

Abstract
In this paper, we present a novel rough-fuzzy clustering (RFC) method to detect overlapping protein complexes in protein-protein interaction (PPI) networks. RFC focuses on fuzzy relation model rather than graph model by integrating fuzzy sets and rough sets, employs the upper and lower approximations of rough sets to deal with overlapping complexes, and calculates the number of complexes automatically. Fuzzy relation between proteins is established and then transformed into fuzzy equivalence relation. Non-overlapping complexes correspond to equivalence classes satisfying certain equivalence relation. To obtain overlapping complexes, we calculate the similarity between one protein and each complex, and then determine whether the protein belongs to one or multiple complexes by computing the ratio of each similarity to maximum similarity. To validate RFC quantitatively, we test it in Gavin, Collins, Krogan and BioGRID datasets. Experiment results show that there is a good correspondence to reference complexes in MIPS and SGD databases. Then we compare RFC with several previous methods, including ClusterONE, CMC, MCL, GCE, OSLOM and CFinder. Results show the precision, sensitivity and separation are 32.4%, 42.9% and 81.9% higher than mean of the five methods in four weighted networks, and are 0.5%, 11.2% and 66.1% higher than mean of the six methods in five unweighted networks. Our method RFC works well for protein complexes detection and provides a new insight of network division, and it can also be applied to identify overlapping community structure in social networks and LFR benchmark networks.
Introduction
In the past several years, large-scale proteomics experiments have produced many PPI data sets from different organisms [1]. These data sets are generally represented as undirected weighted or unweighted networks with proteins as a set of nodes and interactions as a set of edges. Edge weight estimates the reliability of such interaction. Protein-protein interactions play significant roles in cell's structural components and the process ranging from transcription, splicing site and translation to cell cycle control [2]. It is essential to extract overlapping protein complexes or regulatory pathways from PPI networks to investigate disease-related gene and drug target.
Densely connected regions in a graph can be identified by some unsupervised clustering method. However, many clustering methods are not ideal for PPI networks [1]. Some proteins may have multiple functions, hence the corresponding proteins could belong to more than one complex. Recently, a lot of clustering algorithms have been proposed to detect overlapping protein complexes in PPI networks [1], [3], [4], [5], [6], [7]. Each of them has limitations: some algorithms only work in unweighted networks, and can be applied to weighted data sets only after binarizing them by deleting edges whose weights are below a given threshold, while others need to assign the number of complexes firstly [8], [9]. Although the notion of the overlapping protein complexes is easy to understand, constructing an effective algorithm for overlapping protein complexes is highly non-trivial for two reasons: firstly, the number of protein complexes is unknown for a given PPI network; secondly, a protein complex should contain many reliable interactions within its subunit, and it should be well-separated from the rest of the PPI networks [1].
Fuzzy sets and rough sets have been widely applied to many fields, such as fuzzy clustering [10], [11], rough k-means clustering [9], [12], [13], [14], [15], fuzzy c-means clustering [16], [17], rough-fuzzy c-means clustering [18], [19], [20] and dynamic rough clustering [21], [22]. One of the most remarkable attempts to clustering problems may be c-means clustering and its derivatives. However, those algorithms are mainly applied to two dimensional microarray gene data, image data and forest cover rather than three dimensional network data, and mainly adapt rough set and fuzzy set theory to c-means clustering [18]. Those algorithms have the following weaknesses, firstly, the number of clusters c is an input parameter, and an inappropriate choice of c may yield poor results. In most cases, it is difficult to assess the numbers of clusters (c value) in original datasets. Thus, diagnostic checks have to be performed on and on to determine the number of clusters in the data set when performing c-means. Secondly, the choice of the initial cluster centers has a great impact on the clustering results; once the initial value selected is not good, it could not draw effective clustering results. Thirdly, the algorithm requires constant adjustment for sample classification and constantly calculating the adjusted new cluster centers, so when the data is very large, the algorithm time complexity will increase.
In order to solve the three dimensional datasets clustering problems in PPI networks and the weaknesses of c-means clustering, we present a novel method based on rough-fuzzy clustering (RFC) to detect overlapping protein complexes in PPI networks. RFC integrates the merits of fuzzy sets and rough sets, focuses on fuzzy relation model rather than graph model. RFC utilizes fuzzy set to create fuzzy relation between nodes and transform the fuzzy relation into fuzzy equivalence relation, and then create equivalence classes which correspond to non-overlapping protein complexes. The upper and lower approximations of rough sets are used to decide whether one protein belongs to one or more complexes, so we obtain overlapping complexes. RFC can automatically obtain the number of clustering by the number of equivalence classes, removing the limitation of selecting the initial clustering number. RFC also has advantage in datasets with large number of prototypes.
To test RFC's performance, we apply it to identify overlapping and non-overlapping community structure in artificial synthetic networks and social networks. To evaluate RFC quantitatively, we apply it to detect overlapping protein complexes in four weighted yeast data sets [23], [24], [25] and five unweighted yeast data sets [23], [24], [25], [26], and then we execute six other popular clustering methods (ClusterONE [1], CMC [27], MCL [28], GCE [29], OSLOM [30] and CFinder [3]) in the same data sets. Predicted complexes derived by the seven methods are separately compared with reference complexes from the Munich Information Centre for Protein Sequence (MIPS) [31] and the Saccharomyces Genome Database (SGD) [32]. Finally, results derived by the seven methods are compared with some evaluation criteria to assess RFC.
Materials and Methods
The definitions of rough-fuzzy clustering
Prior to providing a detailed description of our algorithm, we introduce some terminologies widely used in the forthcoming sections. Let 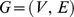 be an undirected graph, where V is a set of nodes, and E is a set of edges.
be an undirected graph, where V is a set of nodes, and E is a set of edges.
Definition 1. Let N(u) be the neighbors of node u. Sim(u, v), similarity for node pair u and v, is 1 if u = v; else 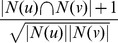 if
if 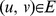 ; 0 otherwise.
; 0 otherwise.
Here, we define similarity between nodes based on their shared neighbors, if u and v are not directly neighbors, 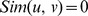 ; if u and v are directly neighbors, the more shared neighbors of u and v, the larger value of
; if u and v are directly neighbors, the more shared neighbors of u and v, the larger value of 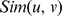 ; if u and v are the same node,
; if u and v are the same node, 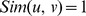 , that is,
, that is, 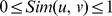 . If two nodes have similar topological structure, they may share similar functions [11]. Similarity in network topological structure decides the degree of similarity between a pair of nodes.
