure through protein-protein docking simulation for ORFs 15 and 16. Other CLFs obtained in the literature by experimental methods were also analyzed to verify the compatibility of the metagenomic KSA model. Analysis of protein-protein docking was performed using the GRAMM-X software (Tovchigrechko and Vakser, 2006).
For this purpose, the three models with the highest identity obtained by ORF 16 submission (metagenomic CLF) in PHYRE were used: the 1TQYH model (47% identity), which is related to the KSA of the actinorhodin polyketide putative beta-ketoacyl; the 1j3nA model (34% identity), which is related to the 3-oxoacyl-(acyl-carrier protein) 2 synthase II from Thermus thermophilus hb8 crystal structure that is associated with FAS; and the 1e5 mA model, which is related to a single chain CLF from Synechocystis sp. (Motche et al., 2001). Figure 20 shows the obtained data.
Figure 20.

Compatibility analysis between the metagenomic KSA against the metagenomic CLF (A), CLF 1tqyH (B), CLF 1j3nA (C), and CLF 1e5mA (D) that was obtained by protein-protein docking simulation (GRAMM-X). Continuous arrows: short and antiparallel beta sheets, dotted arrow: the absence of these structures.
From Figure 20, it is evident that the B and D simulations gave a prediction of contact between proteins that best reflected the situation found for the metagenomic model (A). The CLF used for model B (1tqyH) was found with its origin connected to KSA 1tqyA, forming a dimer that combines with another three identical dimers, making an octahedral structure. The CLF used in model D represents a KS subunit that binds to a KSA, also forming a dimer, though in a single copy. One of the differences between the two CLFs can be observed by the detail indicated with the arrows, showing the absence of antiparallel beta sheet structures in model D, which is indicated for the metagenomic CLF (A) and the CLF 1e5 mA (D).
Simulation C returned a model that was completely different from the metagenomic model arrangements. This situation was expected because the CLF 1J3nA actually belongs to FAS and not KSA, indicating homology by paralogs. This result allows for the visualization of a situation in which there would be no compatibility among the candidate enzymes to a combinatorial biosynthesis assay, representing a rapid diagnosis of the chances of succeeding in enzymatic recombination because there are expectations of using the KS FAS condensation enzyme for the production of new drugs, exploring the relationship between PKS and FAS pathways (Motche et al., 2001).
Metagenomics is undoubtedly a powerful tool for the discovery of new enzymatic pathways, and this work has enabled the advancement of several more steps in the predictive analysis of protein function by homology, thus shedding light on the three-dimensional folding of these molecules. This study allowed for the fast characterization of metagenomic putative enzymes completely in silico, which confirmed the homology between the studied ORFs, detecting the diversity of the 2-D and 3-D structural patterns, and enabling the use of docking to restore KS enzymatic subunits to allow the simulation of KS quaternary structure in a first step towards the study of possible ligands and substrates. It was also possible to simulate recombinant situations among the metagenomic KSA subunits and CLFs in the literature, observing variations among the different recombinant situations.
The results obtained will support future studies of heterologous gene expression and will be useful for determining the most promising experimental design to obtain enzymatic expression products for characterization of pathway enzymology kinetics. The generated 3-D models will be used in enzymatic interaction predictive studies, particularly in protein-protein and protein-ligand docking strategies, to determine enzymatic substrates and to study the compatibility of catalytic subunits with enzymes from other pathways. Once the in silico strategies are validated through in vitro and in vivo experiments (heterologous host), the standardized methodology will be used to generate an internal LBMP database for metagenomic PKS enzymes to facilitate the recognition of promising enzymatic units for the rational design of new drugs. Other pathway enzymes have potential biotechnological application in other areas (e.g., cellulases, laccases, peptidases, and oxidases) and will be studied in parallel employing the approaches described.
Conclusion
Although no longer seen as an “innovation” as a biotechnological tool, the mature metagenomics has presented the fruits of hard work in the search of new gene clusters for the production of antibiotics and other metabolites of many industrial and environmental interests. At the vanguard of new technologies, such as research on “Single Cell” (Prosdocimi et al., 2001; Piel, 2011) and the genetic therapy, its potential is still at the threshold to be fully accessible, especially with regard to the largest extent created by new sequencing techniques, and how to support studies in transcriptome and metabolome.
Regardless, the problematic of the growing demand for new antibiotics face to which new drugs quickly become obsolete, is certainly a topic that can not be ignored, and as such will capture efforts worldwide in all its possibilities, making use of classical and modern pathways, while stimulates the emergence of new tools.
