

Abstract
Antimitotics such as taxanes are being considered as alternatives to conventional cisplatin-based chemotherapy in patients with bladder cancer, but the molecular determinants of sensitivity or resistance to these agents in bladder cancer cells have not been defined. Here we examined the cytotoxic effects of a novel antimitotic, the Eg5 inhibitor AZD4877, in a molecularly diverse panel of human bladder cancer cell lines. The cells displayed heterogeneous responses to the drug that correlated closely with sensitivity to docetaxel but not with sensitivity to cisplatin. Global gene expression profiling identified p63 as the top gene that was differentially expressed between sensitive and resistant cell lines. Stable knockdown of p63 inhibited cell death induced by either AZD4877 or docetaxel and was associated with decreased proliferation and decreased expression of c-myc. Furthermore, c-myc knockdown also rendered cells resistant to AZD4877 or docetaxel. Together, our results implicate p63 and its downstream target c-myc as determinants of sensitivity to anti-mitotics in bladder cancer cells. Our data also suggest that anti-mitotics and cisplatin target different subsets of bladder cancer cells, a conclusion that may have important implications for the therapy of muscle-invasive bladder cancers.
Keywords: apoptosis, c-myc, gene expression profiling, ΔNp63
Introduction
Bladder cancer is currently the fifth most prevalent malignancy in the United States and is the ninth leading cause of cancer deaths in American men.1 Bladder cancers fall into two major pathological subcategories (superficial and muscle-invasive) that pose different challenges for clinical management. While superficial bladder tumors are rarely lethal, they recur frequently, making them the most costly cancers to clinically manage.2 On the other hand, although a significant fraction (approximately 40%) of patients with high-risk muscle-invasive bladder cancers can be cured with surgery and cisplatin-based chemotherapy, progression in patients with cisplatin-resistant disease is extremely rapid and uniformly fatal.2 Therefore, there is a great need to identify therapeutic regimens that target the cisplatin-resistant subset of muscle-invasive tumors.
Some clinical data suggest that antimitotic drugs such as taxanes may also be active in bladder cancer.3 Taxanes work by preventing the depolarization of microtubules, thus interfering with microtubule dynamics necessary during mitosis. This interference results in the activation of the mitotic spindle checkpoint, a critical regulatory mechanism in cells that prevents the onset of anaphase until all chromosomes are properly aligned, and leads to mitotic arrest in cells with intact checkpoint function and apoptosis in cells that possess defects in the checkpoint (i.e., tumors).4,5 However, because microtubules are critical in cellular functions other than mitosis, taxane treatment can result in harmful side effects, such as peripheral neuropathy.6 Therefore, there is interest in developing chemotherapeutic agents that target cells with defects in the mitotic spindle checkpoint more specifically. Inhibitors of the mitotic kinesin Eg5 (kinesin-5, KSP, Kif11) have emerged as potentially specific and effective anticancer agents.7,8 Eg5, active only during mitosis, contributes to the bipolarization of the mitotic spindle. In the absence of Eg5, mitotic cells form a monopolar spindle that activates the spindle checkpoint.9,10
Previous studies have sought to determine the exact mechanisms of antimitotic induced cell death, and to explain why only a subset of tumors shows a clinical response to these drugs. Some investigators have suggested that the effectiveness of the spindle checkpoint itself determines cellular response, as they saw a much weaker apoptotic response to antimitotics in cells lacking critical checkpoint proteins, such as Mad2 or BubR1.11-13 Other studies argue that antimitotic induction of apoptosis is independent of the spindle checkpoint.14-19 Clearly, more research focus must be placed on enhancing our understanding of antimitotics and our ability to pre-determine which tumors will respond to these drugs.
The p63 proteins comprise a subset of the p53 family that play important roles in development and the maintenance of “stemness” in epithelial tissues.20 The p63 proteins can be subdivided into two groups (the TA and ΔN isoforms) based on whether or not they contain a full-length N-terminal transcriptional transactivation domain. TAp63 isoforms are expressed early in development by the basal and intermediate cell layers within the bladder urothelium that contain the stem and transit-amplifying cells, respectively, whereas the ΔN isoforms are only expressed after birth.21,22 p63 ablation in the mouse results in complete loss of these cell populations.22 Comparisons of superficial and muscle-invasive bladder cancers have demonstrated that p63 levels are generally lower in the latter,23 but recent research shows that invasive tumors maintaining ΔNp63 expression represent a more aggressive phenotype than invasive bladder tumors lacking p63.22,24 Thus, p63 appears to play a critical role in bladder cancer that requires further elucidation.
In the current study, we sought to identify the molecular mechanisms that dictate sensitivity to a novel Eg5 inhibitor, AZD4877, in a heterogeneous panel of human bladder cancer cell lines. Using gene expression profiling, we found that ΔNp63 expression correlated with sensitivity to AZD4877 but not with sensivity to the frontline agent, cisplatin. Our functional studies established that p63 expression promotes drug sensitivity via a cell cycle-related mechanism that involves c-myc. Our results suggest that AZD4877 and other anti-mitotics and cisplatin will preferentially target non-overlapping subsets of tumors, and they provide the foundation for a more focused evaluation of anti-mitotics in bladder cancer patients.
Results
A panel of bladder cancer cell lines displays varying sensitivity to AZD4877
In order to characterize sensitivity to the novel antimitotic AZD4877 in vitro, we exposed a panel of 17 bladder cancer cell lines to varying concentrations of the drug and used propidium iodide staining and FACS analysis to quantify the levels of DNA fragmentation characteristic of apoptosis in each cell line (Table 1). We found 10 nM to be an effective drug concentration in vitro, and identified a wide spectrum of responses to the drug at 24 (Fig. 1A) and 48 (Fig. 1B) hour time points in our cell lines. We also assessed cellular responses to cisplatin because it serves as the core of current frontline therapy for muscle-invasive bladder cancer (Fig. 1C). Interestingly, many of the AZD4877-sensitive cell lines were resistant to cisplatin, whereas several of the AZD4877-resistant cell lines were relatively cisplatin-sensitive (Fig. 1C). Therefore, bladder cancer cell lines exhibit complementary patterns of sensitivity and resistance to AZD4877 and cisplatin.