. If two nodes have similar topological structure, they may share similar functions [11]. Similarity in network topological structure decides the degree of similarity between a pair of nodes.
Definition 2. Let V be a nonempty set, and R be an equivalence relation. For each 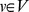 , the equivalence class of object v for R is defined as follows [12]:
, the equivalence class of object v for R is defined as follows [12]:
| (1) |
Definition 3. For set 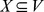 , the upper and lower approximations of X for R are defined as follows, respectively [12]:
, the upper and lower approximations of X for R are defined as follows, respectively [12]:
| (2) |
| (3) |
Here, 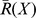 is the upper approximation of X for equivalence relation
is the upper approximation of X for equivalence relation 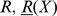 is the lower approximation of X for equivalence relation R. Obviously,
is the lower approximation of X for equivalence relation R. Obviously,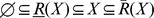 .
. 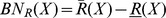 is called as boundary region of X for equivalence relation R, and their relationship is shown in Figure 1.
is called as boundary region of X for equivalence relation R, and their relationship is shown in Figure 1.
Figure 1. The relationship among Set X and its possible lower approximation, upper approximation and boundary region for equivalence relation R.

In the figure, we provide the relationship among set X, lower approximation 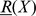 , upper approximation
, upper approximation 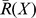 and boundary region
and boundary region 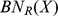 . The internal region of the red curve represents set X, the internal region of the yellow line represents lower approximation
. The internal region of the red curve represents set X, the internal region of the yellow line represents lower approximation 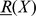 , the green region represents boundary region
, the green region represents boundary region 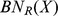 , the internal region of the blue line represents upper approximation
, the internal region of the blue line represents upper approximation 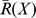 , and the whole region represents universal set.
, and the whole region represents universal set.
Let u be an object of set 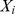 . It is obvious in Figure 1 that the upper and lower approximations of
. It is obvious in Figure 1 that the upper and lower approximations of 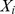 are only a few subsets of V. The family of the k upper and lower approximations of the
are only a few subsets of V. The family of the k upper and lower approximations of the 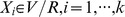 necessarily meet the following basic rough set properties [12]:
necessarily meet the following basic rough set properties [12]:
Property 1: An object u can be a part of at most one lower approximation.
Property 2: 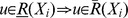 .
.
Property 3: u is not a part of any lower approximation 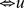 belongs to two or more boundary regions.
belongs to two or more boundary regions.
The next step is how to determine whether an object belongs to boundary region or lower approximation of a set. For each object u, let 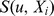 be similarity between u and any set
be similarity between u and any set 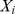 . The definition of
. The definition of 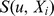 is as follows:
is as follows:
Definition 4. Similarity between node u and set Xi is
| (4) |
Here,  is obtained by Definition 1. The ratio
is obtained by Definition 1. The ratio 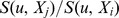 is used to decide the assignment of u as follows [12], [13]:
is used to decide the assignment of u as follows [12], [13]:
If
 is the maximum for
is the maximum for 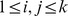 and
and 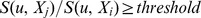 (k denotes the number of sets referring to the number of equivalence classes),
(k denotes the number of sets referring to the number of equivalence classes), 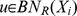 and
and 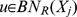 . Furthermore, u is not a part of any lower approximation. This criterion ensures that Property 3 is satisfied.
. Furthermore, u is not a part of any lower approximation. This criterion ensures that Property 3 is satisfied.Otherwise,
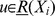 such that
such that 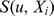 is the maximum for
is the maximum for 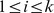 . In addition, by Property 2,
. In addition, by Property 2, 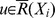 . This criterion also satisfies Property 1.
. This criterion also satisfies Property 1.
The rough-fuzzy clustering method
The RFC consists of the following major steps, as shown in Figure 2.
Figure 2. RFC algorithm flowchart.

In the figure, we briefly give RFC algorithm flowchart to describe the operational process of the algorithm.
- The graph (Figure 3) can be represented by an adjacency matrix N, and then transform the adjacency matrix N into the fuzzy matrix R by calculating the similarities between any two nodes (Definition 1). Obviously, R is reflexive and symmetric.

- Transform the fuzzy matrix R into the fuzzy equivalence relation t(R) by transitive closure [33].

- Choose a threshold
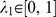 and transform t(R) as a Boolean equivalence relation
and transform t(R) as a Boolean equivalence relation 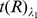 . Let
. Let 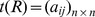 and
and 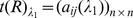 . Here
. Here 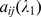 is 1 if
is 1 if 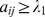 , 0 otherwise. Therefore, different λ
1 corresponds to different equivalence relations and equivalence classes as follows:
, 0 otherwise. Therefore, different λ
1 corresponds to different equivalence relations and equivalence classes as follows:

-
According to different λ 1, S(u, Xi) is computed by Definition 4. Here, each row represents a node, and each column represents an equivalence class which has been obtained in step (3). In the formula
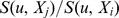 ,
, 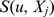 represents the similarity of node u and class Xj, and S(u, Xi) represents the maximum of similarities between node u and each class.
represents the similarity of node u and class Xj, and S(u, Xi) represents the maximum of similarities between node u and each class.
Here,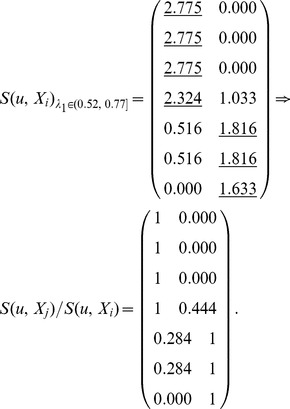
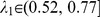 , and these objects are classified into two equivalence classes:
, and these objects are classified into two equivalence classes: 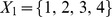 ,
, 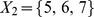 . If
. If 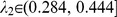 ,
, 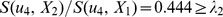 . Therefore, u
4 belongs to the boundary region of X
1 and X
2. In this case, non-overlapping sets,
. Therefore, u
4 belongs to the boundary region of X
1 and X
2. In this case, non-overlapping sets, 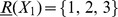 and
and 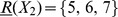 , and overlapping sets
, and overlapping sets 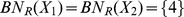 are obtained.The underlined numbers represent the maximum of similarity between each object and each class.
are obtained.The underlined numbers represent the maximum of similarity between each object and each class.
