

In this study, the authors hypothesized that genomic profiling of clinical intrahepatic cholangiocarcinoma samples would identify genomic alterations that are linked to targeted therapies and that could facilitate a personalized approach to therapy. Two-thirds of patients in this study harbored genomic alterations that are associated with targeted therapies and that have the potential to personalize therapy selection individual patients.
Keywords: Intrahepatic cholangiocarcinoma, Next-generation sequencing, Driver mutations, Targeted therapy
Abstract
Background.
Intrahepatic cholangiocarcinoma (ICC) is a subtype of primary liver cancer that is rarely curable by surgery and is rapidly increasing in incidence. Relapsed ICC has a poor prognosis, and current systemic nontargeted therapies are commonly extrapolated from those used in other gastrointestinal malignancies. We hypothesized that genomic profiling of clinical ICC samples would identify genomic alterations that are linked to targeted therapies and that could facilitate a personalized approach to therapy.
Methods.
DNA sequencing of hybridization-captured libraries was performed for 3,320 exons of 182 cancer-related genes and 36 introns of 14 genes frequently rearranged in cancer. Sample DNA was isolated from 40 μm of 28 formalin-fixed paraffin-embedded ICC specimens and sequenced to high coverage.
Results.
The most commonly observed alterations were within ARID1A (36%), IDH1/2 (36%), and TP53 (36%) as well as amplification of MCL1 (21%). Twenty cases (71%) harbored at least one potentially actionable alteration, including FGFR2 (14%), KRAS (11%), PTEN (11%), CDKN2A (7%), CDK6 (7%), ERBB3 (7%), MET (7%), NRAS (7%), BRCA1 (4%), BRCA2 (4%), NF1 (4%), PIK3CA (4%), PTCH1 (4%), and TSC1 (4%). Four (14%) of the ICC cases featured novel gene fusions involving the tyrosine kinases FGFR2 and NTRK1 (FGFR2-KIAA1598, FGFR2-BICC1, FGFR2-TACC3, and RABGAP1L-NTRK1).
Conclusion.
Two thirds of patients in this study harbored genomic alterations that are associated with targeted therapies and that have the potential to personalize therapy selection for to individual patients.
Implications for Practice:
The recent translation of next-generation DNA sequencing technology from the research laboratory to clinical practice has enabled oncologists to personalize therapy decisions for each patient by targeting the genomic alterations driving the disease. For tumors such as primary cholangiocarcinoma of the liver, this new ability to determine all of the major genomic alterations (base substitutions, short insertions and deletions, copy number changes, homozygous deletions, and gene fusions) on very small formalin-fixed paraffin embedded clinical samples holds great promise that less toxic targeted therapies may be available for patients currently being treated with conventional “one size fits all” approaches.
Introduction
Cancer of the bile ducts can arise within the liver as an intrahepatic cholangiocarcinoma (ICC) or originate from extrahepatic bile ducts as a bile duct carcinoma [1–4]. ICC is the second most common primary hepatic malignancy after hepatocellular carcinoma and accounts for 3% of the malignant tumors of the gastrointestinal system and 15% of primary hepatic malignancies [1–4]. In that ICC has a routine histologic appearance of an adenocarcinoma, the diagnosis of ICC on a liver biopsy requires an immunohistochemical study of the tumor and a thorough clinical workup including imaging studies to rule out a metastatic adenocarcinoma to the liver [1–4].
Numerous studies have indicated that the incidence and mortality from ICC are increasing worldwide [1–4]. ICC is associated with primary sclerosing cholangitis, parasitic biliary infection, polycystic disease of the liver, congenital intrahepatic bile duct dilatation (Caroli’s disease), congenital hepatic fibrosis, and choledochal cysts [1–4]. Chronic hepatitis C infection is an established cause of ICC, with some studies describing a more than 300-fold increase in ICC incidence in patients with long-standing hepatitis C infections [5]. ICC has also been associated with cigarette smoking, alcohol consumption, and exposure to a variety of toxins and chemical carcinogens [1–4]. The initial symptoms of ICC are often vague, typically arise late in the course of the disease, and include abdominal pain, anorexia, and palpable abdominal mass lesions [1–4]. The median survival is less than 6 months for inoperable tumors and only 20%–40% for patients who undergo surgery and achieve clear margins [6, 7].
A series of previously published studies using traditional techniques have described a variety of gene mutations and genomic alterations in ICC, including well-known cancer-related genes such as KRAS and BRAF [8–10]. Nevertheless, these studies have focused predominantly on the causation and progression of the disease and not on a search for potential actionable genomic alterations that could lead to targeted therapies. Although several oncogenic alterations are known to influence ICC pathogenesis, the percentage of tumors expressing any given alteration remains low, limiting the ability to develop an effective therapy that would be broadly applicable to the treatment of ICC. For each particular tumor, discovering the limited number of targetable alterations will require a sensitive, specific sequencing assay capable of detecting all categories of genomic alterations in a large number of cancer-related genes. In the following study, ICC DNA extracted from clinical cases has been studied in depth by next-generation sequencing to assess how targeted therapies could be used to treat this devastating disease. More than two thirds of tumors were found to have at least one potentially clinically actionable alteration that suggests sensitivity to targeted therapies.
This updated approach to characterizing ICC tumors has also revealed several key concepts with the potential to guide future research and affect the treatment of ICC [7]. First, the prevalence of mutations within the RAS and PI3K pathways strongly suggests that therapies targeting key components of these signal transduction networks would be valuable for many patients with ICC. Second, these results highlight driver mutations that may facilitate the development of novel therapeutic strategies. Receptor tyrosine kinase fusions previously unidentified in ICC indicate that clinically available, targeted inhibitors more commonly used in other tumor types will be relevant for some patients. Furthermore, a host of mutations in proteins related to cell-cycle control suggest that CDK inhibitors under investigation in clinical trials may provide additional treatment options for nearly a quarter of patients with ICC.