Table 1. AZD4877-induced apoptosis in human bladder cancer cells.
| Cell line | 24 h | S or R | 48 h | S or R |
|---|---|---|---|---|
| UC6 | 58.5 | S | 79.9 | S |
| UC15 | 51.3 | S | 62.6 | S |
| RT4v6 | 45.8 | S | 66.2 | S |
| RT4v1 | 40.5 | S | 55.7 | S |
| UC10 | 37.2 | S | 53.6 | S |
| RT4 | 36.8 | S | 70.8 | S |
| UC5 | 32.5 | S | 63 | S |
| UC17 | 30.9 | S | 63.4 | S |
| UC1 | 30.1 | S | 44.8 | R |
| UC14 | 29.7 | R | 71.8 | S |
| JB | 24.3 | R | 57.4 | S |
| BV | 21.6 | R | 27.3 | R |
| UC9 | 20.5 | R | 20 | R |
| UC13 | 18.8 | R | 46.7 | R |
| UC3 | 17.0 | R | 45.3 | R |
| T24 | 14.2 | R | 28.4 | R |
| UC12 | 10.8 | R | 36.6 | R |
| mean | 30.6 | 52.6 | ||
| median | 30.1 | 55.7 |
Cells were exposed to 10 nM AZD4877 for 24 or 48 h and apoptosis was quantified by propidium iodide staining and FACS analysis as described in Materials and Methods. Background levels of apoptosis were subtracted from the levels observed in drug-exposed cells to derive the values presented in the table. Results were derived from mean values obtained in 3 independent experiments.

Figure 1. Effect of AZD4877 treatment on a panel of bladder cancer cell lines. Propidium iodide staining and FACS analysis were used to determine effects of AZD4877 (10nM) on apoptosis at 24 (A) and 48 (B) h time points. Percentages of subG0/G1 cells in treated and untreated cells is shown on the Y-axis (n = 3). In (C), the effects of AZD4877 (10 nM, 24 h) and cisplatin (10 μM, 48 h) were compared by subtracting background DNA fragmentation levels from the levels observed in drug-exposed cells.
Sensitivity to AZD4877 correlates with p63 expression
To identify molecular markers associated with sensitivity to AZD4877, we performed baseline microarray analyses on five of the more sensitive and five of the more resistant cell lines, and we found that p63 was the top most differentially expressed gene (Table 2). Specifically, AZD4877-sensitive cells generally expressed high levels of p63 mRNA and protein, whereas levels of both were essentially undetectable in the more drug-resistant cells (Fig. 2). The only cell line that did not adhere to this pattern was UC10, which was drug-sensitive but p63-negative (Fig. 2A). In separate studies we determined that the ΔN α isoform is the predominant isoform expressed by these and other human bladder cancer cell lines and in primary patient tumors.24 Interestingly, we noticed that there was also a very close correlation between expression of p63 and epithelial (E- and P-) cadherins in human bladder cancer cell lines (Fig. 2A) and primary tumors,24 suggesting that p63 may also be a marker of epithelial-to-mesenchymal transition (EMT). The UC10 cells expressed high levels of E- and P-cadherin even though they were p63-negative (Fig. 2A).
Table 2. Differences in baseline gene expression in AZD4877-sensitive and -resistant bladder cancer cells.
| p value | FDR | Probe ID | Symbol | GB acc # | Fold-change* |
|---|---|---|---|---|---|
| 1.00E-07 | 0.00473 | ILMN_3305055 | TP63 | NM_001114981 | 0.042 |
| 3.00E-07 | 0.0071 | ILMN_1704294 | CDH3 | NM_001793 | 0.092 |
| 1.50E-06 | 0.0177 | ILMN_2138801 | TP73L | NM_003722 | 0.065 |
| 1.50E-06 | 0.0177 | ILMN_2399016 | MMP28 | NM_001032278 | 0.11 |
| 3.30E-06 | 0.0312 | ILMN_2133205 | GPX2 | NM_002083 | 0.016 |
| 9.90E-06 | 0.0645 | ILMN_2376050 | FXYD3 | NM_005971 | 0.42 |
| 1.06E-05 | 0.0645 | ILMN_1766951 | MSX2 | NM_002449 | 0.48 |
| 1.09E-05 | 0.0645 | ILMN_1696643 | TLN1 | NM_006289 | 1.96 |
| 1.44E-05 | 0.0734 | ILMN_2337955 | ZMAT5 | NM_019103 | 1.56 |
| 1.55E-05 | 0.0734 | ILMN_1664006 | AGL | NM_000642 | 0.58 |
Resistant/sensitive. Baseline gene expression was compared in 5 AZD4877-sensitive (UC5, UC6, UM14, UC15, and RT4v6) and 5 AZD4877-resistant (UC3, UC9, UC12, 253J B-V, and T24) human bladder cancer cell lines using Illumina whole genome arrays as described in Materials and Methods. Note that TP63 and TP73L both refer to p63 and that CDH3 represents P-cadherin.

Figure 2. Differential p63 expression in AZD4877-sensitive and -resistant bladder cancer cell lines. (A) Expression of p63, E-cadherin (CDH1), and P-cadherin (CDH3) were measured using Illumina whole genome mRNA expression profiling. p63 levels were confirmed in a 10 cell line subset by immunoblotting (B) and quantitative RT-PCR (C).