Here,
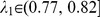 , and these objects are classified into three equivalence classes:
, and these objects are classified into three equivalence classes: 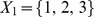 ,
, 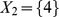 ,
, 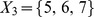 . If
. If 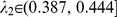 ,
, 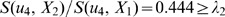 . Therefore, u
4 belongs to the boundary region of X
1 and X
3,
. Therefore, u
4 belongs to the boundary region of X
1 and X
3, 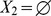 . In this case, non-overlapping sets,
. In this case, non-overlapping sets, 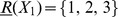 and
and 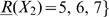 , and overlapping sets
, and overlapping sets 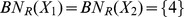 are obtained.
are obtained.
Here,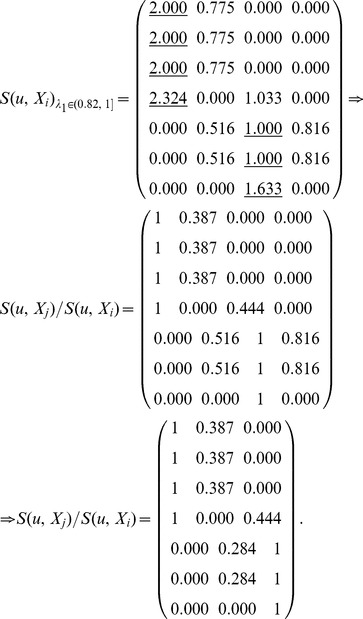
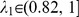 , and these objects are classified into four equivalence classes:
, and these objects are classified into four equivalence classes: 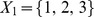 ,
, 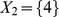 ,
, 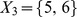 ,
,  . If
. If 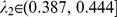 and i = 1, 2 and 4,
and i = 1, 2 and 4, 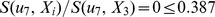 . Therefore, u
7 belongs to the lower approximation of
. Therefore, u
7 belongs to the lower approximation of 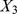

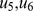 and
and 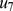 belong to the same equivalence class X
3. If
belong to the same equivalence class X
3. If 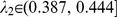 ,
, 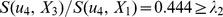 . Therefore, u
4 belongs to the boundary region of X
1 and X
3,
. Therefore, u
4 belongs to the boundary region of X
1 and X
3, 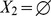 . In this case, non-overlapping sets,
. In this case, non-overlapping sets, 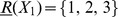 and
and 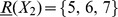 , and overlapping sets
, and overlapping sets 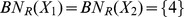 are obtained.
are obtained. Merge the sets with overlapping degree to a very high extent in comparison with their sizes [1]. We evaluate the extent of overlapping between each pair of sets by formula 10 and merge the two sets whose overlapping score is above a specific threshold. Let merging threshold be 0.64, because it shows that the intersection is at least 80% of the size of the set if the two sets are equal in size.
Figure 3. Artificial synthetic graph for illustrating the process of the rough-fuzzy clustering method.
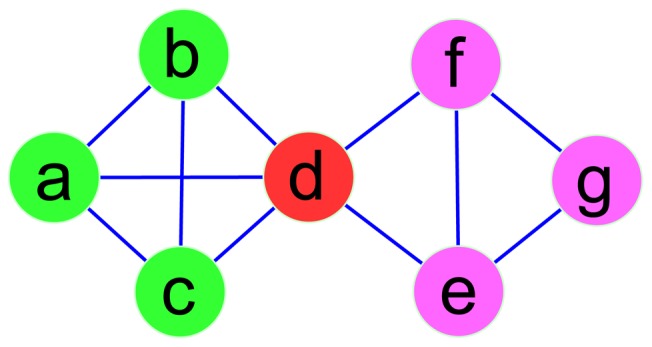
In the figure, the network is made of two communities and node d is overlapping node.
We have discussed the details of RFC. The choice scale of λ is relatively larger and more flexible than fuzzy clustering, and the clustering results are relatively stable for different λ. In the following section, RFC will be applied in artificial synthetic networks, social networks and PPI networks.
Parameter settings
In the algorithm, threshold λ 1 is used to divide networks to get non-overlapping modules. The λ 1 is closely related to the size of similarities of between nodes in all kinds of networks. Based on the analysis of the algorithm and a large number of experiments, we obtain λ 1 according to the following formula:
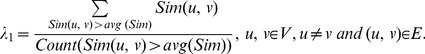 |
(5) |
Here, 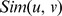 obtained by Definition 1 represents the similarity between nodes,
obtained by Definition 1 represents the similarity between nodes, 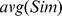 represents the mean of similarities of all pairs of nodes, and
represents the mean of similarities of all pairs of nodes, and 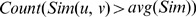 represents the number of the values that are greater than mean
represents the number of the values that are greater than mean 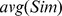 .
.
Threshold λ
2 is applied to determine whether one node belongs to one or multiple modules. In this article, it is set into an adjustable value. Based on a large number of experiments, it is a good choice to set 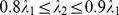 .
.
Evaluation criteria
Different criteria proposed by earlier studies are applied to evaluate RFC. The criteria are defined to assess the similarity between predicted modules and reference modules. The first measure is Normalized Mutual Information (NMI), which is an information theory based on quantifying the closeness of two groups of sets which has been widely used in clustering algorithms and machine learning [30], [34], [35], [36]. It is defined as:
| (6) |
Here, 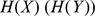 is the entropy of the random variable X(Y), whereas
is the entropy of the random variable X(Y), whereas 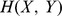 is the joint entropy.
is the joint entropy.
| (7) |
| (8) |
| (9) |
Here, for a random variable X with n outcomes 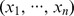 ,
, 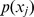 is the probability mass function of outcome
is the probability mass function of outcome 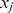 , and
, and 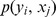 is the probability that
is the probability that 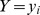 and
and 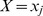 .
.
The Second measure is the overlapping score between predicted and reference complexes, which is shown as follows [37]:
| (10) |
Here, 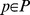 is a predicted complex and
is a predicted complex and  a reference complex. P is the set of predicted complexes and K is the set of reference complexes.
a reference complex. P is the set of predicted complexes and K is the set of reference complexes.
After defining overlapping score 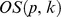 between predicted complex and reference complex, precision, recall and F1 measure are defined as follows [37]:
between predicted complex and reference complex, precision, recall and F1 measure are defined as follows [37]:
| (11) |
| (12) |
| (13) |
| (14) |
| (15) |
Here, 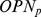 is the number of predicted complexes as
is the number of predicted complexes as 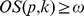 and
and 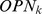 is the number of reference complexes as
is the number of reference complexes as 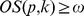 . The overlapping threshold ω = 0.25 is chosen, because it shows that the intersection is at least half of the complex size if the two complexes are equal in size [1]. Precision is the fraction of the predicted complexes that match known complexes. Recall represents the fraction of known complexes that match predicted complexes. F1 measure gives a reasonable combination of both precision and recall.