Methods
Next-generation sequencing was performed on hybridization-captured, adaptor ligation-based libraries using DNA extracted from four formalin-fixed paraffin-embedded (FFPE) sections cut at 10 μm from 28 ICC that had clinically progressed after either surgical resection and/or conventional chemotherapy. The pathologic diagnosis of each case was confirmed on routine hematoxylin and eosin-stained slides, immunohistochemistry, and clinical/imaging evaluations to rule out the possibility of a nonhepatic primary adenocarcinoma. All samples sent for DNA extraction contained a minimum of 20% nuclei derived from tumor cells. Sequence samples were obtained from liver biopsies in 16 cases (59%), from liver resections in 10 cases (37%), a lymph node metastasis in 1 case (4%), and a lung metastasis in 1 case (4%). DNA sequencing was performed for 3,320 exons of 182 cancer-related genes and 36 introns of 14 genes frequently rearranged in cancer on indexed, adaptor-ligated, hybridization-captured fragments (Agilent SureSelect custom kit; Agilent Technologies, Palo Alto, CA, http://www.agilent.com) using 49-base pair paired reads on the Illumina HiSEquation 2000 (Illumina Inc., San Diego, CA, http://www.illumina.com) at an average sequencing depth of 1,115×. Resulting sequence data were evaluated for genomic alterations including point mutations, insertions, deletions, copy number alterations (amplifications and homozygous gene deletions), and select gene fusions/rearrangements, as described previously [11]. To maximize mutation-detection sensitivity in heterogeneous ICC specimens, the test was validated to detect base substitutions as well as short insertions and deletions at a ≥10% mutant allele frequency with ≥99% sensitivity. Publicly available and custom analysis tools (Foundation Medicine, Inc., Cambridge, MA, http://www.foundationmedicine.com) were used in combination to analyze the data and characterize genomic alterations.
Actionability Classification
The genomic alterations detected were further divided into two main classes of actionability: genomic alterations that predict sensitivity or resistance to approved or standard therapies and genomic alterations that are inclusion or exclusion criteria for specific experimental therapies in National Cancer Institute-registered clinical trials.
Results
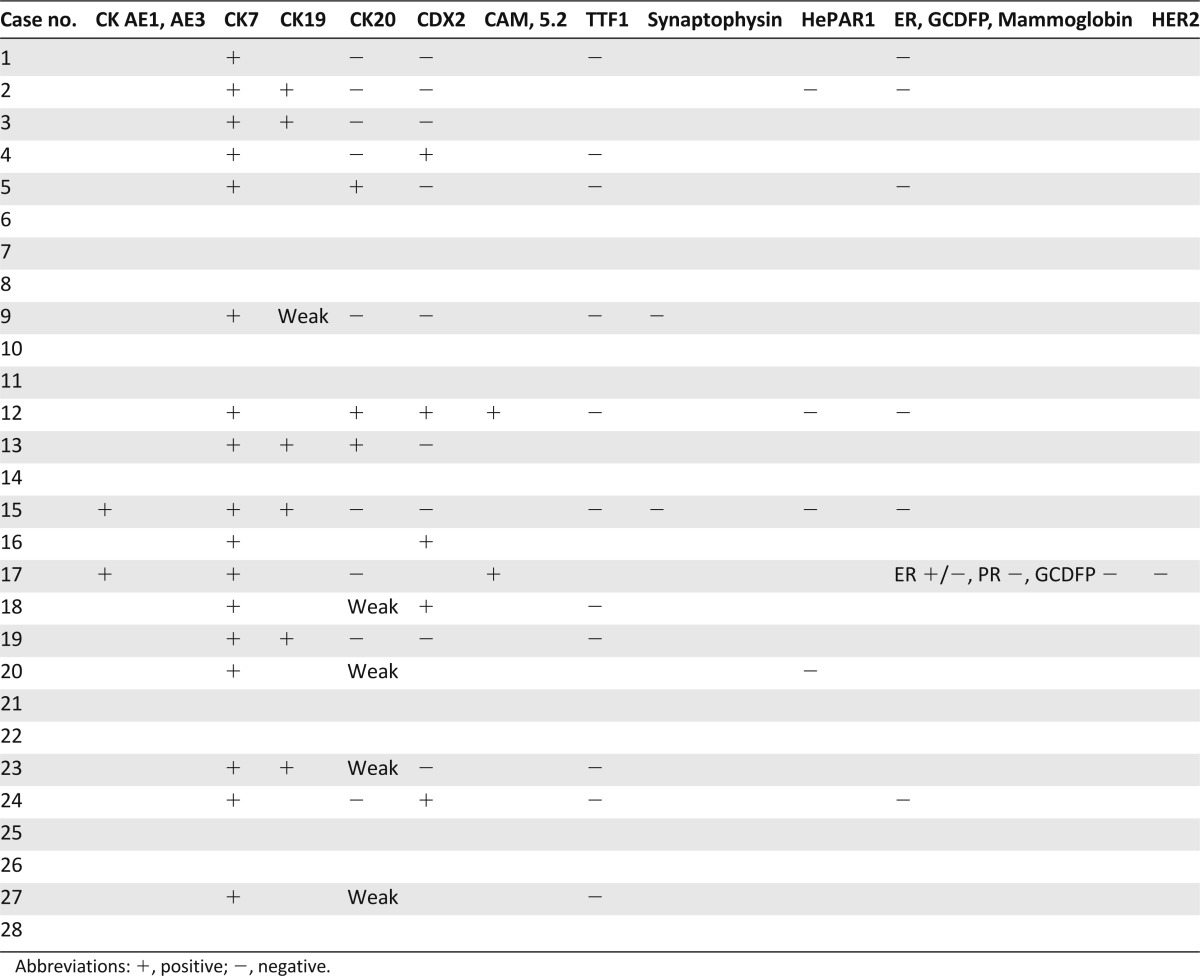
A total of 28 patient samples were analyzed, including tumors from 10 male and 18 female patients with a mean age of 55.9 years (range: 23–75 years) (Table 1). Of these, 10 had undergone an attempted curative hepatic resection operation and 18 underwent only a biopsy procedure (16 liver, 1 lymph node, and 1 lung biopsies). Histology analysis showed 18 of the tumors to be intermediate histologic grade 2 and 10 to be high histologic grade 3. Thirteen of the 28 cases were confined to the liver and without vascular invasion (or pathologic stage I), five were stage II tumors (confined to the liver with vascular invasion), five were stage III tumors (local metastasis), and five were stage IV tumors with the tumor disseminated beyond the liver. Immunohistochemical workup of the ICC was available for review in 19 cases (68%) (Table 2). All 19 cases expressed cytokeratin 7 (CK7), and 6 expressed CK19, whereas only 3 expressed CK20. Of the 16 ICCs stained for the CDX2 marker, 5 were immunopositive. Immunostains to rule out nonhepatic primary tumors including TTF1 for non-small cell lung cancer; ER, PR, HER2, mammoglobin, and GCDFP for breast cancer; prostate-specific antigen for prostate cancer; synaptophysin and chromogrannin for neuroendocrine carcinoma; and calretinin for mesothelioma were uniformly negative in all cases when performed. AFP and HEPAR1 immunostaining to rule out primary hepatocellular carcinoma were used in three cases, and all were negative.