p63 contributes to sensitivity to antimitotic drugs
We next used RNAi to determine whether p63 expression contributed directly to AZD4877 sensitivity. Stable knockdown of TA and ΔN isoforms in UM-UC14 cells was achieved using a lentiviral vector. The UC14 p63 KD cells were markedly resistant to AZD4877 as compared with the parental UC14 cells or cells transduced with a non-targeting shRNA construct (Fig. 3A). To confirm that the effects were dependent on knockdown of the ΔN isoform, were not due to off-target effects of the lentiviral shRNA construct, and were also observed using a different measurement of apoptosis (caspase activation), we transfected the UC14 cells with non-targeting or ΔNp63-specific siRNA constructs and assessed the effects on AZD4877-induced cleavage of the hallmark caspase-3 substrate, poly(ADP-ribose) polymerase (PARP). Again, ΔNp63 silencing strongly attenuated AZD4877-induced apoptosis (Fig. 3B). It has been previously reported that cancer cell lines display similar patterns of sensitivity to different antimitotics in vitro.19 Therefore, we investigated whether p63 contributes to sensitivity to a clinically approved antimitotic drug (docetaxel, also known as Taxotere). Preliminary experiments established that a concentration of 10 ng/ml docetaxel produced maximal levels of apoptosis in the cell lines (data not shown). Again, we found that the UC14 p63KD cells were significantly less sensitive to docetaxel than were the parental UC14 cells or UC14 cells transduced with the non-targeting construct (Fig. 3C). Finally, we assessed the effects of p63 knockdown on sensitivity to cisplatin, gemcitabine, or a combination of the two drugs. Although UC14 cells were markedly resistant to cisplatin alone, cells exposed to gemcitabine plus cisplatin underwent apoptosis, and p63 knockdown also attenuated these responses (Fig. 3D). Therefore, even though p63 expression does not correlate with sensitivity to cisplatin, p63 may still contribute to sensitivity to DNA damaging agents in the cell lines that express it.

Figure 3. Effect of p63 knockdown on drug sensitivity in UC14 cells. (A) Parental UC14 cells, as well as cells transfected with non-targeting and p63 shRNA constructs were exposed to 10 nM AZD4877 for 24 h and apoptosis was measured by propidium iodide staining and FACS analysis. (B) Cells were transfected with non-targeting or ΔNp63-specific siRNAs, and the effects of AZD4877 on PARP cleavage were measured by immunoblotting. Blots were then stripped and reprobed with anti-p63 (4A4) to confirm p63 knockdown and subsequently with anti-tubulin to confirm equal loading. (C) Parental UC14 cells, as well as cells transfected with non-targeting and p63 shRNA constructs were exposed to10 ng/ml docetaxel for 24 h and apoptosis was measured by propidium iodide staining and FACS analysis (n = 3). (D) Cells were exposed to 1 μM gemcitabine or 10 μM cisplatin as single agents or in combination for 48 h, and apoptosis was measured by propidium iodide staining and FACS analysis (n = 3).
p63 regulates cell proliferation and c-myc expression
Previous studies indicate that the process of apoptosis is in fact tightly coupled with cellular proliferation.25 Additionally, it has been shown that antimitotic drugs depend on progression through the cell cycle to mitosis to exert their apoptotic effects.11,26 We noticed that stable p63 silencing appeared to slow the growth of the UM-UC14 cells. To measure this effect directly, we plated equal numbers of parental UM-UC14 cells or the cells stably transduced with non-targeting or p63-specific shRNA constructs and counted the cells at 24, 48 and 72 h after plating. Consistent with our impressions, the UM-UC14 parental cells and the non-targeting shRNA transfected cells displayed significantly faster growth rates than the p63-silenced cells (Fig. 4A). We then used gene expression profiling to explore the molecular mechanisms underlying p63’s growth-promoting effects and found that p63 knockdown significantly inhibited expression of c-myc and several other genes associated with cell cycle progression (M. Tran, unpublished observations). We confirmed that p63 knockdown suppressed c-myc expression at the mRNA level by quantitative real-time PCR (Fig. 4B) and at the protein level by immmunoblotting (Fig. 4C).
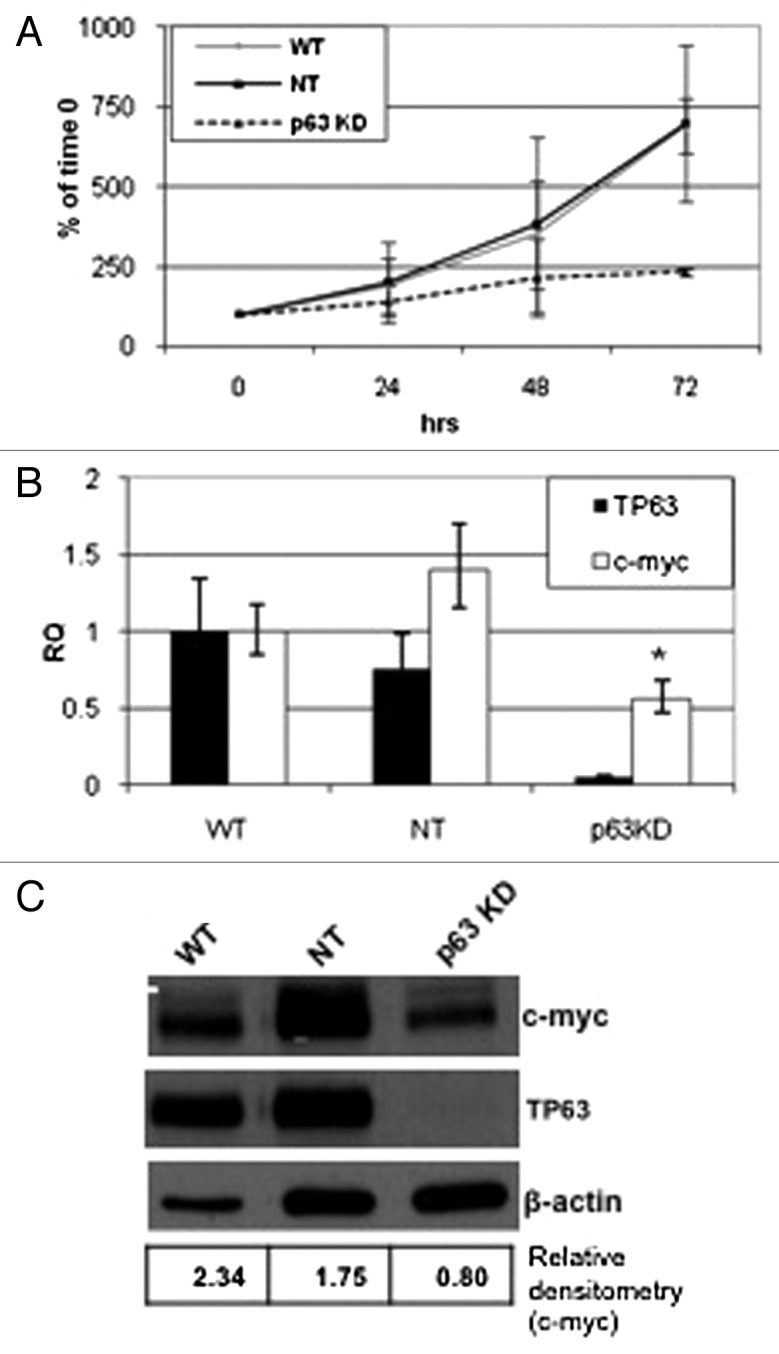
Figure 4. Effect of p63 knockdown on proliferation and c-myc expression. Parental UC14 cells, as well as cells stably transduced with non- targeting and p63 specific shRNAs, were grown in 6-well plates and quantified by trypan blue exclusion at 24, 48 and 72 h time points (A; n = 3). Real-time RT-PCR (B) and western blotting (C) were then used to confirm p63 silencing and to measure Myc expression in the 3 UC14 variants. Myc expression was quantified by densitometry as a ratio to actin expression using ImageJ.