Also, if on the one hand, according Baltz: Molecular Engineering found the way to “Essentially attempts to speed up the process of evolution by many orders of magnitude to compete with natural evolution of new antibiotics” (Baltz, 2006) the evidence of counterpart dates at least 30,000 years ago (D’Costa et al., 2011), considering the existence and evolution of multidrug resistance mechanisms vs. mere 80 years of use of antibiotics on human and animal therapy.
Thus, the dilemma of the roles of hero/villain for the antibiotics takes form while one should bear in mind that we are intensifying the selective pressure on this powerful way to use the plastic raw matter, found in nature, paradoxically acting as irrational use of antimicrobials protagonists’. More than new drugs, we strongly need a new attitude facing the public and environment health.
Acknowledgments
The authors would like to thank Dr. Manoel Victor F. Lemos for comments on this manuscript, and for Dr. Gabriel Padilla, Dr. Luciano Takeshi Kishi and Dr. Silvana Giuliatti for help in to verify bioinformatics analysis. This work was partly supported by grants from FAPESP, CAPES and CNPq. We also would like to thank the Graduate Program on “Microbiologia Agropecuária” from UNESP-FCAV for the support for this research.
List of Abbreviations
- 3-K-ACP-REDUC
3-Ketoacyl ACP-Reductase;
- 3-K-ACP-SYN
3-Keto-acyl ACP- synthase;
- ABC1
cellobiose ABC transporter: solute bind protein;
- ABC2
cellobiose ABC transporter: “transport system inner membrane protein”;
- ABC3
cellobiose transporter permease;
- ACD
Acyl-CoA Deydrogenase;
- ACIL-TRANS
Acyl-transferase (PKSII);
- ACP
Acyl Carrier Protein;
- AT
acyl transferase (PKSI);
- CHITINASE
Hydrolase/chitinase;
- CLF
Chain lenght factor;
- CLP
ATP-dependent Protease Homolog;
- CYC 2/ARO
Polyketide Cyclase I/Aromatase;
- CYC 3
Polyketide Cyclase II;
- CYC1
Polyketide Cyclase I;
- D-HTH
HTH Domain proteina/plu4195;
- DH
dehydratase;
- ER
enoyl reductase;
- GLI-HIDROL
Hydrolase Beta-Glucosidase;
- GLI-TRANS
Glycosyl-transferase;
- HIPOT
Hypothetic/Secreted protein;
- HYDR
Hydroxylase;
- KR
Ketoreductase;
- KS
ketosynthases;
- KSA
β-ketoacyl synthase α;
- MONOX
Monooxygenase;
- MT-TRANS
Methyltransferase;
- MULT-COPP
Multicopper oxidase;
- O-METH
O-Methyltransferase;
- OMT-M
Metagenomic O-Methyltransferase;
- OXD
Oxidoreductase;
- P-450
Cytochrome P450;
- PEP-TRANSP
Peptide Transporter;
- PEPTIDASE
Peptidase/Sortase;
- REG
transcriptional regulator;
- SAN-1
Acyl-coA decarboxylase;
- SAN-2
Acyl-ACP Thioesterase;
- SAN-3
Quinone-forming monooxygenase;
- TRANSP/REG
Generic citation for genes involved in transporter or regulation;
- UNKNOWN
Protein unknown or not cited in the original reference.
References
- Amann RI, Ludwig W, Schleifer KH. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Mol Biol Rev. 1995;59:143–169. doi: 10.1128/mr.59.1.143-169.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann BO, Ravel J. Methods for in silico prediction of microbial polyketide and nonribosomal peptide biosynthetic pathways from DNA sequence data. Methods Enzymol. 2009;458:181–217. doi: 10.1016/S0076-6879(09)04808-3. [DOI] [PubMed] [Google Scholar]
- Baerson SR, Rimando AM. A Plethora of Polyketides: Structures, Biological Activities, and Enzymes. In: Rimando AM, Baerson SR, editors. Polyketides: Biosynthesis, Biological Activity, and Genetic Engineering. American Chemical Society; Washington, USA: 2007. pp. 2–14. [Google Scholar]
- Baltz H. Molecular engineering approaches to peptide, polyketide and other antibiotics. Nat Biotech. 2006;24:1533–1540. doi: 10.1038/nbt1265. [DOI] [PubMed] [Google Scholar]