Table 1.
Clinical features of 28 cases of intrahepatic cholangiocarcinoma
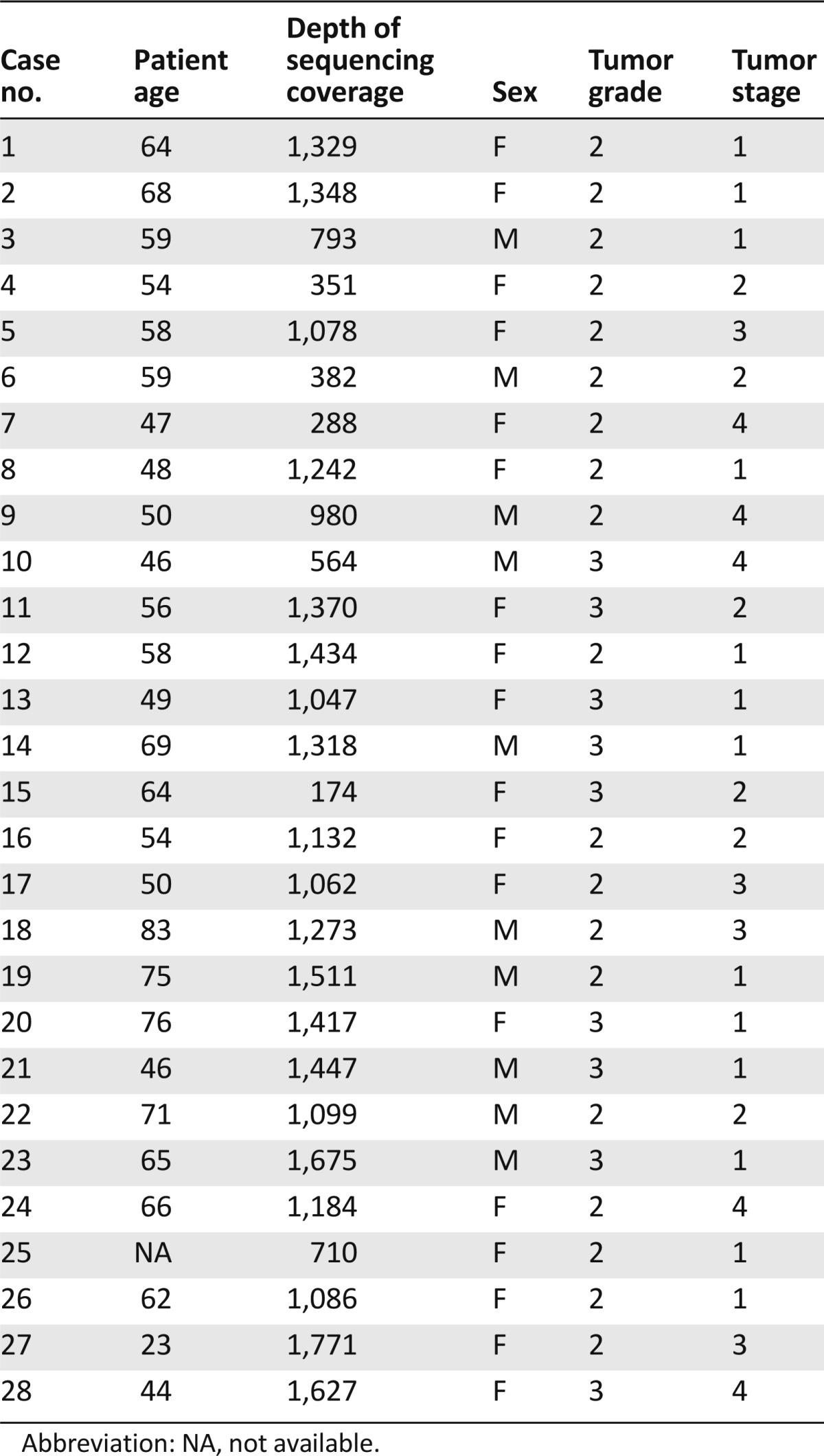
Table 2.