A previous study concluded that ΔNp63 promotes proliferation by inducing nuclear β-catenin accumulation and signaling,27 and it is well established that β-catenin can drive c-myc expression via a TCF/Lef-1-dependent mechanism.28-30 We observed a correlation between p63 and β-catenin expression in 8 out of 10 of our cell lines, but this correlation did not extend to Myc (Fig. 5A). In addition, p63 knockdown had no significant effect on β-catenin mRNA (data not shown) or protein levels in the UC14 cells (Fig. 5B).
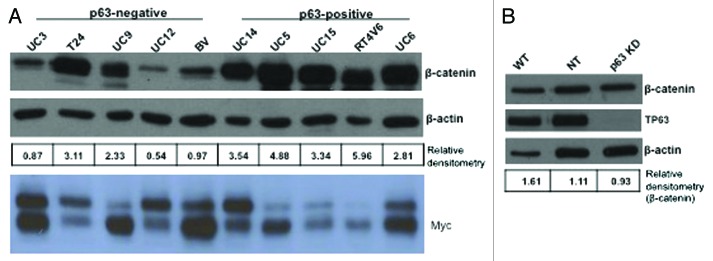
Figure 5. Relationship between β-catenin and p63 expression. Western blotting was used to measure β-catenin and Myc expression levels in a panel of 10 human bladder cancer cell lines (A), and in UC14 cells transfected with non-targeting shRNA and shRNA specific for p63 (B). β-catenin levels were quantified as a ratio to actin expression using ImageJ.
Myc promotes sensitivity to antimitotic drugs
Previous studies have shown that the c-myc gene is of central importance in driving both cell proliferation and apoptosis.25,31,32 To determine whether c-myc contributed directly to AZD4877 and/or docetaxel sensitivity, we used RNAi to knock down c-myc in the UC14 cells. We confirmed that knockdown of c-myc rendered the cells resistant to both drugs (Fig. 6A-B). We then measured cell death in the UC14 cells exposed to gemcitabine, cisplatin, or a combination of the two drugs. Myc knockdown also decreased sensitivity to the GC combination, although the effects were not as dramatic as those observed in the cells that were exposed to AZD4877 or docetaxel (Fig. 6C).

Figure 6. Effect of c-myc silencing on sensitivity to AZD4877 or docetaxel in UC14 cells. Parental UC14 cells, as well as cells transfected with non-targeting or c-myc- specific siRNAs, were exposed to 10 nM AZD4877 for 24 h (A) or to 10 ng/ml docetaxel (B) for 24 h. Additionally, cells were exposed to gemcitabine (1 μM), cisplatin (10 μM) or a combination of the two drugs for 48 h (C). Propidium iodide staining and FACS analysis were used to measure apoptosis. Confirmation of c-myc silencing for all experiments is shown in (D) (n = 3).
Discussion
In 2010, more than 70,000 new cases of bladder cancer were diagnosed in the US.1 Because the high recurrence rate of superficial bladder cancer dictates a long-term medical following, it ranks as the most expensive cancer in the United States.33 While muscle invasive bladder cancers that possess low-risk features can usually be cured with surgery, tumors with high-risk features are usually lethal unless patients are treated with neoadjuvant or adjuvant chemotherapy. Cisplatin-based combination regimens, most notably gemcitabine plus cisplatin (GC) and methotrexate/vinblastine/adriamycin/cisplatin (MVAC), are active in a subset of these tumors, producing cures in nearly 60% of patients.34-36 However, there are currently no active therapies for cisplatin-resistant tumors, and it is not presently possible to prospectively identify them. Methods for predicting patient response to chemotherapy (most notably the COXEN algorithm) are being developed that hopefully will change this situation.37,38
In the present study, we sought to identify molecular markers associated with response to the novel mitotic kinesin inhibitor AZD4877 in bladder cancer. Using a diverse panel of human cell lines, we measured drug-induced apoptosis and used microarray analyses to correlate observed patterns of sensitivity and resistance with baseline gene expression profiles. The results indicate that p63 is a biomarker for sensitivity to AZD4877. In addition, p63 appears to contribute directly to drug sensitivity in these cells via mechanisms that involve Myc. P63 has not previously been shown to play a role in sensitivity to anti-mitotics, nor has the interaction between p63 and Myc been reported. However, our results are consistent with an earlier study that concluded that ΔNp63 regulates proliferation in human head and neck squamous cell carcinoma lines. In that study the effects of ΔNp63 were linked to increased β-catenin expression caused by ΔNp63’s ability to interact with the B56a subunit of protein phosphatase 2A (PP2A) and glycogen synthase kinase-3β (GSK3β), which inhibited β-catenin phosphorylation and increased β-catenin stability.27 Even though we also observed a correlation between p63 and β-catenin expression in most of our cell lines, this correlation did not extend to Myc, and p63 knockdown had no effect on β-catenin levels, strongly suggesting that p63’s effects on Myc were not mediated via β-catenin stabilization. Importantly, we do not yet know whether p63’s effects on Myc and potentially other cell cycle regulators are direct or indirect. Our plan is to use chromatin immunoprecipation and DNA sequencing (ChIP-Seq) along with our gene expression profiling data to obtain a more comprehensive understanding of p63’s effects on Myc, cell cycle progression, and proliferation in bladder cancer cells.