. The overlapping threshold ω = 0.25 is chosen, because it shows that the intersection is at least half of the complex size if the two complexes are equal in size [1]. Precision is the fraction of the predicted complexes that match known complexes. Recall represents the fraction of known complexes that match predicted complexes. F1 measure gives a reasonable combination of both precision and recall.
Giving the known complexes as reference classification, we take sensitivity as the score of members of the ith known complex which are found in the jth predicted complex. Clustering-wise sensitivity (Sn) is defined as follows [1], [37]:
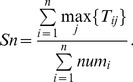 |
(16) |
Here, n is the number of known complexes. 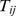 is the number of common proteins between the ith known complex and the jth predicted complex, and
is the number of common proteins between the ith known complex and the jth predicted complex, and 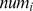 is the number of proteins belonging to the ith known complex.
is the number of proteins belonging to the ith known complex.
The positive predictive value (PPV) is the fraction of members of the jth predicted complex which belongs to the ith known complex. PPV is defined as follows [37]:
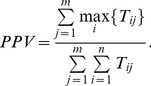 |
(17) |
Here, m is the number of predicted complexes, n is the number of known complexes.
The geometric accuracy (Acc) is the balance of both sensitivity and predictive value. It is obtained by calculating geometrical mean of Sn and PPV [37].
| (18) |
We employ separation to evaluate one-to-one correspondence between predicted complexes and known complexes. Separation of both the ith known complex and the jth predicted complex is shown as follows [1], [2], [37]:
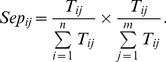 |
(19) |
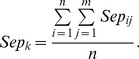 |
(20) |
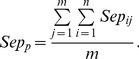 |
(21) |
| (22) |
Here, n is the number of known complexes. m is the number of predicted complexes. 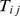 is the number of common proteins between the ith known complex and the jth predicted complex.
is the number of common proteins between the ith known complex and the jth predicted complex.
Results
To validate RFC's feasibility, we apply it in artificial networks, social networks and protein interaction networks. In artificial networks, we compare its performance with those of the best algorithms currently available. The algorithms, GCE [21] and OSLOM [22] are selected for a fair comparison in LFR benchmark networks. To further verify the performance of our method, we apply RFC in Karate club network [38] and Dolphins network [39].
To evaluate RFC quantitatively, we apply it in four weighted and five unweighted large scale yeast PPI datasets (see Table 1), and compare predicted complexes with two reference complexes, MIPS [23] and SGD [24] (see Table 2). We also compare RFC results with those of six other popular methods, MCL [28], CFinder [3], ClusterONE [1], GCE [29], OSLOM [30] and CMC [5], [27] with an immediate purpose to test the performance of extracting overlapping complexes. The similarity in weighted networks is defined by weight of the edge, and the similarity in unweighted networks is calculated by definition 1.
Table 1. Initial datasets.
| Unweighted networks | Weighted networks | Nodes numbers | Edges numbers | Density |
| Gavin [24] | Gavin [24] | 1855 | 7669 | 4.134 |
| Collins [23] | Collins [23] | 1622 | 9074 | 5.594 |
| Krogan_core [25] | Krogan_core [25] | 2708 | 7123 | 2.630 |
| Krogan_extended [25] | Krogan_extended [25] | 3672 | 14317 | 3.899 |
| BioGRID [26] | N/A | 5640 | 59748 | 10.549 |
N/A represents that there is no weighted BioGRID network.
Table 2. Gold standard protein complexes.
Artificial networks
The LFR [36] is a class of benchmark graphs which account for the heterogeneity in the distributions of node degrees and community sizes. It can be applied to overlapping communities, by assigning to each node the same number of neighbors in different communities. To simplify things, we suppose that each node belongs to the same number of communities [30]. Mixing parameter u as independent variable is the ratio of the number of external neighbors of a node by the total degree of the node [30]. Small values of u show well separated communities, whereas large values of u indicate high mixed to each other.
RFC is tested and compared with two recent methods, GCE [29], based on greedy clique expansion, and OSLOM [30], based on local optimization method. The two methods have good performances on LFR benchmark graphs with overlapping communities. The comparison of NMI's changes according to the mixture parameter u by three algorithms is presented in Figure 4
Figure 4. Results comparison of FRC, GCE and OSLOM in LFR benchmark graphs.
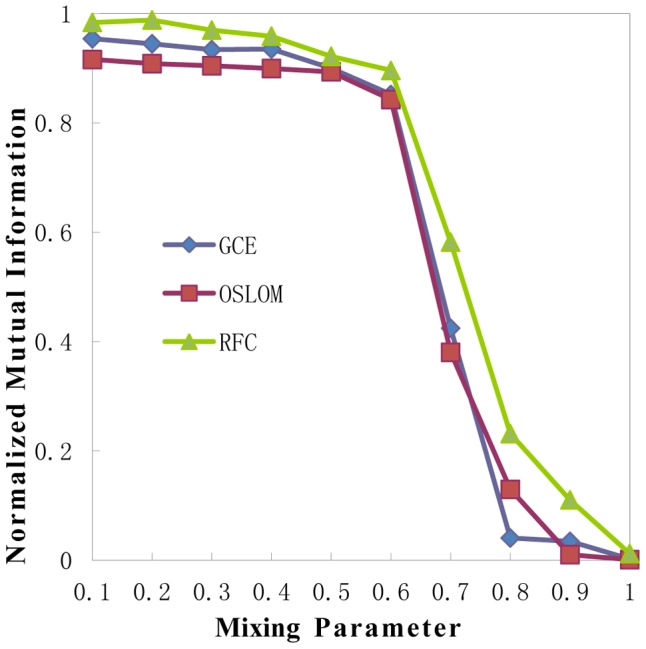
The parameters of the graphs are: network size 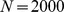 , average degree
, average degree 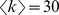 , maximum degree
, maximum degree 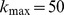 , community size is in the range [20,50].
, community size is in the range [20,50].
In all tests on LFR benchmark graphs, mixing parameter u varies from 0.1 to 0.9 with an interval 0.1 and each point is always 100 realizations, then mean of NMI is obtained as results. By increasing the value of u, communities become more and more fuzzy and it gets harder for any method to correctly detect the modules. We find that RFC performs competitively in comparison with GCE and OSLOM.