Selected immunohistochemical staining results
A total of 81 genomic alterations were identified in 35 genes with an average of 2.9 alterations per tumor (range: 1–9 alterations) (Fig. 1, supplemental online Table 1). The most common alterations were identified in ARID1A (36%), IDH1/2 (36%), TP53 (36%), and MCL1 (21%, all amplifications). In this study, nine (35%) of the ICCs featured mutations in IDH1 and one (4%) harbored a mutation in IDH2. Twenty cases (71%) harbored at least one potentially actionable alteration, with an average of 1.07 actionable alterations per patient including FGFR2 (14%), KRAS (11%), PTEN (11%), CDKN2A (7%), ERBB3 (7%), MET (7%), NRAS (7%), CDKN2A (7%), CDK6 (7%), BRCA1 (4%), BRCA2 (4%), NF1 (4%), PIK3CA (4%), PTCH1 (4%), and TSC1 (4%) (supplemental online Table 1). Of the three KRAS mutations identified in this study (12%), there were two G12D mutations and one G12C mutation. The cell-cycle regulation pathway genes CDKN2A and CDK6 were each altered in 7% of the ICCs in this study. Four gene fusions involving protein kinases were identified, including three fusions between FGFR2 and BICC1, KIAA1598, or TACC3 and one fusion between the kinase NTRK1 and RABGAP1L. Two of the three FGFR2 fusions (Figs. 2, 3) and the one NTRK1 fusion were novel discoveries. Neither the type nor the frequency of gene alteration was associated with patient age or gender. Two of the FGFR2 fusions (67%) occurred in female patients and one (33%) occurred in a male. No information was available with regard to whether patients included in this ICC study also suffered from inflammatory bowel disease or with regard to therapy-specific clinical outcome.
Figure 1.

Tile plot of genomic alterations in 28 cases of intrahepatic cholangiocarcinoma.
Figure 2.

Diagram of FGFR2 gene fusions in three cases of intrahepatic cholangiocarcinoma.
Figure 3.

Histology and list of genomic alterations in three cases of intrahepatic cholangiocarcinoma featuring FGFR2 gene fusions detected by next-generation sequencing.
The FGFR2-BICC1 fusion was identified in a grade 2, stage II ICC from a liver biopsy in a 75-year-old man (case 2). This FGFR2 fusion has been reported previously in cholangiocarcinoma [10]. This tumor also harbored a mutation in IDH1. The second FGFR2 fusion was a novel FGFR2-KIAA1598 fusion identified as the only mutation in a liver resection specimen of a moderately differentiated stage III ICC (case 27) treated with multiple rounds of chemotherapy, from a 23-year-old female patient. The third FGFR2 fusion was a novel FGFR2-TACC3 fusion in a pulmonary metastasis from a grade 3 ICC arising in the liver of a 44-year-old woman. Finally, a third novel fusion, RABGAP1L-NTRK1, was identified in the ICC from a 62-year-old woman on liver biopsy.
Two (7%) of the ICCs featured alterations in the mismatch repair genes MSH2 and MSH6. Case 18 is a liver biopsy of a grade 2, stage II ICC in an 83-year-old male patient with a P1087fs*5 MSH6 mutation associated with additional mutations in EPHB1, EPHA7, CDH1, PIK3CA, and ARID1A, along with an amplification of MCL1. Case 21 is a high-grade advanced stage ICC diagnosed on a lymph node biopsy from a 46-year-old female patient with an MSH2 homozygous deletion. This tumor also featured mutations in INHBA, BRCA2, TSC2, PTCH1, ERBB3, TP53, and ARID1A. The high number of genomic alterations in these two cases are consistent with a hypermutator genotype associated with mismatch repair-deficient tumors.
Discussion
This study identified multiple alterations in FGFR2, including three gene fusions, and is the second report of FGFR2 fusions in primary hepatic cholangiocarcinoma. Amplifications and gain-of-function mutations in FGFR genes have been reported in several cancer types and linked to tumor growth, invasion, and angiogenesis [12, 13]. FGFR2 amplification as seen in case 7 has been reported in several cancer types, most frequently in breast and gastric carcinomas [14, 15]. FGFR2 has been shown to be expressed in cholangiocarcinoma, leading to activation of the MEK1/2 pathway [16]. Amplifications of FGFR2 have been uniformly linked to FGFR2 protein overexpression [14–16]. In a recent study using whole-exome and whole-transcriptome sequencing, FGFR2 fusions were identified in two of four cholangiocarcinomas sequenced (50%) [12]. In both of these cases, an FGFR2-BICC1 fusion was identified [12]. A similar BICC1-FGFR2 fusion was identified in case 19 of this series. The FGFR2-BICC1 fusion results in truncation of the 3′UTR of FGFR2 and likely results in an upregulation of the FGFR2 protein. The FGFR2-KIAA1598 fusion seen in case 27 of this study results in truncation of the 3′UTR of FGFR2 and may also result in upregulation of the FGFR2 protein; however, this rearrangement (Figs. 2, 3) has not been reported in cholangiocarcinoma or other cancers, and the functional consequences are uncertain at this time. The FGFR2-TACC3 fusion found in case 28 of this study is also the first ICC found to harbor this alteration; however, it was recently reported that 3% of glioblastomas feature chromosomal rearrangements that fuse the tyrosine kinase coding domains of FGFR1 or FGFR3 in frame to the transforming acidic coiled-coil coding domains of TACC1 or TACC3, respectively [17]. Regorafenib, which inhibits cellular kinases including FGFR2, has been approved for treatment of some metastatic colorectal cancer patients [18], and clinical trials of multiple FGFR inhibitors are currently under way [19]. Finally, the RABGAP1L-NTRK1 fusion detected in case 2 of this series has not been reported previously (based on database searches of PubMed and COSMIC in January 2013), and NTRK1 alterations have not been analyzed or studied in cholangiocarcinoma. NTRK1 has been recently considered as a potential target for neuroblastoma and metastatic thyroid cancer [20, 21]. Of potential interest is the recent report of a non-small cell lung cancer patient whose tumor featured an MPRIP-NTRK1 fusion and who responded to the kinase inhibitor crizotinib [22].