Previously, investigators have sought to explore in detail exactly how antimitotic drugs cause cell death and how some cells manage to evade their effects. Several studies have found that the effectiveness of the spindle checkpoint is the primary determinant of response to taxanes and other antimitotics, with increased resistance to apoptosis seen in cells with a compromised spindle checkpoint.11-13 However, mutations in spindle checkpoint proteins are rare in human cancers.13 It has also been reported that it is the status of a cell’s apoptotic machinery, not the spindle checkpoint, which determines sensitivity to antimitotics.19 Our data indicate that sensitivity to anti-mitotics is coupled with cell cycle regulation and in particular whether or not p63 is a component of the mechanisms involved in driving Myc expression. They are consistent with previous work by others indicating that Myc plays a central role in coordinating proliferation and apoptosis.25,31,32
In the course of characterizing p63 expression patterns in our cell lines, we noted that there was a very close correlation between expression of p63 and molecular markers of the “epithelial” cellular phenotype, most notably P- and E-cadherin (Fig. 2A).24,39 Subsequently we discovered that p63 stimulates the expression of mir205 by interacting directly with an enhancer element upstream of its transcriptional start site (M. Tran et al., manuscript in preparation). miR205 plays a well-established role in blocking EMT by directly binding to the transcripts encoding the EMT inducers, Zeb-1 and Zeb-2.40 Epithelial-to-mesenchymal transition (EMT) has been implicated in drug resistance,41 so we consider it likely that the effects of p63 in promoting sensitivity to chemotherapy are also linked to its effects on EMT.
While we still have an imperfect understanding of p63’s role in promoting bladder cancer progression, studies have reported that p63 is consistently expressed in superficial bladder tumors but is downregulated in muscle-invasive cancers.22,24,42 Superficial disease rarely leads to cancer-related death,2 so when superficial and muscle-invasive bladder cancers are considered together, p63 expression is associated with good clinical outcomes. However, recent research indicates that patients with ΔNp63-positive muscle-invasive tumors have a worse prognosis than do patients with muscle invasive tumors lacking ΔNp63 expression.22,24 Thus, ΔNp63 expression appears to have very different effects in superficial and muscle-invasive bladder cancers. Importantly, these are probably influenced by the functional status of p53, which is mutated in muscle-invasive but not superficial tumors.43-48 Our findings, which implicate p63 as a determinant of antimitotic response, indicate that it is potentially patients with the lethal, ΔNp63-positive muscle invasive tumors who would most benefit from treatment with antimitotic chemotherapy.
Materials and Methods
Cell culture and reagents
RT4, 253J-P, and T24 were purchased from American Type Culture Collection. 253J B-V cells were generated by orthotopic recycling of 253J-P cells, as described previously.49 RT4V1 and RT4V6 cell lines were generated by Ashish Kamat (Department of Urology, University of Texas MD Anderson Cancer Center) by in vivo recycling of RT4 cells.50 All other cell lines were provided by H Barton Grossman (Department of Urology, University of Texas MD Anderson Cancer Center). Cells were maintained in MEM (Life Technologies, Inc., Rockville, MD) supplemented with 10% fetal bovine serum and 1% each of MEM vitamin solution (Life Technologies, Inc.), sodium pyruvate (BioWhitaker), L-glutamine (BioWhitaker), L-glutamine, penicillin/streptomycin solution, and nonessential amino acids (Life Technologies, Inc.) in a 5% CO2 incubator. The identities of all cell lines were confirmed by STR DNA fingerprinting analysis.
AZD4877 was provided by AstraZeneca. Docetaxel, gemcitabine hydrochloride (Gemzar; Eli Lilly and Company), and cisplatin were purchased from the University of Texas MD Anderson Cancer Center pharmacy. Antibodies used were the TP63 monoclonal antibody 4A4 (Santa Cruz), rabbit anti-human actin (Sigma) and horseradish peroxidase–conjugated sheep anti-mouse or donkey anti-rabbit secondary antibodies (Amersham Biosceinces). Small interfering RNA Smartpoools for c-myc as well as a nonspecific control were purchased from Dharmacon. The ΔNp63 specific siRNA (5′ ACAAUGCCCAGACUCAAUU 3′) was designed based on a previous publication51 and was synthesized by Dhramacon. The non-targeting siRNA is from Dhramacon (Cat# D-00180–10–20). Primers for PPIA, c-myc, and TP63 were purchased from Applied Biosystems.
Propidium iodide-fluorescence-activated cell sorting analysis (PI-FACS)
Cells were seeded in 6-well plates in 10% MEM and were allowed to reach 70% confluence. Cells were then exposed to AZD4877, docetaxel, gemcitabine, cisplatin, or a combination of gemcitabine plus cisplatin for 24 and 48 h. After being washed with phosphate-buffered saline (PBS), cells were harvested by trypsinization and centrifugation. The pellets were then resuspended in PBS containing 50 mg/ml propidium iodine, 0.1% Triton X-100, and 0.1% sodium citrate. Samples were stored at 4°C for 1 h and then analyzed. PI fluorescence was measured by fluorescence-activated cell sorting (FACS) analysis (Beckman-Coulter, FL3 channel). Cells containing hypodiploid DNA were recorded as apoptotic.