Social networks
Although RFC performs well in artificial networks, we have to select two real-world networks for further evaluation.
Karate club network
Zachary observed 34 members of a karate club at a US university in three years [38]. During the course of the time, node 1 (the club's instructor) and node 34 (the club's president) had some different ideas on the price of karate lessons. Ultimately the club was split into two organizations: one group was the supporters of the president and the other group was the supporters of the instructor. In fact, some individuals had friendship between the two groups, that is, these individuals may be overlapping nodes. Here we use an unweighted network version to test RFC and attempt to determine the factions involved in the split of the club. RFC performs well for detecting the two well-known communities which are centered at node 1 and node 34, respectively. The nodes 9, 10, 20, 28 and 29 are shared between the two groups. The communities coincide with overlapping nodes 9, 10, 20 observed by Sun et al. [10] with exception of nodes 28 and 29, which Sun et al. put with the community of the club's president. However, node 28 and node 29 have neighbors 3 and 34, respectively. Neighbor 34 is the club's president in one community, while neighbor 3 in the other community plays a pivotal role in its community. Therefore, it is reasonable that nodes 28 and 29 are overlapping. The detailed community structure of the network is shown in Figure 5.
Figure 5. The RFC results for community structure in Zachary's karate club network.
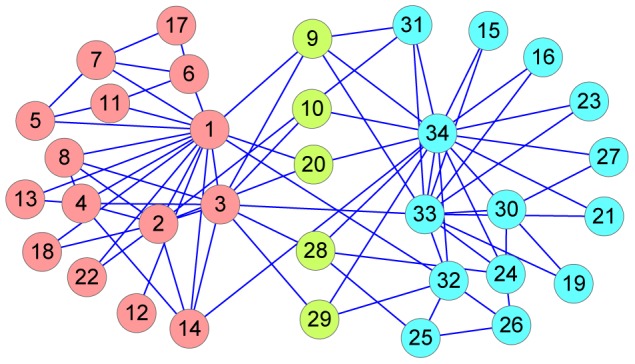
The divided result is shown for 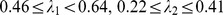 . In the figure, dashed red nodes are fully assigned to the community which is centered at the club's instructor, dashed green nodes are completely assigned to the other community which is centered at the club's president, and dashed yellow nodes are shared between the two communities.
. In the figure, dashed red nodes are fully assigned to the community which is centered at the club's instructor, dashed green nodes are completely assigned to the other community which is centered at the club's president, and dashed yellow nodes are shared between the two communities.
Dolphins network
The second example we discuss is the network studied by the biologist Lusseau [39], who divided a group of dolphins into two groups according to their age. There are 62 nodes and 159 edges in the network. RFC finds two communities with four overlapping nodes (8, 29, 31, 40), which can be seen in Figure 6. The partition of the two communities by RFC agrees with the separation observed by David Lusseau.
Figure 6. The RFC results for community structure in Lusseau's network of bottlenose dolphins.

PPI networks
First, we test the six methods mentioned above in the weighted Gavin, Collins and Krogan datasets. Table 3 indicates the detailed benchmark results in Gavin dataset when the MIPS gold standard dataset is used as gold standard. The detailed benchmark results in Collins and Krogan datasets are provided in Table S1. Figure 7 gives results of a comparison of the six algorithms in the weighted Gavin, Collins, and Krogan datasets using MIPS gold standard. The results by RFC are compared with the ones by ClusterONE, CMC, MCL, OSLOM and CFinder. The precision, sensitivity and separation are 35.8%, 48.3% and 75.9% higher than mean of five other methods in the four weighted networks.
Table 3. Results of six protein complex detection algorithms in weighted Gavin dataset using MIPS gold standard.
| Methods | #Complexes | Precision | F | Sensitivity | Accuracy | Sepk | Sepp | Separation |
| ClusterONE | 196 | 0.536 | 0.526 | 0.358 | 0.374 | 0.274 | 0.283 | 0.278 |
| CMC | 341 | 0.416 | 0.522 | 0.254 | 0.311 | 0.205 | 0.122 | 0.158 |
| CFinder | 262 | 0.591 | 0.666 | 0.162 | 0.247 | 0.215 | 0.167 | 0.189 |
| MCL | 252 | 0.353 | 0.391 | 0.316 | 0.355 | 0.297 | 0.239 | 0.266 |
| OSLOM | 88 | 0.625 | 0.378 | 0.402 | 0.357 | 0.175 | 0.404 | 0.266 |
| RFC | 153 | 0.575 | 0.494 | 0.409 | 0.375 | 0.297 | 0.394 | 0.342 |
Figure 7. Results comparison of the six algorithms in four weighted datasets using MIPS gold standard.

Columns correspond to the following algorithms, ClusterONE, CMC, CFinder, MCL, OSLOM and RFC from left to right in Gavins, Collins, Krogan_core and Krogan_extended weighted datasets, respectively, using MIPS gold standard. Various colors of the same column denote the individual components of the composite score of the algorithm (blue = the clustering-wise sensitivity, purple = geometric accuracy, green = the clustering-wise separation). The total height of each column is the value of the composite score for a special algorithm in a special dataset. Larger scores show the clustering result is better.
Table 4 indicates the detailed benchmark results in Gavin dataset when the SGD gold standard dataset is used as gold standard. The detailed benchmark results in Collins and Krogan datasets are provided in Table S2. Figure 8 gives results of a comparison of the six algorithms in the weighted Gavin, Collins, and Krogan datasets using SGD gold standard. The results by RFC are compared with the ones by ClusterONE, CMC, MCL, OSLOM and CFinder. The precision, sensitivity and separation are 29.7%, 38.9% and 85.9% higher than mean of five other methods in four weighted networks.