MET amplification has rarely been described in ICC but has been associated with adverse clinical outcome [23–25]. In this study, MET amplification of greater than six copies per cell was found in two cases (7%). MET amplification may predict sensitivity to MET inhibitors and has been linked to acquired resistance to EGFR and ERBB2 inhibitors [26]. In this study, 12% of the cases featured a mutation or splicing modification of PTEN, a tumor suppressor that negatively regulates the PI3K/Akt/mTOR pathway [27]. PTEN mutations are rare in ICC (as shown in the COSMIC database in July 2012), although loss of PTEN expression has been associated with increased invasion, advanced tumor stage, and shorter survival [28]. Loss of PTEN may predict sensitivity to inhibitors of PI3K [27], and the mTOR inhibitors temsirolimus and everolimus have been approved for use in some tumor types. Inhibitors of PI3K and Akt are currently in clinical trials in solid tumors, alone or in combination with other therapies.
IDH1 and IDH2 are highly homologous and have similar functions, and mutation hot spots in both genes are conserved [29]. Alterations in IDH1 and IDH2 have been reported previously in cartilagenous tumors, gliomas, and leukemias [29]. Preclinical evidence now suggests that IDH1 and IDH2 alterations are actionable [30]. Almost all (99%) of the somatic mutations found in IDH1 are found at codon R132; this codon is functionally conserved and aligns with R172 of IDH2 [30]. In this study, eight (89%) of the IDH1 mutations were at codon 132, with one mutation (11%) at another locus (G97D). A heterozygous mutation at IDH1 codon R132 alters the activity of the IDH1 enzyme, resulting in a decrease in antioxidant activity in the cell [31–34]. The mutant enzyme promotes the reduction of a-ketoglutarate to 2-hydroxyglutarate, with the coincident conversion of NADPH to NADP+ [30]. Accumulation of d-2-hydroxyglutarate, a potential oncometabolite, has been associated with cancers that possess mutations in the IDH genes [32–35]. Mutations at codon 132 have been identified most frequently in gliomas, acute myeloid leukemia, colon cancer, prostate cancer, and chondrosarcomas (as shown in the COSMIC database in June 2012) [31–33].
IDH1 mutations have been identified in 9% (38 of 436) of biliary tract tumors analyzed in the COSMIC database (in May 2013). Recently, IDH1 and IDH2 mutations were identified in 34 of 326 ICCs (10%) and associated with longer overall survival for the disease in multivariate analysis [35]. IDH1 mutations have been identified in 20% of intrahepatic cholangiocarcinomas, and mutation status correlated with increased IDH1 activity [32]. IDH2 amino acid R172 is a proposed substrate binding site and represents one of two genetic hot spots for cancer mutations in this gene [36]. A majority of somatic IDH2 mutations cluster to two mutation hot spots, R140 and R172, of 841 reported mutations: Mutations at R140 represent 69.9% (588 of 841), and mutations at R172 represent 29.4% (247 of 841) (as shown in the COSMIC database in April 2012). IDH2 mutations have been found in hematopoietic and lymphoid tissue (6%, 730 of 12,303), bone tumors (4%, 18 of 405), tumors of the central nervous system (2%, 90 of 5,033), and skin tumors (2%, 3 of 127) (as shown in the COSMIC database in December 2012). There are no reports of IDH2 mutations in biliary tract cancers or other gastrointestinal tumor types in COSMIC (as of May 2013). However, the IDH2 R172W mutation found in case 1 of this study was also identified in one cholangiocarcinoma of 62 total cases in a previously published study [37]. No therapies targeting this alteration are currently approved, although therapies targeting the altered metabolic pathway resulting from IDH mutations are currently in development.
Loss of the chromosomal region containing CDKN2A and CDKN2B (9p21) has been reported in primary sclerosing cholangitis-associated ICC [38]. Up to 25% of biliary tract tumors harbor CDKN2A mutations (as shown in the COSMIC database in November 2012). Tumors with loss of the CDKN2A/CDKN2B locus may be sensitive to Cdk4/6 inhibitors, and clinical trials of these agents are currently under way for a variety of solid tumors. Of additional interest is the observation that, given the relatively frequent 21% rate of MCL1 focal gene amplification in the ICC cohort, CDK inhibitors may function by reducing MCL1 protein levels as their main mechanism of action [39].
Thirty-eight percent of the ICCs sequenced in this study had mutations in the ARID1A gene. ARID1A encodes the AT-rich interactive domain-containing protein 1A, also known as BAF250a, a member of the SWI/SNF chromatin remodeling complex. ARID1A is believed to function as a tumor suppressor [40]. ARID1A mutations have been reported in endometrial cancer (50%, 2 of 4), head and neck squamous cell carcinoma (50%, 3 of 6), ovarian cancer (34%, 97 of 282), skin squamous and basal cell carcinoma (29%, 2 of 7), gastric cancer (11%, 11 of 101), colorectal cancer (9%, 12 of 131), prostate cancer (9%, 2 of 23), pancreatic cancer (8%, 15 of 178), and a small percentage of lung, breast, and kidney carcinomas and gliomas (as shown in the COSMIC database in April 2012) [41]. Mutations span the length of the ARID1A gene and include point mutations and small deletions and insertions. There are no reports of ARID1A mutation in cholangiocarcinoma (as shown in the COSMIC database in April 2012), and there are no reports of ARID1A mutation in cholangiocarcinoma in the literature. Presently, there are no targeted therapies approved that target ARID1A mutations.