Immunoblot analyses
Cells were scraped from 10 cm tissue culture plates in the presence of lysis buffer [1% Triton X-100, 150 mmol/L NaCl, 25 mmol/L Tris, 1 mmol/L glycerol phosphate, 1 mmol/L sodium orthovanadate, 1mmol/L sodium fluoride, and protease inhibitor cocktail (Sigma)] and were rotated for 1 h at 4°C. The lysates were then centrifuged for 20 min at 4°C and resolved on 10% SDS-PAGE gels. Polypeptides were transferred to nitrocellulose membranes. Membranes were blocked in 5% nonfat milk in TBS containing 0.1% Tween 20 for one hour at room temperature, incubated overnight with relevant antibodies, washed, probed with species specific secondary antibodies and detected by chemiluminescence (Perkin-Elmer Life Sciences). ImageJ analysis was used to measure densitometry relative to actin.
RNA isolation, microarray platform and microarray experiments
All transcriptome data were generated from duplicates of the cell lines. Cells were plated and total RNA was isolated using the mirVana miRNA Isolation Kit (Ambion Inc.). Biotin labeled cRNA was prepared from RNA isolates using the Illumina RNA amplification kit (Ambion, Inc.). Purified cRNA was fragmented and hybridized to Illumina Human-6 v 2 (Illumina, Inc.) chips. The chips were washed and then scanned with Bead station 500X (Illumina, Inc.). Signal intensities were quantified with Bead studio (Illumina, Inc.), and data was normalized using quartile normalization. BRB ArrayTools version 3.6 developed by National Cancer Institute52 was used to analyze the data, as described previously.53
Real-time PCR analysis
For RNA extraction, cells were allowed to reach 70% confluence and the medium was removed. RNA was isolated using the mirVana miRNA Isolation Kit (Ambion Inc.) according to the manufacturer’s protocol. Real-time PCR technology (Step One; Applied Biosystems) was used in conjunction with Assays-on-Demand (Applied Biosystems). The comparative CT method was used to determine relative gene expression levels for each target gene. Cyclophilin A served as an internal control to normalize for the amount of amplifiable RNA in each reaction.
Silencing by siRNA and shRNA
siRNAs were transfected into cells using Lipofectamin RNAiMAX (Invitrogen, Cat# 13778-075) following the Forward Transfection method in the reagent guidelines. Cells were incubated in the transfection reagent at 37°C in a humidified incubator with 5% CO2 for 24 h, cells were incubated with or without drugs for various periods of time, and then they were harvested for analysis of mRNA expression (assayed via real-time RT-PCR), apoptosis, or protein expression (by immunoblotting). The pan p63 targeting lentiviral shRNA construct (Open Biosystems, cat# V3LHS_397885) and pGIPZ lentiviral empty vector (Open Biosystems, cat# RHS4339) were transfected into 293T cells in order to generate lentivirus. UC14 cells were plated on a 6-well plate (12 × 104 cells/well), and medium containing lentiviral particles was added after 24 h. Cells were incubated with lentivirus for 16 h, and were then washed and cultured in fresh medium. Fluorescence-activated cell sorting (FACS) was performed after 4–5 d to isolate GFP positive cells, and these cells were then cultured in medium containing puromycin (4 mg/ml).
Cell proliferation
Cells were seeded in duplicate in 6-well plates at a density of 2 × 105 cells per well. At 24, 48, and 72 h after plating cells were harvested by trypsinization and quantified using trypan blue exclusion.
Note
While this manuscript was under review, a paper describing a link between p63 and Myc in normal keratinocytes was accepted for publication in the Journal of Biological Chemistry (Wu N., Rollin J., Masse I., Lamartine J. and Gidrol X. p63 regulates human keratinocyte proliferation via a MYC-regulated gene network and differentiation through a cell adhesion-related gene network. J Biol Chem 2012; 287:5627-38).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/19590
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Dinney CP, McConkey DJ, Millikan RE, Wu X, Bar-Eli M, Adam L, et al. Focus on bladder cancer. Cancer Cell. 2004;6:111–6. doi: 10.1016/j.ccr.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 3.Galsky MD. The role of taxanes in the management of bladder cancer. Oncologist. 2005;10:792–8. doi: 10.1634/theoncologist.10-10-792. [DOI] [PubMed] [Google Scholar]
- 4.Jordan MA, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer. 2004;4:253–65. doi: 10.1038/nrc1317. [DOI] [PubMed] [Google Scholar]
- 5.Sudakin V, Yen TJ. Targeting mitosis for anti-cancer therapy. BioDrugs. 2007;21:225–33. doi: 10.2165/00063030-200721040-00003. [DOI] [PubMed] [Google Scholar]
- 6.Quasthoff S, Hartung HP. Chemotherapy-induced peripheral neuropathy. J Neurol. 2002;249:9–17. doi: 10.1007/PL00007853. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Y, Xu W. Progress on kinesin spindle protein inhibitors as anti-cancer agents. Anticancer Agents Med Chem. 2008;8:698–704. [PubMed] [Google Scholar]
- 8.Tao W. The mitotic checkpoint in cancer therapy. Cell Cycle. 2005;4:1495–9. doi: 10.4161/cc.4.11.2130. [DOI] [PubMed] [Google Scholar]
- 9.Sawin KE, LeGuellec K, Philippe M, Mitchison TJ. Mitotic spindle organization by a plus-end-directed microtubule motor. Nature. 1992;359:540–3. doi: 10.1038/359540a0. [DOI] [PubMed] [Google Scholar]