- Banikand JJ, Brady SF. Recent application of metagenomic approaches toward the Discovery of antimicrobials and other bioactive small molecules. Current Opinion in Microbiology. 2010;13:603–609. doi: 10.1016/j.mib.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley SD, Chater KF, Cerdeno-Tarraga A-M, Challis GL, Thomson NR, James KD, Harris DE, Quail MA, Kieser H, Harper D, Bateman A, Brown S, Chandra G, Chen CW, Collins M, Cronin A, Fraser A, Goble A, Hidalgo J, Hornsby T, Howarth S, Huang C-H, Kieser T, Larke L, Murphy L, Oliver K, O’Neil S, Rabbinowitsch E, Rajandream M-A, Rutherford K, Rutter S, Seeger K, Saunders D, Sharp S, Squares R, Squares S, Taylor K, Warren T, Wietzorrek A, Woodward J, Barrell BG, Parkhill J, Hopwood DA. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3 (2) Nature. 2002;417:141–147. doi: 10.1038/417141a. [DOI] [PubMed] [Google Scholar]
- Bibb MJ, Ward JM, Hopwood DA. Transformation of plasmid DNA into Streptomyces at high frequence. Nature. 1978;274:398–400. doi: 10.1038/274398a0. [DOI] [PubMed] [Google Scholar]
- Brachmann AO, Joyce SA, Jenke-Kodama H, Schwär G, Clarke DJ, Bode HB. A Type II Polyketide Synthase is Responsible for Anthraquinone Biosynthesis in Photorhabdus luminescens. Chem Bio Chem. 2007;8:1721–1728. doi: 10.1002/cbic.200700300. [DOI] [PubMed] [Google Scholar]
- Brady SF, Chao CJ, Handelsman J, Clard J. Cloning and Heterologous Expression of a Natural Product Biosynthetic Gene Cluster from eDNA. Org Lett. 2001;3:1981–1984. doi: 10.1021/ol015949k. [DOI] [PubMed] [Google Scholar]
- Bull AT, Ward AC, Goodfellow M. Search and discovery strategies for biotechnology: the paradigm shift. Microbiol Mol Biol Rev. 2000;64:573–606. doi: 10.1128/mmbr.64.3.573-606.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castaldo G, Zucko J, Heidelberger S, Vujaklija D, Hranueli D, Cullum J, Wattana-Amorn P, Crump MP, Crosby J, Long PF. Proposed Arrangement of Proteins Forming a Bacterial Type II Polyketide Synthase. Chem Biol. 2008;15:1156–1165. doi: 10.1016/j.chembiol.2008.09.010. [DOI] [PubMed] [Google Scholar]
- Charter KF. Genetics of differentiation in Streptomyces. Annu Rev Microbiol. 1993;47:685–713. doi: 10.1146/annurev.mi.47.100193.003345. [DOI] [PubMed] [Google Scholar]
- Charusanti P, Fong NL, Nagarajan H, Pereira AR, Li HJ, Abate EA, Su Y, Gerwick WH, Palsson BO. Exploiting Adaptive Laboratory Evolution of Streptomyces clavuligerus for Antibiotic Discovery and Overproduction. PLoS ONE. 2012;7:e33727, 1–12. doi: 10.1371/journal.pone.0033727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Committee On Metagenomics, Board of Life Sciences. The New Science of Metagenomics Revealing the Secrets of Our Microbial Planet. The National Academies Press; Washington, DC: 2007. [PubMed] [Google Scholar]
- Courtois S, Cappellano CM, Ball M, Francou F-X, Normand P, Helynck G, Martinez A, Kolvek SJ, Hopke J, Osburne MS, August PR, Nalin R, Guérineau M, Jeannin P, Simonet P, Pernodet J-L. Recombinant environmental libraries provide access to microbial diversity for drug discovery from natural products. Appl Environ Microbiol. 2003;6:49–55. doi: 10.1128/AEM.69.1.49-55.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan D, Meyer Q, Stafford W, Muyanga S, Cameron R, Wittwer P. Metagenomic gene discovery: past, present and future. Trends Biotechnol. 2005;23:321–329. doi: 10.1016/j.tibtech.2005.04.001. [DOI] [PubMed] [Google Scholar]
- D’Costa VM, King CE, Kalan L, Morar M, Sung WWL, Schwarz C, Froese D, Zazula G, Calmels F, Debruyne R, Golding GB, Poinar HN, Wright GD. Antibiotic resistance is ancient. Nature. 2011;477:457–461. doi: 10.1038/nature10388. [DOI] [PubMed] [Google Scholar]
- Da Silveira NJF, Uchôa HB, Pereira JH, Canduri F, Basso LA, Palma MS, Santos DS, Azevedo WF., jr Molecular models of protein targets from Mycobacterium tuberculosis. J Mol Model. 2005;11:160–166. doi: 10.1007/s00894-005-0240-2. [DOI] [PubMed] [Google Scholar]
- Demain AL, Sanchez S. Microbial drug discovery: 80 years of progress. J Antibiot(Tokyo) 2009;62:5–16. doi: 10.1038/ja.2008.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Embrapa. O programa Blast: Guia prático de utilização. Embrapa Recursos Genéticos e Biotecnologia; Brasília, DF, Brazil: 2007. [Google Scholar]