A variety of “one-off” single gene mutation studies have looked at ICC and concluded that TP53 and KRAS are the most frequently mutated genes found in this tumor type [42, 43]. Activating KRAS mutations have been observed in 20%–50% of cholangiocarcinomas (as shown in the COSMIC database in November 2012) [44, 45] and are associated with early recurrence and poor overall survival in ICC [46]. Some investigators have also identified subsets of both extrahepatic cholangiocarcinoma and ICC that appear to be driven by BRAF mutations [47–49]. However, this study did not find any BRAF mutations in the series of 28 ICCs, a finding also reported by others [50]. TP53 alterations have been reported in 39% (232 of 596) of biliary tract cancers and specifically in 41% (106 of 259) of bile duct carcinomas (as shown in the COSMIC database in June 2012). Inactivation of TP53, through mutation, deletion, or LOH, has been observed in 10%–61% of cholangiocarcinomas [51]. In this study of ICC only, the TP53 mutation frequency was 36%.
In this study, “actionable” genomic alterations have been defined as those linked to a drug that is approved for the tumor type in question or another tumor type but that targets the identified genomic alteration or pathway or that is mechanistically linked to an agent in an active registered clinical trial. Currently, there are no approved drugs for the treatment of ICC. It should also be noted that some actionable gene alterations actually are negative selectors that suggest lack of benefit of use of the specific drug when the alteration is present. In this approach, practicing oncologists are given information that can guide therapy selection for their patients in an efficient and straightforward manner.
Conclusion
When ICC was comprehensively genomically profiled with a next-generation sequencing-based diagnostic assay, two thirds of patients harbored potentially actionable genomic alterations that have the potential to influence and personalize therapy and guide the selection of targeted therapies approved or in clinical trials. Given the limited treatment options and the poor prognosis of patients with ICC and the diversity of actionable alterations identified in this study, comprehensive genomic profiling has the potential to maximize the identification of new treatment paradigms and to meet an unmet clinical need.
See http://www.TheOncologist.com for supplemental material available online.
Supplementary Material
Acknowledgments
This work was supported by an NIH K12 award (CA139160-01A), a University of Chicago Comprehensive Cancer Center Award in Precision Oncology, and a Live Like Katie Cholangiocarcinoma Foundation Award (to D.V.T.C.).
Author Contributions
Conception/Design: Jeffrey Ross, Kai Wang, Laurie Gay, Rami Al-Rohil, Janne V. Rand, David M. Jones, Hwa J. Lee, Christine E. Sheehan, Geoff A. Otto, Gary Palmer, Roman Yelensky, Doron Lipson, Deborah Morosini, Matthew Hawryluk, Daniel V. T. Catenacci, Vincent A. Miller, Philip J. Stephens, Siraj Ali
Provision of study material or patients: Jeffrey Ross, Daniel V.T. Catenacci, Chaitanya Churi
Collection and/or assembly of data: Jeffrey Ross, Kai Wang, Laurie Gay, Rami Al-Rohil, Janne V. Rand, David M. Jones, Hwa J. Lee, Christine E. Sheehan, Geoff A. Otto, Gary Palmer, Roman Yelensky, Doron Lipson, Deborah Morosini, Matthew Hawryluk, Daniel V. T. Catenacci, Vincent A. Miller, Chaitanya Churi, Philip J. Stephens, Siraj Ali
Data analysis and interpretation: Jeffrey Ross, Kai Wang, Laurie Gay, Rami Al-Rohil, Janne V. Rand, David M. Jones, Hwa J. Lee, Christine E. Sheehan, Geoff A. Otto, Gary Palmer, Roman Yelensky, Doron Lipson, Deborah Morosini, Matthew Hawryluk, Daniel V.T. Catenacci, Vincent A. Miller, Philip J. Stephens, Siraj Ali
Manuscript writing: Jeffrey Ross, Kai Wang, Laurie Gay, Rami Al-Rohil, Janne V. Rand, David M. Jones, Hwa J. Lee, Christine E. Sheehan, Geoff A. Otto, Gary Palmer, Roman Yelensky, Doron Lipson, Deborah Morosini, Matthew Hawryluk, Daniel V.T. Catenacci, Vincent A. Miller, Philip J. Stephens, Siraj Ali
Final approval of manuscript: Jeffrey Ross, Laurie Gay, Rami Al-Rohil, Janne V. Rand, David M. Jones, Hwa J. Lee, Christine E. Sheehan, Geoff A. Otto, Gary Palmer, Roman Yelensky, Doron Lipson, Deborah Morosini, Matthew Hawryluk, Daniel V.T. Catenacci, Vincent A. Miller, Chaitanya Churi, Philip J. Stephens, Siraj Ali
Disclosures
Jeffrey Ross: Foundation Medicine, Inc. (RF, E, OI); Kai Wang: Foundation Medicine, Inc. (E, OI); Laurie Gay: Foundation Medicine, Inc. (E, OI); Christine E. Sheehan: Foundation Medicine, Inc. (RF); Geoff A. Otto: Foundation Medicine, Inc. (E, OI); Gary Palmer: Foundation Medicine, Inc. (E, OI); Roman Yelensky: Foundation Medicine, Inc. (E, OI); Doron Lipson: Foundation Medicine, Inc. (E, OI); Deborah Morosini: Foundation Medicine, Inc. (C/A, E, OI); Matthew Hawryluk: Foundation Medicine, Inc. (E, OI, IP); Vincent A. Miller: Foundation Medicine, Inc. (E,OI); Philip J. Stephens: Foundation Medicine, Inc. (E, OI, IP); Siraj Ali: Foundation Medicine, Inc. (E, OI, IP). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1.Vasilieva LE, Papadhimitriou SI, Dourakis SP. Modern diagnostic approaches to cholangiocarcinoma. Hepatobiliary Pancreat Dis Int. 2012;11:349–359. doi: 10.1016/s1499-3872(12)60192-1. [DOI] [PubMed] [Google Scholar]