- 10.Blangy A, Lane HA, d’Hérin P, Harper M, Kress M, Nigg EA. Phosphorylation by p34cdc2 regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell. 1995;83:1159–69. doi: 10.1016/0092-8674(95)90142-6. [DOI] [PubMed] [Google Scholar]
- 11.Lee EA, Keutmann MK, Dowling ML, Harris E, Chan G, Kao GD. Inactivation of the mitotic checkpoint as a determinant of the efficacy of microtubule-targeted drugs in killing human cancer cells. Mol Cancer Ther. 2004;3:661–9. [PubMed] [Google Scholar]
- 12.Masuda A, Maeno K, Nakagawa T, Saito H, Takahashi T. Association between mitotic spindle checkpoint impairment and susceptibility to the induction of apoptosis by anti-microtubule agents in human lung cancers. Am J Pathol. 2003;163:1109–16. doi: 10.1016/S0002-9440(10)63470-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sudo T, Nitta M, Saya H, Ueno NT. Dependence of paclitaxel sensitivity on a functional spindle assembly checkpoint. Cancer Res. 2004;64:2502–8. doi: 10.1158/0008-5472.CAN-03-2013. [DOI] [PubMed] [Google Scholar]
- 14.Tang C, Willingham MC, Reed JC, Miyashita T, Ray S, Ponnathpur V, et al. High levels of p26BCL-2 oncoprotein retard taxol-induced apoptosis in human pre-B leukemia cells. Leukemia. 1994;8:1960–9. [PubMed] [Google Scholar]
- 15.Huang Y, Ibrado AM, Reed JC, Bullock G, Ray S, Tang C, et al. Co-expression of several molecular mechanisms of multidrug resistance and their significance for paclitaxel cytotoxicity in human AML HL-60 cells. Leukemia. 1997;11:253–7. doi: 10.1038/sj.leu.2400557. [DOI] [PubMed] [Google Scholar]
- 16.Giannakakou P, Sackett DL, Kang YK, Zhan Z, Buters JT, Fojo T, et al. Paclitaxel-resistant human ovarian cancer cells have mutant beta-tubulins that exhibit impaired paclitaxel-driven polymerization. J Biol Chem. 1997;272:17118–25. doi: 10.1074/jbc.272.27.17118. [DOI] [PubMed] [Google Scholar]
- 17.Yu D, Jing T, Liu B, Yao J, Tan M, McDonnell TJ, et al. Overexpression of ErbB2 blocks Taxol-induced apoptosis by upregulation of p21Cip1, which inhibits p34Cdc2 kinase. Mol Cell. 1998;2:581–91. doi: 10.1016/S1097-2765(00)80157-4. [DOI] [PubMed] [Google Scholar]
- 18.Chin GM, Herbst R. Induction of apoptosis by monastrol, an inhibitor of the mitotic kinesin Eg5, is independent of the spindle checkpoint. Mol Cancer Ther. 2006;5:2580–91. doi: 10.1158/1535-7163.MCT-06-0201. [DOI] [PubMed] [Google Scholar]
- 19.Shi J, Orth JD, Mitchison T. Cell type variation in responses to antimitotic drugs that target microtubules and kinesin-5. Cancer Res. 2008;68:3269–76. doi: 10.1158/0008-5472.CAN-07-6699. [DOI] [PubMed] [Google Scholar]
- 20.Blanpain C, Fuchs E. p63: revving up epithelial stem-cell potential. Nat Cell Biol. 2007;9:731–3. doi: 10.1038/ncb0707-731. [DOI] [PubMed] [Google Scholar]
- 21.Kurzrock EA, Lieu DK, Degraffenried LA, Chan CW, Isseroff RR. Label-retaining cells of the bladder: candidate urothelial stem cells. Am J Physiol Renal Physiol. 2008;294:F1415–21. doi: 10.1152/ajprenal.00533.2007. [DOI] [PubMed] [Google Scholar]
- 22.Karni-Schmidt O, Castillo-Martin M, Shen TH, Gladoun N, Domingo-Domenech J, Sanchez-Carbayo M, et al. Distinct expression profiles of p63 variants during urothelial development and bladder cancer progression. Am J Pathol. 2011;178:1350–60. doi: 10.1016/j.ajpath.2010.11.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Urist MJ, Di Como CJ, Lu ML, Charytonowicz E, Verbel D, Crum CP, et al. Loss of p63 expression is associated with tumor progression in bladder cancer. Am J Pathol. 2002;161:1199–206. doi: 10.1016/S0002-9440(10)64396-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi W, Shah JB, Tran M, Svatek R, Marquis L, Lee IL, et al. p63 expression defines a lethal subset of muscle-invasive bladder cancers. PLoS One. 2012;7:e30206. doi: 10.1371/journal.pone.0030206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Evan GI, Vousden KH. Proliferation, cell cycle and apoptosis in cancer. Nature. 2001;411:342–8. doi: 10.1038/35077213. [DOI] [PubMed] [Google Scholar]
- 26.Donaldson KL, Goolsby GL, Wahl AF. Cytotoxicity of the anticancer agents cisplatin and taxol during cell proliferation and the cell cycle. Int J Cancer. 1994;57:847–55. doi: 10.1002/ijc.2910570614. [DOI] [PubMed] [Google Scholar]
- 27.Patturajan M, Nomoto S, Sommer M, Fomenkov A, Hibi K, Zangen R, et al. DeltaNp63 induces beta-catenin nuclear accumulation and signaling. Cancer Cell. 2002;1:369–79. doi: 10.1016/S1535-6108(02)00057-0. [DOI] [PubMed] [Google Scholar]
- 28.He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, et al. Identification of c-MYC as a target of the APC pathway. Science. 1998;281:1509–12. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 29.Yochum GS, Cleland R, Goodman RH. A genome-wide screen for beta-catenin binding sites identifies a downstream enhancer element that controls c-Myc gene expression. Mol Cell Biol. 2008;28:7368–79. doi: 10.1128/MCB.00744-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yochum GS, Sherrick CM, Macpartlin M, Goodman RH. A beta-catenin/TCF-coordinated chromatin loop at MYC integrates 5′ and 3′ Wnt responsive enhancers. Proc Natl Acad Sci U S A. 2010;107:145–50. doi: 10.1073/pnas.0912294107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, et al. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69:119–28. doi: 10.1016/0092-8674(92)90123-T. [DOI] [PubMed] [Google Scholar]
- 32.Askew DS, Ashmun RA, Simmons BC, Cleveland JL. Constitutive c-myc expression in an IL-3-dependent myeloid cell line suppresses cell cycle arrest and accelerates apoptosis. Oncogene. 1991;6:1915–22. [PubMed] [Google Scholar]