- Ewing B, Green P. Base-calling of automated sequencer traces using Phred II Error probabilities. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- Ewing B, Hilier LD, Wendl MC, Green P. Base-calling of automated sequencer traces using Phred I Accuracy assessment. Genome Res. 1998;8:175–185. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- Feng Z, Kallifidas D, Brady SF. Functional analysis of environmental DNA-derived type II polyketide synthases reveals structurally diverse secondary metabolites. Proc Natl Acad Sci USA. 2011;108:12629–12634. doi: 10.1073/pnas.1103921108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Kim JH, Brady SF. Fluostatins produced by the heterologous expression of a TAR reassembled environmental DNA derived type II PKS gene cluster. J Am Chem Soc. 2010;132:11902–11903. doi: 10.1021/ja104550p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Moreno MA, Caballero JL, Hopwood DA, Malpartida F. The act cluster contains regulatory and antibiotic export genes, direct targets for translational control by the bldA tRNA gene of Streptomyces. Cell. 1991;66:769–780. doi: 10.1016/0092-8674(91)90120-n. [DOI] [PubMed] [Google Scholar]
- Fischbach MA, Walsh CT, Clardy J. The evolution of gene collectives: How natural selection drives chemical innovation. Proc Natl Acad Sci (USA) 2008;105:4601–4608. doi: 10.1073/pnas.0709132105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischbach MA, Walsh CT. Antibiotics for Emerging Pathogens. Science. 2009;325:1089–1093. doi: 10.1126/science.1176667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A, Enkler N, Neudert G, Bocola M, Sterner R, Merkl R. TransCent: computational enzyme design by transferring active sites and considering constraints relevant for catalysis. BMC Bioinformatics. 2009;10:54. doi: 10.1186/1471-2105-10-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Wenzel SC, Perlova O, Wang J, Gross F, Tang Z, Yin Y, Stewart AF, Muller R, Zhang Y. Efficient transfer of two large secondary metabolite pathway gene clusters into heterologous hosts by transposition. Nucleic Acids Res. 2008;36(e113):1–14. doi: 10.1093/nar/gkn499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginolhac A, Jarrin C, Gillet B, Robe P, Pujic P, Tuphile K, Bertrand H, Vogel TM, Perrière G, Simonet P, Nalin R. Phylogenetic analysis of polyketide synthase I domains from soil metagenomic libraries allows selection of promising clones. Appl Environ Microbiol. 2004;70:5522–5527. doi: 10.1128/AEM.70.9.5522-5527.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gladek A, Zabrzewsk AJ. Genome size of Streptomyces. FEMS Microbiol Lett. 1984;24:73–6. [Google Scholar]
- Gomes ES. Graduation thesis. Faculdade de Ciências Agrárias e Veterinárias, UNESP; 2008. Utilização da metagenômica como ferramenta para prospecção de novos genes do sistema PKS tipo II. Jaboticabal, Brazil; p. 76. [Google Scholar]
- Gomes ES. M.Sc Dissertation. Programa de pós-graduação em Microbiologia Agropecuárea, Universidade Estadual Paulista “Júlio de Mesquita Filho”. UNESP; 2011. Análise in silico de um novo “cluster” de PKS II Metagenômico Jaboticabal, Brazil; p. 118. [Google Scholar]
- Gomes ES, Navarrete AA, Lemos EGM, Tsai SM, Moreira FMS. A nova ciência da metagenômica: revelando os segredos do planeta microbiano. Boletim informativo da SBCS. 2010;34:20–22. [Google Scholar]
- Gordon D, Abajian C, Green P. Consed: A graphical tool for sequence finishing. Genome Res. 1998;8:195–202. doi: 10.1101/gr.8.3.195. [DOI] [PubMed] [Google Scholar]
- Gustafsson C, Govindarajan S, Minshull J. Codon bias and heterologous protein expression. Trends Biotechnol. 2004;22:346–353. doi: 10.1016/j.tibtech.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Hamad B. The antibiotics market. Nat Rev Drug Discov. 2010;9:675–676. doi: 10.1038/nrd3267. [DOI] [PubMed] [Google Scholar]
- Hopwood DA. Extrachromosomally determined antibiotic production. Annu Rev Microbiol. 1978;32:373–392. doi: 10.1146/annurev.mi.32.100178.002105. [DOI] [PubMed] [Google Scholar]