- 2.Sempoux C, Jibara G, Ward SC, et al. Intrahepatic cholangiocarcinoma: New insights in pathology. Semin Liver Dis. 2011;31:49–60. doi: 10.1055/s-0031-1272839. [DOI] [PubMed] [Google Scholar]
- 3.Poultsides GA, Zhu AX, Choti MA, et al. Intrahepatic cholangiocarcinoma. Surg Clin North Am. 2010;90:817–837. doi: 10.1016/j.suc.2010.04.011. [DOI] [PubMed] [Google Scholar]
- 4.Bartlett DL. Intrahepatic cholangiocarcinoma: A worthy challenge. Cancer J. 2009;15:255–256. doi: 10.1097/PPO.0b013e3181a7467d. [DOI] [PubMed] [Google Scholar]
- 5.Kobayashi M, Ikeda K, Saitoh S, et al. Incidence of primary cholangiocellular carcinoma of the liver in japanese patients with hepatitis C virus-related cirrhosis. Cancer. 2000;88:2471–2477. doi: 10.1002/1097-0142(20000601)88:11<2471::aid-cncr7>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 6.Yamamoto M, Ariizumi S. Surgical outcomes of intrahepatic cholangiocarcinoma. Surg Today. 2011;41:896–902. doi: 10.1007/s00595-011-4517-z. [DOI] [PubMed] [Google Scholar]
- 7.Geynisman DM, Catenacci DV. Toward personalized treatment of advanced biliary tract cancers. Discov Med. 2012;14:41–57. [PubMed] [Google Scholar]
- 8.Miller G, Socci ND, Dhall D, et al. Genome wide analysis and clinical correlation of chromosomal and transcriptional mutations in cancers of the biliary tract. J Exp Clin Cancer Res. 2009;28:62. doi: 10.1186/1756-9966-28-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Andersen JB, Thorgeirsson SS. Genetic profiling of intrahepatic cholangiocarcinoma. Curr Opin Gastroenterol. 2012;28:266–272. doi: 10.1097/MOG.0b013e3283523c7e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sia D, Hoshida Y, Villanueva A, et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 classes that have different outcomes. Gastroenterology. 2013;144:829–840. doi: 10.1053/j.gastro.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31:1023–1031. doi: 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu YM, Su F, Kalyana-Sundaram S, et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discov. 2013;3:636–647. doi: 10.1158/2159-8290.CD-13-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Powers CJ, McLeskey SW, Wellstein A. Fibroblast growth factors, their receptors and signaling. Endocr Relat Cancer. 2000;7:165–197. doi: 10.1677/erc.0.0070165. [DOI] [PubMed] [Google Scholar]
- 14.Turner N, Lambros MB, Horlings HM, et al. Integrative molecular profiling of triple negative breast cancers identifies amplicon drivers and potential therapeutic targets. Oncogene. 2010;29:2013–2023. doi: 10.1038/onc.2009.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsumoto K, Arao T, Hamaguchi T, et al. FGFR2 gene amplification and clinicopathological features in gastric cancer. Br J Cancer. 2012;106:727–732. doi: 10.1038/bjc.2011.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Narong S, Leelawat K. Basic fibroblast growth factor induces cholangiocarcinoma cell migration via activation of the MEK1/2 pathway. Oncol Lett. 2011;2:821–825. doi: 10.3892/ol.2011.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh D, Chan JM, Zoppoli P, et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science. 2012;337:1231–1235. doi: 10.1126/science.1220834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grothey A, Van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381:303–312. doi: 10.1016/S0140-6736(12)61900-X. [DOI] [PubMed] [Google Scholar]
- 19.Turner N, Grose R. Fibroblast growth factor signalling: From development to cancer. Nat Rev Cancer. 2010;10:116–129. doi: 10.1038/nrc2780. [DOI] [PubMed] [Google Scholar]
- 20.Owens C, Irwin M. Neuroblastoma: The impact of biology and cooperation leading to personalized treatments. Crit Rev Clin Lab Sci. 2012;49:85–115. doi: 10.3109/10408363.2012.683483. [DOI] [PubMed] [Google Scholar]
- 21.Greco A, Miranda C, Pierotti MA. Rearrangements of NTRK1 gene in papillary thyroid carcinoma. Mol Cell Endocrinol. 2010;321:44–49. doi: 10.1016/j.mce.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 22.Doebele RC, Vaishnavi A, Capelletti M, et al. NTRK1 gene fusions as a novel oncogene target in lung cancer. J Clin Oncol. 2013;31(suppl):8023a. [Google Scholar]
- 23.Appleman LJ. MET signaling pathway: A rational target for cancer therapy. J Clin Oncol. 2011;29:4837–4838. doi: 10.1200/JCO.2011.37.7929. [DOI] [PubMed] [Google Scholar]
- 24.Zhu AX. Molecularly targeted therapy for advanced hepatocellular carcinoma in 2012: Current status and future perspectives. Semin Oncol. 2012;39:493–502. doi: 10.1053/j.seminoncol.2012.05.014. [DOI] [PubMed] [Google Scholar]
- 25.Miyamoto M, Ojima H, Iwasaki M, et al. Prognostic significance of overexpression of c-Met oncoprotein in cholangiocarcinoma. Br J Cancer. 2011;105:131–138. doi: 10.1038/bjc.2011.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen CT, Kim H, Liska D, et al. MET activation mediates resistance to lapatinib inhibition of HER2-amplified gastric cancer cells. Mol Cancer Ther. 2012;11:660–669. doi: 10.1158/1535-7163.MCT-11-0754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simpson L, Parsons R. PTEN: Life as a tumor suppressor. Exp Cell Res. 2001;264:29–41. doi: 10.1006/excr.2000.5130. [DOI] [PubMed] [Google Scholar]
- 28.Lee D, Do IG, Choi K, et al. The expression of phospho-AKT1 and phospho-MTOR is associated with a favorable prognosis independent of PTEN expression in intrahepatic cholangiocarcinomas. Mod Pathol. 2012;25:131–139. doi: 10.1038/modpathol.2011.133. [DOI] [PubMed] [Google Scholar]