Table 4. Results of six protein complex detection algorithms in weighted Gavin dataset using SGD gold standard.
| Methods | #Complexes | Precision | F | Sensitivity | Accuracy | Sepk | Sepp | Separation |
| ClusterONE | 196 | 0.642 | 0.485 | 0.412 | 0.513 | 0.284 | 0.469 | 0.365 |
| CMC | 341 | 0.443 | 0.454 | 0.332 | 0.414 | 0.198 | 0.187 | 0.193 |
| CFinder | 262 | 0.687 | 0.615 | 0.221 | 0.366 | 0.222 | 0.274 | 0.247 |
| MCL | 252 | 0.488 | 0.428 | 0.431 | 0.518 | 0.374 | 0.480 | 0.424 |
| OSLOM | 88 | 0.648 | 0.277 | 0.514 | 0.466 | 0.187 | 0.689 | 0.359 |
| RFC | 153 | 0.660 | 0.424 | 0.517 | 0.502 | 0.353 | 0.746 | 0.514 |
Figure 8. Results comparison of the six algorithms in four weighted datasets using SGD gold standard.

Columns correspond to the following algorithms, ClusterONE, CMC, CFinder, MCL, OSLOM and RFC from left to right in Gavins, Collins, Krogan_core and Krogan_extended weighted datasets, respectively, using SGD gold standard. Various colors of the same column denote the individual components of the composite score of the algorithm (blue = the clustering-wise sensitivity, purple = geometric accuracy, green = the clustering-wise separation).
Then we test all the seven methods mentioned above in the unweighted Gavin, Collins, Krogan, and BioGRID datasets. Table 5 indicates the detailed benchmark results in Gavin dataset when the MIPS gold standard dataset is used as gold standard. The detailed benchmark results in Collins, Krogan and Biogrid datasets are provided in Table S3. Figure 9 gives results of a comparison of all the seven algorithms in the unweighted Gavin, Collins, Krogan and Biogrid datasets using MIPS gold standard. RFC results are compared with ClusterONE, CMC, MCL, OSLOM, GCE and CFinder results. The precision, F1 measure, sensitivity, accuracy and separation are 0.1%, 16.1%, 10.5%, 9.6% and 60.5% higher than mean of six other methods in five unweighted networks.
Table 5. Results of seven protein complex detection algorithms in unweighted Gavin dataset using MIPS gold standard.
| Methods | #Complexes | Precision | F | Sensitivity | Accuracy | Sepk | Sepp | Separation |
| ClusterONE | 294 | 0.316 | 0.374 | 0.366 | 0.373 | 0.282 | 0.195 | 0.234 |
| CMC | 156 | 0.532 | 0.462 | 0.381 | 0.363 | 0.221 | 0.288 | 0.252 |
| CFinder | 184 | 0.359 | 0.341 | 0.452 | 0.361 | 0.202 | 0.223 | 0.212 |
| MCL | 228 | 0.364 | 0.385 | 0.292 | 0.338 | 0.291 | 0.259 | 0.275 |
| OSLOM | 105 | 0.552 | 0.377 | 0.388 | 0.356 | 0.181 | 0.350 | 0.252 |
| GCE | 117 | 0.589 | 0.431 | 0.402 | 0.374 | 0.206 | 0.358 | 0.272 |
| RFC | 187 | 0.487 | 0.467 | 0.406 | 0.381 | 0.326 | 0.354 | 0.340 |
Figure 9. Results comparison of all the seven algorithms in five unweighted datasets using MIPS gold standard.

Columns correspond to the various algorithms, ClusterONE, CMC, CFinder, MCL, OSLOM, GCE and RFC from left to right in Gavins, Collins, Krogan_core, Krogan_extended and BioGRID unweighted datasets, respectively, using MIPS gold standard. The two blank columns represent that CFinder algorithm does not give any result within 24 hours for Collins and BioGRID unweighted datasets.
Table 6 indicates the detailed benchmark results in Gavin dataset when the SGD gold standard dataset is used as gold standard. The detailed benchmark results in Collins, Krogan and Biogrid datasets are provided in Table S4. Figure 10 shows results of a comparison of all the seven algorithms in the unweighted Gavin, Collins, and Krogan datasets using SGD gold standard. RFC results are compared with ClusterONE, CMC, MCL, OSLOM, GCE and CFinder results. The precision, F1 measure, sensitivity, accuracy and separation are 2.7%, 26.6%, 11.8%, 10.1% and 69.8% higher than mean of six other methods in five unweighted networks.
Table 6. Results of seven protein complex detection algorithms in unweighted Gavin dataset using SGD gold standard.
| Methods | #Complexes | Precision | F | Sensitivity | Accuracy | Sepk | Sepp | Separation |
| ClusterONE | 294 | 0.395 | 0.376 | 0.492 | 0.518 | 0.360 | 0.395 | 0.377 |
| CMC | 156 | 0.583 | 0.380 | 0.476 | 0.488 | 0.243 | 0.503 | 0.350 |
| CFinder | 184 | 0.446 | 0.323 | 0.532 | 0.456 | 0.240 | 0.421 | 0.318 |
| MCL | 228 | 0.491 | 0.406 | 0.373 | 0.477 | 0.342 | 0.484 | 0.407 |
| OSLOM | 105 | 0.562 | 0.276 | 0.522 | 0.477 | 0.205 | 0.632 | 0.360 |
| GCE | 117 | 0.666 | 0.354 | 0.499 | 0.500 | 0.239 | 0.661 | 0.397 |
| RFC | 187 | 0.626 | 0.459 | 0.533 | 0.519 | 0.412 | 0.711 | 0.541 |
Figure 10. Results comparison of all the seven algorithms in five unweighted datasets using SGD gold standard.

Columns correspond to the following algorithms, ClusterONE, CMC, CFinder, MCL, OSLOM, GCE and RFC from left to right in Gavins, Collins, Krogan_core, Krogan_extended and BioGRID unweighted datasets, respectively, using SGD gold standard. The two blank columns represent that CFinder algorithm does not give any result within 24 hours for Collins and BioGRID unweighted datasets.
Conclusion and Discussion
In this paper, we present a novel method based on rough-fuzzy clustering to detect overlapping and non-overlapping protein complexes in PPI networks. RFC is based on a fuzzy relation model which is transformed into equivalent classes to detect non-overlapping protein complexes. We further apply the upper approximation and lower approximation in rough sets to deal with each node in the network which belongs to one or multiple complexes. Ultimately, each complex corresponds to an overlapping protein complex.
RFC is tested in artificial networks, social networks and PPI networks and it is proved to provide a new insight into network division and to accurately recover communities in artificial networks. To determine whether these results are robust, we perform comparative benchmarks on a range of LFR graphs with overlapping communities, and find RFC performs competitively in comparison with GCE and OSLOM. To complete our evaluation, we test RFC and six other popular clustering algorithms in five unweighted PPI networks and four weighted PPI networks, and compare the results with MIPS and SGD gold standard datasets separately. We discover the three quality scores (accuracy, sensitivity and separation) obtained by RFC are obviously larger than those by six other methods.