- Hopwood DA. Genetic Contribuitions to Understanding Polyketide Syntases. Chem Rev. 1997;97:2465–2497. doi: 10.1021/cr960034i. [DOI] [PubMed] [Google Scholar]
- Hopwood DA. Cracking the polyketide code. Plos Biol. 2004;2(E35):166–169. doi: 10.1371/journal.pbio.0020035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopwood DA, Charter KF, Bibb MJ. Genetics of antibiotic production in Streptomyces coelicolor A3(2), a model Streptomycete. Biotechnology. 1995;28:65–102. doi: 10.1016/b978-0-7506-9095-9.50009-5. [DOI] [PubMed] [Google Scholar]
- Horinouchi S, Beppu T. Autoregulatory factors and communication in actinomycetes. Annu Rev Microbiol. 1992;46:377–398. doi: 10.1146/annurev.mi.46.100192.002113. [DOI] [PubMed] [Google Scholar]
- Hsiao N, Kirby R. Comparative genomics of Streptomyces avermitilis, Streptomyces cattleya, Streptomyces maritimus and Kitasatospora aureofaciens using a Streptomyces coelicolor microarray system. Antonie van Leeuwenhoek. 2008;93:1–25. doi: 10.1007/s10482-007-9175-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson CR. Biosynthetic Studies of Daunorubicin and Tetracenomycin C. Chem Rev. 1997;97:2525–2535. doi: 10.1021/cr960022x. [DOI] [PubMed] [Google Scholar]
- Hutchinson CR, Fujii I. Polyketide synthase gene manipulation: a structure-function approach in engineering novel antibiotics. Annu Rev Microbiol. 1995;49:201–238. doi: 10.1146/annurev.mi.49.100195.001221. [DOI] [PubMed] [Google Scholar]
- Iqbal HA, Feng Z, BRADY SF. Biocatalysts and small molecule products from metagenomic studies. Curr Opin Chem Biol. 2012;16:109–116. doi: 10.1016/j.cbpa.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenke-Kodama H, Elke Dittmann E. Evolution of metabolic diversity: insights from microbial polyketide synthases. Phytochemistry. 2009;70:1858–1866. doi: 10.1016/j.phytochem.2009.05.021. [DOI] [PubMed] [Google Scholar]
- Kaitin K. Deconstructing the Drug Development Process: The New Face of Innovation. Clin Pharmacol Ther. 2010;87:356–361. doi: 10.1038/clpt.2009.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz L. The DEBS paradigm for type I modular polyketide synthases and beyond. Methods Enzymol. 2009;459:113–142. doi: 10.1016/S0076-6879(09)04606-0. [DOI] [PubMed] [Google Scholar]
- Keatinge-Clay AT, Shelat AA, Savage DF, Tsai SC, Miercke LJ, O’Connell JD, III, Khosla C, Stroud RM. Catalysis, specificity, and ACP docking site of Streptomyces coelicolor malonyl-CoA:ACP trans-acylase. Structure. 2003;11:147–154. doi: 10.1016/s0969-2126(03)00004-2. [DOI] [PubMed] [Google Scholar]
- Kim JH, Simmons TL, Brady SF. Comprehensive Natural Products II, Unlocking Environmental DNA Derived Gene Clusters Using a Metagenomics Approach. Chem Biol. 2010;2:455–474. [Google Scholar]
- Kinashi H. Giant linear plasmids in Streptomyces: a treasure trove of antibiotic biosynthetic clusters. J Antibiot (Tokyo) 2011;64:19–25. doi: 10.1038/ja.2010.146. [DOI] [PubMed] [Google Scholar]
- Kinashi H, Shimaji M, Sakai A. Giant linear plasmids in Streptomyces which code for antibiotic biosynthetic genes. Nature. 1987;328:454–56. doi: 10.1038/328454a0. [DOI] [PubMed] [Google Scholar]
- King RW, Bauer JD, Brady SF. An environmental DNA-derived type II polyketide biosynthetic pathway encodes the biosynthesis of the pentacyclic polyketide erdacin. Angew Chem. 2009;48:6257–6261. doi: 10.1002/anie.200901209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kneller R. The importance of new companies for drug discovery: origins of a decade of new drugs. Nat Rev Drug Discov. 2010;9:867–882. doi: 10.1038/nrd3251. [DOI] [PubMed] [Google Scholar]
- Knight V, Sanglier JJ, DiTullio D, Braccili S, Bonner P, Waters J, Hughes D, Zhang L. Diversifyng microbial natural products for drug discovery. Appl Microbiol Biotechnol. 2003;62:446–458. doi: 10.1007/s00253-003-1381-9. [DOI] [PubMed] [Google Scholar]