- 29.Schaap FG, French PJ, Bovée JV. Mutations in the isocitrate dehydrogenase genes IDH1 and IDH2 in tumors. Adv Anat Pathol. 2013;20:32–38. doi: 10.1097/PAP.0b013e31827b654d. [DOI] [PubMed] [Google Scholar]
- 30.Rohle D, Popovici-Muller J, Palaskas N, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340:626–630. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reitman ZJ, Yan H. Isocitrate dehydrogenase 1 and 2 mutations in cancer: Alterations at a crossroads of cellular metabolism. J Natl Cancer Inst. 2010;102:932–941. doi: 10.1093/jnci/djq187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ward PS, Patel J, Wise DR, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gross S, Cairns RA, Minden MD, et al. Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med. 2010;207:339–344. doi: 10.1084/jem.20092506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Amary MF, Bacsi K, Maggiani F, et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol. 2011;224:334–343. doi: 10.1002/path.2913. [DOI] [PubMed] [Google Scholar]
- 35.Wang P, Dong Q, Zhang C, et al. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene. 2013;32:3091–3100. doi: 10.1038/onc.2012.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jin G, Reitman ZJ, Spasojevic I, et al. 2-hydroxyglutarate production, but not dominant negative function, is conferred by glioma-derived NADP-dependent isocitrate dehydrogenase mutations. PLoS One. 2011;6:e16812. doi: 10.1371/journal.pone.0016812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Borger DR, Tanabe KK, Fan KC, et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. The Oncologist. 2012;17:72–79. doi: 10.1634/theoncologist.2011-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DeHaan RD, Kipp BR, Smyrk TC, et al. An assessment of chromosomal alterations detected by fluorescence in situ hybridization and p16 expression in sporadic and primary sclerosing cholangitis-associated cholangiocarcinomas. Hum Pathol. 2007;38:491–499. doi: 10.1016/j.humpath.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 39.Bettayeb K, Baunbæk D, Delehouze C, et al. CDK inhibitors roscovitine and CR8 trigger Mcl-1 down-regulation and apoptotic cell death in neuroblastoma cells. Genes Cancer. 2010;1:369–380. doi: 10.1177/1947601910369817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guan B, Wang TL, Shih IeM. ARID1A, a factor that promotes formation of SWI/SNF-mediated chromatin remodeling, is a tumor suppressor in gynecologic cancers. Cancer Res. 2011;71:6718–6727. doi: 10.1158/0008-5472.CAN-11-1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lowery WJ, Schildkraut JM, Akushevich L, et al. Loss of ARID1A-associated protein expression is a frequent event in clear cell and endometrioid ovarian cancers. Int J Gynecol Cancer. 2012;22:9–14. doi: 10.1097/IGC.0b013e318231f140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen CP, Haas-Kogan D. Neoplasms of the hepatobiliary system: Clinical presentation, molecular pathways and diagnostics. Expert Rev Mol Diagn. 2010;10:883–895. doi: 10.1586/erm.10.76. [DOI] [PubMed] [Google Scholar]
- 43.Nault JC, Zucman-Rossi J. Genetics of hepatobiliary carcinogenesis. Semin Liver Dis. 2011;31:173–187. doi: 10.1055/s-0031-1276646. [DOI] [PubMed] [Google Scholar]
- 44.O’Dell MR, Huang JL, Whitney-Miller CL, et al. Kras(G12D) and p53 mutation cause primary intrahepatic cholangiocarcinoma. Cancer Res. 2012;72:1557–1567. doi: 10.1158/0008-5472.CAN-11-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Andersen JB, Spee B, Blechacz BR, et al. Genomic and genetic characterization of cholangiocarcinoma identifies therapeutic targets for tyrosine kinase inhibitors. Gastroenterology. 2012;142:1021–1031, e15. doi: 10.1053/j.gastro.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nakano H, Yamamoto F, Neville C, et al. Isolation of transforming sequences of two human lung carcinomas: Structural and functional analysis of the activated c-K-ras oncogenes. Proc Natl Acad Sci USA. 1984;81:71–75. doi: 10.1073/pnas.81.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tannapfel A, Sommerer F, Benicke M, et al. Mutations of the BRAF gene in cholangiocarcinoma but not in hepatocellular carcinoma. Gut. 2003;52:706–712. doi: 10.1136/gut.52.5.706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schönleben F, Qiu W, Allendorf JD, et al. Molecular analysis of PIK3CA, BRAF, and RAS oncogenes in periampullary and ampullary adenomas and carcinomas. J Gastrointest Surg. 2009;13:1510–1516. doi: 10.1007/s11605-009-0917-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Andersen JB, Spee B, Blechacz BR, et al. Genomic and genetic characterization of cholangiocarcinoma identifies therapeutic targets for tyrosine kinase inhibitors. Gastroenterology. 2012;142:1021–1031.e15. doi: 10.1053/j.gastro.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu RF, Sun JP, Zhang SR, et al. KRAS and PIK3CA but not BRAF genes are frequently mutated in Chinese cholangiocarcinoma patients. Biomed Pharmacother. 2011;65:22–26. doi: 10.1016/j.biopha.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 51.Della Torre G, Pasquini G, Pilotti S, et al. TP53 mutations and mdm2 protein overexpression in cholangiocarcinomas. Diagn Mol Pathol. 2000;9:41–46. doi: 10.1097/00019606-200003000-00007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.