Our results indicate that RFC outperforms six other popular algorithms in terms of matching more complexes between known complexes and predicted complexes with a higher accuracy, known complexes matching more predicted complexes with a higher sensitivity and providing a better one-to-one mapping with reference complexes with a higher separation. RFC results have a significant comprehensive advantage, especially in the Gavin and Collins datasets whose node numbers are close to the ones of the reference complexes. ClusterONE, OSLOM, GCE and MCL yield the closest score to RFC.
There exist several rough-fuzzy clustering algorithms in previous studies [8], [14], [17], [18], [40], such as rough c-means clustering (RCM) [13], [15], rough-fuzzy c-means clustering (RFCM) [8], [18] and rough-fuzzy possibilistic c-means clustering (RFPCM) [17]. These algorithms are mainly based on rough-fuzzy c-means clustering and its derivatives, and they are used to cluster co-expressed genes or functionally similar genes from microarray gene expression data sets. Recently, fuzzy-rough supervised gene clustering algorithm (FRSAC) has been proposed in [40] to detect groups of co-regulated genes whose expression is strongly associated with sample categories. The research objects of these clustering algorithms are two-dimensional gene expression data, that is, each row represents a gene and each column a sample. In those algorithms, the function of fuzzy sets is to handle overlapping partitions, and rough sets deal with uncertainty, vagueness, and incompleteness in class definition.
To our best knowledge, fuzzy clustering algorithm is firstly proposed in [11] to detect overlapping and non-overlapping community in social networks. In the algorithm, the choice of two thresholds is sensitive and it is difficult to choose accurate thresholds in large social networks and PPI networks. If the first threshold is not precise enough, some nodes supposed to belong to a community may not belong to any equivalence classes, so the nodes will not be allocated to the community. If the second threshold is not accurate enough, the overlapping nodes supposed to belong to two or multiple communities may not be allocated to the communities unless they have to be high correlated with the communities. Therefore, choosing the threshold values may cause some difficulties in large social networks and PPI networks and inaccuracy by excluding some edge nodes.
In order to solve the weaknesses, we propose a new algorithm RFC with different algorithms basis, clustering objects structure and the functions of rough set and fuzzy set. To be more specific, RFC algorithm is not based on c-means clustering, and the research objects of RFC are three-dimensional network data. In RFC, Fuzzy sets are used to create fuzzy equivalence relation and obtain clustering number automatically by calculating the number of equivalence classes. Rough sets are used to determine whether each node belongs to one or multiple complexes. The computing process of RFC indicates that the choice scale of the two thresholds in RFC is relatively larger and more flexible than fuzzy clustering algorithm [11]. It is also easier to detect the edge nodes for a community or a complex by introducing the upper and lower approximation in rough set than fuzzy clustering algorithm. The most significant advantage of RFC is that its separation is larger than the one in other algorithms, thus better evaluating one-to-one correspondence between predicted complexes and known complexes.
Protein complexes are key components to perform cellular functions associated with specific diseases [41], for example, overlapping proteins among multiple complexes tend to be drug targets [41]. In biological networks, some critical genes or motifs participate in multiple biological processes, implying the existence of overlapping modules. Studying the overlapping modules in networks is critical since it helps to confer the relationship between structure and function. In future work, we will focus on detecting human protein complexes to investigate disease related gene and drug target by RFC.
Supporting Information
Results of six protein complex detection algorithms in weighted Collins, Krogan_core and Krogan_extended datasets using MIPS gold standard.
(DOCX)
Results of six protein complex detection algorithms in weighted Collins, Krogan_core and Krogan_extended datasets using SGD gold standard.
(DOCX)
Results of seven protein complex detection algorithms in unweighted Collins, Krogan_core, Krogan_extended and Biogrid datasets using MIPS gold standard.
(DOCX)
Results of seven protein complex detection algorithms in unweighted Collins, Krogan_core, Krogan_extended and Biogrid datasets using SGD gold standard.
(DOCX)
Acknowledgments
We thank Bingbo Wang, Xingli Guo, Feng Li and Xiaoke Ma for their helpful advice and discussions. We also thank the anonymous reviewers for their valuable comments and suggestions.
Funding Statement
This work is supported by the National NSFC (Grant Nos. 60933009 and 91130006, URL: http://www.nsfc.gov.cn/publish/portal1/) and Northwest A&F University Grant (No. 2012RWZX16, URL: http://en.nwsuaf.edu.cn/). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Nepusz T, Yu H, Paccanaro A (2012) Detecting overlapping protein complexes in protein-protein interaction networks. Nature Methods 9: 471–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Qin G, Gao L (2010) Spectral clustering for detecting protein complexes in protein–protein interaction (PPI) networks. Mathematical and Computer Modelling 52: 2066–2074. [Google Scholar]
- 3. Adamcsek B, Palla G, Farkas IJ, Derényi I, Vicsek T (2006) CFinder: locating cliques and overlapping modules in biological networks. Bioinformatics 22: 1021–1023. [DOI] [PubMed] [Google Scholar]
- 4. Bader GD, Hogue CW (2003) An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics 4: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li B, Leal SM (2008) Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. The American Journal of Human Genetics 83: 311–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Macropol K, Can T, Singh A (2009) RRW: repeated random walks on genome-scale protein networks for local cluster discovery. BMC Bioinformatics 10: 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lei X, Wu S, Ge L, Zhang A (2013) Clustering and overlapping modules detection in PPI network based on IBFO. Proteomics 13: 278–290. [DOI] [PubMed] [Google Scholar]