- Kvist T, Ahring BK, Lasken RS, Westermann P. Specific single-cell isolation and genomic amplification of uncultured microorganisms. Appl Microbiol Biotechnol. 2007;74:926–935. doi: 10.1007/s00253-006-0725-7. [DOI] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 20. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Lechevalier HA, Lechevalier MP. Introduction to the order Actinomycetales. In: Starr MP, Stalp H, Tryper HG, Balawi A, Schlegel HG, editors. Prokaryotes A Handbook on Habitats, Isolation and Identification of Bacteria. Springer; New York: 1981. pp. 1915–1922. [Google Scholar]
- Liles MR, Williamson LL, Rodbumrer J, Torsvik V, Goodman RM, Handelsman J. Recovery, purification, and cloning of high molecular-weight DNA from soil microorganisms. Appl Environ Microbiol. 2008;74:3302–3305. doi: 10.1128/AEM.02630-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell SC, Davis IW, Arendall WB, III, Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure Validation by Cα Geometry: ψ and Cβ Deviation. Proteins Struct Funct Genet. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- Malpartida F, Hopwood DA. Molecular cloning of the whole biosynthetic pathway of a Streptomyces antibiotic and its expression in a heterologous host. Nature. 1984;309:462–464. doi: 10.1038/309462a0. [DOI] [PubMed] [Google Scholar]
- Martin JF, Demain AL. Control of antibiotic biosyntesis. Microbiol Rev. 1980;44:230–251. doi: 10.1128/mr.44.2.230-251.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massini KC. Dr.Sc. Thesis. Instituto de Ciências Biomédicas. USP; 2009. Bioprospecção de Genes Biossintéticos de Policetídeos em DNA Metagenômico de Solo de Mata Atlântica São Paulo, Brazil; p. 114. [Google Scholar]
- McCarthy AJ, Williams ST. Actinomycetes as agents of biodegradation in the environment - a review. Gene. 1992;115:189–192. doi: 10.1016/0378-1119(92)90558-7. [DOI] [PubMed] [Google Scholar]
- Méndez C, Salas JA. ABC transporters in antibiotic-producing actinomycetes. FEMS Microbiol Lett. 1998;158:1–8. doi: 10.1111/j.1574-6968.1998.tb12792.x. [DOI] [PubMed] [Google Scholar]
- Metsä-Ketelä M, Salo V, Halo L, Hautala A, Hakala J, Mäntsälä P, Ylihonko K. An eficient approach for screening minimal PKS genes from Streptomyces. FEMS Microbiol Lett. 1999;180:1–6. doi: 10.1111/j.1574-6968.1999.tb08770.x. [DOI] [PubMed] [Google Scholar]
- Moffitt MC, Neilan BA. Evolutionary affiliations within the superfamily of Ketosynthases reflect complex pathway associations. J Mol Evol. 2003;56:446–457. doi: 10.1007/s00239-002-2415-0. [DOI] [PubMed] [Google Scholar]
- Motche M, Dehesh K, Edwards P, Lindqvist Y. The Crystal Structure of β-Ketoacyl-Acyl Carrier Protein Synthase II from Synechocystis sp at 154 Å Resolution and Its Relationship to Other Condensing Enzymes. J Mol Biol. 2001;305:491–503. doi: 10.1006/jmbi.2000.4272. [DOI] [PubMed] [Google Scholar]
- Olano C. Hutchinson’s legacy: keeping on polyketide biosynthesis. J Antibiot (Tokyo) 2011;64:51–57. doi: 10.1038/ja.2010.126. [DOI] [PubMed] [Google Scholar]
- Omura S, Ikeda H, Ishikawa J, Hanamoto A, Takahashi C, Shinose M, Takahashi Y, Horikawa H, Nakazawa H, Osonoe T, Kikuchi H, Shiba T, Sakaki Y, Hattori M. Genome sequence of an industrial Microorganism Streptomyces avermitilis: Deducing the ability of producing secondary metabolites. Proc Natl Acad Sci USA. 2001;98:12215–12220. doi: 10.1073/pnas.211433198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padilla G. In: Biologia molecular de Streptomyces e suas aplicações industriais. Melo IS, Azevedo JL, editors. Ecologia Microbiana; Embrapa, Brazil: 1998. pp. 328–347. [Google Scholar]
- Peirú S, Rodríguez E, Menzella HG, Carney JR, Gramajo H. Metabolically engineered Escherichia coli for efficient production of glycosylated natural products. Microb Biotechnol. 2008;1:476–486. doi: 10.1111/j.1751-7915.2008.00046.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira AL, Pita JR. Alexander Fleming (1881–1955), Da descoberta da penicilina (1928) ao Prémio Nobel (1945) Revista da Faculdade de Letras: História Porto. 2005;6:129–151. [Google Scholar]