- 33.Avritscher EB, Cooksley CD, Grossman HB, Sabichi AL, Hamblin L, Dinney CP, et al. Clinical model of lifetime cost of treating bladder cancer and associated complications. Urology. 2006;68:549–53. doi: 10.1016/j.urology.2006.03.062. [DOI] [PubMed] [Google Scholar]
- 34.Millikan R, Dinney C, Swanson D, Sweeney P, Ro JY, Smith TL, et al. Integrated therapy for locally advanced bladder cancer: final report of a randomized trial of cystectomy plus adjuvant M-VAC versus cystectomy with both preoperative and postoperative M-VAC. J Clin Oncol. 2001;19:4005–13. doi: 10.1200/JCO.2001.19.20.4005. [DOI] [PubMed] [Google Scholar]
- 35.Grossman HB, Natale RB, Tangen CM, Speights VO, Vogelzang NJ, Trump DL, et al. Neoadjuvant chemotherapy plus cystectomy compared with cystectomy alone for locally advanced bladder cancer. N Engl J Med. 2003;349:859–66. doi: 10.1056/NEJMoa022148. [DOI] [PubMed] [Google Scholar]
- 36.Shah JB, McConkey DJ, Dinney CP. New strategies in muscle-invasive bladder cancer: on the road to personalized medicine. Clinical cancer research: an official journal of the American Association for Cancer Research 2011; 17:2608-12. [DOI] [PubMed]
- 37.Smith SC, Baras AS, Dancik G, Ru Y, Ding KF, Moskaluk CA, et al. A 20-gene model for molecular nodal staging of bladder cancer: development and prospective assessment. Lancet Oncol. 2011;12:137–43. doi: 10.1016/S1470-2045(10)70296-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith SC, Baras AS, Lee JK, Theodorescu D. The COXEN principle: translating signatures of in vitro chemosensitivity into tools for clinical outcome prediction and drug discovery in cancer. Cancer Res. 2010;70:1753–8. doi: 10.1158/0008-5472.CAN-09-3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McConkey DJ, Choi W, Marquis L, Martin F, Williams MB, Shah J, et al. Role of epithelial-to-mesenchymal transition (EMT) in drug sensitivity and metastasis in bladder cancer. Cancer Metastasis Rev. 2009;28:335–44. doi: 10.1007/s10555-009-9194-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, et al. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10:593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- 41.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–8. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park BJ, Lee SJ, Kim JI, Lee SJ, Lee CH, Chang SG, et al. Frequent alteration of p63 expression in human primary bladder carcinomas. Cancer Res. 2000;60:3370–4. [PubMed] [Google Scholar]
- 43.Esrig D, Elmajian D, Groshen S, Freeman JA, Stein JP, Chen SC, et al. Accumulation of nuclear p53 and tumor progression in bladder cancer. N Engl J Med. 1994;331:1259–64. doi: 10.1056/NEJM199411103311903. [DOI] [PubMed] [Google Scholar]
- 44.Esrig D, Spruck CH, 3rd, Nichols PW, Chaiwun B, Steven K, Groshen S, et al. p53 nuclear protein accumulation correlates with mutations in the p53 gene, tumor grade, and stage in bladder cancer. Am J Pathol. 1993;143:1389–97. [PMC free article] [PubMed] [Google Scholar]
- 45.Sarkis AS, Dalbagni G, Cordon-Cardo C, Zhang ZF, Sheinfeld J, Fair WR, et al. Nuclear overexpression of p53 protein in transitional cell bladder carcinoma: a marker for disease progression. J Natl Cancer Inst. 1993;85:53–9. doi: 10.1093/jnci/85.1.53. [DOI] [PubMed] [Google Scholar]
- 46.Kompier LC, Lurkin I, van der Aa MN, van Rhijn BW, van der Kwast TH, Zwarthoff EC. FGFR3, HRAS, KRAS, NRAS and PIK3CA mutations in bladder cancer and their potential as biomarkers for surveillance and therapy. PLoS One. 2010;5:e13821. doi: 10.1371/journal.pone.0013821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van Rhijn BW, van der Kwast TH, Vis AN, Kirkels WJ, Boevé ER, Jöbsis AC, et al. FGFR3 and P53 characterize alternative genetic pathways in the pathogenesis of urothelial cell carcinoma. Cancer Res. 2004;64:1911–4. doi: 10.1158/0008-5472.CAN-03-2421. [DOI] [PubMed] [Google Scholar]
- 48.Gui Y, Guo G, Huang Y, Hu X, Tang A, Gao S, et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet. 2011;43:875–8. doi: 10.1038/ng.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dinney CP, Fishbeck R, Singh RK, Eve B, Pathak S, Brown N, et al. Isolation and characterization of metastatic variants from human transitional cell carcinoma passaged by orthotopic implantation in athymic nude mice. J Urol. 1995;154:1532–8. doi: 10.1016/S0022-5347(01)66923-4. [DOI] [PubMed] [Google Scholar]
- 50.Metwalli AR, Khanbolooki S, Jinesh G, Sundi D, Shah JB, Shrader M, et al. Smac mimetic reverses resistance to TRAIL and chemotherapy in human urothelial cancer cells. Cancer Biol Ther. 2010;10:885–92. doi: 10.4161/cbt.10.9.13237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chow SE, Chang YL, Chuang SF, Wang JS. Wogonin induced apoptosis in human nasopharyngeal carcinoma cells by targeting GSK-3β and ΔNp63. Cancer Chemother Pharmacol. 2011;68:835–45. doi: 10.1007/s00280-010-1552-1. [DOI] [PubMed] [Google Scholar]
- 52.Wright GW, Simon RM. A random variance model for detection of differential gene expression in small microarray experiments. Bioinformatics. 2003;19:2448–55. doi: 10.1093/bioinformatics/btg345. [DOI] [PubMed] [Google Scholar]
- 53.Arumugam T, Ramachandran V, Fournier KF, Wang H, Marquis L, Abbruzzese JL, et al. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009;69:5820–8. doi: 10.1158/0008-5472.CAN-08-2819. [DOI] [PMC free article] [PubMed] [Google Scholar]