- 8. Maji P, Paul S (2012) Rough-Fuzzy Clustering for Grouping Functionally Similar Genes from Microarray Data. in Proc 10th Asia Pacific Bioinformatics Conf 2012: 307–320. [DOI] [PubMed] [Google Scholar]
- 9. Peters G (2006) Some refinements of rough k-means clustering. Pattern Recognition 39: 1481–1491. [Google Scholar]
- 10. Dubois D, Prade H (1990) Rough fuzzy sets and fuzzy rough sets. International Journal of General System 17: 191–209. [Google Scholar]
- 11. Sun PG, Gao L, Shan Han S (2011) Identification of overlapping and non-overlapping community structure by fuzzy clustering in complex networks. Information Sciences 181: 1060–1071. [Google Scholar]
- 12. Lingras P, Peters G (2011) Rough clustering. Wiley Interdisciplinary Reviews: Data Mining and Knowledge Discovery 1: 64–72. [Google Scholar]
- 13. Peters G, Lampart M (2006) A partitive rough clustering algorithm. Springer 657–666. [Google Scholar]
- 14. Lingras P (2007) Applications of rough set based k-means, Kohonen SOM, GA clustering. Transactions on rough sets VII: Springer 120–139. [Google Scholar]
- 15. Lingras P, Yan R, West C (2003) Comparison of conventional and rough k-means clustering. Rough Sets, Fuzzy Sets, Data Mining, and Granular Computing: Springer 130–137. [Google Scholar]
- 16. Lingras P, Yan R, West C (2003) Fuzzy C-means clustering of web users for educational sites. Advances in Artificial Intelligence: Springer 557–562. [Google Scholar]
- 17. Maji P, Pal SK (2007) Rough set based generalized fuzzy c-means algorithm and quantitative indices. IEEE Transactions on Systems, Man, and Cybernetics, Part B 37: 1529–1540. [DOI] [PubMed] [Google Scholar]
- 18. Maji P, Pal SK (2007) RFCM: A hybrid clustering algorithm using rough and fuzzy sets. Fundamenta Informaticae 80: 475–496. [Google Scholar]
- 19. Maji P, Pal SK (2008) Maximum class separability for rough-fuzzy c-means based brain mr image segmentation. Transactions on Rough Sets IX: Springer 114–134. [Google Scholar]
- 20. Maji P, Paul S (2011) Microarray time-series data clustering using rough-fuzzy c-means algorithm. IEEE 269–272. [Google Scholar]
- 21. Peters G, Weber R, Nowatzke R (2012) Dynamic rough clustering and its applications. Applied Soft Computing 3193–3207. [Google Scholar]
- 22. Zamir O, Etzioni O (1999) Grouper: a dynamic clustering interface to Web search results. Computer Networks 31: 1361–1374. [Google Scholar]
- 23. Collins SR, Kemmeren P, Zhao X-C, Greenblatt JF, Spencer F, et al. (2007) Toward a comprehensive atlas of the physical interactome of Saccharomyces cerevisiae. Molecular & Cellular Proteomics 6: 439–450. [DOI] [PubMed] [Google Scholar]
- 24. Gavin A-C, Aloy P, Grandi P, Krause R, Boesche M, et al. (2006) Proteome survey reveals modularity of the yeast cell machinery. Nature 440: 631–636. [DOI] [PubMed] [Google Scholar]
- 25. Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, et al. (2006) Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440: 637–643. [DOI] [PubMed] [Google Scholar]
- 26. Stark C, Breitkreutz B-J, Reguly T, Boucher L, Breitkreutz A, et al. (2006) BioGRID: a general repository for interaction datasets. Nucleic Acids Research 34: D535–D539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Guimei Liu LW, Chua Hon Nian (2009) Complex discovery from weighted PPI networks. Bioinformatics 25: 1891–1897. [DOI] [PubMed] [Google Scholar]
- 28.Dongen S (2000) Performance criteria for graph clustering and Markov cluster experiments. Centre for Mathematics and Computer Science (CWI) Report.
- 29.Lee C, Reid F, McDaid A, Hurley N (2010) Detecting highly overlapping community structure by greedy clique expansion. ArXiv Preprint ArXiv:10021827.
- 30. Lancichinetti A, Radicchi F, Ramasco JJ, Fortunato S (2011) Finding statistically significant communities in networks. PloS One 6: e18961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mewes H-W, Amid C, Arnold R, Frishman D, Gueldener U, et al. (2004) MIPS: analysis and annotation of proteins from whole genomes. Nucleic Acids Research 32: D41–D44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hong EL, Balakrishnan R, Dong Q, Christie KR, Park J, et al. (2008) Gene Ontology annotations at SGD: new data sources and annotation methods. Nucleic Acids Research 36: D577–D581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zimmermann HJ (2001) Fuzzy set theory-and its applications: Springer.
- 34.McDaid AF, Greene D, Hurley N (2011) Normalized mutual information to evaluate overlapping community finding algorithms. ArXiv Preprint ArXiv:11102515.
- 35. Lancichinetti A, Fortunato S, Kertész J (2009) Detecting the overlapping and hierarchical community structure in complex networks. New Journal of Physics 11: 033015. [Google Scholar]
- 36. Lancichinetti A, Fortunato S (2009) Benchmarks for testing community detection algorithms on directed and weighted graphs with overlapping communities. Physical Review E 80: 016118. [DOI] [PubMed] [Google Scholar]
- 37. Brohee S, van Helden J (2006) Evaluation of clustering algorithms for protein-protein interaction networks. BMC Bioinformatics 7: 488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zachary WW (1977) An information flow model for conflict and fission in small groups. Journal of Anthropological Research 33 4: 452–473. [Google Scholar]
- 39. Lusseau D (2003) The emergent properties of a dolphin social network. Proceedings of the Royal Society of London Series B: Biological Sciences 270: S186–S188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Maji P (2011) Fuzzy–Rough Supervised Attribute Clustering Algorithm and Classification of Microarray Data. Systems, Man, and Cybernetics, Part B: Cybernetics, IEEE Transactions on 41: 222–233. [DOI] [PubMed] [Google Scholar]
- 41. Wu M, Yu Q, Li X, Zheng J, Huang J-F, Kwoh C-K (2013) Benchmarking Human Protein Complexes to Investigate Drug-Related Systems and Evaluate Predicted Protein Complexes. PloS One 8: e53197. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Results of six protein complex detection algorithms in weighted Collins, Krogan_core and Krogan_extended datasets using MIPS gold standard.
(DOCX)
Results of six protein complex detection algorithms in weighted Collins, Krogan_core and Krogan_extended datasets using SGD gold standard.
(DOCX)
Results of seven protein complex detection algorithms in unweighted Collins, Krogan_core, Krogan_extended and Biogrid datasets using MIPS gold standard.
(DOCX)
Results of seven protein complex detection algorithms in unweighted Collins, Krogan_core, Krogan_extended and Biogrid datasets using SGD gold standard.
(DOCX)