- Pereira RM, Silveira EL, Scaquitto DC, Pedrinho EAN, Val-Moraes SP, Wickert E, Carareto-Alves LM, Lemos EGM. Molecular Characterization of Bacterial Populations of Different Soils. Braz J Microbiol. 2006;37:439–447. [Google Scholar]
- Piel J. Approaches to Capturing and Designing Biologically Active Small Molecules Produced by Uncultured Microbes. Annu Rev Microbiol. 2011;65:431–453. doi: 10.1146/annurev-micro-090110-102805. [DOI] [PubMed] [Google Scholar]
- Prosdocimi F, Cerqueira GC, Binneck E, Silva AF, Reis AN, Junqueira ACM, Santos ACF, Nhani-Júnior A, Wust CI, Camargo-Filho F, Kessedjian JL, Petretski JH, Camargo LP, Ferreira RGM, Lima RP, Pereira RM, Jardim S, Sampaio VS, Folgueras-Flatschart AV. Bioinformática: Manual do usuário. 29. Biotecnologia Ciência & Desenvolvimento; Brasília, Brazil: 2001. [Google Scholar]
- Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, Apweiler R, Lopez R. InterProScan: protein domains identifier. Nucleic Acids Res. 2005;33:W116–W120. doi: 10.1093/nar/gki442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendhran J, Gunasekaran P. Strategies for accessing soil metagenome for desired applications. Biotechnology Advances. 2008;26:576–590. doi: 10.1016/j.biotechadv.2008.08.002. [DOI] [PubMed] [Google Scholar]
- Rivero-Muller A, Lajic S, Huhtaniem I. Assisted large fragment insertion by Red/ET-recombination (ALFIRE) - An alternative and enhanced method for large fragment recombineering. Nucleic Acids Res. 2007;35/78:1–8. doi: 10.1093/nar/gkm250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rix U, Fischer C, Remsing LL, Rohr J. Modification of post-PKS tailoring steps through combinatorial biosynthesis. Nat Prod Rep. 2002;19:542–580. doi: 10.1039/b103920m. [DOI] [PubMed] [Google Scholar]
- Romero D, Traxler MF, López D, Kolter R. Antibiotics as Signal Molecules. Chem Rev. 2011;111:5492–5505. doi: 10.1021/cr2000509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Molecular cloning: A laboratory manual. 3 ed. Cold Spring harbor Laboratory; Cold Spring Harbor, New York: 2001. [Google Scholar]
- Schmieder R, Edwards R. Insights into antibiotic resistance through metagenomic approaches. Future Microbiol. 2012;7:73–89. doi: 10.2217/fmb.11.135. [DOI] [PubMed] [Google Scholar]
- Schuch V. M.Sc. Dissertation. Programa de Pós-graduação em Microbiologia Agropecuárea, Universidade Estadual Paulista “Júlio de Mesquita Filho” UNESP); 2007. Prospecção de genes envolvidos na biossíntese de antibióticos através da abordagem metagenômica Jaboticabal, Brazil; p. 118. [Google Scholar]
- Shen B. Polyketide biosynthesis beyond the type I, II and III polyketide synthase paradigms. Curr Opin Chem Biol. 2003;7:285–295. doi: 10.1016/s1367-5931(03)00020-6. [DOI] [PubMed] [Google Scholar]
- Shen B, Shen Y, Yoon P, Yu T-W, Floss HG, Hopwood D, Moore BS. Ectopic expression of the minimal whiE polyketide synthase generates a library of aromatic polyketides of diverse sizes and shapes. Proc Natl Acad Sci USA. 1999;96:3622–3627. doi: 10.1073/pnas.96.7.3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silakowski B, Schairer HU, Ehret H, Kunze B, Weinig S, Nordsiek G, Brandt P, Blöcker H, Höfle G, Beyer S, Müller R. New lessons for combinatorial biosynthesis from myxobacteria: the myxothiazol biosynthetic gene cluster of Stigmatella aurantiaca DW4/3-1. J Biol Chem. 1999;274:37391–37399. doi: 10.1074/jbc.274.52.37391. [DOI] [PubMed] [Google Scholar]
- Silveira EL, Pereira RM, Scaquitto DC, Pedrinho EAN, Val-Moraes SP, Wickert E, Carareto-Alves LM, Lemos EGM. Bacterial diversity of soil under eucalyptus assessed by 16S rDNA sequencing analysis. Pesq Agropec Bras. 2006;41:1507–1516. [Google Scholar]
- Sjölander K. Phylogenomic inference of protein molecular function: advances and challenges. Bioinformatics. 2004;20:170–179. doi: 10.1093/bioinformatics/bth021. [DOI] [PubMed] [Google Scholar]
- Smith TF, Waterman MS. Identification of common molecular subsequences. J Mol Biol. 1981;147:95–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- Smolke CD. The metabolic pathway engineering handbook: Fundamentals. 1 ed. CRC Press Taylor & Francis Group; Boca Raton, FL: 2010. [Google Scholar]
- Solecka J, Zajko J, Postek M, Rajnisz A. Biologically active secondary metabolites from Actinomycetes. Cent Eur J Biol. 2012;7:373–390. [Google Scholar]
- Starcevic A, Zucko J, Simunkovic J, Long PF, Cullum J, Hranueli D. ClustScan: An integrated program package for the semi-automatic annotation of modular biosynthetic gene clusters and in silico prediction of novel chemical structures. Nucleic Acids Res. 2008;36:6882–6892. doi: 10.1093/nar/gkn685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens DC, Henry MR, Murphy KA, Boddy CN. Heterologous Expression Of The Oxytetracycline Biosynthetic Pathway in Myxococcus xanthus. Appl Environ Microbiol. 2010;76:2681–2683. doi: 10.1128/AEM.02841-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi M, Shin-Ya K. New species of actinomycetes do not always produce new compounds with high frequency. J Antibiot (Tokyo) 2011;64:699–701. doi: 10.1038/ja.2011.66. [DOI] [PubMed] [Google Scholar]
- The Journal of Antibiotics. Special issue of The Journal of Antibiotics dedicated to the late Professor C Richard Hutchinson. J Antibiot (Tokyo) 2011;64:3–5. doi: 10.1038/ja.2010.158. [DOI] [PubMed] [Google Scholar]
- Tovchigrechko A, Vakser IA. Gramm-X Public Web Server For Protein-Protein Docking. Nucleic Acids Res. 2006;34:W310–W314. doi: 10.1093/nar/gkl206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Val-Moraes SP, Marcondes J, Carareto-Alves LM, Lemos EGM. Impact of sewage sludge on the soil bacterial communities by DNA microarray analysis. World J Microbiol Biotechnol. 2011;27:1997–2003. [Google Scholar]
- Vidotti CCF, De Castro LLC, Calil SS. New drugs in Brazil: Do they meet Brazilian public health needs? Rev Panam Salud Publica. 2008;24:36–45. doi: 10.1590/s1020-49892008000700005. [DOI] [PubMed] [Google Scholar]
- Vining LC. Functions of secondary metabolites. Annu Rev Microbiol. 1990;44:395–427. doi: 10.1146/annurev.mi.44.100190.002143. [DOI] [PubMed] [Google Scholar]
- Wadt M. M.Sc. Dissertation. Faculdade de Ciências Farmacêuticas. USP; 2003. Análise Econômica de Novos Fármacos Licenciados no Brasil entre 1998 e 2001. São Paulo, Brazil; p. 236. [Google Scholar]
- Wang GY, Graziani E, Waters B, Pan W, Li X, McDermott J, Meurer G, Saxena G, Andersen RJ, Davies J. Novel natural products from soil DNA libraries in a Streptomycete host. Org Lett. 2000;2:2401–2404. doi: 10.1021/ol005860z. [DOI] [PubMed] [Google Scholar]
- Weissman KJ. Introduction to polyketide biosynthesis. In: Hopwood DA, editor. Complex Enzymes in Microbial Natural Product Biosynthesis, Part B: Polyketides, Aminocoumarins and Carbohydrates Methods in Enzymology. Academic Press; 2009. pp. 3–16. [DOI] [PubMed] [Google Scholar]
- Yadav G, Gokhale RS, Mohanty D. SEARCHPKS: a program for detection and analysis of polyketide synthase domains. Nucleic Acids Res. 2003;31:3654–3658. doi: 10.1093/nar/gkg607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T-W, Shen Y, McDaniel R, Floss HG, Khosla C, Hopwood DA, Moore BS. Engineered Biosynthesis of Novel Polyketides from Streptomyces Spore Pigment Polyketide Synthases. J Am Chem Soc. 1998;120:7749–7759. [Google Scholar]
- Zaehner H, Fieldler HP. The need for new antibiotics: possible ways forward. In: Russel NJ, editor. Fifty years of antimicrobials: past perspectives and future trends. Cambridge University Press; Cambridge, England: 1995. pp. 67–84. [Google Scholar]
- Zaleta-Rivera K, Charkoudian LK, Ridley CP, Khosla C. Cloning, Sequencing, Heterologous Expression, and Mechanistic Analysis of A-74528 Biosynthesis. J Am Chem Soc. 2010;132:9122–9128. doi: 10.1021/ja102519v. [DOI] [PMC free article] [PubMed] [Google Scholar]